 Anna Chiarini
Anna Chiarini Ubaldo Armato
Ubaldo Armato Emanuela Gardenal
Emanuela Gardenal Li Gui
Li Gui Ilaria Dal Prà
Ilaria Dal Prà- 1Human Histology and Embryology Unit, Medical School, University of Verona, Verona, Venetia, Italy
- 2Department of Neurology, Southwest Hospital, Third Military Medical University, Chongqing, China
The two main drivers of Alzheimer's disease (AD), amyloid-β (Aβ) and hyperphosphorylated Tau (p-Tau) oligomers, cooperatively accelerate AD progression, but a hot debate is still ongoing about which of the two appears first. Here we present preliminary evidence showing that Tau and p-Tau are expressed by untransformed cortical adult human astrocytes in culture and that exposure of such cells to an Aβ42 proxy, Aβ25−35, which binds the calcium-sensing receptor (CaSR) and activates its signaling, significantly increases intracellular p-Tau levels, an effect CaSR antagonist (calcilytic) NPS 2143 wholly hinders. The astrocytes also release both Tau and p-Tau by means of exosomes into the extracellular medium, an activity that could mediate p-Tau diffusion within the brain. Preliminary data also indicate that exosomal levels of p-Tau increase after Aβ25−35 exposure, but remain unchanged in cells pre-treated for 30-min with NPS 2143 before adding Aβ25−35. Thus, our previous and present findings raise the unifying prospect that Aβ•CaSR signaling plays a crucial role in AD development and progression by simultaneously activating (i) the amyloidogenic processing of amyloid precursor holoprotein, whose upshot is a surplus production and secretion of Aβ42 oligomers, and (ii) the GSK-3β-mediated increased production of p-Tau oligomers which are next released extracellularly inside exosomes. Therefore, as calcilytics suppress both effects on Aβ42 and p-Tau metabolic handling, these highly selective antagonists of pathological Aβ•CaSR signaling would effectively halt AD's progressive spread preserving patients' cognition and life quality.
Introduction
Late onset (non-familial) Alzheimer's disease (LOAD) is the most common dementia afflicting millions of people worldwide. It is characterized by extracellular deposits of fibrillar Aβ42 peptides (neuritic or senile plaques) and by intracellular pre-tangles and neurofibrillary tangles (NFTs) of phosphorylated Tau (p-Tau) protein (Selkoe, 2008a,b; Grinberg et al., 2009; Braak et al., 2011; Attems et al., 2012; Elobeid et al., 2012; Braak and Del Tredici, 2013). LOAD neuropathology develops stealthily during 20–40 years before its clinical emergence (Masdeu et al., 2012). It is thought to be driven by the tandem toxic activities of oligomers of amyloid β (Aβ-os) and p-Tau (p-Tau-os) let out from affected cell processes via exocytosis and/or exosomes (or extracellular vesicles) (Saman et al., 2012). Both released Aβ-os and p-Tau-os thus reach adjacent or connected cells, inducing them to release in their turn newly produced Aβ-os and p-Tau-os. Thus, LOAD spreads from entorhinal cortex layer II to upper cognitive cortical areas killing unreplaceable neurons and disconnecting their networks in its path (Morrison and Hof, 1997; Selkoe, 2008a,b; Khan et al., 2014). Notably, p-Tau can be neurotoxic all by itself too in advanced AD and in tauopathies caused by mechanisms independent of Aβ-os or senile plaques (Medeiros et al., 2013). Which of the two main AD toxic drivers appears first is controversial. According to some, a very early surfacing and spread of intraneuronal p-Tau pathology (i.e., pre-tangles, NFTs, and neuropil threads) from the brainstem to the cerebral cortex occurs in the total absence of extra-neuronal Aβ42 accumulation (Braak et al., 2013; see also below). However, others hold that poorly detectable soluble Aβ42-os are the earliest LOAD drivers (Selkoe, 2008a,b; Crimins et al., 2013; Kayed and Lasagna-Reeves, 2013), bringing about p-Tau-os, NFTs, and synaptic pathology in the total absence of senile plaques (reviewed by Klein, 2013). Indeed, the para-hippocampal and inferior temporal gyri of 8-year-old Down's syndrome children already exhibited Aβ deposits (Leverenz and Raskind, 1998). In fact, they had a chromosome 21 tri-ploidy and three copies of the Aβ precursor holoprotein (hAPP) gene which made them susceptible to develop an early AD neuropathology. In long-term in vitro three-dimensional cultures of neural cells, Aβ-os build-up preceded any p-Tau-os detection further strengthening the view Aβ-os are the first AD drivers (Choi et al., 2014) while also stressing the usefulness of preclinical in vitro models to elucidate molecular mechanisms underlying AD development.
Accordingly, p-Tau-os seem to occupy the second tier in the hierarchy of AD drivers (Clavaguera et al., 2009, 2013a,b; Gerson and Kayed, 2013). Under physiological conditions, Tau is a soluble microtubule-associated phosphoprotein (MAP) strongly expressed in neurons (Goedert, 1993) and human astrocytes (Ferrer et al., 2002; Tanji et al., 2003; Wakabayashi et al., 2006, and present results). Tau moiety encompasses a microtubule-binding C-terminal repeat domain, a central proline-rich domain, and an N-terminal domain interacting with membranes and/or other proteins. In human adult brain, an alternatively spliced single gene allows the expression of six Tau isoforms, of which 4RTau and 3RTau are the most intensely produced and phosphorylated ones (Hanger et al., 1998; Hasegawa, 2006). Soluble Tau monomers are physiologically gathered within neurons' axons where they tightly bind, stabilize, and help elongate microtubules, besides associating with the plasma membrane (Pooler and Hanger, 2010). They partake in the fast anterograde transport (FAT) of various cargos (e.g., mitochondria, synaptic vesicles) on kinesin motors linked to microtubule trackways. Tau is rapidly and reversibly phosphorylated by several protein kinases and phosphatases. Soluble Tau purified from normal human brains is phosphorylated at about 10 sites only (Hanger et al., 2007; Sergeant et al., 2008). Yet, Tau is endowed with 85 serine and threonine phosphorylable sites, and glycogen synthase kinase (GSK)-3β is the main kinase for 45 of them in poorly soluble p-Tau (Buée et al., 2000; Sergeant et al., 2008; Tavares et al., 2013). When GSK-3β hyper-phosphorylates Tau, the latter's ability to promote normal microtubule assembly wanes (Utton et al., 1997). Then p-Tau detaches from tubulin, destabilizing and disassembling microtubules (Lindwall and Cole, 1984; Drechsel et al., 1992). Hence, increases in p-Tau due to GSK-3β activity surges are typical marks of blunted physiological functions (e.g., axonal transport, etc.) in neurons (LaPointe et al., 2009). In AD and various tauopathies, p-Tau accumulates intracellularly as filaments, pre-tangles, and insoluble NFTs, and hyper-reacts to anti-p-Tau-specific antibodies (Greenberg and Davies, 1990; Ballatore et al., 2007; Gendron and Petrucelli, 2009). Not surprisingly, GSK-3β colocalizes with NFTs in AD and AD-related disorders (Hanger et al., 1998; Ferrer et al., 2002; Hanger and Noble, 2011). Notably, p-Tau from AD brains coimmunoprecipitates with a fraction of Tau, revealing that AD's p-Tau-os are Tau/p-Tau mixtures (Köpke et al., 1993) just as AD's Tau filaments or fibrils are (Alonso et al., 1997). The pathological role of GSK-3β-phosphorylated Tau is supported by results in mouse transgenic AD or tauopathy models, in which GSK-3β inhibition lessened Tau phosphorylation and aggregation and axonal degeneration (Serenó et al., 2009; Leroy et al., 2010). And, SB-415286, a specific inhibitor of GSK-3β activity, decreased p-Tau levels and kept cultured primary neurons viable (Gross et al., 2001).
According to Braak et al. (2011) and Braak and Del Tredici (2012, 2013), abnormal p-Tau-os in non-fibrillar form were seen within proximal axons and AT-8 antibody-positive pre-tangles were observed within the somata and dendrites of projection neurons of brainstem locus coeruleus/ subcoeruleus of young boys well before they became manifest in the hippocampal trans-entorhinal region, the putative site of AD onset (Khan et al., 2014). The authors posited AD begins from brainstem neurons which inject neurotoxic p-Tau-os into higher cortical regions (Hertz, 1989; Agnati et al., 1995)—a process starting AD's “Braak stages” (Braak et al., 2011; Braak and Del Tredici, 2012, 2013). Concurrently, others set forth the concept of trans-synaptically transmittable, prion-like, soluble Tau-os which by destroying first synapses, then axons, and finally neurons would disconnect neuronal networks (Clavaguera et al., 2009, 2013a,b; Lasagna-Reeves et al., 2012; de Calignon et al., 2012; Gerson and Kayed, 2013). However, p-Tau-os cannot cross synaptic terminals as prions do (Stranahan and Mattson, 2010).
Hitherto, as with Aβs, neurons were held as the main source of Tau/p-Tau (Wu et al., 2013; Avila et al., 2014). But what about other neural cell types? Wakabayashi et al. (2006) reported the co-localization of Aβ and p-Tau in the subiculum and entorhinal cortex astrocytes of a patient with corticobasal degeneration. They interpreted this finding as follows: “the phagocytosis of Aβ coincides with production of phospho-Tau in the same reactive astrocytes.” However, as we previously showed, untreated cortical untransformed adult human astrocytes produce basal amounts of Aβ42 and, once challenged with exogenous fibrillar (f)Aβ25−35, an Aβ42 proxy, make and release significantly greater amounts of endogenous Aβ42/Aβ42-os (Armato et al., 2013; Dal Prà et al., 2015; Chiarini et al., 2016). We also demonstrated exogenous fAβ25−35-os bind the astrocytes' and neurons' calcium-sensing receptors (CaSRs) (Dal Prà et al., 2014a,b) and activate their signaling pathways heightening the production and secretion of endogenous Aβ42/Aβ42-os. In fact, a specific CaSR agonist (calcimimetic), NPS R-568 (Nemeth and Goodman, 2016), mimicked the enhancing effect of exogenous fAβ25−35-os on Aβ42/Aβ42-os secretion (Armato et al., 2013). Conversely, a highly selective CaSR antagonist (calcilytic), NPS 2143 (Nemeth and Goodman, 2016), fully quelled the fAβ25−35-os-induced surplus de novo production and secretion of Aβ42/Aβ42-os in both human neurons and astrocytes (Armato et al., 2013; Dal Prà et al., 2015; Chiarini et al., 2016). Interestingly, human MIC neuroblasts secrete Tau enclosed within exosomes which are found in human cerebrospinal fluid too (Saman et al., 2012). Even plasma astrocyte-derived exosomes contain p-Tau proteins (Goetzl et al., 2016). And, neurons uptake exogenous Tau proteins via endocytosis into the somatodentritic compartments or axon termini from which they are conveyed to various cell sites (Wu et al., 2013). Altogether, these data indicated the urgent need to reassess the relationship between Aβ peptides exposure and p-Tau production and release in adult human astrocytes and neurons. Therefore, we undertook a pilot study using as model cultured human astrocytes (Armato et al., 2013) whose preliminary results we herein report. Details on materials and methods we used are in Supplementary Materials.
Results
Aβ•CaSR Signaling Increases GSK-3β Tau Kinase Activity in Normal Adult Human Astrocytes
An ongoing balance between phosphorylation and de-phosphorylation of some of its serine (Ser) and tyrosine (Tyr) residues controls GSK-3β enzymatic activity: relative increases in Tyr216 phosphorylation upregulate and, conversely, of Ser9 phosphorylation downregulate it (Forde and Dale, 2007) heightening or reducing, respectively, p-Tau levels (Qian et al., 2010). In human adult astrocyte lysates an exposure to exogenous fAβ25−35 nearly doubles between 0 and 48-h the p-Tyr216GSK-3β/total GSK-3 ratio values (Figures 1A,B) while simultaneously curtailing p-Ser9GSK-3β/total GSK-3 ratio values (Figures 1A,C). As a consequence, the p-Tyr216/p-Ser9 ratio values and hence GSK-3β activity increase up to 8-fold in fAβ25−35-exposed astrocytes (Figure 1D) as the latter does in hippocampal neurons (Takashima et al., 1998). Remarkably, a 30 min pre-treatment with calcilytic NPS 2143 totally quells the raise in fAβ25−35-induced p-Tyr216GSK-3β levels; contrariwise, the p-Tyr216GSK-3β/total GSK-3 ratio values fall below control values (Figures 1A,B). Concurrently, NPS 2143 increases the p-Ser9GSK-3β/total GSK-3 ratio values well above control ones (Figures 1A,C). As a result, the p-Tyr216/p-Ser9 ratio values and hence activity levels fall below basal values (Figure 1D). These results constitute the first evidence that pathological Aβ•CaSR signaling directly intensifies GSK-3β activity besides rising endogenous Aβ42/Aβ42-os production/release from the cortical adult human astrocytes.
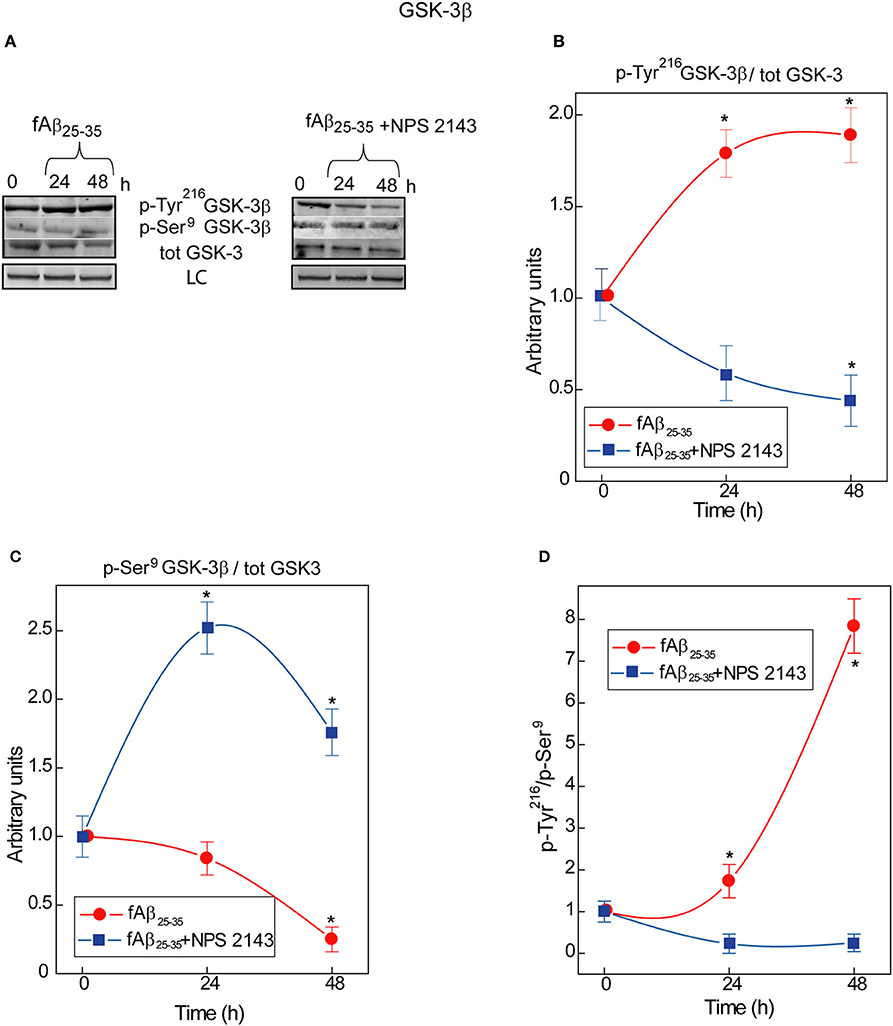
Figure 1. Time course of GSK-3β phosphorylations in human adult astrocytes exposed to fAβ25−35 ±NPS 2143. (A) Typical immunoblots of human astrocytes total protein lysates illustrating the changes in the specific bands corresponding to p-Tyr216GSK-3β, p-Ser9GSK-3β, and total (tot) GSK-3 total in untreated (control) cells and in cells exposed to fAβ25−35± a short (30-min) treatment with calcilytic NPS 2143. LC, loading control (lamin B1). (B) p-Tyr216GSK-3β/GSK-3 ratio increased in fAβ25−35 (20 μM)-treated cells (red line), an effect a short NPS 2143 pre-treatment completely prevented (blue line). (C) p-Ser9GSK-3β/GSK-3 ratio decreased under the stimulus of fAβ25−35 alone whereas it increased when of fAβ25−35 administration was preceded by a 30-min pre-treatment with calcilytic NPS 2143 (blue line). (D) As indicated by the augmented pTyr216GSK-3β/pSer9GSK-3β ratio, the activity of GSK-3β hugely increased in the astrocytes exposed to exogenous fAβ25−35 alone (red line), but was significantly downregulated when calcilytic NPS 2143 was given for 30-min before fAβ25−35 to the astrocyte cultures (blue line). Points in the curves express the mean ratios between the specific phosphorylated sites and total GSK-3 ± SEMs from 3 distinct experiments. *P < 0.01 vs. control (0-h) values.
Expression of Tau Protein Isoforms in Adult Human Astrocytes
First, we examined Tau proteins expression in astrocytes by means of immunofluorescence staining and observed a diffuse granular Tau-immunoreactivity pattern mostly in the cytoplasm (Figure 2A). Cytoplasmic granular Tau aggregates were previously reported (Ward et al., 2013). Then, to identify the several Tau isoforms involved, we analyzed via Western blotting whole astrocyte lysates from both untreated and fAβ25−35-treated cells (Figure 2B). We used a pan-Tau antibody which recognizes all Tau isoforms (Tran et al., 2011), and confirmed the identity of each specific isoform band by using a commercially available Tau protein ladder composed of the six known Tau isoforms. Thus, in both untreated and fAβ25−35-treated astrocytes, the pan-Tau antibody recognized three resolvable Tau bands in the size range between 45 and 60 kDa, corresponding to Tau isoforms 2N4R, 1N3R, and 0N4R, which are those involved in the formation of pre-tangles and NFTs (Espinoza et al., 2008). Total Tau levels were alike in untreated and fAβ25−35-treated cultures, suggesting no changes in total Tau isoforms expression were elicited by fAβ25−35-exposure vs. no treatment in the astrocytes at least during 72-h of treatment.
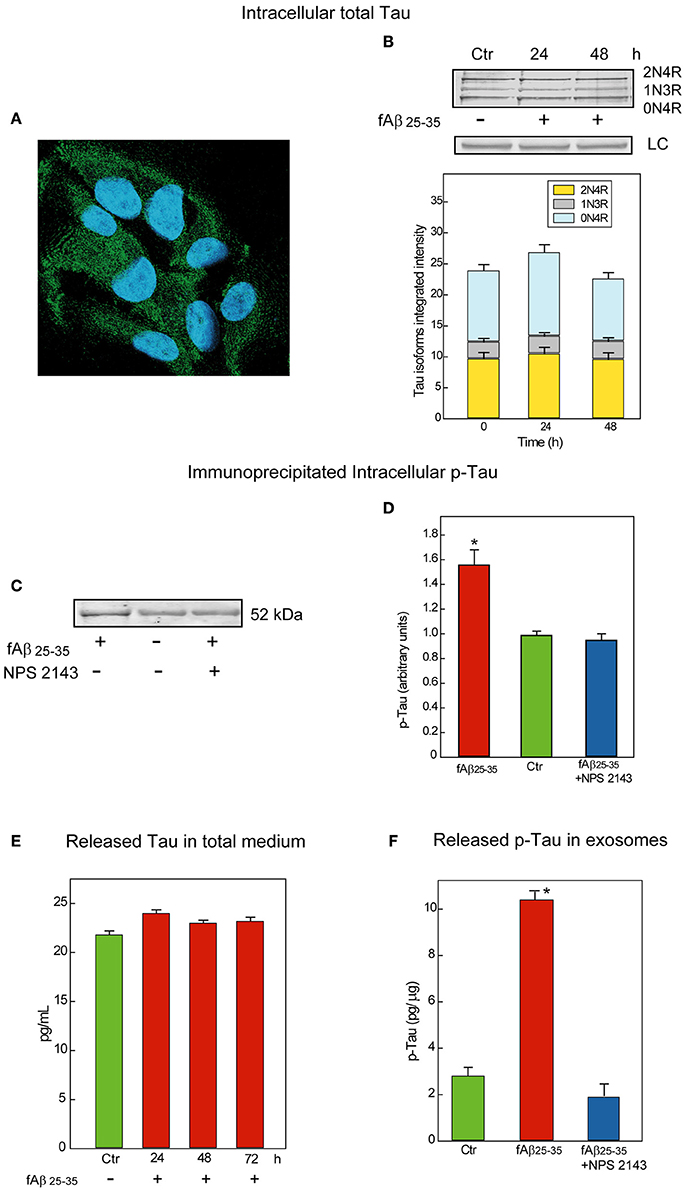
Figure 2. Characterization and release of Tau/p-Tau from human adult astrocytes. (A) Immunofluorescence staining of total Tau (antibody HT7) in untreated astrocytes as a diffuse granular green labeling of the cytoplasms. Nuclei are stained with DAPI. Merged picture. Magnification, 640X. (B) Top: Typical immunoblot analysis of the Tau isoforms astrocytes express when untreated (Ctr) or exposed for 24 or 48-h to fAβ25−35 alone treatment. Notably, 1N3R, 0N4R, and 2N4R are known as the Tau isoforms involved in the formation of pre-tangles and NFTs (Espinoza et al., 2008). LC, load controls (lamin B1). Bottom: Densitometric assessment of the three Tau isoforms integrated intensities. No significant changes are detectable (n = 3). (C) Typical immunoblot analysis of immunoprecipitated p-Tau in lysates from untreated and 48-h fAβ25−35±NPS 2143-treated astrocytes. Adding NPS 2143 pretreatment totally blocks any increase in p-Tau levels elicited fAβ25−35 alone which remain at basal values. (D) Densitometric data corresponding to p-Tau specific bands are shown as bars which are the means ± SEMs expressed as arbitrary units (n = 3), and normalized taking as 1.0 the values of untreated astrocytes. *P < 0.001 vs. control values. (E) Time course of Tau protein release in the growth medium of un-treated and fAβ25−35-exposed astrocytes. The ELISA values of total Tau protein detected in 24 to 72-h astrocytes treated with fAβ fAβ25−35 do not significantly differ from control ones at each time point examined. Bars are the mean values ± SEMs of three experiments in duplicate. (F) p-Tau is released in exosomes under physiological conditions and its amount remarkably increases in fAβ25−35 treated astrocytes, but adding NPS 2143 for 30 min prior to exposing astrocytes to fAβ25−35 prevents any increase in exosomal p-Tau from occurring. Bars are means ± SEMs of three experiments in duplicate. *P < 0.001 vs. control values and vs. fAβ25−35±NPS 2143-treated values.
fAβ25−35-Treated Astrocytes Have Increased Intracellular P-Tau Levels NPS 2143 Suppresses
Via Kinex Antibody Microarray™ we analyzed the p-Tau pattern in total cellular lysates of untreated and fAβ25−35-treated astrocytes. Using phospho-site-specific antibodies this analysis tracked the main Tau phospho-sites regulated by GSK-3β activity (Hanger and Noble, 2011) and demonstrated that the phosphorylation levels of Ser199, Ser396, and Ser422 of the Tau molecule were remarkably increased in 24-h fAβ25−35−exposed astrocytes (not shown). This preliminary evaluation invited further investigations in order to specifically establish the amount of increased p-Tau proteins.
Therefore, we first immunoprecipitated the phosphorylated proteins from whole lysates of untreated and fAβ25−35-exposed astrocytes—the latter pre-treated or not pre-treated with calcilytic NPS 2143 since, as we just saw, Aβ•CaSR-signaling regulates GSK-3β activity (Figure 1). Next, we probed the immunoblots of the immunoprecipitated total phospho-proteins with a specific anti-Tau antibody. As shown in Figure 2C, the levels of p-Tau markedly increased in the fAβ25−35-treated astrocytes as compared to untreated cells. Importantly, NPS 2143 pre-treatment kept p-Tau at physiological (untreated control) levels in the fAβ25−35-exposed cells (Figures 2C,D).
Exosome-Associated Tau and P-Tau Releases From Human Adult Astrocytes
Untreated and Aβ-exposed cortical adult human astrocytes also release Tau proteins into the growth medium. In preliminary experiments, by using an ELISA assay with a sensitivity <10 pg/mL we found total Tau protein levels of 21.8 pg/mL in 72-h untreated (control) astrocytes medium samples. In the medium of 24–72-h fAβ25−35-treated astrocytes, we detected unchanging values of the total Tau proteins which did not differ from control ones, suggesting the operation of a steady balance between Tau release and Tau re-uptake (Figure 2E).
But, is Tau/p-Tau secreted free into the growth medium or is it enclosed within exosomes? To answer this question, we started analyzing Tau release under physiological conditions. We purified exosomes from media conditioned for 72-h by untreated astrocytes and then quantified Tau by means of a specific ELISA kit in exosome fractions purified from them and in exosome-depleted media samples. This analysis showed Tau proteins associated with the exosome fractions and the exosomal Tau levels did not significantly differ from those found in whole media samples (~24.3 pg/mL). Conversely, under the same conditions, Tau could not be detected at all in exosome-depleted media samples. Therefore, all the Tau human astrocytes release is enclosed within exosomes.
Next, we investigated whether endogenous Tau released from astrocytes within exosomes was phosphorylated. By means of a p-Tau-specific ELISA kit we could demonstrate that under physiological conditions p-Tau secretion occurred inside exosomes too (Figure 2F, Ctr). Finally, we asked whether an exposure to fAβs25−35 ±NPS 2143 affected the amount of p-Tau released via exosomes. Our pilot results (n = 3) hint that this is indeed the case. In fact, using the same p-Tau-specific ELISA kit we observed that exosome-associated p-Tau increased markedly with fAβ25−35-treated astrocytes as compared to untreated ones, but a 30-min pretreatment with NPS 2143 of the fAβ25−35-exposed cells wholly quelled any exosomal p-Tau surge keeping it at controls' levels (Figure 2F). Further in depth studies will validate and extend these pilot findings.
Conclusions and Future Perspectives
AD is a complex human illness which is only partially modeled in rodents. Using as paradigm cultured cortical untransformed adult human astrocytes, which differ from rodents' ones from both morphological and functional standpoints (Ogata and Kosaka, 2002; Tsai et al., 2012; Robertson, 2014) and are not killed by accumulating Aβs (Armato et al., 2013; Dal Prà et al., 2015; Chiarini et al., 2016) has brought to light molecular mechanisms which likely partake in AD's onset and progression. Previous work showed exogenous Aβs bind the plasma membrane CaSRs of human astrocytes and neurons (Dal Prà et al., 2014a,b). The thus triggered pathological Aβ•CaSR signaling increases the amyloidogenic processing of hAPP which entails a surplus extracellular secretion of endogenous Aβ42 from both cell types (Armato et al., 2013; Dal Prà et al., 2015; Chiarini et al., 2016). Additionally, Aβ•CaSR signaling elicits neurotoxic surpluses of nitric oxide and VEGF-A production and release from human astrocytes (Dal Prà et al., 2005, 2014b; Armato et al., 2013). Under these multiple neurotoxic insults human cortical neurons start progressively dying (Armato et al., 2013; Chiarini et al., 2015). And the Aβ42-os accumulating in the neuropil spread to bind and activate the CaSRs of adjacent neurons and astrocytes, thus promoting further Aβ42-os production and diffusion (Dal Prà et al., 2015; Chiarini et al., 2016). Remarkably, a highly selective CaSR antagonist (calcilytic), NPS 2143, effectively blocks the Aβ•CaSR signaling and all of its neurotoxic consequences, preserving human neurons' viability notwithstanding a persisting Aβ-os presence. Thus, from the Aβs standpoint calcilytics would be effective as anti-AD therapeutics (Armato et al., 2013; Dal Prà et al., 2014b, 2015; Chiarini et al., 2015, 2016).
However, we cannot ignore the main drivers of AD are both Aβ-os and p-Tau-os. It has been argued that p-Tau-os advent precedes Aβ42-os' (Braak et al., 2011; Elobeid et al., 2012). But, evidence also exists that Aβ42-os manifestation antecedes p-Tau-os' (Leverenz and Raskind, 1998; Klein, 2013; Choi et al., 2014). Beyond question is only that when both Aβ-os and p-Tau-os are present, AD course toward patient's demise briskly accelerates (Ittner and Gotz, 2011). So how this drivers' antinomy might be solved? Our findings show that besides stimulating the pathological amyloidogenic processing of hAPP into Aβ42, Aβ•CaSR signaling increases the activity of GSK-3β and hence the intracellular accumulation of p-Tau in human astrocytes. Next, mixtures of both Tau and p-Tau are enclosed within exosomes and released into the extracellular environment. In the static in vitro system we used, a balance is kept between release and reuptake of Tau/p-Tau-containing exosomes. In vivo, such Tau/p-Tau-containing exosomes would spread into the neuropil to be uptaken by adjacent neurons and astrocytes. Given astrocytes' higher numbers, a persistent Aβ-elicited exosomal p-Tau overrelease would exacerbate the neurons' toxic accumulation of p-Taues favoring their aggregation into pre-tangles and NFTs.
Therefore, our present results raise the enticing prospect that pathological Aβ•CaSR signaling would simultaneously trigger both the Aβ-mediated and the p-Tau-mediated neurotoxic mechanisms driving AD neuropathology. The other exciting facet of these findings is that calcilytic NPS 2143 can fully suppress all the neurotoxic effects Aβ•CaSR signaling wakes up, including the intracellular accumulation and exosomal release of p-Tau surpluses from human astrocytes. Further work will assess whether calcilytics similarly hinder excess p-Tau production/release from Aβ-exposed human cortical neurons. However, NPS 2143 does suppress the Aβ42 surplus production and secretion from Aβ-exposed human neurons (Armato et al., 2013). Therefore, it seems feasible that NPS 2143 would block neurons' GSK-3β's Tau hyperphosphorylating activity too.
In conclusion, with all the advisable caution our preclinical findings deserve, the present perspective suggests CaSR antagonists would block the intracerebral seeding of both AD main drivers, the Aβs and p-Taues, besides accessory neurotoxic factors like NO and VEGF-A surpluses. Accordingly, if administered early enough, calcilytics would freeze AD progression and preserve patients' ongoing cognitive abilities and quality of life.
Ethics Statement
This research work was approved by the Ethical Committee of the Integrated Verona University-Hospital Co., Prog. No. CE118CESC.
Author Contributions
AC, UA, and IDP conceived the research and designed the experiments. AC, IDP, and EG performed the experiments and collected the results. UA and LG statistically analyzed the data. AC, UA, and IDP interpreted the results. The manuscript was principally written and revised by UA, AC, and IDP. All the authors critically reviewed the manuscript for important intellectual content and approved the final submitted manuscript.
Funding
This work was supported in part by the Italian Ministry for University and Research (F.U.R. 2014 and 2015 allotments to AC, and IDP).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnins.2017.00217/full#supplementary-material
References
Agnati, L. F., Zoli, M., Strömberg, I., and Fuxe, K. (1995). Intercellular communication in the brain: wiring versus volume transmission. Neuroscience 69, 711–726. doi: 10.1016/0306-4522(95)00308-6
Alonso, A. D., Grundke-Iqbal, I., Barra, H. S., and Iqbal, K. (1997). Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. U.S.A. 94, 298–303.
Armato, U., Chiarini, A., Chakravarthy, B., Chioffi, F., Pacchiana, R., Colarusso, E., et al. (2013). Calcium-sensing receptor antagonist (calcilytic) NPS 2143 specifically blocks the increased secretion of endogenous Aβ42 prompted by exogenous fibrillary or soluble Aβ25-35 in human cortical astrocytes and neurons–Therapeutic relevance to Alzheimer's disease. Biochim. Biophys. Acta 1832, 1634–1652. doi: 10.1016/j.bbadis.2013.04.020
Attems, J., Thal, D. R., and Jellinger, K. A. (2012). The relationship between subcortical Tau pathology and Alzheimer's disease. Biochem. Soc. Trans. 40, 711–715. doi: 10.1042/BST20120034
Avila, J., Simón, D., Díaz-Hernández, M., Pintor, J., and Hernández, F. (2014). Sources of extracellular tau and its signaling. J. Alzheimers Dis. 40, S7–S15. doi: 10.3233/JAD-131832
Ballatore, C., Lee, V. M., and Trojanowski, J. Q. (2007). Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 8, 663–672. doi: 10.1038/nrn2194
Braak, H., and Del Tredici, K. (2012). Where, when, and in what form does sporadic Alzheimer's disease begin? Curr. Opin. Neurol. 25, 708–714. doi: 10.1097/WCO.0b013e32835a3432
Braak, H., and Del Tredici, K. (2013). Evolutional aspects of Alzheimer's disease pathogenesis. J. Alzheimers Dis. 33, S155–S161. doi: 10.3233/JAD-2012-129029
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi: 10.1097/NEN.0b013e318232a379
Braak, H., Zetterberg, H., Del Tredici, K., and Blennow, K. (2013). Intraneuronal Tau aggregation precedes diffuse plaque deposition, but amyloid-β changes occur before increases of Tau in cerebrospinal fluid. Acta Neuropathol. 126, 631–641. doi: 10.1007/s00401-013-1139-0
Buée, L., Bussière, T., Buée-Scherrer, V., Delacourte, A., and Hof, P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33, 95–130. doi: 10.1016/S0165-0173(00)00019-9
Chiarini, A., Armato, U., Liu, D., and Dal Prà, I. (2016). Calcium-sensing receptors of human neural cells play crucial roles in Alzheimer's disease. Front. Physiol. 7:134. doi: 10.3389/fphys.2016.00134
Chiarini, A., Gardenal, E., Whitfield, J. F., Chakravarthy, B., Armato, U., and Dal Pra, I. (2015). Preventing the spread of Alzheimer's disease neuropathology: a role for calcilytics? Curr. Pharm. Biotechnol. 16, 696–706. doi: 10.2174/1389201016666150505123813
Choi, S. H., Kim, Y. H., Hebisch, M., Sliwinski, C., Lee, S., D'Avanzo, C., et al. (2014). A three-dimensional human neural cell culture model of Alzheimer's disease. Nature 515, 274–278. doi: 10.1038/nature13800
Clavaguera, F., Akatsu, H., Fraser, G., Crowther, R. A., Frank, S., Hench, J., et al. (2013a). Brain homogenates from human tauopathies induce Tau inclusions in mouse brain. Proc. Natl. Acad. Sci. U.S.A. 110, 9535–9540. doi: 10.1073/pnas.1301175110
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell. Biol. 11, 909–913. doi: 10.1038/ncb1901
Clavaguera, F., Lavenir, I., Falcon, B., Frank, S., Goedert, M., and Tolnay, M. (2013b). “Prion-like” templated misfolding in tauopathies. Brain Pathol. 23, 342–349. doi: 10.1111/bpa.12044
Crimins, J. L., Pooler, A., Polydoro, M., Luebke, J. I., and Spires-Jones, T. L. (2013). The intersection of amyloid beta and Tau in glutamatergic synaptic dysfunction and collapse in Alzheimer's disease. Ageing Res. Rev. 12, 757–763. doi: 10.1016/j.arr.2013.03.002
Dal Prà, I., Armato, U., Chioffi, F., Pacchiana, R., Whitfield, J. F., Chakravarthy, B., et al. (2014b). The Aβ peptides-activated calcium-sensing receptor stimulates the production and secretion of vascular endothelial growth factor-A by normoxic adult human cortical astrocytes. Neuromolecular Med. 16, 645–657. doi: 10.1007/s12017-014-8315-9
Dal Prà, I., Chiarini, A., Gui, L., Chakravarthy, B., Pacchiana, R., Gardenal, E., et al. (2015). Do astrocytes collaborate with neurons in spreading the “infectious” Aβ and Tau drivers of Alzheimer's disease? Neuroscientist 21, 9–29. doi: 10.1177/1073858414529828
Dal Prà, I., Chiarini, A., Nemeth, E. F., Armato, U., and Whitfield, J. F. (2005). Roles of Ca2+ and the Ca2+-sensing receptor (CASR) in the expression of inducible NOS (nitric oxide synthase)-2 and its BH4 (tetrahydrobiopterin)-dependent activation in cytokine-stimulated adult human astrocyes. J. Cell. Biochem. 96, 428–438. doi: 10.1002/jcb.20511
Dal Prà, I., Chiarini, A., Pacchiana, R., Gardenal, E., Chakravarthy, B., Whitfield, J. F., et al. (2014a). Calcium-sensing receptors of human astrocyte-neuron teams: amyloid-β-driven mediators and therapeutic targets of Alzheimer's disease Curr. Neuropharmacol. 12, 353–364. doi: 10.2174/1570159X12666140828214701
de Calignon, A., Polydoro, M., Suarez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., et al. (2012). Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697. doi: 10.1016/j.neuron.2011.11.033
Drechsel, D. N., Hyman, A. A., Cobb, M. H., and Kirschner, M. W. (1992). Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell. 3, 1141–1154. doi: 10.1091/mbc.3.10.1141
Elobeid, A., Soininen, H., and Alafuzoff, I. (2012). Hyperphosphorylated Tau in young and middle-aged subjects. Acta Neuropathol. 123, 97–104. doi: 10.1007/s00401-011-0906-z
Espinoza, M., de Silva, R., Dickson, D. W., and Davies, P. (2008). Differential incorporation of tau isoforms in Alzheimer's disease. J. Alzheimers Dis. 14, 1–16. doi: 10.3233/JAD-2008-14101
Ferrer, I., Barrachina, M., and Puig, B. (2002). Anti-tau phospho-specific Ser262 antibody recognizes a variety of abnormal hyperphosphorylated tau deposits in tauopathies including Pick bodies and argyrophilic grains. Acta Neuropathol. 104, 658–664. doi: 10.1007/s00401-002-0600-2
Forde, J. E., and Dale, T. C. (2007). Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol. Life Sci. 64, 1930–1944. doi: 10.1007/s00018-007-7045-7
Gendron, T. F., and Petrucelli, L. (2009). The role of tau in neurodegeneration. Mol. Neurodegener. 4:13. doi: 10.1186/1750-1326-4-13
Gerson, J. E., and Kayed, R. (2013). Formation and propagation of tau oligomeric seeds. Front. Neurol. 4:93. doi: 10.3389/fneur.2013.00093
Goedert, M. (1993). Tau protein and the neurofibrillary pathology of Alzheimer's disease. Trends Neurosci. 16, 460–465.
Goetzl, E. J., Mustapic, M., Kapogiannis, D., Eitan, E., Lobach, I. V., Goetzl, L., et al. (2016). Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer's disease. FASEB J. 30, 3853–3859. doi: 10.1096/fj.201600756R
Greenberg, S. G., and Davies, P. (1990). A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc. Natl. Acad. Sci. U.S.A. 87, 5827–5831.
Grinberg, L. T., Rueb, U., Ferretti, R. E., Nitrini, R., Farfel, J. M., Polichiso, L., et al. (2009). The dorsal raphe nucleus shows phospho-Tau neurofibrillary changes before the transentorhinal region in Alzheimer's disease. A precocious onset? Neuropathol. Appl. Neurobiol. 35, 406–416. doi: 10.1111/j.1365-2990.2008.00997.x
Gross, D. A., Culbert, A. A., Chalmers, K. A., Facci, L., Skaper, S. D., and Reith, A. D. (2001). Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J. Neurochem. 77, 94–102. doi: 10.1046/j.1471-4159.2001.00251.x
Hanger, D. P., Betts, J. C., Loviny, T. L., Blackstock, W. P., and Anderton, B. H. (1998). New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer's disease brain using nanoelectrospray mass spectrometry. J. Neurochem. 71, 2465–2476. doi: 10.1046/j.1471-4159.1998.71062465.x
Hanger, D. P., Byers, H. L., Wray, S., Leung, K. Y., Saxton, M. J., Seereeram, A., et al. (2007). Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J. Biol. Chem. 282, 23645–23654. doi: 10.1074/jbc.M703269200
Hanger, D. P., and Noble, W. (2011). Functional implications of glycogen synthase kinase-3-mediated tau phosphorylation. Int. J. Alzheimers Dis. 2011:352805. doi: 10.4061/2011/352805
Hasegawa, M. (2006). Biochemistry and molecular biology of tauopathies. Neuropathology 26, 484–490. doi: 10.1111/j.1440-1789.2006.00666.x
Hertz, L. (1989). Is Alzheimer's disease an anterograde degeneration, originating in the brainstem, and disrupting metabolic and functional interactions between neurons and glial cells? Brain Res. Brain Res. Rev. 14, 335–353.
Ittner, L. M., and Gotz, J. (2011). Amyloid-β and Tau—a toxic pas de deux in Alzheimer's disease. Nat. Rev. Neurosci. 12, 65–72. doi: 10.1038/nrn2967
Kayed, R., and Lasagna-Reeves, C. A. (2013). Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimers Dis. 33, S67–S78. doi: 10.3233/JAD-2012-129001
Khan, U. A., Liu, L., Provenzano, F. A., Berman, D. E., Profaci, C. P., Sloan, R., et al. (2014). Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer's disease. Nat. Neurosci. 17, 304–311. doi: 10.1038/nn.3606
Klein, W. L. (2013). Synaptotoxic amyloid-β oligomers: a molecular basis for the cause, diagnosis, and treatment of Alzheimer's disease? J. Alzheimers Dis. 33, S49–S65. doi: 10.3233/JAD-2012-129039
Köpke, E., Tung, Y. C., Shaikh, S., Alonso, A. C., Iqbal, K., and Grundke-Iqbal, I. (1993). Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 268, 24374–24384.
LaPointe, N. E., Morfini, G., Pigino, G., Gaisina, I. N., Kozikowski, A. P., Binder, L. I., et al. (2009). The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J. Neurosci. Res. 87, 440–451. doi: 10.1002/jnr.21850
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Guerrero-Munoz, M. J., Kiritoshi, T., Neugebauer, V., et al. (2012). Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2:700. doi: 10.1038/srep00700
Leroy, K., Ando, K., Héraud, C., Yilmaz, Z., Authelet, M., Boeynaems, J. M., et al. (2010). Lithium treatment arrests the development of neurofibrillary tangles in mutant tau transgenic mice with advanced neurofibrillary pathology. J. Alzheimers Dis. 19, 705–719. doi: 10.3233/JAD-2010-1276
Leverenz, J. B., and Raskind, M. A. (1998). Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: a regional quantitative analysis. Exp. Neurol. 150, 296–304. doi: 10.1006/exnr.1997.6777
Lindwall, G., and Cole, R. D. (1984). Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 259, 5301–5305.
Masdeu, J. C., Kreisl, W. C., and Berman, K. F. (2012). The neurobiology of Alzheimer disease defined by neuroimaging. Curr. Opin. Neurol. 25, 410–420. doi: 10.1097/WCO.0b013e3283557b36
Medeiros, R., Chabrier, M. A., and LaFerla, F. M. (2013). Elucidating the triggers, progression, and effects of Alzheimer's disease. J. Alzheimers Dis. 33, S195–S210. doi: 10.3233/JAD-2012-129009
Morrison, J. H., and Hof, P. R. (1997). Life and death of neurons in the aging brain. Science 278, 412–419. doi: 10.1126/science.278.5337.412
Nemeth, E. F., and Goodman, W. G. (2016). Calcimimetic and calcilytic drugs: feats, flops, and futures. Calcif. Tissue Int. 98, 341–358. doi: 10.1007/s00223-015-0052-z
Ogata, K., and Kosaka, T. (2002). Structural and quantitative analysis of astrocytes in the mouse hippocampus. Neuroscience 113, 221–233. doi: 10.1016/S0306-4522(02)00041-6
Pooler, A. M., and Hanger, D. P. (2010). Functional implications of the association of tau with the plasma membrane. Biochem. Soc. Trans. 38, 1012–1015. doi: 10.1042/BST0381012
Qian, W., Shi, J., Yin, X., Iqbal, K., Grundke-Iqbal, I., Gong, C. X., et al. (2010). PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3beta. J. Alzheimers Dis. 19, 1221–1229. doi: 10.3233/JAD-2010-1317
Robertson, J. M. (2014). Astrocytes and the evolution of the human brain. Med. Hypotheses. 82, 236–239. doi: 10.1016/j.mehy.2013.12.004
Saman, S., Kim, W., Raya, M., Visnick, Y., Miro, S., Saman, S., et al. (2012). Exosome-associated Tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849. doi: 10.1074/jbc.M111.277061
Selkoe, D. J. (2008a). Biochemistry and molecular biology of amyloid beta-protein and the mechanism of Alzheimer's disease. Handb. Clin. Neurol. 89, 245–260. doi: 10.1016/S0072-9752(07)01223-7
Selkoe, D. J. (2008b). Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav. Brain. Res. 192, 106–113. doi: 10.1016/j.bbr.2008.02.016
Serenó, L., Coma, M., Rodríguez, M., Sánchez-Ferrer, P., Sánche, M. B., Gich, I., et al. (2009). A novel GSK-3beta inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 35, 359–367. doi: 10.1016/j.nbd.2009.05.025
Sergeant, N., Bretteville, A., Hamdane, M., Caillet-Boudin, M. L., Grognet, P., Bomboi, S., et al. (2008). Biochemistry of Tau in Alzheimer's disease and related neurological disorders. Expert Rev. Proteomics. 5, 207–224. doi: 10.1586/14789450.5.2.207
Stranahan, A. M., and Mattson, M. P. (2010). Selective vulnerability of neurons in layer II of the entorhinal cortex during aging and Alzheimer's disease. Neural Plast. 2010:108190. doi: 10.1155/2010/108190
Takashima, A., Honda, T., Yasutake, K., Michel, G., Murayama, O., Murayama, M., et al. (1998). Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25-35) enhances phosphorylation of tau in hippocampal neurons. Neurosci. Res. 31, 317–323. doi: 10.1016/S0168-0102(98)00061-3
Tanji, K., Mori, F., Imaizumi, T., Yoshida, H., Satoh, K., and Wakabayashi, K. (2003). Interleukin-1 induces tau phosphorylation and morphological changes in cultured human astrocytes. Neuroreport 14, 413–417. doi: 10.1097/01.wnr.0000059783.23521.7c
Tavares, I. A., Touma, D., Lynham, S., Troakes, C., Schober, M., Causevic, M., et al. (2013). Prostate-derived sterile 20-like kinases (PSKs/TAOKs) phosphorylate tau protein and are activated in tangle-bearing neurons in Alzheimer disease. J. Biol. Chem. 288, 15418–15429. doi: 10.1074/jbc.M112.448183
Tran, T. H., LaFerla, F. M., Holtzman, D. M., and Brody, D. L. (2011). Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-β accumulation and independently accelerates the development of tau abnormalities. J. Neurosci. 31, 9513–9525. doi: 10.1523/JNEUROSCI.0858-11.2011
Tsai, H. H., Li, H., Fuentealba, L. C., Molofsky, A. V., Taveira-Marques, R., Zhuang, H., et al. (2012). Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science 337, 358–362. doi: 10.1126/science.1222381
Utton, M. A., Vandecandelaere, A., Wagner, U., Reynolds, C. H., Gibb, G. M., Miller, C. C., et al. (1997). Phosphorylation of tau by glycogen synthase kinase 3beta affects the ability of tau to promote microtubule self-assembly. Biochem J. 323 (Pt 3), 741–747. doi: 10.1042/bj3230741
Wakabayashi, K., Mori, F., Hasegawa, M., Kusumi, T., Yoshimura, I., Takahashi, H., et al. (2006). Co-localization of beta-peptide and phosphorylated tau in astrocytes in a patient with corticobasal degeneration. Neuropathology. 26, 66–71. doi: 10.1111/j.1440-1789.2006.00635.x
Ward, S. M., Himmelstein, D. S., Lancia, J. K., Fu, Y., Patterson, K. R., and Binder, L. I. (2013). TOC1: characterization of a selective oligomeric tau antibody. J. Alzheimers Dis. 37, 593–602. doi: 10.3233/JAD-131235
Keywords: amyloid-β, Tau, adult human astrocytes, exosome, calcium-sensing receptor, calcilytics, Alzheimer's disease, GSK-3β
Citation: Chiarini A, Armato U, Gardenal E, Gui L and Dal Prà I (2017) Amyloid β-Exposed Human Astrocytes Overproduce Phospho-Tau and Overrelease It within Exosomes, Effects Suppressed by Calcilytic NPS 2143—Further Implications for Alzheimer's Therapy. Front. Neurosci. 11:217. doi: 10.3389/fnins.2017.00217
Received: 27 December 2016; Accepted: 31 March 2017;
Published: 20 April 2017.
Edited by:
Diana K. Sarko, Southern Illinois University Carbondale, USAReviewed by:
Alberto Granzotto, CeSI-MeT - Centro Scienze dell'Invecchiamento e Medicina Traslazionale, ItalyAlexey P. Bolshakov, Institute of Higher Nervous Activity and Neurophysiology, Russia
Copyright © 2017 Chiarini, Armato, Gardenal, Gui and Dal Prà. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Chiarini, YW5jaGlhcmlAZ21haWwuY29t
Ilaria Dal Prà, aXBwZGFscHJhQGdtYWlsLmNvbQ==