 Xi Wang
Xi Wang Xiao-Gang Zhang1†
Xiao-Gang Zhang1† Shao Li
Shao Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurosci., 09 March 2016
Sec. Neurodegeneration
Volume 10 - 2016 | https://doi.org/10.3389/fnins.2016.00094
This article is part of the Research TopicMolecular Diagnostics in the Detection of Neurodegenerative DisordersView all 10 articles
Aberrant increases in neuronal network excitability may contribute to the cognitive deficits in Alzheimer's disease (AD). However, the mechanisms underlying hyperexcitability are not fully understood. Such overexcitation of neuronal networks has been detected in the brains of APP/PS1 mice. In the present study, using current-clamp recording techniques, we observed that 12 days in vitro (DIV) primary cultured pyramidal neurons from P0 APP/PS1 mice exhibited a more prominent action potential burst and a lower threshold than WT littermates. Moreover, after treatment with Aβ1−42 peptide, 12 DIV primary cultured neurons showed similar changes, to a greater degree than in controls. Voltage-clamp recordings revealed that the voltage-dependent sodium current density of neurons incubated with Aβ1−42 was significantly increased, without change in the voltage-dependent sodium channel kinetic characteristics. Immunohistochemistry and western blot results showed that, after treatment with Aβ1−42, expressions of Nav and Nav1.6 subtype increased in cultured neurons or APP/PS1 brains compared to control groups. The intrinsic neuronal hyperexcitability of APP/PS1 mice might thus be due to an increased expression of voltage-dependent sodium channels induced by Aβ1−42. These results may illuminate the mechanism of aberrant neuronal networks in AD.
Alzheimer's disease (AD) is the most frequent neurodegenerative disease and a common cause of dementia in elderly individuals. Various evidence suggests, that β-amyloid (Aβ) peptides play a causal role in AD's pathogenesis, but the underlying mechanisms remain unclear (Palop and Mucke, 2010). In prodromal AD patients, functional MRI has revealed increased activities in neural networks, rather than loss of activity (Putcha et al., 2011). Other evidence supports the view, that Aβ-induced excitotoxicity could be critically involved in the pathogenesis of AD (Ong et al., 2013). In Aβ-induced excitotoxicity, high levels of glutamate overexcite neurons and cause cell death (Choi, 1988; Olney et al., 1997). Aβ also enhances the sensitivity of neuron to glutamate, which increases the activity of neuronal networks, resulting in excitatory potentials and Ca2+ influx (Brorson et al., 1995). However, these changes cannot explain patients with early AD frequently alternating between sober and confused, because synaptic disruption, or regeneration cannot repeatedly occur within such short time periods. This phenomenon is more likely due to the abnormal neural network excitability.
Voltage-gated sodium channels (Nav) play an essential role in excitable cells. They are necessary components required to generate and propagate action potentials (Goldin et al., 2000; Yu and Catterall, 2003; Catterall et al., 2005). The 260 kDa α subunit is the main component of the voltage-gated sodium channel. Nine α subtypes, named Nav1.1-Nav1.9, are expressed in excitable cells (Goldin et al., 2000; Ragsdale, 2008). Among them, the Nav1.1, Nav1.2, and Nav1.6 subtypes are expressed in the adult brain and regulate voltage-dependent sodium currents across the plasma membrane. Nav1.1 is primarily localized in the neuronal somata of GABAergic neurons (Yu et al., 2006; Ogiwara et al., 2007). Nav1.2 shows preferentially high expression in unmyelinated fibers (Ragsdale, 2008). The Nav1.6 subtype, encoded by the gene SCN8A, is conspicuously expressed at the nodes of Ranvier and axon initial segments (Trimmer and Rhodes, 2004). Unique features of Nav1.6 include its contribution to the persistent current, resurgent current, and repetitive neuronal firing.
Our previous results showed that Nav1.6 interacts with amyloid precursor protein (APP), which undergoes abnormal proteolytic processing to generate Aβ (Xu et al., 2014). APP also increases the surface expression of sodium channels through a Go protein-coupled JNK pathway (Liu et al., 2015). Due to this effect of APP, we hypothesized that the upregulatory effect of Aβ on neuronal excitability might be partially based on modulating the expression of sodium channels. As we expected, in cultured cortical neurons, Aβ1−42 increased the expression of sodium channels, particularly the Nav1.6 subtype. The increased voltage-dependent sodium current could decrease action potential threshold and increase the probability of action potential generation in response to synaptic excitation, thus increasing the excitability of the neuron.
Newborn C57BL/6J mice were obtained from the Animal Center of Dalian Medical University. APP/PS1 transgenic mice were purchased from Jackson Laboratory (stock number 004462) and were maintained on a C57BL/6J background by crossing heterozygous transgenic mice with C57BL/6J breeders. All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize the number of animals used and their suffering.
Lyophilized Aβ1−42 peptide (SIGMA, A9810) and reverse peptide Aβ42−1 (SIGMA, SCP0048) were diluted to 1 mg/ml using sterile PBS and incubated at 37°C, 220 rpm for 48 h allowed to aggregate as described (Jones et al., 2013). For concrete forms of Aβ1−42 peptide, see Supplementary Figure S1. In a further set of experiments primary cultured neurons were incubated with Aβ1−42 (5.0 μM) or reverse peptide Aβ42−1 (5.0 μM) for 24 h at 37°C.
P0 (post-natal day 0–1) C57BL/6J mice or APP/PS1 mice and their littermates were sacrificed by CO2 inhalation and the cortices were rapidly dissected under sterile conditions in cold PBS. The tissue was digested with 0.125% trypsin-EDTA at 37°C for 30 min. The trypsin solution was replaced with 2 ml 10% DMEM (DMEM with 10% FBS, 1% L-glutamine, and 1% penicillin/streptomycin solution). The digested tissue was gently triturated by suction using a glass pipette flamed on the tip to avoid cellular damage. The cell suspension was filtered through a 74 μm screen mesh, plated on poly-L-lysine-coated coverslips or 6/12-well-plates and incubated in a 37°C, 5% CO2 incubator. After 2 h, the medium was changed to Neurobasal (Gibco, 21103–049) supplemented with 2% B27, 1% L-glutamine, and 1% penicillin/streptomycin. In accordance with the routine culture, the medium was changed every 2 days until use.
Cell viability of neurons was determined by the MTT assay. Neurons were plated in 96 well-plates. Aβ42−1 and Aβ1−42 peptides were added to wells for 24 h. After cell treatments the medium was removed and the cells were incubated with red free medium and MTT solution (0.5 mg/ml) for 4 h at 37°C. Finally, the medium was removed and formazan particles were dissolved in DMSO. Cell viability, defined as the relative amount of MTT reduction was determined by spectrophotometry at 570 nm.
Electrophysiological measurements were performed on pyramidal cells. Action potentials or sodium currents were recorded at room temperature using whole-cell patch-clampings. The extracellular solution contained the following (in mM): NaCl 150, KCl 5, MgCl2 1.1, CaCl2 2.6, HEPES 10, glucose 10, pH adjusted to 7.4 with NaOH. Patch pipettes were made from borosilicate glass capillaries (1.5 mm outer diameter, 0.8 mm inner diameter) using a micropipette puller (Narishige, PP 830, Japan). Pipette resistance ranged from 3 to 5 MΩ. Stimulation and data acquisition were performed using the EPC-10 patch-clamp amplifier and Pulse program (HEKA Electronik, Germany). Membrane currents were filtered at 2 kHz and digitized at 10 kHz.
Action potentials recordings were made using the current-clamp mode. The intracellular solution contained the following (in mM): KCl 65, KOH 5.0, KF 80, HEPES 10, EGTA 10, Na2ATP 2, pH adjusted to 7.2 with KOH. Cells were held at −70 mV, then peak amplitude (80 pA, 10 ms), threshold (80 pA, 10 ms), and action potential firing (200 pA, 500 ms) were recorded and measured.
Sodium currents were recorded using the voltage-clamp mode. The intracellular solution contained the following (in mM): CsCl 140, MgCl2 2, Na2ATP 2, EGTA 10, HEPES 20, pH adjusted to 7.2 with Tris-HCl. Cells were held at −70 mV and stepped to a range of potentials (−60 to +60 mV in 10 mV increments) for 12 ms each. Peak inward currents (I) were plotted as a function of depolarizing potential to generate I–V curves. Activation curves were obtained by converting current (I) to conductance (G) at each voltage (V) using the equation G = I/(V − Vrev), where Vrev is the reversal potential, which was determined for each cell individually. Activation curves were then fit with the Boltzmann function in the form of G/Gmax = 1/{1 + exp [(V1∕2 − V)/κ]}, where Gmax is the maximal sodium conductance, V1∕2 is the half-maximal activation potential, V is the test potential, and κ is the slope factor. Steady-state fast inactivation was achieved with a series of 500 ms prepulses (−120 to −10 mV in 10 mV increments), and the remaining available channels were activated by a 12 ms test pulse to 0 mV. Peak inward currents obtained from steady-state fast-inactivation protocols were normalized to the maximal peak current (Imax) and fit with Boltzmann functions: I/Imax = 1/{1 + exp [(V − V1∕2)/κ]}, where V represents the inactivating prepulse potential, and V1∕2 represents the mid-point of inactivation curve.
Data were analyzed using Pulsefit 8.6 and Origin 7.5, and presented as means ± SEM. The Kruskal–Wallis non-parametric test was used to analyze current density data. One-way ANOVA was used to assess the statistical significance of changes in characteristics of channel activation and inactivation. Statistical comparisons were performed by Student's t-test.
All cells were lysed in RIPA buffer (with 1% PMSF), and total protein concentrations were determined with a BCA Protein Assay Kit (TransGen). Total protein (10–20 mg) for each sample was loaded into precast 8% SDS-PAGE gels and run with running buffer. Gels were transferred onto PVDF membranes (Millipore). Antigen-specific primary antibodies (Pan sodium channel, Chemicon-AB5210; Nav1.6, Chemicon-AB5580 and Abcam-ab65166; β-tubulin, Abcam-ab6046; β-actin, Abcam-ab6276) were incubated overnight at 4°C and detected with species-specific horseradish-peroxidase-labeled secondary antibodies. An ECL Western Blotting Detection kit (TIANGEN) was used to obtain a chemiluminescence signal, which was detected using Gel Imaging System (Bio-Rad). Band quantification was performed using a Gel-Pro software. Bands of interest were normalized to actin- or tubulin- for a loading control.
Total RNA of cultured neurons was prepared by using the Trizol Reagent (Invitrogen, USA). The Superscript TM-III kit (Invitrogen, USA) was used for reverse-transcribed with oligo dT and 2.5 mg total RNA. Primer sequences were as follows: NM_019266 (SCN8A), and NM_031144 (β-actin). SCN8A forward 5′-CTG GAG AAT GGA GGC ACA CAC-3′, reverse 5′-ACG CTG CTG CTT CTC CTT GTC-3′; and β-actin forward 5′-CGT TGA CAT CCG TAA AGA CCT-3′, reverse 5′-TCA GGA GGA GCA ATG ATC TTG-3′. The resulting cDNA PCR amplification was performed by using the following protocol: 95°C for 10 s followed by 50 cycles of 95°C for 5 s and 60°C for 31 s, and verified by 2.0% agarose gel electrophoresis. Images were captured by using Gel Imaging System (Bio-Rad). The amplicon size of each gene was108 and 144 bp, respectively.
Cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton-x-100 for 5 min, and incubated with 5% BSA for 60 min at room temperature to block non-specific binding. Without washing, the diluted primary antibodies (MAP2, Abcam-ab32454; Nav1.6, Abcam-ab65166) were added and incubated at 4°C overnight. After three washes with PBS, cells were incubated with the corresponding secondary antibodies at room temperature for 1 h. For immunohistochemistry, cells were stained with DAB kit (Vector Laboratories) according to the instructions of the manufacturer for peroxidase labeling. Images were acquired from a fluorescence microscopy (Leica Microsystems DM400B, Germany).
Quantitative analysis of mean fluorescence intensities (MFIs) of immunoreactive neurons was performed using Image J software (National Institutes of Health). MFI per square micrometer was calculated by dividing the MFI units by the area of outlined regions.
To investigate change in intrinsic excitability in APP/PS1 mice, whole-cell patch-clamp recordings were performed on 12 DIV (days in vitro) primary cultured neurons obtained from P0 APP/PS1 mice and littermate wide type mice in current-clamp mode. Frequency, threshold, and peak amplitude of action potentials (AP) were examined by different protocols. AP firing frequencies (f) elicited by increasing depolarizing currents (200 pA, 500 ms) were significantly increased in neurons from APP/PS1 mice compared to WT mice (Figures 1A,D; fWT = 15.375 ± 3.428 Hz, n = 12; fAPP∕PS1 = 23.25 ± 7.264 Hz, n = 10; p = 0.0128). We used a depolarizing current (80 pA, 10 ms) to induce a single firing of an AP. Threshold potential (Vthreshold) and amplitude of action potential (Vpeak) were recorded and compared with WT. We found that thresholds of neurons from APP/PS1 mice were significantly decreased compared to WT mice (Figures 1B,E; Vthreshold, WT = −29.001 ± 2.304 mV, n = 9; Vthreshold, APP∕PS1 = −41.601 ±1.965 mV, n = 9; p = 0.0012), but the peak amplitude was not (Figures 1C,F; Vpeak, WT = 46.159 ± 2.663 mV, n = 9; Vpeak, APP∕PS1 = 47.236 ± 3.849 mV, n = 9; p = 0.6475). These results suggested that the excitability of mature neurons obtained from APP/PS1 mice was increased.
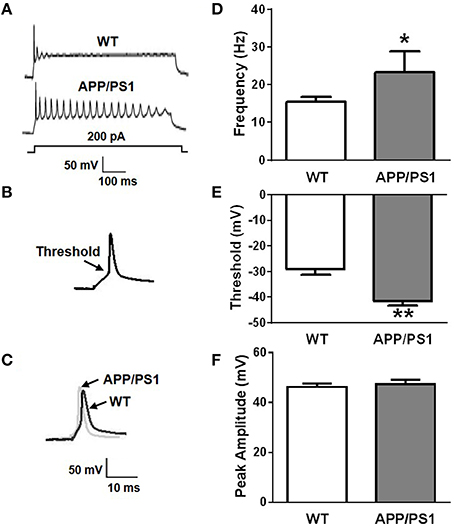
Figure 1. Increased neuronal excitability in APP/PS1 mice. (A) Representative recording traces of action potential repetitive firing. (B) The schematic of threshold. (C) Representative recording traces of action potential peak amplitude. (D) The mean number of APs elicited by 200 pA depolarizing current. Neurons from APP/PS1 mice (n = 10) fired significantly more APs than WT (n = 12). (E) AP threshold in APP/PS1 mice cultured pyramidal neurons was significantly lower than WT mice (n = 9 per group). (F) The peak amplitude of APs in WT and APP/PS1 (n = 9 per group) mice cultured pyramidal neurons didn't show significantly difference. Mean ± SEM was displayed. *Presents p < 0.05; **presents p < 0.01.
It is likely that diverse factors contribute to the pathogenesis of AD patients or mice (Blennow et al., 2006; Bertram and Tanzi, 2008; Mucke, 2009). Among them, Aβ stands out on the basis of overwhelming genetic evidence and strong experimental data (Farrer et al., 1997; Hardy and Selkoe, 2002; Tanzi and Bertram, 2005; Mahley and Huang, 2006). Accordingly, we attempted to investigate whether Aβ1−42 contributed to intrinsic excitability. Cell viability was firstly determined by MTT assay in primary cultured neurons treated with 5 μM Aβ42−1 or 5 μM Aβ1−42 for 24 h. We found that Aβ1−42 slightly induced loss of neuron viability (Supplementary Figure S2; n = 3, means three independent experiments; p = 0.047). As described previously, whole-cell patch-clamp recordings were performed in current-clamp mode on normal morphological primary neurons which were incubated with Aβ1−42 or Aβ42−1 for 24 h. Frequency, threshold, and peak amplitude of AP were examined. We found AP firing frequencies were significantly increased in neurons after treatment with Aβ1−42 peptide compared to controls (Figures 2A,D; fcontrol = 20.25 ± 4.742 Hz, n = 8, p = 0.0050; fAβ42−1 = 20.75 ± 4.644 Hz, n = 8, p = 0.0054; fAβ1−42 = 43.25 ± 5.028 Hz, n = 8). Aβ1−42 also significantly decreased the threshold (Figures 2B,E; Vthreshold, control = −33.83 ± 1.207 mV, n = 7, p = 0.0126; Vthreshold, Aβ42−1 = −32.55 ± 0.6600 mV, n = 7, p = 0.0035; Vthreshold, Aβ1−42 = −41.37 ± 1.771 mV, n = 7). But it had no effect on the peak amplitude (Figures 2C,F; Vpeak, control = 42.83 ± 0.9437 mV, n = 7, p = 0.6579; Vpeak, Aβ42−1 = 44.60 ± 1.051 mV, n = 7, p = 0.5633; Vpeak, Aβ1−42 = 43.58 ± 1.305 mV, n = 7). These results suggested that incubation in Aβ1−42 increases neuronal excitability in vitro.
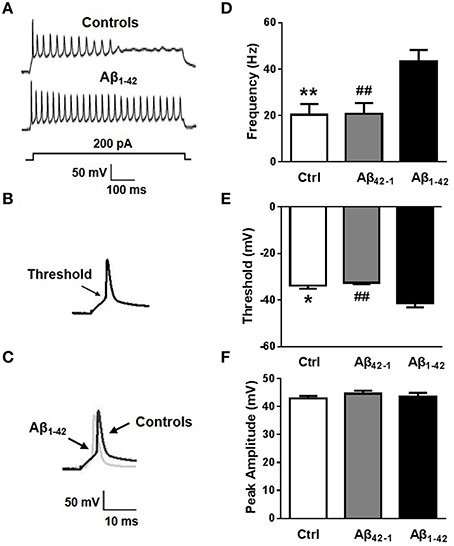
Figure 2. Aβ affects neuronal excitability. (A) Representative recording traces of action potential repetitive firing. (B) The schematic of threshold. (C) Representative recording traces of action potential peak amplitude. (D) The mean number of APs elicited by 200 pA depolarizing current. Neurons after Aβ1−42 treatment (n = 8) fired significantly more APs than controls (both negative control group and reverse peptide Aβ42−1 group, n = 8). (E) AP threshold in pyramidal neurons treated with Aβ1−42 (n = 7) was significantly lower than controls (n = 7). (F) The peak amplitude of APs in pyramidal neurons treated with Aβ1−42 (n = 7) didn't show significantly difference with controls (n = 7). Mean ± SEM was displayed. *Presents p < 0.05 vs. control group; **presents p < 0.01 vs. control group; ##presents p < 0.01 vs. Aβ1−42 group.
In mammalian neurons, dense clusters of voltage-gated sodium channels (Nav) at the axonal initial segment and nodes of Ranvier underlie action potential generation and fast propagation (Leterrier et al., 2011). In addition, non-inactivating persistent sodium currents support maintained depolarization during and between action potentials. Finally, the resurgent persistent sodium current is triggered upon repolarization and supports repetitive firing in some types of neurons (Raman et al., 1997). We therefore hypothesized that the increased excitability induced by Aβ1−42 could be due, at least in part, to an upregulation of Nav current. To test this hypothesis, we evaluated the magnitude of Nav currents in voltage-clamp mode after incubation with Aβ1−42 or reverse peptide for 24 h. Representative traces of the total inward current recorded in response to voltage steps from −60 to +60 mV are shown in Figure 3A. Current density curves (Figure 3B) and peak current densities (Figure 3C) were significantly increased after Aβ1−42 treatment compared to controls (Peakcontrol = 79.18 ± 11.60 pA/pF, n = 12, p = 0.0028; PeakAβ42−1 = 74.14 ± 13.54 pA/pF, n = 12, p = 0.0031; PeakAβ1−42 = 146.3 ± 10.70pA/pF, n = 12).
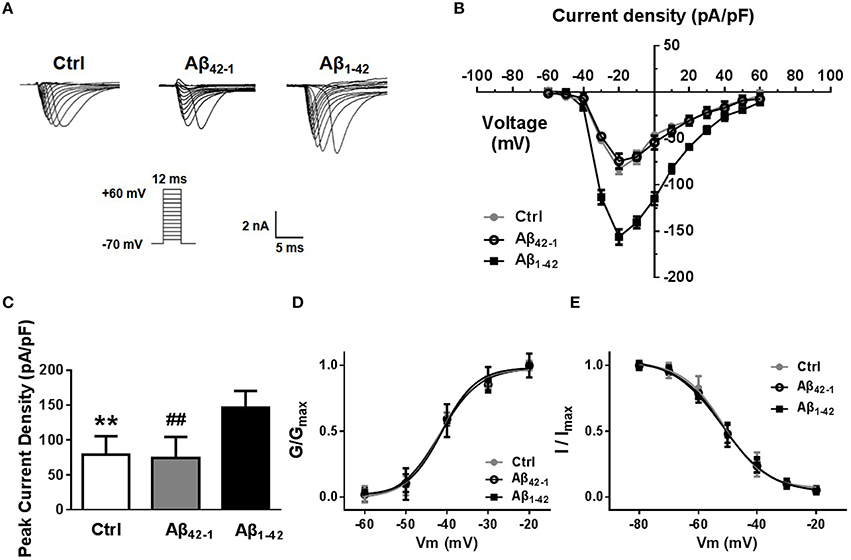
Figure 3. Aβ1−42 up-regulates voltage-dependent sodium current. (A) Representative traces of voltage gated sodium currents recorded in response to voltage steps from −60 to +60 mV. (B) Current density-voltage relationship, illustrates that Na+ current density recorded from Aβ1−42 treatment pyramidal neurons was significantly bigger than counterparts (negative control group and reverse peptide Aβ42−−1 group, n = 12). (C) Graph, data showed peak current density recorded from Aβ1−42 treatment neurons was significantly bigger than counterparts (n = 12). (D) Normalized steady-state activation curves for Na+ currents recorded from cultured pyramidal neurons after Aβ1−42 treatment and counterparts (n = 12). The activation curves were not significantly shift after Aβ1−42 treatment. (E) Normalized steady-state inactivation curves for Na+ currents recorded from these three groups, and the inactivation curve was not significantly shift after Aβ1−42 treatment either. Mean ± SEM was displayed. **Presents p < 0.01 vs. Aβ1−42 group; ##presents p < 0.01 vs. Aβ1−42 group.
We next investigated the effect of Aβ1−42 on sodium channel kinetic characteristics (Figures 3D,E). We found that Aβ1−42 did not significantly shift the voltage-dependent sodium activation curve (Figure 3D). Single Boltzmann distribution fits showed a V1∕2, control = −41.62 ± 1.462 mV, and a κ of 4.165 ± 1.548 mV (n = 12); a V1∕2, Aβ42−1 = −41.71 ± 1.768 mV and a κ of 4.795 ± 1.825 mV (n = 12); and a V1∕2, Aβ1−42 = −41.19 ± 1.052 mV and a κ of 4.155 ± 1.184 mV (n = 12). For steady-state inactivation curves (Figure 3E), the V1∕2 and κ of inactivation also did not change significantly (V1∕2, control = −51.47 ± 1.040 mV and κ = 6.992 ± 1.017 mV, n = 12; V1∕2, Aβ42−1 = −51.90 ± 0.8201 mV and κ = 8.384 ± 0.8552 mV, n = 12; V1∕2, Aβ1−42 = −51.40 ± 0.4131 mV and κ = 7.723 ± 0.4178 mV, n = 12).
Overall, these results demonstrated that exposure of neurons to Aβ1−42 leads to an increased neuronal excitability, likely through a Nav current-mediated mechanism.
Although, the experiments described above revealed that Aβ1−42 increased neuronal excitability, perhaps dues to increasing Nav currents, it remained unclear how the Nav currents increased. There was no observable difference in sodium channel kinetic characteristics after Aβ1−42 incubation (Figures 3D,E). We then examined the expression of Nav in neurons after Aβ1−42 treatment. Using Western blot analysis of cultured cortical neurons, we found that incubation with Aβ1−42 significantly increased the expression of Nav [Figures 4A,B; n = 3; pCtrl vs. Aβ1−42 = 0.0146; pAβ42−1 vs. Aβ1−42 = 0.0358]. We also investigated the Nav1.6 subtype, which plays a major role in the transmission of subthreshold currents, namely the persistent and resurgent currents (Raman et al., 1997), and the electrophysiological properties of Nav1.6 make these channels especially suited for the sustained repetitive firing of neurons (Van Wart and Matthews, 2006). As shown in Figures 4C,D, Nav1.6 displayed a significant increase after Aβ1−42 treatment [n = 3; pCtrl vs. Aβ1−42 = 0.0372; pAβ42−1 vs. Aβ1−42 = 0.0354]. To further identify the increased expression of Nav1.6 after Aβ1−42 treatment, mRNA expression levels were detected too. Paralleled with western blot analysis, Nav1.6 mRNA obtained from cultured neurons treated with Aβ1−42 showed significantly increased than control groups [Figures 4E,F, n = 3; pCtrl vs. Aβ1−42 = 0.0257; pAβ42−1 vs. Aβ1−42 = 0.017].
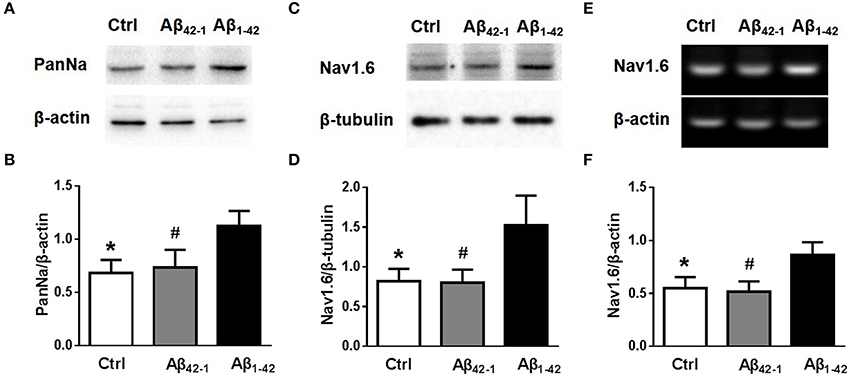
Figure 4. Aβ1−42 increases the expression of sodium channels. (A) Equal amounts of collected protein samples obtained from 12 DIV neurons after different treatments were analyzed by western blot to detect the protein expression level of sodium channel (n = 3 per group). (B) Quantification of protein levels of sodium channel. β-actin was used as an internal control (n = 3 per group). (C) Western blots of 12 DIV neurons after different treatments to detect the expression of Nav1.6 protein (n = 3 per group). (D) Quantification of protein levels of Nav1.6. β-tubulin was used as an internal control (n = 3 per group). (E) RT-PCR of Nav1.6 mRNA levels of 12 DIV neurons after different treatments (n = 3 per group). (F) Quantification of mRNA levels of Nav1.6. β-actin was used as an internal control (n = 3 per group). Mean ± SEM was displayed. *Presents p < 0.05 vs. Aβ1−42 group; #presents p < 0.05 vs. Aβ1−42 group.
Morphological methods were also used to test the expression of Nav1.6. The localization of Nav1.6 in neurons was revealed by immunohistochemistry (Figure 5A). Consistent with the result from western blotting, most neurons incubated with Aβ1−42 showed significantly deepening stain [Figures 5A,C; n = 3; pCtrl vs. Aβ1−42 = 0.0162]. After obtaining these results, we wondered if the expression of Nav1.6 in APP/PS1 mouse brains would have changed. Coronal brain sections from 9 month old APP/PS1 mice and WT littermates were stained for MAP2, a maker of mature neurons, and Nav1.6 (Figure 5B; n = 3). We found, that APP/PS1 mice exhibited more Nav1.6 immunoreactivity than WT mice in cortex regions (Figure 5D; n = 3; p = 0.0286).
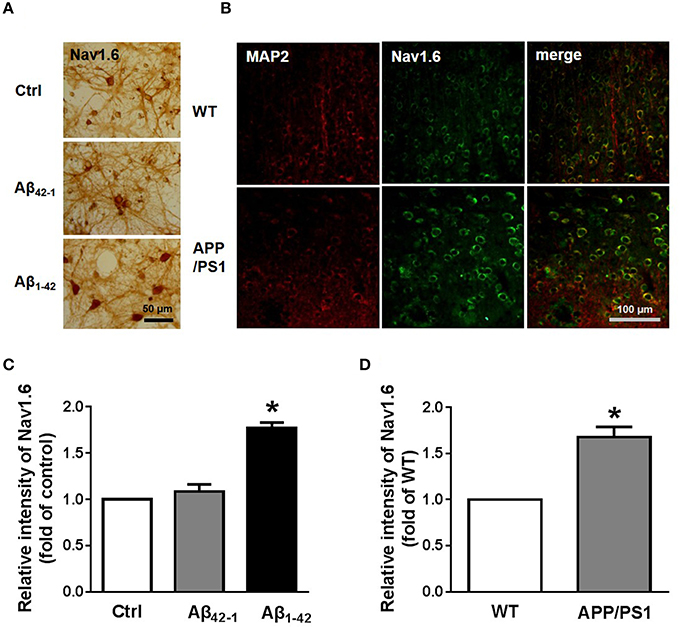
Figure 5. Elevated expression of Nav1.6 in cultured neurons and APP/PS1 mice. (A) Immunohistochemistry images of 12 DIV primary cortical culture neurons after 24 h Aβ42−1 or Aβ1−42 treatments stained with anti-Nav1.6 antibody. Scale bar = 50 μm (n = 3 per group). (B) Immunostaining images of temporal lobe cortex sections obtained from 9 months APP/PS1 mice and its littermates co-stained with anti-Nav1.6 antibody and anti-MAP2 antibody (n = 3 per group). (C) Quantification of Nav1.6 immunohistochemistry images by using intensity analysis (n = 3 per group). (D) Quantification of fluorescence intensity of Nav1.6 (n = 3 per group). Mean ± SEM was displayed. *Presents p < 0.05 vs. control group.
Taken together, our findings and previous studies suggest that Aβ1−42 increased the excitability of cultured cortical neurons, and this effect was mediated by overexpression of Nav, with the Nav1.6 subtype perhaps accounting for much of this increase.
In addition to cognitive deficits, AD patients have an increased incidence of epileptic seizures. This incidence is even higher in patients with early-onset AD who overexpress human APP, the proteolysis of which generates Aβ (Palop and Mucke, 2009). Hyperexcitability is also detected in the brains of various AD transgenic mice (Tamagnini et al., 2015), including the APP/PS1 mice used here. Such aberrant increases in network excitability and compensatory inhibitory mechanisms in the hippocampus may contribute to the cognitive deficits in AD (Palop et al., 2007; Sanchez et al., 2012; Verret et al., 2012). However, the mechanisms underlying the hyperexcitability detected in AD brains are not fully understood.
In the present study, we investigated the excitability in cultured pyramidal neurons of APP/PS1 mice using patch clamp techniques. We found that the excitability of 12 DIV cultured pyramidal neurons from APP/PS1 mice was significantly greater than in WT littermates. Additionally, AP firing frequencies increased and Vthreshold decreased in neurons from APP/PS1 mice compared to WT mice. These results confirm previous studies that showed neuronal hyperexcitability in APP/PS1 mice by patch clamp methods. In recent years, many studies demonstrated that Aβ is associated with increased excitability of neurons in vitro and in animal models, leading to hypersynchronous network activity and higher risk for seizures (Minkeviciene et al., 2009; Busche et al., 2012; Born et al., 2014; Davis et al., 2014). In our current work, we observed the altered excitability of pyramidal neurons after treatment with Aβ. The higher firing frequency and lower threshold indicated an increased intrinsic excitability of pyramidal neurons.
Aβ-induced aberrant excitatory activity might occur through many different mechanisms. Previous studies have shown that Aβ can downregulate A-type K+ currents, thereby increasing excitability of hippocampal pyramidal neurons (Good and Murphy, 1996; Chen, 2005). Elevated Aβ also causes GABAergic dysfunction and attenuates excitatory synaptic transmission by decreasing the number of surface AMPA and NMDA receptors (Kamenetz et al., 2003; Hsieh et al., 2006; Shankar et al., 2007), as well as disrupting the development of aberrant synchrony in neural networks, damaging cognitive functions. Subsequent studies proved that neuronal activity regulates Aβ production (Kamenetz et al., 2003; Cirrito et al., 2005). Blocking neuronal electrical activity with TTX, a sodium channel blocker, decreased the cleavage of APP by β-secretase (Kamenetz et al., 2003). Recent studies found that blocking the network hyperactivity with the anti-epileptic drug lamotrigine, a voltage-dependent sodium channel inhibitor, reversed synaptic disorder and cognitive dysfunction in APP transgenic mice (Bakker et al., 2012; Sanchez et al., 2012; Zhang et al., 2014). These results indicate that Aβ, or other AD-related factors, plays a significant role in regulating neuronal activity at specific types of neurons as well as in wider neuronal networks, and Aβ and sodium channels have a certain relationship.
Voltage-dependent sodium currents play a critical role in action potential depolarization and firing frequency in many types of neurons (Kim et al., 2005; Baroni et al., 2013). We therefore hypothesized that the increased excitability induced by Aβ1−42 could be due, at least in part, to an upregulation of the Nav current. As we expected, the peaks of the voltage-dependent sodium current were significantly increased after Aβ1−42 treatment. The increased voltage-dependent sodium current could decrease action potential threshold and increase the probability of action potential generation in response to synaptic excitation, thus increasing the excitability of the neuron. However, Aβ upregulated sodium currents without significantly altering the voltage-dependence of activation and inactivation. We found that Aβ increased the expression of Nav and Nav1.6 in cultured neurons, indicating that the number of Nav channels could be altered by Aβ1−42. These results provide a possible mechanism for the increased excitability of pyramidal neurons previously observed after Aβ treatment. Confirming the causal relationship between the aberrant excitatory activity induced by Aβ and cognitive decline in AD patients would be an important insight into the pathogenesis of AD and provide new therapeutic avenues.
Conceived and designed the experiments: XW, SL. Performed the experiments: XW, XZ, and CJ. Analyzed the data: XW, XZ, ZX, and QM. Contributed reagents/materials/analysis tools: TZ, NL. Wrote the paper: XW, XZ.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by Grant (81571061, 81371223, and 30871006) from the National Natural Science Foundation of China and the Research Fund for the Doctoral Program of Higher Education of China (20122105110010).
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnins.2016.00094
Supplementary Figure S1. Biochemical characterization of Aβ1−42. Eighty nanogram aged Aβ1−42 and Aβ1−42 monomer samples were analyzed by SDS-PAGE and detected with anti-6E10 antibody. After 48 h aggregated Aβ1−42 with different molecular weight forms were displayed. aged presents Aβ1−42 with aggregation treatment; mono presents Aβ1−42 monomer.
Supplementary Figure S2. Cell viability measurement of neuron treated with Aβ1−42. Cell viability was determined by MTT assay in primary cultured neurons treated with 5 μM Aβ42−1 or 5 μM Aβ1−42 for 24 h. Aβ1−42 induced slightly loss of cell viability in neurons. (n = 3, means 3 independent experiments). Mean ± SEM was displayed. MTT, 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide. *Presents p < 0.05 vs. control group.
Bakker, A., Krauss, G. L., Albert, M. S., Speck, C. L., Jones, L. R., Stark, C. E., et al. (2012). Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74, 467–474. doi: 10.1016/j.neuron.2012.03.023
Baroni, D., Barbieri, R., Picco, C., and Moran, O. (2013). Functional modulation of voltage-dependent sodium channel expression by wild type and mutated C121W-beta1 subunit. J. Bioenerg. Biomembr. 45, 353–368. doi: 10.1007/s10863-013-9510-3
Bertram, L., and Tanzi, R. E. (2008). Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat. Rev. Neurosci. 9, 768–778. doi: 10.1038/nrn2494
Blennow, K., de Leon, M. J., and Zetterberg, H. (2006). Alzheimer's disease. Lancet 368, 387–403. doi: 10.1016/S0140-6736(06)69113-7
Born, H. A., Kim, J. Y., Savjani, R. R., Das, P., Dabaghian, Y. A., Guo, Q., et al. (2014). Genetic suppression of transgenic APP rescues Hypersynchronous network activity in a mouse model of Alzeimers disease. J. Neurosci. 34, 3826–3840. doi: 10.1523/JNEUROSCI.5171-13.2014
Brorson, J. R., Bindokas, V. P., Iwama, T., Marcuccilli, C. J., Chisholm, J. C., and Miller, R. J. (1995). The Ca2+ influx induced by beta-amyloid peptide 25-35 in cultured hippocampal neurons results from network excitation. J. Neurobiol. 26, 325–338. doi: 10.1002/neu.480260305
Busche, M. A., Chen, X., Henning, H. A., Reichwald, J., Staufenbiel, M., Sakmann, B., et al. (2012). Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 109, 8740–8745. doi: 10.1073/pnas.1206171109
Catterall, W. A., Goldin, A. L., and Waxman, S. G. (2005). International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409. doi: 10.1124/pr.57.4.4
Chen, C. (2005). beta-Amyloid increases dendritic Ca2+ influx by inhibiting the A-type K+ current in hippocampal CA1 pyramidal neurons. Biochem. Biophys. Res. Commun. 338, 1913–1919. doi: 10.1016/j.bbrc.2005.10.169
Choi, D. W. (1988). Glutamate neurotoxicity and diseases of the nervous system. Neuron 1, 623–634. doi: 10.1016/0896-6273(88)90162-6
Cirrito, J. R., Yamada, K. A., Finn, M. B., Sloviter, R. S., Bales, K. R., May, P. C., et al. (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. doi: 10.1016/j.neuron.2005.10.028
Davis, K. E., Fox, S., and Gigg, J. (2014). Increased hippocampal excitability in the 3xTgAD mouse model for Alzheimer's disease in vivo. PLoS ONE 9:e91203. doi: 10.1371/journal.pone.0091203
Farrer, L. A., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., et al. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease meta analysis consortium. JAMA 278, 1349–1356. doi: 10.1001/jama.1997.03550160069041
Goldin, A. L., Barchi, R. L., Caldwell, J. H., Hofmann, F., Howe, J. R., Hunter, J. C., et al. (2000). Nomenclature of voltage-gated sodium channels. Neuron 28, 365–368. doi: 10.1016/S0896-6273(00)00116-1
Good, T. A., and Murphy, R. M. (1996). Effect of beta-amyloid block of the fast-inactivating K+ channel on intracellular Ca2+ and excitability in a modeled neuron. Proc. Natl. Acad. Sci. U.S.A. 93, 15130–15135. doi: 10.1073/pnas.93.26.15130
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hsieh, H., Boehm, J., Sato, C., Iwatsubo, T., Tomita, T., Sisodia, S., et al. (2006). AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843. doi: 10.1016/j.neuron.2006.10.035
Jones, R. S., Minogue, A. M., Connor, T. J., and Lynch, M. A. (2013). Amyloid-beta-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. J. Neuroimmune Pharmacol. 8, 301–311. doi: 10.1007/s11481-012-9427-3
Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., Iwatsubo, T., et al. (2003). APP processing and synaptic function. Neuron 37, 925–937. doi: 10.1016/S0896-6273(03)00124-7
Kim, D. Y., Ingano, L. A., Carey, B. W., Pettingell, W. H., and Kovacs, D. M. (2005). Presenilin/gamma-secretase-mediated cleavage of the voltage-gated sodium channel beta2-subunit regulates cell adhesion and migration. J. Biol. Chem. 280, 23251–23261. doi: 10.1074/jbc.M412938200
Leterrier, C., Brachet, A., Dargent, B., and Vacher, H. (2011). Determinants of voltage-gated sodium channel clustering in neurons. Semin. Cell Dev. Biol. 22, 171–177. doi: 10.1016/j.semcdb.2010.09.014
Liu, C., Tan, F. C., Xiao, Z. C., and Dawe, G. S. (2015). Amyloid precursor protein enhances Nav1.6 sodium channel cell surface expression. J. Biol. Chem. 290, 12048–12057. doi: 10.1074/jbc.M114.617092
Mahley, R. W., and Huang, Y. (2006). Apolipoprotein (apo) E4 and Alzheimer's disease: unique conformational and biophysical properties of apoE4 can modulate neuropathology. Acta Neurol. Scand. Suppl. 185, 8–14. doi: 10.1111/j.1600-0404.2006.00679.x
Minkeviciene, R., Rheims, S., Dobszay, M. B., Zilberter, M., Hartikainen, J., Fulop, L., et al. (2009). Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. 29, 3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009
Ogiwara, I., Miyamoto, H., Morita, N., Atapour, N., Mazaki, E., Inoue, I., et al. (2007). Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 27, 5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007
Olney, J. W., Wozniak, D. F., and Farber, N. B. (1997). Excitotoxic neurodegeneration in Alzheimer disease. New hypothesis and new therapeutic strategies. Arch. Neurol. 54, 1234–1240. doi: 10.1001/archneur.1997.00550220042012
Ong, W. Y., Tanaka, K., Dawe, G. S., Ittner, L. M., and Farooqui, A. A. (2013). Slow excitotoxicity in Alzheimer's disease. J. Alzheimers. Dis. 35, 643–668. doi: 10.3233/JAD-121990
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 55, 697–711. doi: 10.1016/j.neuron.2007.07.025
Palop, J. J., and Mucke, L. (2009). Epilepsy and cognitive impairments in Alzheimer's disease. Arch. Neurol. 66, 435–440. doi: 10.1001/archneurol.2009.15
Palop, J. J., and Mucke, L. (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818. doi: 10.1038/nn.2583
Putcha, D., Brickhouse, M., O'Keefe, K., Sullivan, C., Rentz, D., Marshall, G., et al. (2011). Hippocampal hyperactivation associated with cortical thinning in Alzheimer's disease signature regions in non-demented elderly adults. J. Neurosci. 31, 17680–17688. doi: 10.1523/JNEUROSCI.4740-11.2011
Ragsdale, D. S. (2008). How do mutant Nav1.1 sodium channels cause epilepsy? Brain Res. Rev. 58, 149–159. doi: 10.1016/j.brainresrev.2008.01.003
Raman, I. M., Sprunger, L. K., Meisler, M. H., and Bean, B. P. (1997). Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 19, 881–891. doi: 10.1016/S0896-6273(00)80969-1
Sanchez, P. E., Zhu, L., Verret, L., Vossel, K. A., Orr, A. G., Cirrito, J. R., et al. (2012). Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc. Natl. Acad. Sci. U.S.A. 109, E2895–E2903. doi: 10.1073/pnas.1121081109
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007
Tamagnini, F., Scullion, S., Brown, J. T., and Randall, A. D. (2015). Intrinsic excitability changes induced by acute treatment of hippocampal CA1 pyramidal neurons with exogenous amyloid beta peptide. Hippocampus 25, 786–797. doi: 10.1002/hipo.22403
Tanzi, R. E., and Bertram, L. (2005). Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. doi: 10.1016/j.cell.2005.02.008
Trimmer, J. S., and Rhodes, K. J. (2004). Localization of voltage-gated ion channels in mammalian brain. Annu. Rev. Physiol. 66, 477–519. doi: 10.1146/annurev.physiol.66.032102.113328
Van Wart, A., and Matthews, G. (2006). Impaired firing and cell-specific compensation in neurons lacking nav1.6 sodium channels. J. Neurosci. 26, 7172–7180. doi: 10.1523/JNEUROSCI.1101-06.2006
Verret, L., Mann, E. O., Hang, G. B., Barth, A. M., Cobos, I., Ho, K., et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. doi: 10.1016/j.cell.2012.02.046
Xu, D. E., Zhang, W. M., Yang, Z. Z., Zhu, H. M., Yan, K., Li, S., et al. (2014). Amyloid precursor protein at node of Ranvier modulates nodal formation. Cell Adh. Migr. 8, 396–403. doi: 10.4161/cam.28802
Yu, F. H., and Catterall, W. A. (2003). Overview of the voltage-gated sodium channel family. Genome Biol. 4:207. doi: 10.1186/gb-2003-4-3-207
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., et al. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. doi: 10.1038/nn1754
Keywords: Alzheimer's disease, beta-amyloid peptide, excitability, voltage-gated sodium channel, neurodegeneration
Citation: Wang X, Zhang X-G, Zhou T-T, Li N, Jang C-Y, Xiao Z-C, Ma Q-H and Li S (2016) Elevated Neuronal Excitability Due to Modulation of the Voltage-Gated Sodium Channel Nav1.6 by Aβ1−42. Front. Neurosci. 10:94. doi: 10.3389/fnins.2016.00094
Received: 21 December 2015; Accepted: 24 February 2016;
Published: 09 March 2016.
Edited by:
William Cho, Queen Elizabeth Hospital, Hong KongReviewed by:
Peng Lei, Sichuan University, ChinaCopyright © 2016 Wang, Zhang, Zhou, Li, Jang, Xiao, Ma and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shao Li, bGlzaGFvODlAaG90bWFpbC5jb20=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.