
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 16 May 2017
Sec. Neurodegeneration
Volume 11 - 2017 | https://doi.org/10.3389/fnins.2017.00254
This article is part of the Research Topic Molecular Chaperones and Neurodegeneration View all 15 articles
 Rachel E. Lackie1,2
Rachel E. Lackie1,2 Andrzej Maciejewski1,3
Andrzej Maciejewski1,3 Valeriy G. Ostapchenko1
Valeriy G. Ostapchenko1 Jose Marques-Lopes1
Jose Marques-Lopes1 Wing-Yiu Choy3
Wing-Yiu Choy3 Martin L. Duennwald4
Martin L. Duennwald4 Vania F. Prado1,2,5,6
Vania F. Prado1,2,5,6 Marco A. M. Prado1,2,5,6*
Marco A. M. Prado1,2,5,6*The accumulation of misfolded proteins in the human brain is one of the critical features of many neurodegenerative diseases, including Alzheimer's disease (AD). Assembles of beta-amyloid (Aβ) peptide—either soluble (oligomers) or insoluble (plaques) and of tau protein, which form neurofibrillary tangles, are the major hallmarks of AD. Chaperones and co-chaperones regulate protein folding and client maturation, but they also target misfolded or aggregated proteins for refolding or for degradation, mostly by the proteasome. They form an important line of defense against misfolded proteins and are part of the cellular quality control system. The heat shock protein (Hsp) family, particularly Hsp70 and Hsp90, plays a major part in this process and it is well-known to regulate protein misfolding in a variety of diseases, including tau levels and toxicity in AD. However, the role of Hsp90 in regulating protein misfolding is not yet fully understood. For example, knockdown of Hsp90 and its co-chaperones in a Caenorhabditis elegans model of Aβ misfolding leads to increased toxicity. On the other hand, the use of Hsp90 inhibitors in AD mouse models reduces Aβ toxicity, and normalizes synaptic function. Stress-inducible phosphoprotein 1 (STI1), an intracellular co-chaperone, mediates the transfer of clients from Hsp70 to Hsp90. Importantly, STI1 has been shown to regulate aggregation of amyloid-like proteins in yeast. In addition to its intracellular function, STI1 can be secreted by diverse cell types, including astrocytes and microglia and function as a neurotrophic ligand by triggering signaling via the cellular prion protein (PrPC). Extracellular STI1 can prevent Aβ toxic signaling by (i) interfering with Aβ binding to PrPC and (ii) triggering pro-survival signaling cascades. Interestingly, decreased levels of STI1 in C. elegans can also increase toxicity in an amyloid model. In this review, we will discuss the role of intracellular and extracellular STI1 and the Hsp70/Hsp90 chaperone network in mechanisms underlying protein misfolding in neurodegenerative diseases, with particular focus on AD.
A major requirement for cellular growth, function, and survival is the proper folding, maturation, and degradation of proteins. These activities are carried out by molecular chaperones, many of which are heat shock proteins (Hsps). The heat shock response was first discovered in the early 1960s in Drosophila that displayed changes in salivary gland transcriptional activity in response to different incubation temperatures (Ritossa, 1962). It was not until 1974 that Hsps were discovered and interest in this field of biology became widespread (Tissieres et al., 1974). Transcription of heat shock genes is mostly regulated by heat shock factor 1 (HSF1). Inactive HSF1 is localized in the cytosol, but upon heat stress translocates to the nucleus and binds to promoters of heat shock elements, inducing transcription and leading to an increase in Hsp expression (Morimoto, 1998). Activation of HSF1 and subsequent shuttling to the nucleus is a typical stress response and also allows for control of cell cycle, protein translation and glucose metabolism (Dai et al., 2007). It is now well-accepted that Hsps not only aid in mediating cellular responses to stress, but are also critical in general protein quality control. Some of the major roles of molecular chaperones include the regulation of the unfolding protein response due to stress, degradation of misfolded or aggregated proteins, regulation of macromolecular complexes, and protein-protein interactions.
There are several major classes of Hsps involved in the protein quality control machinery: Hsp60, Hsp70 and Hsp90, Hsp40, Hsp100, Hsp110, as well as the ATP-independent small heat shock proteins (sHsps) such as Hsp20, αA-crystallin, and αB-crystallin. Hsp40, also known as DnaJ, is commonly found acting as a co-chaperone for Hsp70 and regulates ATP-dependent polypeptide binding to Hsp70, prevention of premature polypeptide folding, and ATPase activity of Hsp70 (Cyr et al., 1992; Frydman et al., 1994; Tsai and Douglas, 1996). In yeast, the family of Hsp100 proteins protect cells from extreme physiological and environmental stress (Sanchez et al., 1992; Glover and Lindquist, 1998) and have the unique ability to re-solubilize aggregated insoluble proteins (Parsell et al., 1994). In metazoans disaggregase activity is carried out by the tricomplex of Hsp70, a J Protein and Hsp110 (Shorter, 2011; Rampelt et al., 2012; Gao et al., 2015). For the purpose of this review, we will focus mainly on the roles of Hsp70 and Hsp90 as well as of the critical co-chaperone stress-inducible phosphoprotein I (STI1, STIP1) and their regulation of protein misfolding and signaling in neurodegenerative diseases. Comprehensive discussion of different chaperones including their roles in the ER can be found in excellent recent reviews elsewhere (McLaughlin and Vandenbroeck, 2011; Marzec et al., 2012; Melnyk et al., 2015; Ellgaard et al., 2016).
Hsp70 and Hsp90 and homologs are both widely expressed in some lower order prokaryotes and in all eukaryotes, with Hsp90 constituting ~1% of all cellular proteins in eukaryotes (Borkovich et al., 1989). Hsp90 activity is regulated through interactions with a large network of co-chaperones providing quality control of a wide range of client proteins. Initially, client proteins are recruited by Hsp40 and Hsp70 and then transferred to Hsp90 by the co-chaperone STI1 (the human homolog is also known as Hsp-organizing protein or HOP; Lassle et al., 1997; Chen and Smith, 1998; Johnson et al., 1998; Taipale et al., 2010). Recent studies suggest that Hsp90 has an important role in neurodegeneration. Pharmacological inhibition of Hsp90 results in Hsp70 and Hsp40 upregulation, which can control the expression of several synaptic proteins, but it can also channel misfolded protein for degradation by the proteasome (Luo et al., 2007; Chen et al., 2014; Wang et al., 2016). Protein aggregation is a major hallmark of several neurodegenerative diseases, including Alzheimer's disease (AD), Amyotrophic lateral sclerosis (ALS), Parkinson's disease (PD), Huntington's disease (HD), and Creutzfeldt-Jakob disease (CJD). Therefore, the chaperone machinery is becoming a major therapeutic target across these diseases.
The Hsp70 family of proteins is a class of highly abundant and ubiquitously expressed chaperones that participate in many biological processes, including protein trafficking, early stages of nascent polypeptide folding and the refolding or degradation of aggregated peptide products (Bukau et al., 2006). There are several eukaryotic Hsp70 isoforms (reviewed in Kabani and Martineau, 2008). Hsp70 is composed of two distinct domains, a 40 kDa N-terminal nucleotide-binding domain (NBD) that regulates client association and a 25 kDa C-terminal substrate-binding domain (SBD), which recognizes exposed hydrophobic stretches in the early stages of client protein folding (Rudiger et al., 1997; Bukau et al., 2006). A short, flexible hydrophobic linker joins both domains (Jiang et al., 2005). ATP binding and hydrolysis is coupled to allosteric changes in Hsp70, which influence protein-client interactions in the Hsp70 chaperone cycle (Mayer et al., 2000). Post-translational modification of Hsp70 by phosphorylation at T504 and/or acetylation at several serine residues promotes dimerization of Hsp70 (Morgner et al., 2015). Furthermore, presence of a client and Hsp40 supports dimerization of Hsp70 and substrate binding (Morgner et al., 2015).
ATP binding to the NBD results in the opening of a SBD α-helical lid, stimulating binding of substrate proteins through interactions with the NBD and SBD (Jiang et al., 2005). The ADP-bound state results in the closing of the α-helical lid over the substrate-binding cleft and stabilizes client association (Schlecht et al., 2011). The Hsp70 chaperone cycle is inherently slow due to the low ATPase activity of Hsp70 (Swain et al., 2007). Thus, a family of J proteins recruits the client to Hsp70 and stimulates the Hsp70 ATPase activity (Misselwitz et al., 1998). The J protein then dissociates from the ternary complex and a nucleotide-exchange factor releases the bound ADP from Hsp70 returning it to the apo-conformation. This leaves the NBD available for recruitment of ATP, upon which the α-helical lid can “open” and release the client peptide (Misselwitz et al., 1998; Schlecht et al., 2011). The cycle repeats in an interactive process until the client peptide adopts its native structure or is passed on to another part of the chaperone machinery.
Hsp70 class of chaperones typically recognizes client proteins in the early stages of folding through short hydrophobic sequences rich in leucine residues (Rudiger et al., 1997). Such hydrophobic stretches are greatly exposed during the early stages of protein translation and folding, leading to unfavorable intra- and inter-molecular interactions (Hartl and Hayer-Hartl, 2009). Hsp70 binding to client proteins in the early stages of protein folding controls the availability of such regions, facilitating formation of the proper protein fold, while inhibiting aggregate formation. If proper folding of the client is not possible, Hsp70 association with additional co-chaperones promotes degradation of the misfolded protein (Meacham et al., 2001; Petrucelli et al., 2004; Jana et al., 2005; Dickey et al., 2007; Muller et al., 2008). The abundance of exposed hydrophobic stretches in proteins prone to self-association in neurodegenerative diseases draws parallel to proteins in early stages of folding, which indicates a potential role of the Hsp70/Hsp90 machinery in modulating pathogenic aggregate formation (Muchowski and Wacker, 2005; Hartl and Hayer-Hartl, 2009).
Hsp90 is a highly conserved molecular chaperone from yeast to mammals and it is essential for proper folding and maintenance of its client proteins in eukaryotic cells (see Figure 1). Over 500 physical and genetic interactions have been identified in yeast spanning diverse families of cellular proteins, which include transcription factors, steroid hormone receptors, and protein kinases (Zhao et al., 2005; McClellan et al., 2007). Through concerted efforts involving other chaperones and co-chaperones it drives the final maturation of client proteins. There are two major isoforms of Hsp90 identified in the cytosol, inducible Hsp90α and constitutively expressed Hsp90β, as well as Grp94 in the endoplasmic reticulum (Csermely et al., 1998).
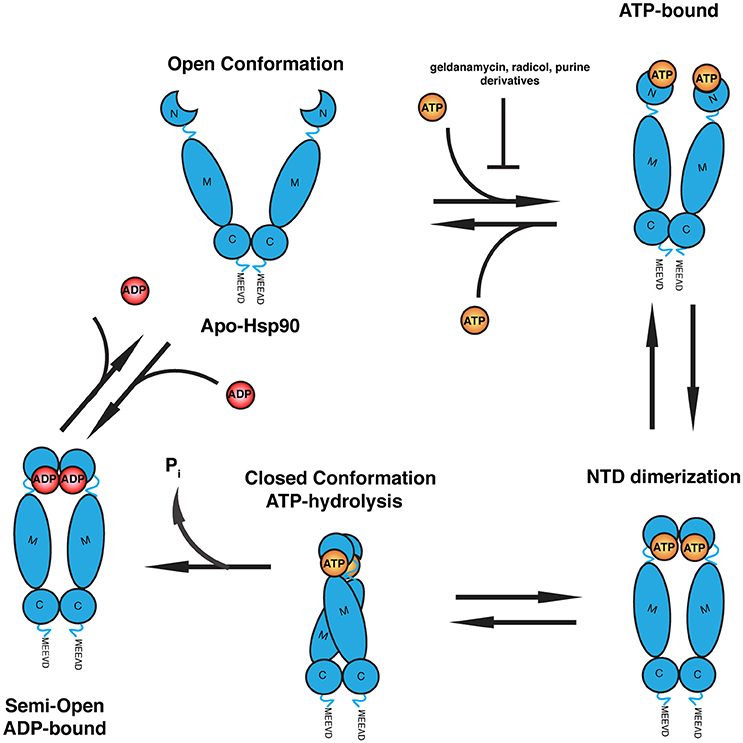
Figure 1. Schematic of the ATPase cycle of Hsp90. Hsp90 homodimer initially adopts an open V-shaped conformation. Binding of ATP to the N-terminal ATPase domain induces a conformational change where the N-terminal lids close and ATP is cradled in the nucleotide-binding pocket. This induces dimerization of the N-terminal domains of each homodimer followed by closure of Hsp90 and recruitment of the M domain for ATP hydrolysis. The dimers dissociate into a semi-open intermediate state with ADP bound. Release of ADP dissociates the N-termini to allow repetition of the ATPase cycle. Common Hsp90 inhibitors (geldanamycin, radicicol, and purine derivatives) bind to the N-terminus of Hsp90 and compete with ATP for binding.
Hsp90 forms a homodimeric biologically functional unit composed of three distinct regions connected by flexible linker. Each protomer contains a highly-conserved N-terminal domain (NTD) responsible for nucleotide binding, a middle domain (MD) important for client recognition and ATP hydrolysis and a C-terminal domain (CTD), which is the primary site responsible for dimerization (Pearl and Prodromou, 2006; Taipale et al., 2010). Additionally, the CTD contains a conserved “MEEVD” sequence used for interactions with the large tetratricopeptide repeat domain class of co-chaperone proteins, which regulate Hsp90 protein activity (Young et al., 1998).
The NTD contains a deep ATP/ADP binding pocket evolutionarily conserved amongst the GHKL (gyrase, Hsp90, histidine kinase, MutL) ATPase superfamily (Meyer et al., 2003). The binding pocket is composed of multiple α-helices and a β-sheet flanking the bound ATP molecule (Prodromou et al., 1997). Intramolecular recruitment of the MD on each protomer brings critical residues into contact forming the required split ATPase active site necessary for ATP hydrolysis (Ali et al., 2006). The N-terminal region contains an additional molecular lid mechanism, which crosses over the nucleotide-binding pocket in the ATP-bound state and holds the nucleotide in place, but remains in the open conformation upon ADP-binding (Ali et al., 2006). See Figure 1 for an overview of the Hsp90 conformations when bound to ADP or ATP.
Nucleotide binding and hydrolysis are directly coupled to large structural rearrangements in Hsp90 that are regulated through interactions with client and co-chaperone proteins (Csermely et al., 1993; Kirschke et al., 2014; Lavery et al., 2014). Apo-Hsp90 presents an “open” conformation where the two protomers form a V-shaped dimer (Vaughan et al., 2010). Nucleotide binding induces an intermediate-state where the N-terminal lid cradles ATP within the nucleotide-binding pocket. This results in a second repositioning of the N-terminal region favoring its dimerization and recruitment of the MD for ATP hydrolysis through a conserved arginine (R380 in yeast) that contacts the γ-phosphate of ATP (Cunningham et al., 2012). Finally, ATP is hydrolyzed and Hsp90 reverts to the “open” conformation where the N-terminal domains dissociate, allowing for the repetition of the cycle. ATP hydrolysis and the structural rearrangements in Hsp90 during client refolding are influenced and regulated by a diverse set of co-chaperones (Zuehlke and Johnson, 2010). These co-chaperones have varying effects on the ATPase activity and conformational rearrangements in Hsp90. Post-translational modifications of Hsp90 include acetylation, nitrosylation, phosphorylation, and methylation, which has been elegantly reviewed by Li and Buchner (2013).
While ATPase induced conformational rearrangements in Hsp90 are well-understood, their influence on client protein folding remains enigmatic. Hsp90-directed folding occurs late during the folding of nascent peptides. Structural studies involving binding to intrinsically disordered tau suggest that substrate recognition occurs through low-affinity hydrophobic interactions between the client protein and a large substrate-binding interface on Hsp90 (Zuehlke and Johnson, 2010; Karagoz et al., 2014). This mechanism allows for detection of scattered exposed hydrophobic patches in protein folding intermediates. Interestingly, Hsp90 affinity for tau appears to be independent of ATP binding as both the apo and ATP-bound forms possess equal affinities for tau (Karagoz et al., 2014).
The prominent role and poor prognosis of Hsp90 overexpression in various cancers has led to the development of a number of Hsp90 inhibitors (Roe et al., 1999; Trepel et al., 2010). Hsp90 inhibition results in the downregulation of many oncogenic client proteins that require Hsp90 for maturation. The most prominent drugs inhibit the ATPase activity of Hsp90 and are based on the geldanamycin, radicicol, or purine derivatives, which function as competitive inhibitors of ATP binding to Hsp90 (Roe et al., 1999; Sidera and Patsavoudi, 2014). Initial geldanamycin and radicicol derivatives proved to be potent inhibitors of Hsp90; however, their therapeutic value is low due to severe toxicity by targeting Hsp90 in normal cells (Jhaveri et al., 2012; Trendowski, 2015).
Co-chaperones along with individual Hsps comprise a chaperone network that is altered in malignancy and neurodegeneration (Workman et al., 2007; Moulick et al., 2011; Lindberg et al., 2015; Rodina et al., 2016). Targeting specific co-chaperones or chaperone complexes may help to avoid cytotoxicity by directly targeting Hsp90 activity (Yi and Regan, 2008; Moulick et al., 2011; Rodina et al., 2016). The purine derivative PU-H71 possesses unique selectivity amongst Hsp90 inhibitors, preferentially targeting high-molecular-weight complexes composed of Hsp70/90 and various co-chaperones and client proteins, which are enriched in numerous malignant cell models, but absent in non-oncogenic tissue (Moulick et al., 2011; Rodina et al., 2016). The formation of these large stable chaperone species appears to be cancer specific and diagnostic proteomic approaches may serve as a method to clinically screen patients that are most likely to benefit from targeting such species. Whether large and stable chaperone complexes with misfolded proteins occur in different neurodegenerative diseases is currently unclear. Indeed, therapeutic approaches targeting chaperones in neurodegeneration still fall behind from those in cancer cells.
Hsp70 and Hsp90 both interact with many co-chaperones containing tetratricopeptide repeat (TPR) domains, which consist of three or more 34-amino acid residues (Lamb et al., 1995). These motifs form anti-parallel α-helices (Allan and Ratajczak, 2011) that bind to the C-terminus of the chaperone and are the main interaction site for co-chaperones (Smith, 2004), along the EEVD peptide motif on Hsp70 and Hsp90 (Kajander et al., 2009). Proteins containing TPR domains typically share no other sequence homology, but are commonly found to be involved in regulation of cell cycle, protein trafficking, phosphate turnover, and transcriptional events (Blatch and Lassle, 1999). TPR domain-containing co-chaperones regulate the ATP cycle of chaperones and aid in client transport to binding pockets, where they are folded. Hsp40 may help coordinate other co-chaperones in binding Hsp70, such as Hsp70-interacting protein (Hip; Hohfeld et al., 1995) in the early stages of the chaperone cycle, as well as STI1 and SGT (Allan and Ratajczak, 2011). STI1 is also a co-chaperone for Hsp90, along with p23, Cdc37, and the immunophilins peptidyl-prolyl cis-trans (PPIases) isomerases FKBP51 and FKBP52, phosphatase PP5 and the cyclophillin Cyp40 (Allan and Ratajczak, 2011). Some of these co-chaperones inhibit Hsp90 ATP turnover (Rehn and Buchner, 2015). C-terminal Hsp70-interacting protein (CHIP) is also a co-chaperone for both Hsp70 and Hsp90. In this review, we will focus mainly on the roles of Hsp70, Hsp90, and the co-chaperone STI1 in protein misfolding. We will also discuss the unique cytokine-like activities of STI1. Importantly, both the extracellular and intracellular activities of STI1 seem to converge to increase cellular resilience (Beraldo et al., 2013). Moreover, we will briefly describe some of the co-chaperones that may also have a role in protein misfolding diseases, such as CHIP and high molecular weight immunophilins.
There is a number of Hsp70 and Hsp90 co-chaperones that have implications for neurodegenerative diseases. High molecular weight FK506-binding proteins (FKBPs) FKBP51 and FKBP52 impose a variety of effects on tau structure and function, which will be discussed further in the AD subsection of this review. Another co-chaperone, a high molecular weight PPIase Cyp40, can bind Aβ and regulate import of Aβ into mitochondria. Interestingly, inhibiting Cyp40 was found to be protective against Aβ-toxicity in mitochondria and neurons in an amyloid-precursor protein (APP) transgenic mouse model (Du and Yan, 2010). This suggests that Aβ may be regulated by an Hsp90/PPIase complex, but further investigation is required. For a more extensive review on PPIases in regulating the levels and toxicity of proteins in AD, see excellent review by Blair et al. (2015).
The Hsp90 co-chaperone p23 typically comes into play in a mature Hsp90-client complex (Felts and Toft, 2003), whereby it inhibits ATP turnover on Hsp90 (Rehn and Buchner, 2015). In the context of AD, p23 has been found to bind γ-secretases and to promote the non-amyloidogenic pathway of amyloid precursor protein (ultimately reducing production of Aβ species; Vetrivel et al., 2008) and silencing of p23 gene expression reduced the levels of total tau and phosphorylated tau (Dickey et al., 2007).
Hsp70 and Hsp90 are critical to folding and maturation of a number of clients, but they are also key players in regulation of the proteasome, a simplified representation of this activity can be found in Figure 2. CHIP, a Hsp70-Hsp90 co-chaperone, is an E3 ubiquitin ligase (Ballinger et al., 1999; McDonough and Patterson, 2003) that promotes client degradation via the proteasome. CHIP contains three TPR motifs at its N-terminus, a middle domain of charged residues (Ballinger et al., 1999) and a C-terminus containing a U-Box domain (Zhang et al., 2005). U-Box domains are characteristic of proteins involved in ubiquitination (Aravind and Koonin, 2000). Within this pathway, ubiquitin molecules are covalently attached to the protein of interest, and then recruited to the proteasome complex for degradation. CHIP interacts with Hsp70 and Hsp90 via TPR domain and then can bind with the substrate protein of interest, promoting substrate ubiquitination. Of note, CHIP is capable of binding tau and is responsible for its ubiquitination (Petrucelli et al., 2004), a critical activity that may be of importance for tauopathies.
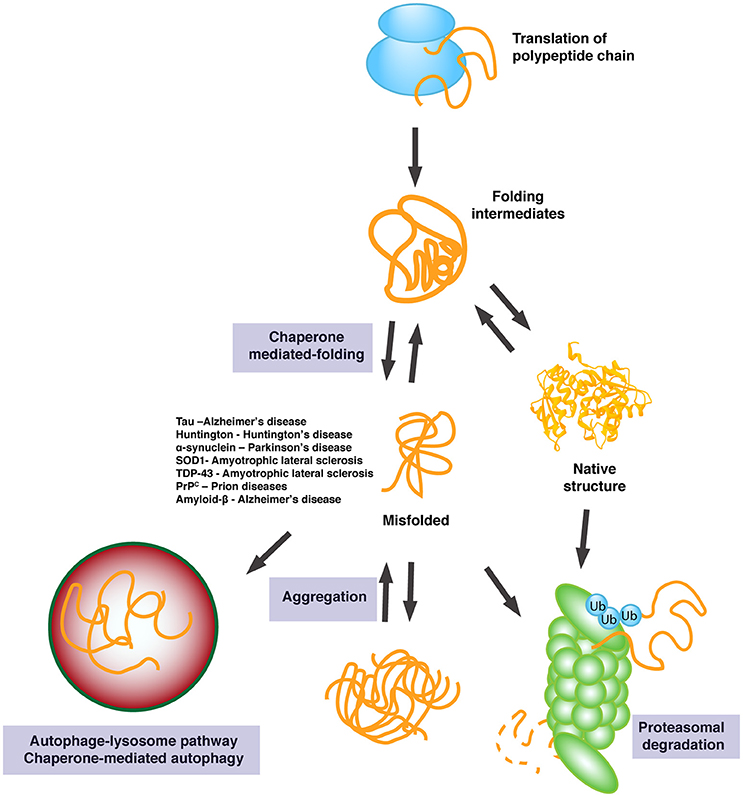
Figure 2. Outline of the chaperone response in protein folding. Chaperones facilitate proper folding of a diverse array of client proteins and prevent oligomer and aggregate formation. If folding is not possible, misfolded proteins are targeted for protein degradation to maintain proper protein homeostasis. Degradation is achieved through the ubiquitin-proteasome system (UPS) or the autophagy-lysosome pathway. A list of diseases and the associated aggregate discussed in this review are outlined.
Transcriptional regulation of the heat shock response is mediated by HSF1. HSF1 activity is dependent upon levels of chaperones and misfolded proteins, and other environmental stressors such as heat, aging, and changes in osmosis. Alternatively, Hsp70 and Hsp90 can negatively regulate the activation of HSF1, suppressing the heat shock response, allowing for a recovery period after the stressor is no longer present. Once in the nucleus, HSF1 trimerizes and binds Hsp gene promoters to activate transcription. Dai et al. (2003) further showed that this trimerization of HSF1 is mediated by CHIP, since both localize to the nucleus in response to cellular stress and form a complex once HSF1 is bound to DNA. Moreover, once degradation of misfolded proteins is complete, CHIP begins to degrade Hsp70 (unbound to any client). Therefore, CHIP stimulates HSF1 upregulation of Hsp70 in stress conditions and mediates its degradation to basal levels in the recovery period from stress.
STI1 was discovered in 1989, as a protein that is upregulated during cellular stress in yeast (Nicolet and Craig, 1989). STI1 is a modular protein composed of three TPR domains (TPR1, TPR2A and TPR2B) and two domains rich in aspartate and proline residues (DP1 and DP2, see Figure 3). The TPR domains of STI1 bind Hsp70 and Hsp90 to facilitate client protein transfer. While the C-terminal domain of Hsp90 is critical for the binding of TPR domains, additional contacts are made with the middle domain of Hsp90 (Lee et al., 2012; Schmid et al., 2012). Binding of Hsp90 by STI1 results in non-competitive inhibition of its ATPase activity through interaction with TPR2A-TPR2B fragment and stabilizes Hsp90 in an open conformation. However, the human homolog of STI1, HOP, appears to be ~10-fold less potent as an inhibitor of the Hsp90 ATPase activity (Prodromou et al., 1999; Richter et al., 2008). Hsp70 and Hsp90 engagement is facilitated through sequential interactions with the individual TPR domains of STI1 (Rohl et al., 2015b). The function of the DP domains (DP1 and DP2) is less clear. The minimal fragment of STI1 that supports client activation is composed of TPR2A-TPR2B-DP2 (Schmid et al., 2012; Rohl et al., 2015b). The Caenorhabditis elegans STI1 homolog, CeHOP, contains only the TPR2A-TPR2B-DP2 module, but is still capable of binding Hsp70 and Hsp90, though not simultaneously (Gaiser et al., 2009). Deletion of CeHOP is not lethal, but mutant worms have deficits in sexual development and reduced resistance to heat stress (Gaiser et al., 2009; Song et al., 2009). Functionality of CeHOP suggests the TPR1-DP1 module may be dispensable for client activation in vivo (Gaiser et al., 2009).
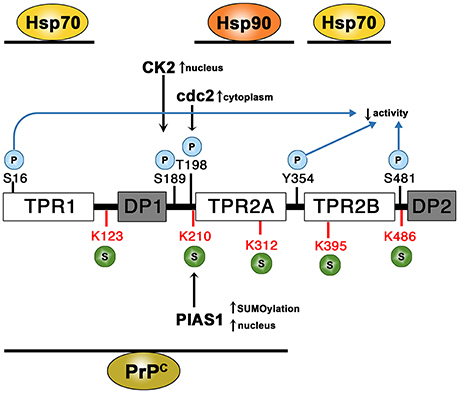
Figure 3. Domain structure of STI1 and sites of post-translational modifications (PTM). STI1 is composed of three structurally similar tetratricopeptide repeat domains (TPR1, TPR2A, and TPR2B) and two regions rich in aspartate and proline residues (DP1 and DP2). The protein is subject to phosphorylation (S16, S189, T198, Y354, and S481). CK2 phosphorylation at S189 induces STI1 accumulation in the nucleus. In contrast phosphorylation by cdc2 at T198 localizes STI1 to the cytoplasm. Five possible SUMOylation sites have been identified (K123, K210, K312, K395, and K486). SUMOylation by PIAS1 at K210 may stimulate SUMOylation at the alternate sites. Association of PIAS1 with STI1 by a SUMO-independent mechanism increases STI1 nuclear accumulation. Regions that bind HSP70, HSP90, and PrPC are illustrated.
Interestingly, STI1-based constructs lacking the DP2 domain did not support glucocorticoid receptor activation, one of STI1 functions in yeast cells (Schmid et al., 2012). X-ray crystallographic structures of the TPR2A-TPR2B domain revealed the linker between these domains is quite rigid. As a result, the TPR domains adopt an S-shaped form with their hydrophobic clefts responsible for binding the C-terminal Hsp90 residues oriented in opposite directions (Schmid et al., 2012). Complementary NMR spectroscopy experiments revealed that additional inter-domain contacts are formed between the C-terminal helix of TPR2B, the linker connecting TPR2B to the DP2 domain and α-helices 1 and 2 of the DP2 domain (Rohl et al., 2015b). These additional contacts form a rigid C-terminus composed of TPR2A-TPR2B-DP2. These results indicate that DP2 contributes to the quaternary structure of STI1, underlying its importance in STI1 function.
STI1 possesses two Hsp70 binding sites located in TPR1 and TPR2B; however, it binds to Hsp70 in a 1:1 stoichiometry (Scheufler et al., 2000; Rohl et al., 2015b). The current model for STI1 function in Hsp70 and Hsp90 coordination proposes that in the absence of Hsp90, the TPR2B domain represents the high-affinity binding site for Hsp70, these interaction sites are shown in Figure 3. Hsp90 binding induces a more “open” conformation between TPR1-DP1 fragment and the functional C-terminal TPR2A-TPR2B-DP2 domains. Hsp90 binding to TPR2A reduces accessibility of Hsp70 binding to TPR2B, thus the TPR1 domain becomes the predominant binding site for Hsp70 in the ternary complex (Rohl et al., 2015b). Binding of Hsp90 may also reorient the TPR1-DP1 module into close proximity to TPR2B, presumably facilitating transfer of client protein (Rohl et al., 2015b). Thus, the length of the linker bridging TPR1-DP1 to TPR2A-TPR2B-DP2 impacts STI1 function in client refolding (Rohl et al., 2015b). Deletion of the linker results in decreased formation of ternary complexes of STI1-Hsp70-Hsp90 and decreased protein client activation in vivo (Rohl et al., 2015b).
STI1 is widely expressed in most tissues and is typically localized in the cytoplasm, but can be found associated with the Golgi (Honore et al., 1992) and in the nucleus (Longshaw et al., 2004; Beraldo et al., 2013; Soares et al., 2013). Nuclear accumulation of STI1 is characteristic of stressed cells (Beraldo et al., 2013).
STI1 is subject to posttranslational modifications, which regulate its co-chaperone activity (Rohl et al., 2015a). Five different phosphorylation sites have been identified in the human STI1 homolog HOP corresponding to S16, S189, T198, Y354, and S481, see Figure 3 for simplified STI1 structure and respective phosphorylation sites. Phosphomimetic mutations resulted in decreased glucocorticoid receptor activation in vivo and Hsp70 binding affinities indicating that phosphorylation regulates STI1 co-chaperone function (Rohl et al., 2015a). Interestingly, Y354E phosphomimetic variant located in the loop joining TPR2A-TPR2B appeared to disrupt the rigid linker joining the two domains and promotes a more dynamic flexibility causing loss of function (Rohl et al., 2015a).
STI1 is typically found in the cytosol, but it can shuttle between the nucleus and cytoplasm (Longshaw et al., 2004), due to the presence of a nuclear localization signal (NLS) at amino acids 222–239 (of mouse STI1). Specifically, phosphorylation of STI1 by casein kinase II (CKII) and cell division cycle kinase II (cdc2) at S189 and T198 (respectively), contiguous to STI1 NLS (Longshaw et al., 2000) regulates nuclear localization of STI1 (Longshaw et al., 2004). CKII stimulates cellular growth by promoting cells to enter G1 phase of cell cycle (Pyerin, 1994), whereas cdc2 favors cell division (Matsumoto and Fujimoto, 1990). The rate of STI1 export from the nucleus is much higher than import, which can be inhibited with leptomycin B, a nuclear export inhibitor (Longshaw et al., 2004). Of particular interest, STI1 interacts with the nuclear small ubiquitin-like modifier (SUMO) E3 ligase family protein inhibitor of activated STAT (PIAS) and many other components of the SUMOylation machinery (Soares et al., 2013), suggesting potential STI1 regulation of genotoxic stress responses. STI1 was retained in the nucleus of astrocytes overexpressing PIAS1 (Soares et al., 2013) and Hsp90 was also found to be appreciably colocalized with STI1 and PIAS1 in the nucleus. Specifically, PIAS1 can poly-SUMOylate STI1 at several lysine residues: K123, K210, K312, K395, and K486 (depicted in Figure 3; Soares et al., 2013) and it is proposed that K210 hierarchically regulates SUMOylation of the other sites. Co-transfection of HEK293 cells with SUMO, PIAS1, and STI1 increased SUMOylation of STI1, but it was the interaction between PIAS1 and STI1 that supported nuclear retention, not SUMOylation. This suggests that nuclear localization of STI1 may alter its co-chaperone activities with Hsp90 and can help with nuclear protein crowding in particular sites that facilitate protein-protein interactions, such as PML nuclear bodies (Soares et al., 2013). However, STI1 has also been shown to function as a scaffold to recruit Hsp90 for other nuclear functions, including regulation of canalization or developmental robustness, by controlling Piwi and regulating Piwi-interacting RNA and impacting transposons (Gangaraju et al., 2011; Karam et al., 2017).
Deletion of STI1 is not lethal in yeast (Flom et al., 2007) and in C. elegans elimination of STI1 caused reduced lifespan (Song et al., 2009). Interestingly, knockout of STI1 in mice is embryonically lethal by E10.5 (Beraldo et al., 2013). Half of STI1-null blastocysts also failed to thrive, suggesting a key role for STI1 early in development (Beraldo et al., 2013). Homozygous embryos have a 50% reduction in Hsp90 client protein expression (p53, GRK2, STAT3—all clients that when knocked out are embryonically lethal; Beraldo et al., 2013). Mouse embryonic fibroblast lacking STI1 also failed to thrive in culture. Moreover, astrocytes derived from STI1 haplo-sufficient mice are also less resilient to irradiation (Soares et al., 2013), and neurons are less resistant to oxygen-glucose deprivation (Beraldo et al., 2013) or β-amyloid toxicity (Ostapchenko et al., 2013). This indicates that in higher organisms, STI1 is essential for regulating cellular resilience.
STI1 is a critical co-chaperone in the functionality of the Hsp70/Hsp90 machinery in cells. However, there is also a large body of literature that focuses on the extracellular effects of STI1, specifically the consequences of interaction with the cellular prion protein (PrPC; Zanata et al., 2002; Lopes et al., 2005; Caetano et al., 2008; Arantes et al., 2009; Beraldo et al., 2010, 2013; Roffe et al., 2010; Hajj et al., 2013; Ostapchenko et al., 2013; Maciejewski et al., 2016), which is illustrated in Figure 4. PrPC is anchored to cell membranes by a glycosylphosphatidylinositol (GPI) moiety and it is thought to serve as a molecular scaffolding protein organizing signaling complexes (Linden et al., 2008).
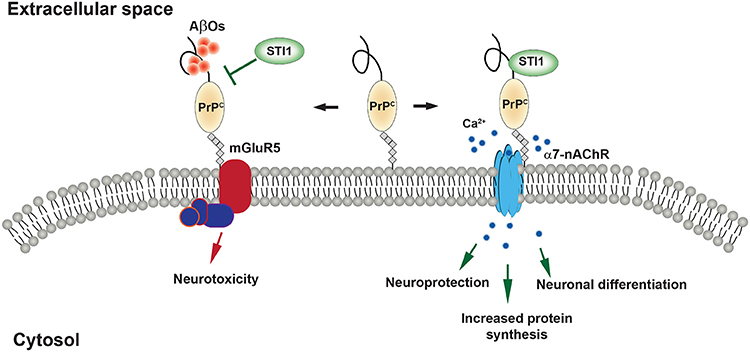
Figure 4. STI1 signaling mediated by the cellular prion protein (PrPC). (Right) PrPC binding to extracellular STI1 induces neuroprotective and neuro- differentiation through Ca2+ influx via α7-nAChR. (Left) Aβ oligomers transmit toxic signaling events through PrPC. STI1 inhibits Aβ oligomer binding to PrPC and/or activate protective signaling events.
STI1 must be secreted in order to interact with PrPC at the cell surface. Like Hsp70 and Hsp90 (Clayton et al., 2005; Lancaster and Febbraio, 2005), STI1 can be secreted via exosomes (Hajj et al., 2013). PrPC is also secreted by exosomes and both PrPC and STI1 can be found on the surface of the exosomes derived from astrocytes (Hajj et al., 2013). Secreted STI1 can then interact with and bind to the surface of neurons (Hajj et al., 2013), having a variety of effects on cell growth and survival, in a PrPC-dependent manner.
Martins, Linden and Brentani (Chiarini et al., 2002; Zanata et al., 2002) were the first to report the interaction between STI1 and PrPC and to describe STI1 as a signaling molecule. Notably, interaction between STI1 and PrPC reduced apoptotic cell death induced by the protein synthesis inhibitor anisomycin. These effects were confirmed to be PrPC-dependent using PrP-null neurons or exogenous treatment with a truncated form of STI1 lacking a critical PrPC binding site (STI1Δ230–245). Lopes et al. (2005) found that cultured neurons treated with exogenous STI1 are more resilient to protein synthesis inhibitors, but this increased neuronal resilience required activation of cAMP-PKA pathway. STI1-PrPC engagement is also capable of stimulating neuronal differentiation and this was dependent upon activation of the mitogen-activated protein kinase (MAPK/ERK) signaling pathway (Lopes et al., 2005). Roffe et al. (2010) showed that STI1-PrPC interaction increased protein synthesis in hippocampal neurons via mTOR and this was dependent upon the MAPK/ERK and PI3K signaling pathways. STI1 and PrPC interact briefly at the cell surface and are quickly internalized by distinct cellular pathways, limiting the levels of signaling activation by STI1 (Caetano et al., 2008). Activation of PKA and ERK signaling cascades by STI1-PrPC interaction is in part due to calcium signaling (Beraldo et al., 2010). STI1-PrPC signaling in hippocampal neurons requires α7 nicotinic acetylcholine receptors and inhibition of these receptors with α-bungarotoxin or use of knockout neurons eliminates STI1-PrPC neuroprotection (Beraldo et al., 2010; Ostapchenko et al., 2013). Finally, STI1-PrPC association supports proliferation and pluripotency of neural stem cells and promotes neurosphere formation (Santos et al., 2011). These findings altogether suggest that STI1 is critical for proper growth, development, and resilience to cellular stress.
Decreased STI1 levels significantly reduces cellular tolerance and cells display stress phenotypes, such as increased STI1 nuclear localization and nuclear labeling for γ-H2AX, a marker for double stranded DNA breaks, as seen in germ-line knockdown of STI1 in Drosophila (Karam et al., 2017) and in mouse embryonic fibroblasts (Beraldo et al., 2013). Astrocytes derived from STI1 haplo-insufficent mice also secrete 50% less STI1, which has consequences on PrPC-dependent resilience. Exogenous treatment with recombinant STI1 in neuronal cultures subjected to oxygen-glucose deprivation (OGD) reduced levels of cellular death in a PrPC and α7 nicotinic receptor-dependent manner (Beraldo et al., 2013), further supporting the notion that extracellular STI1 is responsible for inducing some of these neuroprotective effects. Neuronal cultures from PrP-null animals were not protected from OGD upon STI1 treatment, further indicating that these effects are indeed PrPC-dependent.
Middle cerebral artery occlusion, a model for ischemic stroke was conducted on STI1-haplosufficient mice (Beraldo et al., 2013), which had increased mortality and a reliably larger infarct volume compared to control littermates. Additionally, Lee et al. (2013) found an upregulation of STI1 immunoreactivity in brains from MCAO rats and post-mortem tissue of stroke patients. STI1 contains a hypoxia response element within its promoter region, which can be activated by hypoxia-inducible factor 1-α (HIF1-α; Lee et al., 2013). This binding event is responsible for the elevated STI1 levels post-stroke, as knockout for HIF1-α or lentiviral shRNA administration to mice inhibited the increase in STI1 immunoreactivity around the infarct. Lee et al. (2013) also showed that STI1 increases proliferation of bone-marrow derived cells and recruits these cells to the areas of damage, as a means to promote and accelerate recovery. Together, these studies provide strong evidence that extracellular STI1 is required for recovery of post-ischemic insult and that exogenous treatment with STI1 could potentiate the recovery process.
It is educational also to learn how STI1 can signal in cancer cells, as some of the signaling pathways may be similar to those in neurons. In the context of cancer, increased proliferative capacity due to STI1 is a major problem. Wang et al. (2010) found that levels of STI1 were much higher in malignant vs. benign ovarian tumors. Additionally, serum levels of STI1 were ~6 times higher in ovarian cancer patients (Wang et al., 2010) and were being secreted by the cancerous cells. Activation of the ERK signaling pathway by secreted STI1 was responsible for the increased proliferative capacity of these cancerous cells. Elevated levels of STI1 were correlated with worsened prognosis in ovarian cancer patients (Chao et al., 2013), making this a useful biomarker for this type of cancer. Recently, Wang et al. (2017) found increased intracellular and extracellular levels of STI1 in renal cell carcinoma (RCCs) tumor cells. Increased proliferation was mediated by the activin A receptor, type II-like kinase 2 (ALK2), and the receptor regulated SMAD1/5 protein signaling cascade, independent of PrPC. Only differentiation of the osteoclasts from the RCCs was dependent upon STI1-PrPC signaling. These studies provide further evidence that STI1 acts as a cytokine-like signaling molecule, promoting cellular growth by activating PrPC dependent and independent pathways.
Protein misfolding can lead to the formation of aggregates in diverse neurodegenerative diseases, such as AD, PD, ALS, frontotemporal dementia (FTD), HD, and prion diseases (Knowles et al., 2014). Protein aggregates are formed by highly ordered filamentous inclusions with β-sheet conformation in the core. Deposits can be fibrillar and insoluble (Chiti and Dobson, 2006), fibrillary but with some degree of solubility (Han et al., 2012), or amorphous. The composition of these deposits is often specific for each disease and composed of one predominant protein in each of these diseases, such as β-amyloid, tau, huntingtin, α-synuclein or prion protein (Goedert and Spillantini, 2006). These proteins will be further discussed in following subsections with relation to the disease the misfolded species are typically found in.
Degradation of misfolded proteins by the ubiquitin-proteasome system (UPS) or the autophagy-lysosome pathway (ALP; Labbadia and Morimoto, 2015; Yerbury et al., 2016) have been regarded as potential therapeutic targets for the treatment of neurodegenerative diseases, as aging leads to decreased efficiency of protein quality control (Ben-Zvi et al., 2009; Wang J. et al., 2009). Ubiquitination of misfolded proteins ultimately leads to degradation of proteins by the 26S proteasome and release of the ubiquitin chain (Pickart, 2001).
Proteins with longer half-life are predominantly degraded via the ALP. The role of chaperone-independent macroautophagy (which leads to the formation of autophagosomes) in the central nervous system of mammals is well-documented (Grant and Donaldson, 2009; He and Klionsky, 2009). In addition, chaperone-mediated autophagy (CMA) has been shown to protect against accumulation of tau, α-synuclein, and polyQ-huntingtin (Htt) in models of tauopathy, PD, and HD, respectively (Wang Y. et al., 2009; Qi et al., 2012; Xilouri et al., 2013). As shown by Agarraberes and Dice (2001), CMA requires the formation of complexes of chaperones and co-chaperones, including Hsp40, Hsp70, STI1/HOP, Hsp70-interacting protein (Hip), and Bcl2-associated athanogene 1 protein (BAG-1). Increasing evidence has shown that disrupted autophagy—either through autophagosomes or CMA—and lysosomal mechanisms contribute to the pathogenesis of AD, PD, HD, ALS, and FTD (Anglade et al., 1997; Nagata et al., 2004; Yu et al., 2005; Morimoto et al., 2007; Hu et al., 2010; Cuervo and Wong, 2014; Nah et al., 2015).
The following subsections will briefly discuss PD, HD, ALS, prion diseases and AD, and the effects of Hsp70, Hsp90, and STI1 (if investigated) on the misfolded protein species in each of these diseases and it is also summarized briefly in Table 1. We will also discuss how modulating the levels or activities of these chaperones affects protein toxicity and aggregative-capacity.
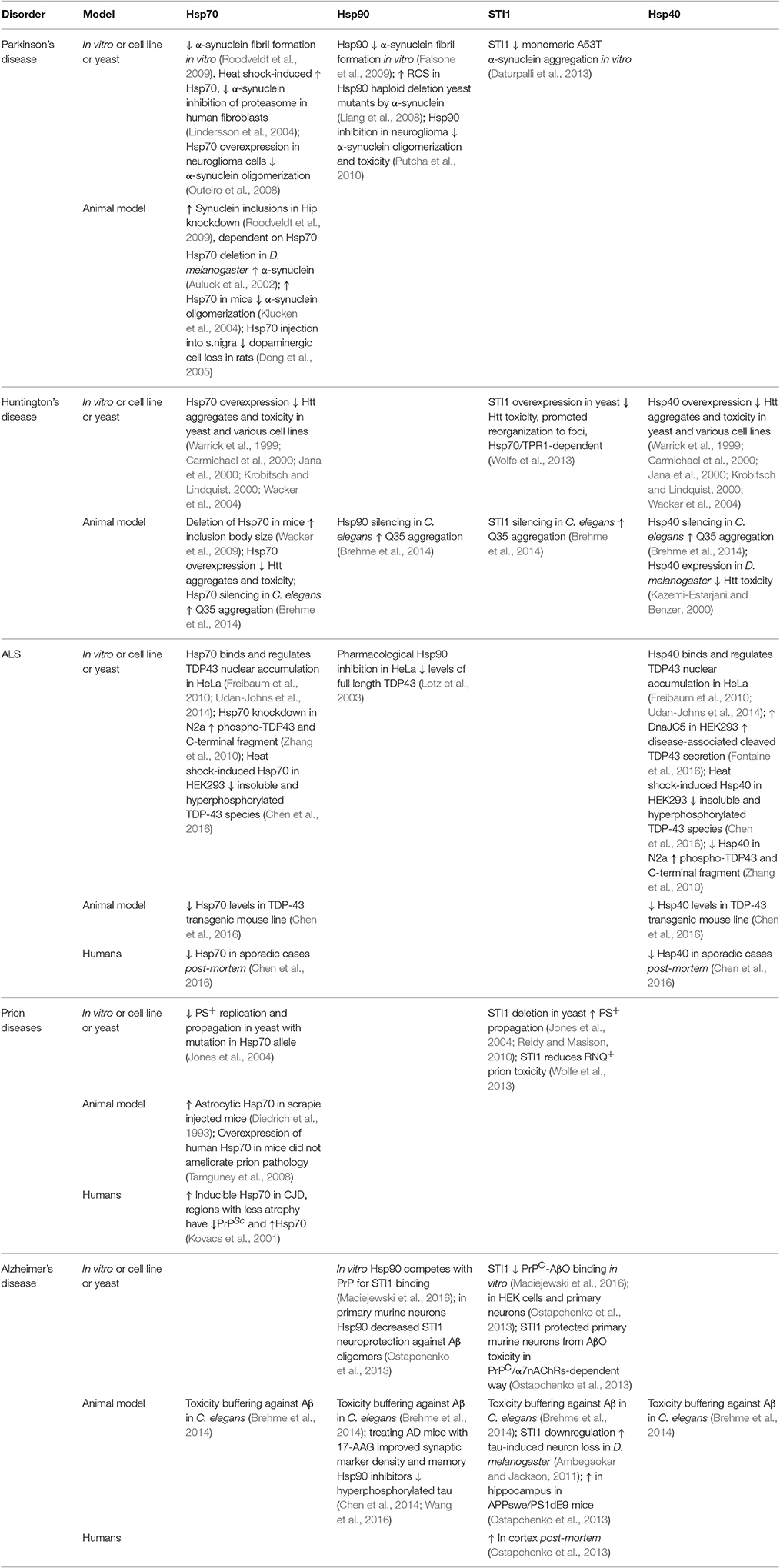
Table 1. Overview comparing Hsp70, Hsp90, STI1, and/or Hsp40 protein quality control in various model organisms of neurodegenerative disease.
PD is the second most common neurodegenerative disease characterized mainly by loss of dopaminergic neurons that project from the Substantia nigra, although several other neuronal groups are also affected (Davie, 2008). Several genes have been linked to genetic forms of PD, and amongst them SNCA, that codes for α-synuclein is of particular interest. α-synuclein, a protein associated with neuronal membranes (Maroteaux and Scheller, 1991), can aggregate in plaques in AD, termed the non-amyloid component (Ueda et al., 1993). However, α-synuclein is more commonly associated with PD, where it forms filamentous intraneuronal inclusions composed of ubiquitinated and phosphorylated α-synuclein, a component of Lewy bodies (Trojanowski et al., 1998; Goedert, 1999). This results in loss of neurons, and of importance to symptoms in PD, dopaminergic neurons in the Substantia nigra are particularly affected (de Lau and Breteler, 2006; Dickson, 2012). Augmented levels of α-synuclein or α-synuclein-containing aggregates are also characteristic of other neurodegenerative diseases including Lewy body dementia, multiple system atrophy and AD (Halliday et al., 2011; Serrano-Pozo et al., 2011; Ingelsson, 2016), forming a group of diseases termed “synucleinopathies.” The involvement of molecular chaperones in PD was first suggested by the observation that Hsp90, Hsp70, Hsp60, Hsp40, and Hsp27 were localized in Lewy bodies (McLean et al., 2002; Uryu et al., 2006; Leverenz et al., 2007). Increasing evidence has shown protective actions of molecular chaperones against α-synuclein-induced toxicity both in vitro and in vivo. Lindersson et al. (2004) showed that filaments of α-synuclein are able to bind to a component of the proteasome (the 20S subunit) and selectively impede the chymotrypsin-like activities of the proteasome in vitro. Recombinant human Hsp70 was capable of binding these α-synuclein filaments and attenuating their chymotrypsin-like inhibitory activity. Furthermore, heat shock of fibroblasts expressing α-synuclein lead to a significant increase in Hsp70 levels, which also reduced the inhibitory effects of α-synuclein on the proteasome (Lindersson et al., 2004). Roodveldt et al. (2009) found that Hsp70 and Hsp70-interacting protein (Hip) prevented formation of α-synuclein fibrils in vitro and in C. elegans knockdown of Hip increased synuclein inclusions in an Hsp70-dependent manner. Interestingly, Hsp70 overexpression in human neuroglioma cells transfected with mutant α-synuclein led to 50% less oligomeric α-synuclein species (Outeiro et al., 2008). Transgenic expression of familial PD mutations (A30P and A53T in α-synuclein) in fruit flies causes a significant degeneration of dopaminergic neurons, but co-expression of human Hsp70 abrogated this loss (Auluck et al., 2002). Overexpression of Hsp70 in yeast and mouse models, and Hsp90 inhibition with geldanamycin in human cell lines has been shown to counteract formation and accumulation of α-synuclein oligomers and alleviate α-synuclein-induced toxicity (Klucken et al., 2004; McLean et al., 2004; Flower et al., 2005; Luk et al., 2008). Conversely, Auluck et al. (2002) also reported acceleration of α-synuclein toxicity after inducing a dominant negative mutation of fruit fly Hsp70, which further confirmed the critical role of Hsp70 in α-synuclein regulation. Similarly, injection of Hsp70 into the Substantia nigra of MPTP-treated rats (a toxin that results in similar pathologies to those seen in PD) prevented dopaminergic cell loss (Dong et al., 2005).
Much less is known about the role of Hsp90 in regulating α-synuclein aggregation. In vitro experiments show that Hsp90 can both abolish α-synuclein binding to vesicles and promote fibril formation in an ATP-dependent manner (Falsone et al., 2009). More recent in vitro experiments investigating Hsp90 interaction with the A53T mutant of α-synuclein revealed that all three Hsp90 domains bind to and prevent A53T α-synuclein aggregation. However, Hsp90 could not bind to monomeric or fibrillary synuclein species in this model (Daturpalli et al., 2013). Interestingly, haploid deletion of yeast Hsp90 (Hsp82) enhanced α-synuclein toxicity, specifically, by increasing reactive oxygen species accumulation (Liang et al., 2008). However, Putcha et al. (2010) showed that Hsp90 inhibition with 17-AAG (a geldanamycin derivative), which leads to upregulation of Hsp70, prevented α-synuclein oligomer formation and toxicity in the H4 neuroglioma cell line. Due to STI1 ability to modulate Hsp70/Hsp90 activity, Daturpalli et al. (2013) conducted an in vitro experiment to assess if STI1 could have its own effect on α-synuclein aggregation. STI1 could only attenuate monomeric A53T α-synuclein aggregation in vitro (Daturpalli et al., 2013). This suggests that STI1 is capable of having some of its own chaperone-like activity, but interaction with Hsp70 or Hsp90 would have a greater effect on reorganization of toxic α-synuclein species. The literature thus far suggests that increasing Hsp70 levels by activating the heat shock response or by genetic manipulation would be a suitable method for reducing α-synuclein toxicity. This could prove beneficial in reducing toxicity-related symptoms.
Excess CAG repeats in the IT15 gene is a heritable mutation that causes HD and leads to the accumulation of huntingtin protein [Huntingtons's Disease Collaborative Research Group (1993)]. Protein deposits of mutated huntingtin form inclusion bodies within motor neurons in the spinal cord, as well as neurons in the cerebellum, cortex and striatum (Davies et al., 1997). Recent work suggests that the sequestering of toxic huntingin (Htt) into inclusion bodies may be a way to remove this toxic species as increased formation correlated with a reduction in levels of toxicity and neuronal death (Arrasate et al., 2004; Miller et al., 2010).
HD is part of the polyQ group of neurodegenerative diseases, which includes spinocerebellar ataxias, spinal and bulbar muscular atrophy (also known as Kennedy's disease), and dentatorubral-pallidoluysian atrophy (Williams and Paulson, 2008). In a fly model, Kazemi-Esfarjani and Benzer (2000) showed transgenic vector-mediated suppression of Htt toxicity by the molecular chaperones dHDJ1, a homolog of human Hsp40, and dTPR2, a homolog of human tetratricopeptide repeat protein 2. Likewise, deletion of Hsp70 in mice increased the size of polyQ inclusion bodies (Wacker et al., 2009). In addition, overexpression of Hsp40 and/or Hsp70 suppressed polyQ-dependent aggregation and neurodegeneration in cell cultures, yeast, fly, and mouse models (Warrick et al., 1999; Carmichael et al., 2000; Jana et al., 2000; Krobitsch and Lindquist, 2000; Wacker et al., 2004). STI1 overexpression in yeast suppressed Htt toxicity and drove the re-organization of small Htt103Q foci into larger assemblies through interaction with Hsp70, whereas STI1 deletion aggravated Htt toxicity and hampered foci formation (Wolfe et al., 2013). Specifically, point mutations (A31T or G76N) in the TPR1 domain of yeast STI1 (Hsp70 interacting domain) prevented reorganization of Htt to STI1 foci, which resulted in a significant reduction in cell growth. This indicates that functional STI1 interaction with Hsp70 is required for Htt103Q reorganization and toxicity buffering. In C. elegans expressing the Q35 aggregate prone-protein, siRNA for Hsp40, Hsp70, Hsp90, or STI1 significantly increased the number of Htt aggregates (Brehme et al., 2014). Therefore, there is strong evidence for an important role of chaperones and co-chaperones as therapeutic targets in HD.
ALS is a fatal neurodegenerative disorder that affects motor neurons of the brainstem, cortex and spinal cord, and results in weakness and atrophy of voluntary skeletal muscles (Paez-Colasante et al., 2015). ALS can be divided into two major classes—either the disease presents sporadically, which is 90% of cases, or it can be inherited. There are a number of proteins, RNAs and miRNAs dysregulated in ALS. The first aggregated protein to be identified was Cu/Zn superoxide dismutase (SOD1; Rosen et al., 1993), then trans-active DNA binding protein-43 (TDP-43; Arai et al., 2006; Neumann et al., 2006), along with fused in sarcoma/translocated in liposarcoma (FUS; Kwiatkowski et al., 2009; Vance et al., 2009), see Blokhuis et al. (2013) for a more extensive review on toxic protein accumulation in ALS.
However, in most familial or sporadic cases of ALS, the RNA binding protein TDP-43 shows signs of mislocalization and aggregation. TDP-43 is capable of binding to DNA and RNA, making it a key regulator of transcription, translation and cellular growth (Ayala et al., 2011; Polymenidou et al., 2011). TDP-43 can mislocalize to the cytoplasm, be ubiquitinated, hyperphosphorylated and ultimately form aggregates (Neumann et al., 2006; Mackenzie et al., 2007; Sreedharan et al., 2008; Brettschneider et al., 2013). Mutations in TDP-43 have been linked to ALS and FTD (Arai et al., 2006; Neumann et al., 2006). All together these results suggest the need to better understand the relationship between TDP-43 and chaperones.
TDP-43 carries out most of its functions in the nucleus, but it can be transported to the cytosol due to the nuclear export sequence near its N-terminus (Ayala et al., 2008). TDP-43 contains two RNA recognition motifs in the core of the protein and a C-terminal domain that contains a glutamine/asparagine-rich prion-like region that cooperates in protein-protein interactions (Ou et al., 1995; Budini et al., 2012a,b; Mackness et al., 2014). This prion-like domain allows for association with other TDP-43 molecules (Budini et al., 2012b) and is becoming a major area of research in neurodegenerative diseases involving TDP-43 associated pathology. Deletion of the prion-like domain of TDP-43 in HeLa cells eliminated heat shock induced nuclear aggregation, and further deletion of a glycine-rich region in this domain significantly reduced cytosolic mislocalization of the toxic 25 kDa TDP-43 variant (Udan-Johns et al., 2014). Recent studies in HeLa cells showed that Hsp40 and Hsp70 constitutively bind to and regulate the nuclear aggregation of TDP-43 (Freibaum et al., 2010; Udan-Johns et al., 2014).
Using HEK293T cells Fontaine et al. (2016) investigated the roles of the constitutively expressed Hsp70 homolog Hsc70 and its co-chaperone DnaJC5 in the secretion of neurodegenerative-disease associated proteins. Secretion of these proteins by unconventional mechanisms is thought to contribute to their spreading in the brain. DnaJC5 supports secretion via the Soluble NSF Attachment Protein Receptor (SNARE) complex at synapses in a calcium-dependent manner (Jacobsson and Meister, 1996; Chamberlain and Burgoyne, 1997, 1998; Umbach and Gundersen, 1997; Weng et al., 2009; Sharma et al., 2012). Overexpression of DnaJC5 lead to significant secretion of WT and disease associated mutants of TDP-43 from HEK cells (Fontaine et al., 2016) and this was dependent upon functional Hsc70. Interference with this mechanism could potentially regulate the spreading of misfolded TDP-43 in the brain. However, to date there has been limited experiments in neuronal cells, neurons or in animal models to test these findings obtained in non-neuronal cells.
Both Hsp70 and Hsp90 can be co-immunoprecipitated with TDP-43. Moreover, knockdown of Hsp70 or Hsp90 in human neuroblastoma cells lead to a significant increase in C-terminal and phosphorylated TDP-43, which are toxic TDP-43 species known to aggregate in the cytoplasm (Zhang et al., 2010). Treating HeLa cells with celastrol, an Hsp90 inhibitor, reduced levels of full length TDP-43, specifically by impairing Cdc37 (an Hsp90 co-chaperone which aids in client docking; Lotz et al., 2003)—Hsp90 interaction with TDP-43 (Jinwal et al., 2012). Recent work by Chen et al. (2016) further supported the role of Hsps in TDP-43 regulation, whereby activation of the heat shock response, by overexpression of HSF1 in HEK cells increased levels of Hsp70 and Hsp40, which lead to increased clearance of insoluble and hyperphosphorylated TDP-43. Interestingly, TDP-43 misregulation has also been found in a proportion of patients with AD (Amador-Ortiz et al., 2007; Wilson et al., 2011). hnRNP A2/B1 and A1, which are RNA-binding proteins that interact with TDP-43, have been shown to be decreased in AD, due to abnormal regulation of cholinergic signaling (Berson et al., 2012; Kolisnyk et al., 2013, 2016a,b). Although we have started to understand how chaperones and co-chaperones may regulate TDP-43, their role in neurons and other brain cells has not yet been examined in detail.
A significant reduction in Hsp70 and Hsp40 protein levels is observed in the brains of TDP-43Q331K transgenic mouse model of ALS and patients with sporadic ALS (Chen et al., 2016). HSF1 protein levels were also reduced in mice, but not in human brains (Chen et al., 2016). This suggests that, in disease, the heat shock response may be compromised and thus contribute to the accumulation of insoluble TDP-43 protein aggregates.
In prion diseases, PrPC is converted into PrPSc, which can work via template-mediated misfolding to further convert host PrPC protein into a variety of misfolded forms that aggregate and accumulate within the nervous tissue (Will and Ironside, 1999; Budka, 2003; Soto and Castilla, 2004; Linden et al., 2008). Misfolding of PrPC results in a class of diseases called transmissible spongiform encephalopathies (TSEs). Prion disease can arise sporadically, from genetic mutation or through transmission, such as by consumption of prion-infected tissues. TSEs include bovine spongiform encephalopathy in cattle, as well as sheep scrapie and variant CJD in humans (Linden et al., 2008). PrPC contains a disordered N-terminal domain and a globular C-terminal domain that is largely α-helical (Riek et al., 1996, 1997).
In order to better understand prion propagation and chaperone regulation in a simple eukaryotic model, yeast prions have been extensively studied. Yeast prions are self-propagating amyloid forms of soluble proteins that can function as protein-based inheritable elements. The yeast prion, PSI+, is a transmissible, self-replicating, and aggregation prone mutant of yeast translation termination factor Sup35p (Glover et al., 1997; King et al., 1997). Jones et al. (2004) found that a mutation in the SSA1 Hsp70 allele (SSA1–21p) significantly impaired PSI+ self-replication and propagation. Interestingly, in this cell line, deletion of yeast STI1 regenerated PSI+ propagation. Conversely, overexpression of STI1 reduced the mitotic capacity of PSI+ prions (Jones et al., 2004). Additionally, overexpression of Hsp104 is capable of eliminating PSI+ prions (Chernoff et al., 1995), but this is dependent upon expression of STI1 (Reidy and Masison, 2010). Deleting STI1 had no effect on levels of Hsp104, but eliminated Hsp104 “curing” activity (Reidy and Masison, 2010). Specifically, STI1 coordination of Hsp70 and Hsp90 was responsible for this prion elimination activity, as mutations in the TPR1 and TPR2 domains of STI1 lead to a drastic increase in PSI+ propagation. This suggests that STI1 coordination of Hsp70-Hsp90 as well as Hsp104 activity is required for disaggregation of yeast prions. Furthermore, STI1 expression and activity was also found to reduce toxicity of Rnq1 (a yeast protein with a glutamine-rich prion domain) prions, RNQ+ (Wolfe et al., 2013). STI1 recruited RNQ+ prions to foci containing Hsp104, amyloid like proteins and Hsp40, ultimately buffering toxicity by these prions.
As with many of the misfolded proteins that cause neurodegenerative diseases, knowledge of the physiological functions of PrPC is still not complete. Knockout of PrPC in mice affects synaptic transmission (Maglio et al., 2006), causes gross demyelination in the sciatic nerve specifically due to PrPC depletion in neurons (Bremer et al., 2010), and alterations in sleep pattern (Tobler et al., 1996). Elimination of PrPC also protects mice against infection with PrPSc (Bueler et al., 1993).
C57BL6 mice injected with 22L strain of scrapie had a significant increase in protein levels of inducible Hsp70 in active astrocytes (Diedrich et al., 1993). Similarly, mice infected with forms of scrapie known to induce plaques and increased vacuolation, had a significant increase in Hsp70 RNA expression toward the terminal phases of infection (Kenward et al., 1994). Kovacs et al. (2001) found increased immunoreactivity of inducible Hsp70 in Purkinje cells from CJD patients, and regions with higher levels of Hsp70 had less spongiform-like atrophy and increased levels of PrPC rather than PrPSc. This suggests a potential neuroprotective effect of Hsp70.
Tamguney et al. (2008) conducted a seminal study on 20 potential gene candidates that could regulate the replication of prions in mice infected with scrapie or cow 301V prions. Genes were selected based upon known interactions with PrPC in a diseased or non-diseased state, significant upregulation in prion disease, post-translational modification of PrP, or involvement in PrPC-related signal transduction (Tamguney et al., 2008). Interestingly, overexpression of human Hsp70 had no effect on prion disease onset.
To our knowledge there have been almost no studies investigating the role of Hsp90 and its co-chaperones in prion diseases. STI1 can signal via the prion protein as discussed above, and prion infection in cells abolishes STI1 signaling via the prion protein (Roffe et al., 2010). Interestingly, interaction of Hsp90 with STI1 also decreases PrPC-dependent STI1 neuroprotection (Maciejewski et al., 2016), which suggests that secreted Hsp90 may interfere with STI1 interaction with PrPC. Given that STI1 regulates protein aggregates via its co-chaperone activity (Wolfe et al., 2013), and also has extracellular cytokine-like neurotrophic function, it is likely that its effects on prion diseases and other neurodegenerative diseases are complex. By further understanding the cause and mechanism of the aggregation of these proteins, interventions targeting the chaperone machinery toward refolding or degradation could be utilized.
AD is the most common form of dementia, particularly affecting the aging population. Pathologically, it is defined by accumulation of two types of protein aggregates in the forebrain; extracellular plaques of Aβ, and intraneuronal neurofibrillary tangles (NFT) of microtubule-associated protein tau (Selkoe, 1991). As is the case with other diseases associated with misfolded proteins, analysis of AD brains and AD animal models revealed increased levels of Hsps and their co-chaperones, including Hsp27 (Renkawek et al., 1994), Hsp70 (Perez et al., 1991), and STI1 (Ostapchenko et al., 2013). A significant effort was made by a number of researchers to test whether chaperone system participates, directly or indirectly, in the pathogenic processes of Aβ and tau misfolding.
Aβ peptides consisting of around 39–43 residues are formed by proteolytic cleavage of its precursor, APP, by beta-site APP cleaving enzyme (BACE, β-secretase) and by a γ-secretase complex, formed by several proteins including presenilins (O'Brien and Wong, 2011). Aβ peptide toxicity was originally thought to be related mainly to the amyloid plaques that form throughout cortex and hippocampus in AD. However, during the last two decades it has also become recognized that soluble Aβ oligomers (AβOs) are toxic to synapses (Lambert et al., 1998; Walsh et al., 2002; Ferreira and Klein, 2011; Benilova et al., 2012; Mucke and Selkoe, 2012; Lesne et al., 2013). Aβ oligomers are thought to increase early before plaque formation and correlate with the onset of the neurotoxic events, such as excitotoxicity, synaptic loss as well as impairment of LTP and learning/memory in rodent models (Lambert et al., 1998; Klein et al., 2001; Klein, 2002; Walsh et al., 2002; Wang et al., 2005). Synaptotoxicity by AβOs depends on their interaction and corruption of multiple neuronal receptors, an effect that seems to depend on the initial interaction with PrPC (Lauren et al., 2009; Gimbel et al., 2010; Caetano et al., 2011; Kudo et al., 2012; Um et al., 2012; Ostapchenko et al., 2013; Beraldo et al., 2016). AβO/PrPC can engage metabotropic glutamate receptor 5 (Um et al., 2012, 2013; Beraldo et al., 2016) to activate pathogenic intracellular pathway that leads to activation of Fyn kinase, NMDA receptor mistrafficking, excitotoxicity and LTP inhibition.
Significant effort by several research groups were aimed to prevent AβO toxicity, employing anti-Aβ42/AβO (reviewed in Wisniewski and Drummond, 2016) and anti-PrPC (Chung et al., 2010; Barry et al., 2011) antibodies and N-terminal fragment of PrPC (Beland et al., 2014). However, to our knowledge, none of these potential therapies passed or even reached clinical trials yet. Remarkably, extracellular STI1, can bind to PrPC and activate α7 nicotinic acetylcholine receptor (nAChR), which mediate STI1-PrPC neurotrophic effects, efficiently preventing the binding of AβOs to PrPC on the neuronal surface, as well as general binding of these oligomers to neurons (Ostapchenko et al., 2013). Due to this effect, as well as protective signaling via α7 nAChR/PrPC complex, STI1 completely blocks AβO/PrPC toxicity in vitro (Ostapchenko et al., 2013). Whether Hsp70/Hsp90/STI1 exist extracellularly in AD brain separately or as a complex is unknown, but one may expect complex effects of extracellular chaperones on Aβ aggregation and toxicity in AD brain. To start with, we found in a biochemical assay that Hsp90 modulates formation of the STI1/PrPC complex, possibly resulting in decreased STI1 neurotrophic signals (Maciejewski et al., 2016).
Many in vivo AD models, including those that employ invertebrate and mammal species, are based on Aβ toxicity. In C. elegans Aβ expression leads to formation of peptide deposits and decreased motility (Link, 1995). This toxicity can be rescued by blocking the insulin growth factor-like signaling pathway, with a major role being played by HSF1 (Cohen et al., 2006). In this study, Cohen and colleagues showed that treatment with HSF1 RNAi increased Aβ toxicity in worms, probably due to increased amount of neurotoxic Aβ aggregates. The question remains, which mechanism activated by HSF1, plays a role in increased Aβ toxicity. Morimoto and colleagues approached this question by analyzing the various chaperones in worms expressing Aβ (Brehme et al., 2014). Systematic knockdown of Hsps and co-chaperones showed that Hsp40, Hsc70, Hsp90, and STI1, while not affecting motility in young animals, seem to normally buffer Aβ toxicity in C. elegans, as well as to alleviate age-related decrease in worm motility. Of interest, these chaperones and co-chaperones form an expression network in human brains, but the connecting links are significantly weakened in both AD and normal aging (Brehme et al., 2014), suggesting dysfunctional chaperone activity with age and disease.
The role of HSF1 in AD suggested by the results in C. elegans was recently supported by work with AD mouse models studying the effect of Hsp90 inhibitors on Aβ synaptotoxicity and behavioral impairment. Treatment of AD mice with 17-AAG (Chen et al., 2014) or OS47720 (Wang et al., 2016), Hsp90 inhibitors improved synaptic markers and density, in vivo LTP and memory loss and these effects were mediated by HSF1 activation and upregulation of synaptic genes. In contrast to the effect of HSF1, it is remarkable that whereas in C. elegans knockdown of Hsp90 is deleterious, in mammals inhibition of Hsp90 can actually improve Aβ-mediated toxicity.
Interestingly HSF1, which under stress conditions induces expression of Hsp70, Hsp90 and other chaperones, also upregulates production of APP (Dewji and Do, 1996). HSF1, besides its role in upregulation of heat shock machinery, is known as a major factor facilitating synaptic fidelity (Hooper et al., 2016). It is possible that HSF1-induced APP upregulation may be due to the pro-synaptogenic activity of this transcription factor. Indeed, APP has been shown to serve as a cell adhesion molecule (Small et al., 1999). On the other hand, synaptic activity by itself affects APP trafficking, routing it toward synapses (Tampellini et al., 2009). Besides, the same study showed that in neurons overexpressing APP with the Swedish familial mutation, synaptic activity also decreases intraneuronal Aβ. This result is paralleled by findings that activation or de-activation of synaptic activity, increases or decreases Aβ secretion, respectively (Kamenetz et al., 2003; Cirrito et al., 2005; Bero et al., 2011; Li et al., 2013; Yuan and Grutzendler, 2016). Considering the deleterious effect of Aβ on synaptic activity and integrity, HSF1, APP, and Aβ may form a self-regulating mechanism for controlling neuronal function.
Significant influence of Hsp70/90 machinery on AD pathology is implemented via microtubule associated protein tau. Physiologically, tau acts as a major regulator of microtubule formation (Weingarten et al., 1975) and in the CNS, tau is typically found in the cytoplasm or axons (Binder et al., 1985), where it promotes outgrowth and stabilizes microtubule formation. Tau is abnormally phosphorylated in AD due to increased activity of GSK-3 and other tau kinases (Alvarez et al., 1999; Avila et al., 2010; Tremblay et al., 2010; Cavallini et al., 2013), likely as a result of initial Aβ toxicity (Tamagno et al., 2003; Ryan et al., 2009; Hernandez et al., 2010). Hyperphosphorylated tau forms paired helical filaments, which are the main component of neurofibrillary tangles (Grundke-Iqbal et al., 1986a,b; Cao and Konsolaki, 2011), a critical pathological hallmark in AD. A number of studies have shown that these hyperphosphorylated tau species can be recognized by Hsps and their co-chaperones, including Hsp27, Hsp70, CHIP, and αB crystalline, in order to repair malignant tau or proceed with its recycling (Dou et al., 2003; Dabir et al., 2004; Petrucelli et al., 2004; Shimura et al., 2004; Luo et al., 2007). Recently, the structure of Hsp90-tau complex has been resolved (Karagoz et al., 2014). It explained how Hsp70 and Hsp90 can simultaneously bind to the intrinsically unstructured tau, making use of the atypically large substrate-binding site on Hsp90, which is rather open and accessible to clients such as tau. Of interest, low affinity hydrophobic connections in the Hsp90 substrate binding site could explain a general principle of Hsp90 interaction with disordered substrates or folded proteins.
Interestingly, inhibitors of Hsp90 decrease levels of phosphorylated tau, suggesting that Hsp90 may protect hyperphosphorylated tau from degradation (Dickey et al., 2006). Inhibition of Hsp90 in HeLa cells transfected with mutant tau (P301L) increased CHIP complex formation with phosphorylated tau (p-tau) and CHIP selectively degraded these p-tau species, essentially preventing aggregation of p-tau (Dickey et al., 2007). CHIP is also highly colocalized with p-tau and neurofibrillary tangles (aggregates of hyperphosphorylated tau; Dickey et al., 2007). These findings make CHIP a suitable candidate for modulating tau activity in neurodegenerative tauopathies, especially due to its ubiquitin enzyme activity. On the other hand, a complex of Hsp90 with the co-chaperone FKBP51 protected tau from proteasomal degradation and correlated with the neurotoxic tau species (Jinwal et al., 2010; Blair et al., 2013). FKBP51 overexpression decreased the amount of tau tangles in P301L tau transgenic mice, but increased soluble tau, including oligomeric and hyperphosphorylated species. This in turn led to increased tau toxicity, reflected in neuronal loss in P301L mice hippocampus and in decreased proliferation of tau-expressing neuronal cultures (Blair et al., 2013). Dickey and colleagues also found that FKBP51 expression is increased with age and in AD (Blair et al., 2013). This led them to hypothesize that Hsp90 interaction with FKBP51 is altered in aging and AD brains, allowing for the preservation of soluble, but possibly neurotoxic protein species. Another member of FKBP family, FKBP52, may also be involved in tau-related neurodegeneration. Recent evidence suggests that FKBP52 is a key regulator of tau association with microtubules, specifically in inhibiting this function (Chambraud et al., 2010). Moreover, a significant reduction in tau-mediated neurite outgrowth was observed in cells overexpressing FKBP52 (Chambraud et al., 2010).
Alternatively, Hsp70 promotes tau stability and association with microtubules at high levels of expression (Dou et al., 2003; Jinwal et al., 2009). STI1 may also be important for protection against aberrant tau species, as its downregulation in fruit flies worsened tau-induced retinal degeneration (Ambegaokar and Jackson, 2011). Upregulation of both Hsp70 and Hsp90 increases tau association with microtubules (Dou et al., 2003). Of note, this study used geldanamycin-induced Hsp90 inhibition, which resulted in increased Hsp70/90 expression due to HSF1 activation. As HSF1 activates multiple members of Hsp machinery, it is difficult to draw conclusions as to which particular chaperone affected tau-microtubule coupling. Soluble levels of tau correlate with those of Hsps and their co-chaperones, while in tauopathies where total levels of tau increase, Hsp70/90 decrease (Dou et al., 2003). Overall, tau regulation by the Hsp machinery is very complex and careful analysis of all possible effects on tau is needed when considering an anti-AD therapy that modulates this machinery.
In summary, the common observation of misfolded and aggregated proteins in neurodegenerative disease suggests dysregulation of chaperone activity. The balance between levels of Hsp70 and Hsp90 are becoming a major area of investigation, as both upregulation of Hsp70 and inhibition of Hsp90 in mammals reduce protein aggregation and toxicity. STI1 should be further investigated in models of protein aggregation, as STI1-PrPC interaction results in neuroprotection, attenuates AβO toxicity, and STI1 is an irreplaceable co-chaperone for the Hsp70/Hsp90 machinery. Ultimately, much is still unknown about how to effectively control protein misfolding and prevent aggregation by targeting chaperones and co-chaperones in neurodegenerative disease. Further, investigation of chaperones and their partners using new mouse models, could help to elucidate the underlying mechanisms of these proteinopathies and allow for generation of effective and unambiguous pharmacological therapies.
RL, AM, JML, and VO literature review, wrote the manuscript; WC, MD, and VP edited the manuscript; MP Literature review, wrote, and provide editing and scientific direction for the manuscript.
Work supported by Canadian Institute of Health Research (MOP 93651, MOP 136930, MOP 126000, MOP 89919), NSERC (402524-2013), Brain Canada, Weston Brain Institute—Canada, Canadian Foundation for Innovation, and Ontario Research Fund (MP and VP). RL and AM received support from OGS.
MP is an Associated Editor for the Frontiers Section on Neurodegeneration.
The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
(1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-E
Agarraberes, F. A., and Dice, J. F. (2001). A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 114(Pt 13), 2491–2499.
Ali, M. M., Roe, S. M., Vaughan, C. K., Meyer, P., Panaretou, B., Piper, P. W., et al. (2006). Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 440, 1013–1017. doi: 10.1038/nature04716
Allan, R. K., and Ratajczak, T. (2011). Versatile TPR domains accommodate different modes of target protein recognition and function. Cell Stress Chaperones 16, 353–367. doi: 10.1007/s12192-010-0248-0
Alvarez, G., Munoz-Montano, J. R., Satrustegui, J., Avila, J., Bogonez, E., and Diaz-Nido, J. (1999). Lithium protects cultured neurons against beta-amyloid-induced neurodegeneration. FEBS Lett. 453, 260–264. doi: 10.1016/S0014-5793(99)00685-7
Amador-Ortiz, C., Lin, W. L., Ahmed, Z., Personett, D., Davies, P., Duara, R., et al. (2007). TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann. Neurol. 61, 435–445. doi: 10.1002/ana.21154
Ambegaokar, S. S., and Jackson, G. R. (2011). Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from tau phosphorylation. Hum. Mol. Genet. 20, 4947–4977. doi: 10.1093/hmg/ddr432
Anglade, P., Vyas, S., Javoy-Agid, F., Herrero, M. T., Michel, P. P., Marquez, J., et al. (1997). Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol. Histopathol. 12, 25–31.
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Arantes, C., Nomizo, R., Lopes, M. H., Hajj, G. N., Lima, F. R., and Martins, V. R. (2009). Prion protein and its ligand stress inducible protein 1 regulate astrocyte development. Glia 57, 1439–1449. doi: 10.1002/glia.20861
Aravind, L., and Koonin, E. V. (2000). The U box is a modified RING finger—a common domain in ubiquitination. Curr. Biol. 10, R132–R134. doi: 10.1016/S0960-9822(00)00398-5
Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., and Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810. doi: 10.1038/nature02998
Auluck, P. K., Chan, H. Y., Trojanowski, J. Q., Lee, V. M., and Bonini, N. M. (2002). Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science 295, 865–868. doi: 10.1126/science.1067389
Avila, J., Wandosell, F., and Hernandez, F. (2010). Role of glycogen synthase kinase-3 in Alzheimer's disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev. Neurother. 10, 703–710. doi: 10.1586/ern.10.40
Ayala, V., Granado-Serrano, A. B., Cacabelos, D., Naudi, A., Ilieva, E. V., Boada, J., et al. (2011). Cell stress induces TDP-43 pathological changes associated with ERK1/2 dysfunction: implications in ALS. Acta Neuropathol. 122, 259–270. doi: 10.1007/s00401-011-0850-y
Ayala, Y. M., Zago, P., D'Ambrogio, A., Xu, Y. F., Petrucelli, L., Buratti, E., et al. (2008). Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 121(Pt 22), 3778–3785. doi: 10.1242/jcs.038950
Ballinger, C. A., Connell, P., Wu, Y., Hu, Z., Thompson, L. J., Yin, L.-Y., et al. (1999). Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 19, 4535–4545. doi: 10.1128/MCB.19.6.4535
Barry, A. E., Klyubin, I., Mc Donald, J. M., Mably, A. J., Farrell, M. A., Scott, M., et al. (2011). Alzheimer's disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 31, 7259–7263. doi: 10.1523/JNEUROSCI.6500-10.2011
Beland, M., Bedard, M., Tremblay, G., Lavigne, P., and Roucou, X. (2014). Abeta induces its own prion protein N-terminal fragment (PrPN1)-mediated neutralization in amorphous aggregates. Neurobiol. Aging 35, 1537–1548. doi: 10.1016/j.neurobiolaging.2014.02.001
Benilova, I., Karran, E., and De Strooper, B. (2012). The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. doi: 10.1038/nn.3028
Ben-Zvi, A., Miller, E. A., and Morimoto, R. I. (2009). Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl. Acad. Sci. U.S.A. 106, 14914–14919. doi: 10.1073/pnas.0902882106
Beraldo, F. H., Arantes, C. P., Santos, T. G., Queiroz, N. G., Young, K., Rylett, R. J., et al. (2010). Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J. Biol. Chem. 285, 36542–36550. doi: 10.1074/jbc.M110.157263
Beraldo, F. H., Ostapchenko, V. G., Caetano, F. A., Guimaraes, A. L., Ferretti, G. D., Daude, N., et al. (2016). Regulation of Amyloid beta Oligomer Binding to Neurons and Neurotoxicity by the Prion Protein-mGluR5 Complex. J. Biol. Chem. 291, 21945–21955. doi: 10.1074/jbc.M116.738286
Beraldo, F. H., Soares, I. N., Goncalves, D. F., Fan, J., Thomas, A. A., Santos, T. G., et al. (2013). Stress-inducible phosphoprotein 1 has unique cochaperone activity during development and regulates cellular response to ischemia via the prion protein. FASEB J. 27, 3594–3607. doi: 10.1096/fj.13-232280
Bero, A. W., Yan, P., Roh, J. H., Cirrito, J. R., Stewart, F. R., Raichle, M. E., et al. (2011). Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat. Neurosci. 14, 750–756. doi: 10.1038/nn.2801
Berson, A., Barbash, S., Shaltiel, G., Goll, Y., Hanin, G., Greenberg, D. S., et al. (2012). Cholinergic-associated loss of hnRNP-A/B in Alzheimer's disease impairs cortical splicing and cognitive function in mice. EMBO Mol. Med. 4, 730–742. doi: 10.1002/emmm.201100995
Binder, L. I., Frankfurter, A., and Rebhun, L. I. (1985). The distribution of tau in the mammalian central nervous system. J. Cell Biol. 101, 1371–1378. doi: 10.1083/jcb.101.4.1371
Blair, L. J., Baker, J. D., Sabbagh, J. J., and Dickey, C. A. (2015). The emerging role of peptidyl-prolyl isomerase chaperones in tau oligomerization, amyloid processing, and Alzheimer's disease. J. Neurochem. 133, 1–13. doi: 10.1111/jnc.13033
Blair, L. J., Nordhues, B. A., Hill, S. E., Scaglione, K. M., O'Leary, J. C. III., Fontaine, S. N., et al. (2013). Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Invest. 123, 4158–4169. doi: 10.1172/JCI69003
Blatch, G. L., and Lassle, M. (1999). The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays 21, 932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N
Blokhuis, A. M., Groen, E. J., Koppers, M., van den Berg, L. H., and Pasterkamp, R. J. (2013). Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 777–794. doi: 10.1007/s00401-013-1125-6
Borkovich, K. A., Farrelly, F. W., Finkelstein, D. B., Taulien, J., and Lindquist, S. (1989). hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol. Cell. Biol. 9, 3919–3930. doi: 10.1128/MCB.9.9.3919
Brehme, M., Voisine, C., Rolland, T., Wachi, S., Soper, J. H., Zhu, Y., et al. (2014). A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 9, 1135–1150. doi: 10.1016/j.celrep.2014.09.042
Bremer, J., Baumann, F., Tiberi, C., Wessig, C., Fischer, H., Schwarz, P., et al. (2010). Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 13, 310–318. doi: 10.1038/nn.2483
Brettschneider, J., Del Tredici, K., Toledo, J. B., Robinson, J. L., Irwin, D. J., Grossman, M., et al. (2013). Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 74, 20–38. doi: 10.1002/ana.23937
Budini, M., Buratti, E., Stuani, C., Guarnaccia, C., Romano, V., De Conti, L., et al. (2012a). Cellular model of TAR DNA-binding protein 43 (TDP-43) aggregation based on its C-terminal Gln/Asn-rich region. J. Biol. Chem. 287, 7512–7525. doi: 10.1074/jbc.M111.288720
Budini, M., Romano, V., Avendano-Vazquez, S. E., Bembich, S., Buratti, E., and Baralle, F. E. (2012b). Role of selected mutations in the Q/N rich region of TDP-43 in EGFP-12xQ/N-induced aggregate formation. Brain Res. 1462, 139–150. doi: 10.1016/j.brainres.2012.02.031
Bueler, H., Aguzzi, A., Sailer, A., Greiner, R. A., Autenried, P., Aguet, M., et al. (1993). Mice devoid of PrP are resistant to scrapie. Cell 73, 1339–1347. doi: 10.1016/0092-8674(93)90360-3
Bukau, B., Weissman, J., and Horwich, A. (2006). Molecular chaperones and protein quality control. Cell 125, 443–451. doi: 10.1016/j.cell.2006.04.014
Caetano, F. A., Beraldo, F. H., Hajj, G. N., Guimaraes, A. L., Jurgensen, S., Wasilewska-Sampaio, A. P., et al. (2011). Amyloid-beta oligomers increase the localization of prion protein at the cell surface. J. Neurochem. 117, 538–553. doi: 10.1111/j.1471-4159.2011.07225.x
Caetano, F. A., Lopes, M. H., Hajj, G. N., Machado, C. F., Pinto Arantes, C., Magalhaes, A. C., et al. (2008). Endocytosis of prion protein is required for ERK1/2 signaling induced by stress-inducible protein 1. J. Neurosci. 28, 6691–6702. doi: 10.1523/JNEUROSCI.1701-08.2008
Cao, W., and Konsolaki, M. (2011). FKBP immunophilins and Alzheimer's disease: a chaperoned affair. J. Biosci. 36, 493–498. doi: 10.1007/s12038-011-9080-7
Carmichael, J., Chatellier, J., Woolfson, A., Milstein, C., Fersht, A. R., and Rubinsztein, D. C. (2000). Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington's disease. Proc. Natl. Acad. Sci. U.S.A. 97, 9701–9705. doi: 10.1073/pnas.170280697
Cavallini, A., Brewerton, S., Bell, A., Sargent, S., Glover, S., Hardy, C., et al. (2013). An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J. Biol. Chem. 288, 23331–23347. doi: 10.1074/jbc.M113.463984
Chamberlain, L. H., and Burgoyne, R. D. (1997). The molecular chaperone function of the secretory vesicle cysteine string proteins. J. Biol. Chem. 272, 31420–31426. doi: 10.1074/jbc.272.50.31420
Chamberlain, L. H., and Burgoyne, R. D. (1998). Cysteine string protein functions directly in regulated exocytosis. Mol. Biol. Cell 9, 2259–2267. doi: 10.1091/mbc.9.8.2259
Chambraud, B., Sardin, E., Giustiniani, J., Dounane, O., Schumacher, M., Goedert, M., et al. (2010). A role for FKBP52 in Tau protein function. Proc. Natl. Acad. Sci. U.S.A. 107, 2658–2663. doi: 10.1073/pnas.0914957107
Chao, A., Lai, C. H., Tsai, C. L., Hsueh, S., Hsueh, C., Lin, C. Y., et al. (2013). Tumor stress-induced phosphoprotein1 (STIP1) as a prognostic biomarker in ovarian cancer. PLoS ONE 8:e57084. doi: 10.1371/journal.pone.0057084
Chen, H. J., Mitchell, J. C., Novoselov, S., Miller, J., Nishimura, A. L., Scotter, E. L., et al. (2016). The heat shock response plays an important role in TDP-43 clearance: evidence for dysfunction in amyotrophic lateral sclerosis. Brain 139(Pt 5), 1417–1432. doi: 10.1093/brain/aww028
Chen, S., and Smith, D. F. (1998). Hop as an adaptor in the heat shock protein 70 (Hsp70) and hsp90 chaperone machinery. J. Biol. Chem. 273, 35194–35200. doi: 10.1074/jbc.273.52.35194
Chen, Y., Wang, B., Liu, D., Li, J. J., Xue, Y., Sakata, K., et al. (2014). Hsp90 chaperone inhibitor 17-AAG attenuates Abeta-induced synaptic toxicity and memory impairment. J. Neurosci. 34, 2464–2470. doi: 10.1523/JNEUROSCI.0151-13.2014
Chernoff, Y. O., Lindquist, S. L., Ono, B., Inge-Vechtomov, S. G., and Liebman, S. W. (1995). Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268, 880–884. doi: 10.1126/science.7754373
Chiarini, L. B., Freitas, A. R., Zanata, S. M., Brentani, R. R., Martins, V. R., and Linden, R. (2002). Cellular prion protein transduces neuroprotective signals. EMBO J. 21, 3317–3326. doi: 10.1093/emboj/cdf324
Chiti, F., and Dobson, C. M. (2006). Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366. doi: 10.1146/annurev.biochem.75.101304.123901
Chung, E., Ji, Y., Sun, Y., Kascsak, R. J., Kascsak, R. B., Mehta, P. D., et al. (2010). Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci. 11:130. doi: 10.1186/1471-2202-11-130
Cirrito, J. R., Yamada, K. A., Finn, M. B., Sloviter, R. S., Bales, K. R., May, P. C., et al. (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. doi: 10.1016/j.neuron.2005.10.028
Clayton, A., Turkes, A., Navabi, H., Mason, M. D., and Tabi, Z. (2005). Induction of heat shock proteins in B-cell exosomes. J. Cell Sci. 118(Pt 16), 3631–3638. doi: 10.1242/jcs.02494
Cohen, E., Bieschke, J., Perciavalle, R. M., Kelly, J. W., and Dillin, A. (2006). Opposing activities protect against age-onset proteotoxicity. Science 313, 1604–1610. doi: 10.1126/science.1124646
Csermely, P., Kajtar, J., Hollosi, M., Jalsovszky, G., Holly, S., Kahn, C. R., et al. (1993). ATP induces a conformational change of the 90-kDa heat shock protein (hsp90). J. Biol. Chem. 268, 1901–1907.
Csermely, P., Schnaider, T., Soti, C., Prohaszka, Z., and Nardai, G. (1998). The 90-kDa molecular chaperone family: structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 79, 129–168. doi: 10.1016/S0163-7258(98)00013-8
Cuervo, A. M., and Wong, E. (2014). Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 24, 92–104. doi: 10.1038/cr.2013.153
Cunningham, C. N., Southworth, D. R., Krukenberg, K. A., and Agard, D. A. (2012). The conserved arginine 380 of Hsp90 is not a catalytic residue, but stabilizes the closed conformation required for ATP hydrolysis. Protein Sci. 21, 1162–1171. doi: 10.1002/pro.2103
Cyr, D. M., Lu, X., and Douglas, M. G. (1992). Regulation of Hsp70 function by a eukaryotic DnaJ homolog. J. Biol. Chem. 267, 20927–20931.
Dabir, D. V., Trojanowski, J. Q., Richter-Landsberg, C., Lee, V. M., and Forman, M. S. (2004). Expression of the small heat-shock protein alphaB-crystallin in tauopathies with glial pathology. Am. J. Pathol. 164, 155–166. doi: 10.1016/S0002-9440(10)63106-9
Dai, C., Whitesell, L., Rogers, A. B., and Lindquist, S. (2007). Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130, 1005–1018. doi: 10.1016/j.cell.2007.07.020
Dai, Q., Zhang, C., Wu, Y., McDonough, H., Whaley, R. A., Godfrey, V., et al. (2003). CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 22, 5446–5458. doi: 10.1093/emboj/cdg529
Daturpalli, S., Waudby, C. A., Meehan, S., and Jackson, S. E. (2013). Hsp90 inhibits alpha-synuclein aggregation by interacting with soluble oligomers. J. Mol. Biol. 425, 4614–4628. doi: 10.1016/j.jmb.2013.08.006
Davie, C. A. (2008). A review of Parkinson's disease. Br. Med. Bull. 86, 109–127. doi: 10.1093/bmb/ldn013
Davies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp, A. H., Ross, C. A., et al. (1997). Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548. doi: 10.1016/S0092-8674(00)80513-9
de Lau, L. M., and Breteler, M. M. (2006). Epidemiology of Parkinson's disease. Lancet Neurol. 5, 525–535. doi: 10.1016/S1474-4422(06)70471-9
Dewji, N. N., and Do, C. (1996). Heat shock factor-1 mediates the transcriptional activation of Alzheimer's beta-amyloid precursor protein gene in response to stress. Brain Res. Mol. Brain Res. 35, 325–328. doi: 10.1016/0169-328X(95)00214-D
Dickey, C. A., Dunmore, J., Lu, B., Wang, J. W., Lee, W. C., Kamal, A., et al. (2006). HSP induction mediates selective clearance of tau phosphorylated at proline-directed Ser/Thr sites but not KXGS (MARK) sites. FASEB J. 20, 753–755. doi: 10.1096/fj.05-5343fje
Dickey, C. A., Kamal, A., Lundgren, K., Klosak, N., Bailey, R. M., Dunmore, J., et al. (2007). The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Invest. 117, 648–658. doi: 10.1172/JCI29715
Dickson, D. W. (2012). Parkinson's disease and parkinsonism: neuropathology. Cold Spring Harb. Perspect. Med. 2:a009258. doi: 10.1101/cshperspect.a009258
Diedrich, J. F., Carp, R. I., and Haase, A. T. (1993). Increased expression of heat shock protein, transferrin, and beta 2-microglobulin in astrocytes during scrapie. Microb. Pathog. 15, 1–6. doi: 10.1006/mpat.1993.1051
Dong, Z., Wolfer, D. P., Lipp, H. P., and Bueler, H. (2005). Hsp70 gene transfer by adeno-associated virus inhibits MPTP-induced nigrostriatal degeneration in the mouse model of Parkinson disease. Mol. Ther. 11, 80–88. doi: 10.1016/j.ymthe.2004.09.007
Dou, F., Netzer, W. J., Tanemura, K., Li, F., Hartl, F. U., Takashima, A., et al. (2003). Chaperones increase association of tau protein with microtubules. Proc. Natl. Acad. Sci. U.S.A. 100, 721–726. doi: 10.1073/pnas.242720499
Du, H., and Yan, S. S. (2010). Mitochondrial permeability transition pore in Alzheimer's disease: cyclophilin D and amyloid beta. Biochim. Biophys. Acta 1802, 198–204. doi: 10.1016/j.bbadis.2009.07.005
Ellgaard, L., McCaul, N., Chatsisvili, A., and Braakman, I. (2016). Co- and Post-translational protein folding in the ER. Traffic 17, 615–638. doi: 10.1111/tra.12392
Falsone, S. F., Kungl, A. J., Rek, A., Cappai, R., and Zangger, K. (2009). The molecular chaperone Hsp90 modulates intermediate steps of amyloid assembly of the Parkinson-related protein alpha-synuclein. J. Biol. Chem. 284, 31190–31199. doi: 10.1074/jbc.M109.057240
Felts, S. J., and Toft, D. O. (2003). p23, a simple protein with complex activities. Cell Stress Chaperones 8, 108–113. doi: 10.1379/1466-1268(2003)008<0108:PASPWC>2.0.CO;2
Ferreira, S. T., and Klein, W. L. (2011). The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer's disease. Neurobiol. Learn. Mem. 96, 529–543. doi: 10.1016/j.nlm.2011.08.003
Flom, G., Behal, R. H., Rosen, L., Cole, D. G., and Johnson, J. L. (2007). Definition of the minimal fragments of Sti1 required for dimerization, interaction with Hsp70 and Hsp90 and in vivo functions. Biochem. J. 404, 159–167. doi: 10.1042/BJ20070084
Flower, T. R., Chesnokova, L. S., Froelich, C. A., Dixon, C., and Witt, S. N. (2005). Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson's disease. J. Mol. Biol. 351, 1081–1100. doi: 10.1016/j.jmb.2005.06.060
Fontaine, S. N., Zheng, D., Sabbagh, J. J., Martin, M. D., Chaput, D., Darling, A., et al. (2016). DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 35, 1537–1549. doi: 10.15252/embj.201593489
Freibaum, B. D., Chitta, R. K., High, A. A., and Taylor, J. P. (2010). Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 9, 1104–1120. doi: 10.1021/pr901076y
Frydman, J., Nimmesgern, E., Ohtsuka, K., and Hartl, F. U. (1994). Folding of nascent polypeptide chains in a high molecular mass assembly with molecular chaperones. Nature 370, 111–117. doi: 10.1038/370111a0
Gaiser, A. M., Brandt, F., and Richter, K. (2009). The non-canonical Hop protein from Caenorhabditis elegans exerts essential functions and forms binary complexes with either Hsc70 or Hsp90. J. Mol. Biol. 391, 621–634. doi: 10.1016/j.jmb.2009.06.051
Gangaraju, V. K., Yin, H., Weiner, M. M., Wang, J., Huang, X. A., and Lin, H. (2011). Drosophila Piwi functions in Hsp90-mediated suppression of phenotypic variation. Nat. Genet. 43, 153–158. doi: 10.1038/ng.743
Gao, X., Carroni, M., Nussbaum-Krammer, C., Mogk, A., Nillegoda, N. B., Szlachcic, A., et al. (2015). Human Hsp70 disaggregase reverses Parkinson's-linked alpha-synuclein amyloid fibrils. Mol. Cell 59, 781–793. doi: 10.1016/j.molcel.2015.07.012
Gimbel, D. A., Nygaard, H. B., Coffey, E. E., Gunther, E. C., Lauren, J., Gimbel, Z. A., et al. (2010). Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010
Glover, J. R., and Lindquist, S. (1998). Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82. doi: 10.1016/S0092-8674(00)81223-4
Glover, J. R., Kowal, A. S., Schirmer, E. C., Patino, M. M., Liu, J. J., and Lindquist, S. (1997). Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 89, 811–819. doi: 10.1016/S0092-8674(00)80264-0
Goedert, M. (1999). Filamentous nerve cell inclusions in neurodegenerative diseases: tauopathies and alpha-synucleinopathies. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 1101–1118. doi: 10.1098/rstb.1999.0466
Goedert, M., and Spillantini, M. G. (2006). A century of Alzheimer's disease. Science 314, 777–781. doi: 10.1126/science.1132814
Grant, B. D., and Donaldson, J. G. (2009). Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 10, 597–608. doi: 10.1038/nrm2755
Grundke-Iqbal, I., Iqbal, K., Quinlan, M., Tung, Y. C., Zaidi, M. S., and Wisniewski, H. M. (1986a). Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089.
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986b). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917. doi: 10.1073/pnas.83.13.4913
Hajj, G. N., Arantes, C. P., Dias, M. V., Roffe, M., Costa-Silva, B., Lopes, M. H., et al. (2013). The unconventional secretion of stress-inducible protein 1 by a heterogeneous population of extracellular vesicles. Cell. Mol. Life Sci. 70, 3211–3227. doi: 10.1007/s00018-013-1328-y
Halliday, G. M., Holton, J. L., Revesz, T., and Dickson, D. W. (2011). Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 122, 187–204. doi: 10.1007/s00401-011-0852-9
Han, T. W., Kato, M., Xie, S., Wu, L. C., Mirzaei, H., Pei, J., et al. (2012). Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149, 768–779. doi: 10.1016/j.cell.2012.04.016
Hartl, F. U., and Hayer-Hartl, M. (2009). Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 16, 574–581. doi: 10.1038/nsmb.1591
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93. doi: 10.1146/annurev-genet-102808-114910
Hernandez, F., Gomez de Barreda, E., Fuster-Matanzo, A., Lucas, J. J., and Avila, J. (2010). GSK3: a possible link between beta amyloid peptide and tau protein. Exp. Neurol. 223, 322–325. doi: 10.1016/j.expneurol.2009.09.011
Hohfeld, J., Minami, Y., and Hartl, F. U. (1995). Hip, a novel cochaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. Cell 83, 589–598. doi: 10.1016/0092-8674(95)90099-3
Honore, B., Leffers, H., Madsen, P., Rasmussen, H. H., Vandekerckhove, J., and Celis, J. E. (1992). Molecular cloning and expression of a transformation-sensitive human protein containing the TPR motif and sharing identity to the stress-inducible yeast protein STI1. J. Biol. Chem. 267, 8485–8491.
Hooper, P. L., Durham, H. D., Torok, Z., Hooper, P. L., Crul, T., and Vigh, L. (2016). The central role of heat shock factor 1 in synaptic fidelity and memory consolidation. Cell Stress Chaperones 21, 745–753. doi: 10.1007/s12192-016-0709-1
Hu, F., Padukkavidana, T., Vaegter, C. B., Brady, O. A., Zheng, Y., Mackenzie, I. R., et al. (2010). Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68, 654–667. doi: 10.1016/j.neuron.2010.09.034
Ingelsson, M. (2016). Alpha-synuclein oligomers-neurotoxic molecules in Parkinson's disease and other lewy body disorders. Front. Neurosci. 10:408. doi: 10.3389/fnins.2016.00408
Jacobsson, G., and Meister, B. (1996). Molecular components of the exocytotic machinery in the rat pituitary gland. Endocrinology 137, 5344–5356.
Jana, N. R., Dikshit, P., Goswami, A., Kotliarova, S., Murata, S., Tanaka, K., et al. (2005). Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 280, 11635–11640. doi: 10.1074/jbc.M412042200
Jana, N. R., Tanaka, M., Wang, G., and Nukina, N. (2000). Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet. 9, 2009–2018. doi: 10.1093/hmg/9.13.2009
Jhaveri, K., Taldone, T., Modi, S., and Chiosis, G. (2012). Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta 1823, 742–755. doi: 10.1016/j.bbamcr.2011.10.008
Jiang, J., Prasad, K., Lafer, E. M., and Sousa, R. (2005). Structural basis of interdomain communication in the Hsc70 chaperone. Mol. Cell 20, 513–524. doi: 10.1016/j.molcel.2005.09.028
Jinwal, U. K., Abisambra, J. F., Zhang, J., Dharia, S., O'Leary, J. C., Patel, T., et al. (2012). Cdc37/Hsp90 protein complex disruption triggers an autophagic clearance cascade for TDP-43 protein. J. Biol. Chem. 287, 24814–24820. doi: 10.1074/jbc.M112.367268
Jinwal, U. K., Koren, J. III., Borysov, S. I., Schmid, A. B., Abisambra, J. F., Blair, L. J., et al. (2010). The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules. J. Neurosci. 30, 591–599. doi: 10.1523/JNEUROSCI.4815-09.2010
Jinwal, U. K., Miyata, Y., Koren, J. III., Jones, J. R., Trotter, J. H., Chang, L., et al. (2009). Chemical manipulation of hsp70 ATPase activity regulates tau stability. J. Neurosci. 29, 12079–12088. doi: 10.1523/JNEUROSCI.3345-09.2009
Johnson, B. D., Schumacher, R. J., Ross, E. D., and Toft, D. O. (1998). Hop modulates Hsp70/Hsp90 interactions in protein folding. J. Biol. Chem. 273, 3679–3686. doi: 10.1074/jbc.273.6.3679
Jones, G., Song, Y., Chung, S., and Masison, D. C. (2004). Propagation of Saccharomyces cerevisiae [PSI+] prion is impaired by factors that regulate Hsp70 substrate binding. Mol. Cell. Biol. 24, 3928–3937. doi: 10.1128/MCB.24.9.3928-3937.2004
Kabani, M., and Martineau, C. N. (2008). Multiple hsp70 isoforms in the eukaryotic cytosol: mere redundancy or functional specificity? Curr. Genomics 9, 338–248. doi: 10.2174/138920208785133280
Kajander, T., Sachs, J. N., Goldman, A., and Regan, L. (2009). Electrostatic interactions of Hsp-organizing protein tetratricopeptide domains with Hsp70 and Hsp90: computational analysis and protein engineering. J. Biol. Chem. 284, 25364–25374. doi: 10.1074/jbc.M109.033894
Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., Iwatsubo, T., et al. (2003). APP processing and synaptic function. Neuron 37, 925–937. doi: 10.1016/S0896-6273(03)00124-7
Karagoz, G. E., Duarte, A. M., Akoury, E., Ippel, H., Biernat, J., Moran Luengo, T., et al. (2014). Hsp90-Tau complex reveals molecular basis for specificity in chaperone action. Cell 156, 963–974. doi: 10.1016/j.cell.2014.01.037
Karam, J. A., Parikh, R. Y., Nayak, D., Rosenkranz, D., and Gangaraju, V. K. (2017). Co-chaperone Hsp70/Hsp90 organizing protein (Hop) is required for transposon silencing and piRNA biogenesis. J. Biol. Chem. 292, 6039–6046. doi: 10.1074/jbc.C117.777730
Kazemi-Esfarjani, P., and Benzer, S. (2000). Genetic suppression of polyglutamine toxicity in Drosophila. Science 287, 1837–1840. doi: 10.1126/science.287.5459.1837
Kenward, N., Hope, J., Landon, M., and Mayer, R. J. (1994). Expression of polyubiquitin and heat-shock protein 70 genes increases in the later stages of disease progression in scrapie-infected mouse brain. J. Neurochem. 62, 1870–1877. doi: 10.1046/j.1471-4159.1994.62051870.x
King, C. Y., Tittmann, P., Gross, H., Gebert, R., Aebi, M., and Wuthrich, K. (1997). Prion-inducing domain 2-114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc. Natl. Acad. Sci. U.S.A. 94, 6618–6622. doi: 10.1073/pnas.94.13.6618
Kirschke, E., Goswami, D., Southworth, D., Griffin, P. R., and Agard, D. A. (2014). Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 157, 1685–1697. doi: 10.1016/j.cell.2014.04.038
Klein, W. L. (2002). ADDLs & protofibrils–the missing links? Neurobiol. Aging 23, 231–235. doi: 10.1016/S0197-4580(01)00312-8
Klein, W. L., Krafft, G. A., and Finch, C. E. (2001). Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 24, 219–224. doi: 10.1016/S0166-2236(00)01749-5
Klucken, J., Shin, Y., Masliah, E., Hyman, B. T., and McLean, P. J. (2004). Hsp70 reduces alpha-synuclein aggregation and toxicity. J. Biol. Chem. 279, 25497–25502. doi: 10.1074/jbc.M400255200
Knowles, T. P., Vendruscolo, M., and Dobson, C. M. (2014). The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396. doi: 10.1038/nrm3810
Kolisnyk, B., Al-Onaizi, M. A., Hirata, P. H., Guzman, M. S., Nikolova, S., Barbash, S., et al. (2013). Forebrain deletion of the vesicular acetylcholine transporter results in deficits in executive function, metabolic, and RNA splicing abnormalities in the prefrontal cortex. J. Neurosci. 33, 14908–14920. doi: 10.1523/JNEUROSCI.1933-13.2013
Kolisnyk, B., Al-Onaizi, M. A., Xu, J., Parfitt, G. M., Ostapchenko, V. G., Hanin, G., et al. (2016b). Cholinergic regulation of hnRNPA2/B1 translation by M1 muscarinic receptors. J. Neurosci. 36, 6287–6296. doi: 10.1523/JNEUROSCI.4614-15.2016
Kolisnyk, B., Al-Onaizi, M., Soreq, L., Barbash, S., Bekenstein, U., Haberman, N., et al. (2016a). Cholinergic surveillance over hippocampal RNA metabolism and Alzheimer's-like pathology. Cereb. Cortex. doi: 10.1093/cercor/bhw177. [Epub ahead of print].
Kovacs, G. G., Kurucz, I., Budka, H., Adori, C., Muller, F., Acs, P., et al. (2001). Prominent stress response of Purkinje cells in Creutzfeldt-Jakob disease. Neurobiol. Dis. 8, 881–889. doi: 10.1006/nbdi.2001.0418
Krobitsch, S., and Lindquist, S. (2000). Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 1589–1594. doi: 10.1073/pnas.97.4.1589
Kudo, W., Lee, H. P., Zou, W. Q., Wang, X., Perry, G., Zhu, X., et al. (2012). Cellular prion protein is essential for oligomeric amyloid-beta-induced neuronal cell death. Hum. Mol. Genet. 21, 1138–1144. doi: 10.1093/hmg/ddr542
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Labbadia, J., and Morimoto, R. I. (2015). The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464. doi: 10.1146/annurev-biochem-060614-033955
Lamb, J. R., Tugendreich, S., and Hieter, P. (1995). Tetratrico peptide repeat interactions: to TPR or not to TPR? Trends Biochem. Sci. 20, 257–259. doi: 10.1016/S0968-0004(00)89037-4
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Lancaster, G. I., and Febbraio, M. A. (2005). Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J. Biol. Chem. 280, 23349–23355. doi: 10.1074/jbc.M502017200
Lassle, M., Blatch, G. L., Kundra, V., Takatori, T., and Zetter, B. R. (1997). Stress-inducible, murine protein mSTI1. Characterization of binding domains for heat shock proteins and in vitro phosphorylation by different kinases. J. Biol. Chem. 272, 1876–1884. doi: 10.1074/jbc.272.3.1876
Lauren, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132. doi: 10.1038/nature07761
Lavery, L. A., Partridge, J. R., Ramelot, T. A., Elnatan, D., Kennedy, M. A., and Agard, D. A. (2014). Structural asymmetry in the closed state of mitochondrial Hsp90 (TRAP1) supports a two-step ATP hydrolysis mechanism. Mol. Cell 53, 330–343. doi: 10.1016/j.molcel.2013.12.023
Lee, C. T., Graf, C., Mayer, F. J., Richter, S. M., and Mayer, M. P. (2012). Dynamics of the regulation of Hsp90 by the co-chaperone Sti1. EMBO J. 31, 1518–1528. doi: 10.1038/emboj.2012.37
Lee, S. D., Lai, T. W., Lin, S. Z., Lin, C. H., Hsu, Y. H., Li, C. Y., et al. (2013). Role of stress-inducible protein-1 in recruitment of bone marrow derived cells into the ischemic brains. EMBO Mol. Med. 5, 1227–1246. doi: 10.1002/emmm.201202258
Lesne, S. E., Sherman, M. A., Grant, M., Kuskowski, M., Schneider, J. A., Bennett, D. A., et al. (2013). Brain amyloid-beta oligomers in ageing and Alzheimer's disease. Brain 136(Pt 5), 1383–1398. doi: 10.1093/brain/awt062
Leverenz, J. B., Umar, I., Wang, Q., Montine, T. J., McMillan, P. J., Tsuang, D. W., et al. (2007). Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathol. 17, 139–145. doi: 10.1111/j.1750-3639.2007.00048.x
Li, J., and Buchner, J. (2013). Structure, function and regulation of the hsp90 machinery. Biomed. J. 36, 106–117. doi: 10.4103/2319-4170.113230
Li, X., Uemura, K., Hashimoto, T., Nasser-Ghodsi, N., Arimon, M., Lill, C. M., et al. (2013). Neuronal activity and secreted amyloid beta lead to altered amyloid beta precursor protein and presenilin 1 interactions. Neurobiol. Dis. 50, 127–134. doi: 10.1016/j.nbd.2012.10.002
Liang, J., Clark-Dixon, C., Wang, S., Flower, T. R., Williams-Hart, T., Zweig, R., et al. (2008). Novel suppressors of alpha-synuclein toxicity identified using yeast. Hum. Mol. Genet. 17, 3784–3795. doi: 10.1093/hmg/ddn276
Lindberg, I., Shorter, J., Wiseman, R. L., Chiti, F., Dickey, C. A., and McLean, P. J. (2015). Chaperones in neurodegeneration. J. Neurosci. 35, 13853–13859. doi: 10.1523/JNEUROSCI.2600-15.2015
Linden, R., Martins, V. R., Prado, M. A., Cammarota, M., Izquierdo, I., and Brentani, R. R. (2008). Physiology of the prion protein. Physiol. Rev. 88, 673–728. doi: 10.1152/physrev.00007.2007
Lindersson, E., Beedholm, R., Hojrup, P., Moos, T., Gai, W., Hendil, K. B., et al. (2004). Proteasomal inhibition by alpha-synuclein filaments and oligomers. J. Biol. Chem. 279, 12924–12934. doi: 10.1074/jbc.M306390200
Link, C. D. (1995). Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 92, 9368–9372. doi: 10.1073/pnas.92.20.9368
Longshaw, V. M., Chapple, J. P., Balda, M. S., Cheetham, M. E., and Blatch, G. L. (2004). Nuclear translocation of the Hsp70/Hsp90 organizing protein mSTI1 is regulated by cell cycle kinases. J. Cell Sci. 117(Pt 5), 701–710. doi: 10.1242/jcs.00905
Longshaw, V. M., Dirr, H. W., Blatch, G. L., and Lassle, M. (2000). The in vitro phosphorylation of the co-chaperone mSTI1 by cell cycle kinases substantiates a predicted casein kinase II-p34cdc2-NLS (CcN) motif. Biol. Chem. 381, 1133–1138. doi: 10.1515/BC.2000.139
Lopes, M. H., Hajj, G. N., Muras, A. G., Mancini, G. L., Castro, R. M., Ribeiro, K. C., et al. (2005). Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 25, 11330–11339. doi: 10.1523/JNEUROSCI.2313-05.2005
Lotz, G. P., Lin, H., Harst, A., and Obermann, W. M. (2003). Aha1 binds to the middle domain of Hsp90, contributes to client protein activation, and stimulates the ATPase activity of the molecular chaperone. J. Biol. Chem. 278, 17228–17235. doi: 10.1074/jbc.M212761200
Luk, K. C., Mills, I. P., Trojanowski, J. Q., and Lee, V. M. (2008). Interactions between Hsp70 and the hydrophobic core of alpha-synuclein inhibit fibril assembly. Biochemistry 47, 12614–12625. doi: 10.1021/bi801475r
Luo, W., Dou, F., Rodina, A., Chip, S., Kim, J., Zhao, Q., et al. (2007). Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc. Natl. Acad. Sci. U.S.A. 104, 9511–9516. doi: 10.1073/pnas.0701055104
Maciejewski, A., Ostapchenko, V. G., Beraldo, F. H., Prado, V. F., Prado, M. A., and Choy, W. Y. (2016). Domains of STIP1 responsible for regulating PrPC-dependent amyloid-beta oligomer toxicity. Biochem. J. 473, 2119–2130. doi: 10.1042/BCJ20160087
Mackenzie, I. R., Bigio, E. H., Ince, P. G., Geser, F., Neumann, M., Cairns, N. J., et al. (2007). Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 61, 427–434. doi: 10.1002/ana.21147
Mackness, B. C., Tran, M. T., McClain, S. P., Matthews, C. R., and Zitzewitz, J. A. (2014). Folding of the RNA recognition motif (RRM) domains of the amyotrophic lateral sclerosis (ALS)-linked protein TDP-43 reveals an intermediate state. J. Biol. Chem. 289, 8264–8276. doi: 10.1074/jbc.M113.542779
Maglio, L. E., Martins, V. R., Izquierdo, I., and Ramirez, O. A. (2006). Role of cellular prion protein on LTP expression in aged mice. Brain Res. 1097, 11–18. doi: 10.1016/j.brainres.2006.04.056
Maroteaux, L., and Scheller, R. H. (1991). The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res. Mol. Brain Res. 11, 335–343. doi: 10.1016/0169-328X(91)90043-W
Marzec, M., Eletto, D., and Argon, Y. (2012). GRP94: an HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta 1823, 774–787. doi: 10.1016/j.bbamcr.2011.10.013
Matsumoto, M., and Fujimoto, H. (1990). Cloning of a hsp70-related gene expressed in mouse spermatids. Biochem. Biophys. Res. Commun. 166, 43–49. doi: 10.1016/0006-291X(90)91909-C
Mayer, M. P., Schroder, H., Rudiger, S., Paal, K., Laufen, T., and Bukau, B. (2000). Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat. Struct. Biol. 7, 586–593. doi: 10.1038/76819
McClellan, A. J., Xia, Y., Deutschbauer, A. M., Davis, R. W., Gerstein, M., and Frydman, J. (2007). Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 131, 121–135. doi: 10.1016/j.cell.2007.07.036
McDonough, H., and Patterson, C. (2003). CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones 8, 303–308.
McLaughlin, M., and Vandenbroeck, K. (2011). The endoplasmic reticulum protein folding factory and its chaperones: new targets for drug discovery? Br. J. Pharmacol. 162, 328–345. doi: 10.1111/j.1476-5381.2010.01064.x
McLean, P. J., Kawamata, H., Shariff, S., Hewett, J., Sharma, N., Ueda, K., et al. (2002). TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. J. Neurochem. 83, 846–854. doi: 10.1046/j.1471-4159.2002.01190.x
McLean, P. J., Klucken, J., Shin, Y., and Hyman, B. T. (2004). Geldanamycin induces Hsp70 and prevents alpha-synuclein aggregation and toxicity in vitro. Biochem. Biophys. Res. Commun. 321, 665–669. doi: 10.1016/j.bbrc.2004.07.021
Meacham, G. C., Patterson, C., Zhang, W., Younger, J. M., and Cyr, D. M. (2001). The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 3, 100–105. doi: 10.1038/35050509
Melnyk, A., Rieger, H., and Zimmermann, R. (2015). Co-chaperones of the mammalian endoplasmic reticulum. Subcell. Biochem. 78, 179–200. doi: 10.1007/978-3-319-11731-7_9
Meyer, P., Prodromou, C., Hu, B., Vaughan, C., Roe, S. M., Panaretou, B., et al. (2003). Structural and functional analysis of the middle segment of hsp90: implications for ATP hydrolysis and client protein and cochaperone interactions. Mol. Cell 11, 647–658. doi: 10.1016/S1097-2765(03)00065-0
Miller, J., Arrasate, M., Shaby, B. A., Mitra, S., Masliah, E., and Finkbeiner, S. (2010). Quantitative relationships between huntingtin levels, polyglutamine length, inclusion body formation, and neuronal death provide novel insight into Huntington's disease molecular pathogenesis. J. Neurosci. 30, 10541–10550. doi: 10.1523/JNEUROSCI.0146-10.2010
Misselwitz, B., Staeck, O., and Rapoport, T. A. (1998). J proteins catalytically activate Hsp70 molecules to trap a wide range of peptide sequences. Mol. Cell 2, 593–603. doi: 10.1016/S1097-2765(00)80158-6
Morgner, N., Schmidt, C., Beilsten-Edmands, V., Ebong, I.-o., Patel, N. A., Clerico, E. M., et al. (2015). Hsp70 forms antiparallel dimers stabilized by post-translational modifications to position clients for transfer to Hsp90. Cell Rep. 11, 759–769. doi: 10.1016/j.celrep.2015.03.063
Morimoto, N., Nagai, M., Ohta, Y., Miyazaki, K., Kurata, T., Morimoto, M., et al. (2007). Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1167, 112–117. doi: 10.1016/j.brainres.2007.06.045
Morimoto, R. I. (1998). Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 12, 3788–3796. doi: 10.1101/gad.12.24.3788
Moulick, K., Ahn, J. H., Zong, H., Rodina, A., Cerchietti, L., Gomes DaGama, E. M., et al. (2011). Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat. Chem. Biol. 7, 818–826. doi: 10.1038/nchembio.670
Muchowski, P. J., and Wacker, J. L. (2005). Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 6, 11–22. doi: 10.1038/nrn1587
Mucke, L., and Selkoe, D. J. (2012). Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2:a006338. doi: 10.1101/cshperspect.a006338
Muller, P., Hrstka, R., Coomber, D., Lane, D. P., and Vojtesek, B. (2008). Chaperone-dependent stabilization and degradation of p53 mutants. Oncogene 27, 3371–3383. doi: 10.1038/sj.onc.1211010
Nagata, E., Sawa, A., Ross, C. A., and Snyder, S. H. (2004). Autophagosome-like vacuole formation in Huntington's disease lymphoblasts. Neuroreport 15, 1325–1328. doi: 10.1097/01.wnr.0000127073.66692.8f
Nah, J., Yuan, J., and Jung, Y. K. (2015). Autophagy in neurodegenerative diseases: from mechanism to therapeutic approach. Mol. Cells 38, 381–389. doi: 10.14348/molcells.2015.0034
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nicolet, C. M., and Craig, E. A. (1989). Isolation and characterization of STI1, a stress-inducible gene from Saccharomyces cerevisiae. Mol. Cell. Biol. 9, 3638–3646. doi: 10.1128/MCB.9.9.3638
O'Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer's disease. Annu. Rev. Neurosci. 34, 185–204. doi: 10.1146/annurev-neuro-061010-113613
Ostapchenko, V. G., Beraldo, F. H., Mohammad, A. H., Xie, Y. F., Hirata, P. H., Magalhaes, A. C., et al. (2013). The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-beta oligomer toxicity. J. Neurosci. 33, 16552–16564. doi: 10.1523/JNEUROSCI.3214-13.2013
Ou, S. H., Wu, F., Harrich, D., Garcia-Martinez, L. F., and Gaynor, R. B. (1995). Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 69, 3584–3596.
Outeiro, T. F., Putcha, P., Tetzlaff, J. E., Spoelgen, R., Koker, M., Carvalho, F., et al. (2008). Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS ONE 3:e1867. doi: 10.1371/annotation/9282f173-df82-4b70-9120-b4e62b3dacb1
Paez-Colasante, X., Figueroa-Romero, C., Sakowski, S. A., Goutman, S. A., and Feldman, E. L. (2015). Amyotrophic lateral sclerosis: mechanisms and therapeutics in the epigenomic era. Nat. Rev. Neurol. 11, 266–279. doi: 10.1038/nrneurol.2015.57
Parsell, D. A., Kowal, A. S., Singer, M. A., and Lindquist, S. (1994). Protein disaggregation mediated by heat-shock protein Hsp104. Nature 372, 475–478. doi: 10.1038/372475a0
Pearl, L. H., and Prodromou, C. (2006). Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem. 75, 271–294. doi: 10.1146/annurev.biochem.75.103004.142738
Perez, N., Sugar, J., Charya, S., Johnson, G., Merril, C., Bierer, L., et al. (1991). Increased synthesis and accumulation of heat shock 70 proteins in Alzheimer's disease. Brain Res. Mol. Brain Res. 11, 249–254. doi: 10.1016/0169-328X(91)90033-T
Petrucelli, L., Dickson, D., Kehoe, K., Taylor, J., Snyder, H., Grover, A., et al. (2004). CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 13, 703–714. doi: 10.1093/hmg/ddh083
Pickart, C. M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. doi: 10.1146/annurev.biochem.70.1.503
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Huelga, S. C., Moran, J., Liang, T. Y., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468. doi: 10.1038/nn.2779
Prodromou, C., Roe, S. M., O'Brien, R., Ladbury, J. E., Piper, P. W., and Pearl, L. H. (1997). Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90, 65–75. doi: 10.1016/S0092-8674(00)80314-1
Prodromou, C., Siligardi, G., O'Brien, R., Woolfson, D. N., Regan, L., Panaretou, B., et al. (1999). Regulation of Hsp90 ATPase activity by tetratricopeptide repeat (TPR)-domain co-chaperones. EMBO J. 18, 754–762. doi: 10.1093/emboj/18.3.754
Putcha, P., Danzer, K. M., Kranich, L. R., Scott, A., Silinski, M., Mabbett, S., et al. (2010). Brain-permeable small-molecule inhibitors of Hsp90 prevent alpha-synuclein oligomer formation and rescue alpha-synuclein-induced toxicity. J. Pharmacol. Exp. Ther. 332, 849–857. doi: 10.1124/jpet.109.158436
Pyerin, W. (1994). Human casein kinase II: structures, genes, expression and requirement in cell growth stimulation. Adv. Enzyme Regul. 34, 225–246. doi: 10.1016/0065-2571(94)90018-3
Qi, L., Zhang, X. D., Wu, J. C., Lin, F., Wang, J., DiFiglia, M., et al. (2012). The role of chaperone-mediated autophagy in huntingtin degradation. PLoS ONE 7:e46834. doi: 10.1371/journal.pone.0046834
Rampelt, H., Kirstein-Miles, J., Nillegoda, N. B., Chi, K., Scholz, S. R., Morimoto, R. I., et al. (2012). Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 31, 4221–4235. doi: 10.1038/emboj.2012.264
Rehn, A. B., and Buchner, J. (2015). p23 and Aha1. Subcell. Biochem. 78, 113–131. doi: 10.1007/978-3-319-11731-7_6
Reidy, M., and Masison, D. C. (2010). Sti1 regulation of Hsp70 and Hsp90 is critical for curing of Saccharomyces cerevisiae [PSI+] prions by Hsp104. Mol. Cell. Biol. 30, 3542–3552. doi: 10.1128/MCB.01292-09
Renkawek, K., Bosman, G. J., and de Jong, W. W. (1994). Expression of small heat-shock protein hsp 27 in reactive gliosis in Alzheimer disease and other types of dementia. Acta Neuropathol. 87, 511–519. doi: 10.1007/BF00294178
Richter, K., Soroka, J., Skalniak, L., Leskovar, A., Hessling, M., Reinstein, J., et al. (2008). Conserved conformational changes in the ATPase cycle of human Hsp90. J. Biol. Chem. 283, 17757–17765. doi: 10.1074/jbc.M800540200
Riek, R., Hornemann, S., Wider, G., Billeter, M., Glockshuber, R., and Wuthrich, K. (1996). NMR structure of the mouse prion protein domain PrP(121-231). Nature 382, 180–182. doi: 10.1038/382180a0
Riek, R., Hornemann, S., Wider, G., Glockshuber, R., and Wuthrich, K. (1997). NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett. 413, 282–288. doi: 10.1016/S0014-5793(97)00920-4
Ritossa, F. (1962). A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia 18, 571–573. doi: 10.1007/BF02172188
Rodina, A., Wang, T., Yan, P., Gomes, E. D., Dunphy, M. P., Pillarsetty, N., et al. (2016). The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538, 397–401. doi: 10.1038/nature19807
Roe, S. M., Prodromou, C., O'Brien, R., Ladbury, J. E., Piper, P. W., and Pearl, L. H. (1999). Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 42, 260–266. doi: 10.1021/jm980403y
Roffe, M., Beraldo, F. H., Bester, R., Nunziante, M., Bach, C., Mancini, G., et al. (2010). Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc. Natl. Acad. Sci. U.S.A. 107, 13147–13152. doi: 10.1073/pnas.1000784107
Rohl, A., Tippel, F., Bender, E., Schmid, A. B., Richter, K., Madl, T., et al. (2015a). Hop/Sti1 phosphorylation inhibits its co-chaperone function. EMBO Rep. 16, 240–249. doi: 10.15252/embr.201439198
Rohl, A., Wengler, D., Madl, T., Lagleder, S., Tippel, F., Herrmann, M., et al. (2015b). Hsp90 regulates the dynamics of its cochaperone Sti1 and the transfer of Hsp70 between modules. Nat. Commun. 6, 6655. doi: 10.1038/ncomms7655
Roodveldt, C., Bertoncini, C. W., Andersson, A., van der Goot, A. T., Hsu, S.-T., Fernandez-Montesinos, R., et al. (2009). Chaperone proteostasis in Parkinson's disease: stabilization of the Hsp70/α-synuclein complex by Hip. EMBO J. 28, 3758–3770. doi: 10.1038/emboj.2009.298
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rudiger, S., Germeroth, L., Schneider-Mergener, J., and Bukau, B. (1997). Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 16, 1501–1507. doi: 10.1093/emboj/16.7.1501
Ryan, S. D., Whitehead, S. N., Swayne, L. A., Moffat, T. C., Hou, W., Ethier, M., et al. (2009). Amyloid-beta42 signals tau hyperphosphorylation and compromises neuronal viability by disrupting alkylacylglycerophosphocholine metabolism. Proc. Natl. Acad. Sci. U.S.A. 106, 20936–20941. doi: 10.1073/pnas.0905654106
Sanchez, Y., Taulien, J., Borkovich, K. A., and Lindquist, S. (1992). Hsp104 is required for tolerance to many forms of stress. EMBO J. 11, 2357–2364.
Santos, T. G., Silva, I. R., Costa-Silva, B., Lepique, A. P., Martins, V. R., and Lopes, M. H. (2011). Enhanced neural progenitor/stem cells self-renewal via the interaction of stress-inducible protein 1 with the prion protein. Stem Cells 29, 1126–1136. doi: 10.1002/stem.664
Scheufler, C., Brinker, A., Bourenkov, G., Pegoraro, S., Moroder, L., Bartunik, H., et al. (2000). Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101, 199–210. doi: 10.1016/S0092-8674(00)80830-2
Schlecht, R., Erbse, A. H., Bukau, B., and Mayer, M. P. (2011). Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat. Struct. Mol. Biol. 18, 345–351. doi: 10.1038/nsmb.2006
Schmid, A. B., Lagleder, S., Grawert, M. A., Rohl, A., Hagn, F., Wandinger, S. K., et al. (2012). The architecture of functional modules in the Hsp90 co-chaperone Sti1/Hop. EMBO J. 31, 1506–1517. doi: 10.1038/emboj.2011.472
Selkoe, D. J. (1991). The molecular pathology of Alzheimer's disease. Neuron 6, 487–498. doi: 10.1016/0896-6273(91)90052-2
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Sharma, M., Burre, J., and Sudhof, T. C. (2012). Proteasome inhibition alleviates SNARE-dependent neurodegeneration. Sci. Transl. Med. 4:147ra113. doi: 10.1126/scitranslmed.3004028
Shimura, H., Miura-Shimura, Y., and Kosik, K. S. (2004). Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J. Biol. Chem. 279, 17957–17962. doi: 10.1074/jbc.M400351200
Shorter, J. (2011). The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 6:e26319. doi: 10.1371/journal.pone.0026319
Sidera, K., and Patsavoudi, E. (2014). HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat. Anticancer. Drug Discov. 9, 1–20. doi: 10.2174/15748928113089990031
Small, D. H., Clarris, H. L., Williamson, T. G., Reed, G., Key, B., Mok, S. S., et al. (1999). Neurite-outgrowth regulating functions of the amyloid protein precursor of Alzheimer's disease. J. Alzheimers. Dis. 1, 275–285. doi: 10.3233/JAD-1999-14-508
Smith, D. F. (2004). Tetratricopeptide repeat cochaperones in steroid receptor complexes. Cell Stress Chaperones 9, 109–121. doi: 10.1379/CSC-31.1
Soares, I. N., Caetano, F. A., Pinder, J., Rodrigues, B. R., Beraldo, F. H., Ostapchenko, V. G., et al. (2013). Regulation of stress-inducible phosphoprotein 1 nuclear retention by protein inhibitor of activated STAT PIAS1. Mol. Cell. Proteomics 12, 3253–3270. doi: 10.1074/mcp.M113.031005
Song, H. O., Lee, W., An, K., Lee, H. S., Cho, J. H., Park, Z. Y., et al. (2009). C. elegans STI-1, the homolog of Sti1/Hop, is involved in aging and stress response. J. Mol. Biol. 390, 604–617. doi: 10.1016/j.jmb.2009.05.035
Soto, C., and Castilla, J. (2004). The controversial protein-only hypothesis of prion propagation. Nat. Med. 10(Suppl.), S63–S67. doi: 10.1038/nm1069
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Swain, J. F., Dinler, G., Sivendran, R., Montgomery, D. L., Stotz, M., and Gierasch, L. M. (2007). Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol. Cell 26, 27–39. doi: 10.1016/j.molcel.2007.02.020
Taipale, M., Jarosz, D. F., and Lindquist, S. (2010). HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 11, 515–528. doi: 10.1038/nrm2918
Tamagno, E., Robino, G., Obbili, A., Bardini, P., Aragno, M., Parola, M., et al. (2003). H2O2 and 4-hydroxynonenal mediate amyloid beta-induced neuronal apoptosis by activating JNKs and p38MAPK. Exp. Neurol. 180, 144–155. doi: 10.1016/S0014-4886(02)00059-6
Tamguney, G., Giles, K., Glidden, D. V., Lessard, P., Wille, H., Tremblay, P., et al. (2008). Genes contributing to prion pathogenesis. J. Gen. Virol. 89(Pt 7), 1777–1788. doi: 10.1099/vir.0.2008/001255-0
Tampellini, D., Rahman, N., Gallo, E. F., Huang, Z., Dumont, M., Capetillo-Zarate, E., et al. (2009). Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J. Neurosci. 29, 9704–9713. doi: 10.1523/JNEUROSCI.2292-09.2009
Tissieres, A., Mitchell, H. K., and Tracy, U. M. (1974). Protein synthesis in salivary glands of Drosophila melanogaster: relation to chromosome puffs. J. Mol. Biol. 84, 389–398. doi: 10.1016/0022-2836(74)90447-1
Tobler, I., Gaus, S. E., Deboer, T., Achermann, P., Fischer, M., Rulicke, T., et al. (1996). Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 380, 639–642. doi: 10.1038/380639a0
Tremblay, M. A., Acker, C. M., and Davies, P. (2010). Tau phosphorylated at tyrosine 394 is found in Alzheimer's disease tangles and can be a product of the Abl-related kinase, Arg. J. Alzheimers. Dis. 19, 721–733. doi: 10.3233/JAD-2010-1271
Trendowski, M. (2015). PU-H71: an improvement on nature's solutions to oncogenic Hsp90 addiction. Pharmacol. Res. 99, 202–216. doi: 10.1016/j.phrs.2015.06.007
Trepel, J., Mollapour, M., Giaccone, G., and Neckers, L. (2010). Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10, 537–549. doi: 10.1038/nrc2887
Trojanowski, J. Q., Goedert, M., Iwatsubo, T., and Lee, V. M. (1998). Fatal attractions: abnormal protein aggregation and neuron death in Parkinson's disease and Lewy body dementia. Cell Death Differ. 5, 832–837. doi: 10.1038/sj.cdd.4400432
Tsai, J., and Douglas, M. G. (1996). A conserved HPD sequence of the J-domain is necessary for YDJ1 stimulation of Hsp70 ATPase activity at a site distinct from substrate binding. J. Biol. Chem. 271, 9347–9354. doi: 10.1074/jbc.271.16.9347
Udan-Johns, M., Bengoechea, R., Bell, S., Shao, J., Diamond, M. I., True, H. L., et al. (2014). Prion-like nuclear aggregation of TDP-43 during heat shock is regulated by HSP40/70 chaperones. Hum. Mol. Genet. 23, 157–170. doi: 10.1093/hmg/ddt408
Ueda, K., Fukushima, H., Masliah, E., Xia, Y., Iwai, A., Yoshimoto, M., et al. (1993). Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 11282–11286. doi: 10.1073/pnas.90.23.11282
Um, J. W., Kaufman, A. C., Kostylev, M., Heiss, J. K., Stagi, M., Takahashi, H., et al. (2013). Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79, 887–902. doi: 10.1016/j.neuron.2013.06.036
Um, J. W., Nygaard, H. B., Heiss, J. K., Kostylev, M. A., Stagi, M., Vortmeyer, A., et al. (2012). Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235. doi: 10.1038/nn.3178
Umbach, J. A., and Gundersen, C. B. (1997). Evidence that cysteine string proteins regulate an early step in the Ca2+-dependent secretion of neurotransmitter at Drosophila neuromuscular junctions. J. Neurosci. 17, 7203–7209.
Uryu, K., Richter-Landsberg, C., Welch, W., Sun, E., Goldbaum, O., Norris, E. H., et al. (2006). Convergence of heat shock protein 90 with ubiquitin in filamentous alpha-synuclein inclusions of alpha-synucleinopathies. Am. J. Pathol. 168, 947–961. doi: 10.2353/ajpath.2006.050770
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Vaughan, C. K., Neckers, L., and Piper, P. W. (2010). Understanding of the Hsp90 molecular chaperone reaches new heights. Nat. Struct. Mol. Biol. 17, 1400–1404. doi: 10.1038/nsmb1210-1400
Vetrivel, K. S., Kodam, A., Gong, P., Chen, Y., Parent, A. T., Kar, S., et al. (2008). Localization and regional distribution of p23/TMP21 in the brain. Neurobiol. Dis. 32, 37–49. doi: 10.1016/j.nbd.2008.06.012
Wacker, J. L., Huang, S. Y., Steele, A. D., Aron, R., Lotz, G. P., Nguyen, Q., et al. (2009). Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington's disease. J. Neurosci. 29, 9104–9114. doi: 10.1523/JNEUROSCI.2250-09.2009
Wacker, J. L., Zareie, M. H., Fong, H., Sarikaya, M., and Muchowski, P. J. (2004). Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat. Struct. Mol. Biol. 11, 1215–1222. doi: 10.1038/nsmb860
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi: 10.1038/416535a
Wang, B., Liu, Y., Huang, L., Chen, J., Li, J. J., Wang, R., et al. (2016). A CNS-permeable Hsp90 inhibitor rescues synaptic dysfunction and memory loss in APP-overexpressing Alzheimer's mouse model via an HSF1-mediated mechanism. Mol. Psychiatry. doi: 10.1038/mp.2016.104. [Epub ahead of print].
Wang, J., Farr, G. W., Zeiss, C. J., Rodriguez-Gil, D. J., Wilson, J. H., Furtak, K., et al. (2009). Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc. Natl. Acad. Sci. U.S.A. 106, 1392–1397. doi: 10.1073/pnas.0813045106
Wang, J., Ho, L., Qin, W., Rocher, A. B., Seror, I., Humala, N., et al. (2005). Caloric restriction attenuates beta-amyloid neuropathology in a mouse model of Alzheimer's disease. FASEB J. 19, 659–661. doi: 10.1096/fj.04-3182fje
Wang, J., You, H., Qi, J., Yang, C., Ren, Y., and Cheng, H. (2017). Autocrine and paracrine STIP1 signaling promote osteolytic bone metastasis in renal cell carcinoma. Oncotarget 8, 17012–17026. doi: 10.18632/oncotarget.15222
Wang, T. H., Chao, A., Tsai, C. L., Chang, C. L., Chen, S. H., Lee, Y. S., et al. (2010). Stress-induced phosphoprotein 1 as a secreted biomarker for human ovarian cancer promotes cancer cell proliferation. Mol. Cell. Proteomics 9, 1873–1884. doi: 10.1074/mcp.M110.000802
Wang, Y., Martinez-Vicente, M., Kruger, U., Kaushik, S., Wong, E., Mandelkow, E. M., et al. (2009). Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet. 18, 4153–4170. doi: 10.1093/hmg/ddp367
Warrick, J. M., Chan, H. Y., Gray-Board, G. L., Chai, Y., Paulson, H. L., and Bonini, N. M. (1999). Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 23, 425–428. doi: 10.1038/70532
Weingarten, M. D., Lockwood, A. H., Hwo, S. Y., and Kirschner, M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862. doi: 10.1073/pnas.72.5.1858
Weng, N., Baumler, M. D., Thomas, D. D., Falkowski, M. A., Swayne, L. A., Braun, J. E., et al. (2009). Functional role of J domain of cysteine string protein in Ca2+-dependent secretion from acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G1030–G1039. doi: 10.1152/ajpgi.90592.2008
Will, R. G., and Ironside, J. W. (1999). Oral infection by the bovine spongiform encephalopathy prion. Proc. Natl. Acad. Sci. U.S.A. 96, 4738–4739. doi: 10.1073/pnas.96.9.4738
Williams, A. J., and Paulson, H. L. (2008). Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci. 31, 521–528. doi: 10.1016/j.tins.2008.07.004
Wilson, A. C., Dugger, B. N., Dickson, D. W., and Wang, D. S. (2011). TDP-43 in aging and Alzheimer's disease - a review. Int. J. Clin. Exp. Pathol. 4, 147–155. Available online at: http://www.ijcep.com/files/IJCEP1010002.pdf
Wisniewski, T., and Drummond, E. (2016). Developing therapeutic vaccines against Alzheimer's disease. Expert Rev. Vaccines 15, 401–415. doi: 10.1586/14760584.2016.1121815
Wolfe, K. J., Ren, H. Y., Trepte, P., and Cyr, D. M. (2013). The Hsp70/90 cochaperone, Sti1, suppresses proteotoxicity by regulating spatial quality control of amyloid-like proteins. Mol. Biol. Cell 24, 3588–3602. doi: 10.1091/mbc.E13-06-0315
Workman, P., Burrows, F., Neckers, L., and Rosen, N. (2007). Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann. N.Y. Acad. Sci. 1113, 202–216. doi: 10.1196/annals.1391.012
Xilouri, M., Brekk, O. R., Landeck, N., Pitychoutis, P. M., Papasilekas, T., Papadopoulou-Daifoti, Z., et al. (2013). Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain 136(Pt 7), 2130–2146. doi: 10.1093/brain/awt131
Yerbury, J. J., Ooi, L., Dillin, A., Saunders, D. N., Hatters, D. M., Beart, P. M., et al. (2016). Walking the tightrope: proteostasis and neurodegenerative disease. J. Neurochem. 137, 489–505. doi: 10.1111/jnc.13575
Yi, F., and Regan, L. (2008). A novel class of small molecule inhibitors of Hsp90. ACS Chem. Biol. 3, 645–654. doi: 10.1021/cb800162x
Young, J. C., Obermann, W. M., and Hartl, F. U. (1998). Specific binding of tetratricopeptide repeat proteins to the C-terminal 12-kDa domain of hsp90. J. Biol. Chem. 273, 18007–18010. doi: 10.1074/jbc.273.29.18007
Yu, W. H., Cuervo, A. M., Kumar, A., Peterhoff, C. M., Schmidt, S. D., Lee, J. H., et al. (2005). Macroautophagy–a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J. Cell Biol. 171, 87–98. doi: 10.1083/jcb.200505082
Yuan, P., and Grutzendler, J. (2016). Attenuation of beta-amyloid deposition and neurotoxicity by chemogenetic modulation of neural activity. J. Neurosci. 36, 632–641. doi: 10.1523/JNEUROSCI.2531-15.2016
Zanata, S. M., Lopes, M. H., Mercadante, A. F., Hajj, G. N., Chiarini, L. B., Nomizo, R., et al. (2002). Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 21, 3307–3316. doi: 10.1093/emboj/cdf325
Zhang, M., Windheim, M., Roe, S. M., Peggie, M., Cohen, P., Prodromou, C., et al. (2005). Chaperoned ubiquitylation–crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol. Cell 20, 525–538. doi: 10.1016/j.molcel.2005.09.023
Zhang, Y. J., Gendron, T. F., Xu, Y. F., Ko, L. W., Yen, S. H., and Petrucelli, L. (2010). Phosphorylation regulates proteasomal-mediated degradation and solubility of TAR DNA binding protein-43 C-terminal fragments. Mol. Neurodegener. 5:33. doi: 10.1186/1750-1326-5-33
Zhao, R., Davey, M., Hsu, Y. C., Kaplanek, P., Tong, A., Parsons, A. B., et al. (2005). Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell 120, 715–727. doi: 10.1016/j.cell.2004.12.024
Keywords: STIP1, HOP, Alzheimer's disease, tau, ALS, Parkinson's disease, Huntington's disease, TDP-43
Citation: Lackie RE, Maciejewski A, Ostapchenko VG, Marques-Lopes J, Choy W-Y, Duennwald ML, Prado VF and Prado MAM (2017) The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 11:254. doi: 10.3389/fnins.2017.00254
Received: 02 February 2017; Accepted: 20 April 2017;
Published: 16 May 2017.
Edited by:
Cintia Roodveldt, Centro Andaluz de Biología Molecular y Medicina Regenerativa, SpainReviewed by:
Toshihide Takeuchi, Osaka University, JapanCopyright © 2017 Lackie, Maciejewski, Ostapchenko, Marques-Lopes, Choy, Duennwald, Prado and Prado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco A. M. Prado, bXByYWRvQHJvYmFydHMuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.