 Davide Tampellini
Davide Tampellini
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Neurosci. , 04 November 2015
Sec. Neurodegeneration
Volume 9 - 2015 | https://doi.org/10.3389/fnins.2015.00423
Synapses have been known for many years to be the crucial target of pathology in different forms of dementia, in particular Alzheimer's disease (AD). Synapses and their appropriate activation or inhibition are fundamental for the proper brain function. Alterations in synaptic/neuronal activity and brain metabolism are considered among the earliest symptoms linked to the progression of AD, and lead to a central question in AD research: what is the role played by synaptic activity in AD pathogenesis? Intriguingly, in the last decade, important studies demonstrated that the state of activation of synapses affects the homeostasis of beta-amyloid (Aβ) and tau, both of which aggregate and accumulate during AD, and are involved in neuronal dysfunction. In this review we aim to summarize the up-to-date data linking synaptic/neuronal activity with Aβ and tau; moreover, we also intend to provide a critical overview on brain activity alterations in AD, and their role in the disease's pathophysiology.
Synapses are considered to be an early site of dysfunction/pathology in AD (Selkoe, 2002), and loss of synapses is the best pathologic correlate of cognitive impairment in AD patients (Terry et al., 1991). For many years it has been known that Aβ peptide, one of the main players in AD pathology derived by the β- and γ-secretase cleavage of the amyloid precursor protein (APP), can induce morphological and functional alterations to synapses and synaptic plasticity (Selkoe, 2002; Coleman and Yao, 2003; Almeida et al., 2005). Surprisingly, elegant studies from Roberto Malinow's and David Holtzman's groups demonstrated that, in turn, synaptic activity affects Aβ: increased activity enhances secretion of Aβ, while reduced activity inhibits it (Kamenetz et al., 2003; Cirrito et al., 2005). This discovery represented an important breakthrough in the field. For the first time it was shown that Aβ homeostasis was controlled by the main target of Aβ itself: synapses and their state of activation.
Since inhibition of synaptic activity reduces Aβ secretion, reduced activity appears to be positive for AD. Thus, our and other groups investigated the effect of chronic inhibition of synaptic activity on transgenic AD mice to explore whether it could indeed protect synapses. Chronic reduction of synaptic activity by unilateral whisker ablation, diminishes plaque burden in the deafferented barrel cortex of AD mice (Tampellini et al., 2010; Bero et al., 2011). On the contrary, chronic unilateral activation of the perforant pathway by optogenetic light activation (an experimental approach which also induced epileptic seizure in the studied AD mouse cohort), increases the amount of amyloid plaques (Yamamoto et al., 2015). Despite the reduction of amyloid plaques, we found that levels of synaptophysin and number of synapses were also reduced by chronic inhibition of activity in AD, but not wild-type, mouse brains (Tampellini et al., 2010). In addition, reduced activity worsened memory impairments in AD mice compared to controls (Tampellini et al., 2010). Intriguingly, in brain areas where plaques were reduced intraneuronal Aβ was increased, and presented an inverse correlation with levels of synaptophysin (Tampellini et al., 2010). Intraneuronal Aβ accumulation has been observed in human AD brains, in several rodent models of AD (Gouras et al., 2000; D'Andrea et al., 2001; Busciglio et al., 2002; Mori et al., 2002; Oddo et al., 2003; Cataldo et al., 2004; Echeverria et al., 2004; Cruz et al., 2006; Oakley et al., 2006; LaFerla et al., 2007), and, more recently, also in brains of aged mouse lemur primates (Roy et al., 2014). Within neurons, Aβ accumulates on the outer membrane of multivesicular bodies, both in somas and neurites (Takahashi et al., 2002; Casas et al., 2004; Cataldo et al., 2004), and is associated with early pathological alterations in dendrites, axonal terminals and synapses (Takahashi et al., 2004; Bayer and Wirths, 2010; Gouras et al., 2010). Clearance of intraneuronal Aβ by immunotherapy was shown to protect synapses and improve memory in in vitro and in vivo models of AD (Billings et al., 2005; Tampellini et al., 2007).
On the other hand, synaptic activity induced by specific activation of synaptic (but not extra synaptic) NMDA receptors (Lu et al., 2001), produced beneficial effects on AD transgenic neurons by reducing levels of intraneuronal Aβ and increasing levels of synaptic proteins (Tampellini et al., 2009, 2011). These outcomes are in line with the protection exerted by environmental enrichment, which has been demonstrated to enhance synaptic activity and plasticity (Eckert and Abraham, 2013), in AD mouse models (Lazarov et al., 2005; Briones et al., 2009; Gerenu et al., 2013). Activity-dependent decrease of intraneuronal Aβ might be explained with the relocation of Aβ from the inside to the outside of neurons (enhanced secretion); however, we demonstrated that degradation is also involved. The activity-dependent reduction of Aβ42, one of the most pathologic isoforms of Aβ, have been shown to occur via neprilysin (Tampellini et al., 2009), a neutral endopeptidase which is the most efficient Aβ degrading enzyme (Iwata et al., 2000). During activation, neprilysin relocates to the cell surface and shows increased colocalization with Aβ42, suggesting enhanced Aβ degradation (Tampellini et al., 2011). We are inclined to think that this pool of Aβ42 might derive from APP processing in synaptic endosomes with activation (as further discussed), and might then be transported to the neuronal surface (Rajendran et al., 2006).
During synaptic activity APP traffics anterogradly toward synapses, where it is endocytosed (Tampellini et al., 2009). This last observation complements data showing that enhanced Aβ secretion upon synaptic activation requires endocytosis (Cirrito et al., 2008). Therefore, a production of Aβ might occur at synapses with activity, as also supported by increased levels of β-C-terminal fragments (βCTFs; Kamenetz et al., 2003; Tampellini et al., 2009). Activity-dependent Aβ secretion has been observed in patients after brain injury: Aβ levels were reduced in the interstitial fluid (ISF) with worsened neurological status, and increased with improved neurological condition (Brody et al., 2008). Since, Aβ has been experimentally shown to inhibit synapses and impair synaptic plasticity (Hsieh et al., 2006; Shankar et al., 2008), one hypothesis on the physiologic role of activity-dependent Aβ secretion suggests that it might serve as feedback mechanism to prevent synaptic hyperactivation and excitotoxicity (Kamenetz et al., 2003). Intriguingly, further studies demonstrated that low concentrations (in the range of picomoles) of Aβ enhance LTP, and are involved in memory formation (Puzzo et al., 2008, 2011; Garcia-Osta and Alberini, 2009), providing evidence for a physiological function of secreted Aβ.
Altogether, the reported data suggest that, despite promoting Aβ secretion, synaptic activity might have a protective role against AD.
Tau is one of the microtubule-associated proteins that bind and stabilize neuronal microtubules during development of neuronal processes, establishment of cell polarity and intracellular transport (Binder et al., 1985; Drechsel et al., 1992; Mandelkow and Mandelkow, 1998). When phosphorylated, tau detaches from microtubules; abnormal tau phosphorylation in neurons is a hallmark of AD and other neurodegenerative diseases (including frontotemporal dementia, and progressive supranuclear palsy), and is accompanied by aggregation, and progressive intraneuronal tau accumulation. In addition to its buildup within neurons, more recent studies demonstrated that tau is also released in the extracellular space (Gómez-Ramos et al., 2006; Avila, 2010); and that increased levels of tau (total and phosphorylated) in the human's cerebrospinal fluid (CSF) are associated with an increased risk of developing AD (Blennow et al., 2010).
Tau protein is traditionally considered to be localized in axons; however, when neurons are exposed to Aβ oligomers, tau relocates to somatodendritic compartments in association with loss of spines and microtubule breakdown (Zempel et al., 2010). More recent data demonstrated the presence of tau at synapses in physiologic and pathological conditions (Pooler et al., 2014). Tau localizes in both pre and post-synaptic compartments, and the number of synaptosomes containing tau did not differ between control and AD human brains; however, a particular form of phosphorylated-tau (pS396/pS404) and tau oligomers were specifically found in AD synaptosomes (Tai et al., 2012).
Little is known on the link between tau and synaptic activity. Recent studies showed that synaptic activation enhances secretion of tau in vitro and in vivo (Pooler et al., 2013; Yamada et al., 2014). Synaptic activity was also shown to induce tau translocation to excitatory synapses, precisely in dendritic spines and post-synaptic compartments, in wild-type neurons (Frandemiche et al., 2014). In the same study, authors demonstrated that also Aβ oligomers induce tau localization to synapses; intriguingly, such translocation requires the residue S404 of tau to be phosphorylated, the same observed specifically in AD synaptosomes (Tai et al., 2012). Synaptic activation induces tau phosphorylation on residue T205; however, this phosphorylation is not mandatory for tau translocation to synapses (Frandemiche et al., 2014).
The localization of tau in dendrites is considered to be pathologic, because it is associated with loss of spines, as mentioned above (Zempel et al., 2010), and because it targets the kinase Fyn to post-synaptic compartments (Ittner et al., 2010). Fyn mediates Aβ toxicity, and its reduction or its overexpression attenuated or enhanced, respectively, synaptic alterations and cognitive impairments in AD transgenic mice (Chin et al., 2004, 2005). Fyn entry to post-synaptic compartment is tau dependent: in mice overexpressing the tau amino-terminal projection domain (PD) only (from amino acid 1 to 255, excluding the microtubule binding domain and the carboxy-terminal tail region), the tau-PD fragment does not enter dendrites, and Fyn post-synaptic targeting is diminished (Ittner et al., 2010). As result, AD transgenic mice crossed with tau-PD overexpressing mice showed decreased susceptibility to excitotoxic seizure, and improved memory (Ittner et al., 2010), suggesting protective effects when tau (and Fyn) access to dendrites is reduced. A recent paper shows that tau localization into spines is enhanced by phosphorylation. Low levels of endogenous tau are observed in dendritic spines of hippocampal neurons: when specific phosphorylation sites are replaced with glutamic acid to mimic phosphorylation (including the S404 site), tau localization in spines increases (Xia et al., 2015).
In the light of what reported so far, the translocation of tau to spines and post-synaptic compartments with synaptic activity might have negative implications for AD. However, more studies are required before ending to this conclusion. For example, what phosphorylated forms of tau are specifically present in spines and synapses during activity? Synaptic activity and Aβ seem to induce phosphorylation on different sites of tau (Frandemiche et al., 2014): perhaps, activity-induced tau phosphorylation might be more physiologic, while Aβ-induced tau phosphorylation might be more toxic. In addition, the observed activity-induced tau phosphorylation on residue T205 was reported to not be mandatory for tau targeting to synapses (Frandemiche et al., 2014), raising the possibility that tau translocation to spines with activity might occur without phosphorylation; and this would also be an intriguing subject to explore. Finally, another important question to answer is whether synaptic activation increases Fyn targeting to spines and post-synaptic compartments. Synaptic localization of Fyn has been shown to worsen the phenotype in models of β-amyloidosis; however, it has also been reported that, despite total levels of Fyn are unchanged between human AD and control, in AD brains Fyn levels are increased in somas, where it colocalizes with tau tangles, and are decreased in synaptic compartments (Ho et al., 2005), suggesting a physiologic role of Fyn at synapses.
In the last decade several studies reported that functional alterations are common in the brain of AD patients. Even more intriguingly, neuronal dysfunction has been observed in non-demented older subjects with amyloid deposition before memory impairments (Sperling et al., 2009), which suggests it to be an early event in AD pathophysiology. Data have been provided for both increased and decreased neuronal excitability in AD patients, and in animal models of AD. Hippocampal hyperactivity has been observed in MCI patients (Bakker et al., 2012), and in young presenilin 1 (PSEN1) E280A mutation carriers (Reiman et al., 2012). Some mouse models of AD present episodes of epileptic seizure (Palop et al., 2007; Marchetti and Marie, 2011), and enhancement of GABAergic inhibitory transmission, or use of antiepileptic drugs showed protection (Sanchez et al., 2012; Verret et al., 2012; Levenga et al., 2013; Hall et al., 2015). On the other hand, reduced hippocampal activation has been observed to correlate with clinical decline in elderly (O'Brien et al., 2010). Glucose metabolism in young subject with predisposition to develop AD (ApoE4 carriers) is reduced several decades before the appearance of the first symptoms (Reiman et al., 2004), suggesting reduced brain activity. In addition, synaptic plasticity is decreased in several mouse models of AD (Trinchese et al., 2004; Shankar et al., 2008; Marchetti and Marie, 2011; Warmus et al., 2014; Menkes-Caspi et al., 2015) and by exposure to Aβ (Hu et al., 2009; Tu et al., 2014). How to interpret these mixed outcomes, and to reconcile these apparently contradictory results of reduced synaptic transmission and increased excitability in AD is still debated, as recently reviewed (Stargardt et al., 2015).
There is evidence for a protective effect of synaptic activity against AD (Swaab and Bao, 2010; Tampellini and Gouras, 2010). Higher educational attainment or participation in intellectually stimulating activities is associated with reduced risk of developing AD (Stern et al., 1994; Stern, 2006); in addition, in memory disorder clinics is common practice to encourage patients to be involved in brain stimulating activities (solving puzzles, crossed words, among others). Deep brain stimulation of the fornix in AD patients resulted in better outcomes in cognition, memory, and quality of life (Smith et al., 2012). The higher activity observed in early stages of AD might be an adaptive response boosting neuroprotection. A recent study compared brain activity of young subjects with cognitively normal older people having brain Aβ deposition: the study outcome revealed that older people had Aβ-related hyperactivation, which resulted to be a compensatory/protective mechanism reflecting neural plasticity (Elman et al., 2014).
Synaptic activity has been demonstrated to be important for neuronal survival: it is involved in the activation of survival pathways, including transcription of Activity-regulated Inhibitors of Death (AID) (a set of pro-survival genes), and resistance to apoptosis-inducing compounds (Bas-Orth and Bading, 2013). Importantly, local ATP synthesis at synapses is activity-driven, and it is fundamental for correct synaptic efficacy (Rangaraju et al., 2014). Enhanced synaptic activity was recently shown to be part of the protective mechanism exerted by rapamycin in models of AD. Rapamycin treatment increases levels of the presynaptic protein SV2 and the frequency of excitatory postsynaptic currents reducing Aβ oligomers synaptotoxicity (Ramírez et al., 2014).
The activity-dependent Aβ secretion might also work as protective mechanism to prevent Aβ buildup in the brain. One of the most accepted biomarker for AD risk/diagnosis is the decrease of Aβ42 in the CSF, which can be observed up to 10 years before conversion of MCI to AD (Buchhave et al., 2012). As observed in a study on PSEN1 mutation carriers, when Aβ42 begins to accumulate in the brain, its release in the CSF declines (Potter et al., 2013). In line with these finding, also levels of Aβ42 in the ISF are reduced in AD transgenic mouse models with aging (Cirrito et al., 2003; Hong et al., 2011). One proposed mechanism suggests that ISF Aβ42 is progressively sequestered within forming plaques in young mice; in old plaque-rich mice, ISF Aβ42 (which is less concentrated than in young mice ISF) would not derive from new biosynthesis but rather from less soluble Aβ42 deposits present in the brain parenchyma (Hong et al., 2011). Another possible mechanism to explain reduction of Aβ42 in ISF and CSF is its progressive reduced secretion by neurons. We demonstrated that AD transgenic neurons, but not wild-type neurons, secrete less Aβ1-42 in the medium, and accumulate it in distal neurites with time in culture (Tampellini et al., 2011). Thus, activity-enhanced Aβ secretion might be a physiologic event to enhance efflux of Aβ42 to the CSF. In young PSEN1 E280A mutation carriers, Aβ1-42 levels in the CSF are increased compared to control (Reiman et al., 2012). As mentioned above, the same subjects showed increased hippocampal activity; however, no differences in dementia ratings, and neuropsychological test scores were found in comparison with the control group, suggesting compensatory/protective effect of enhanced activation.
Finally, activity-induced Aβ secretion might avoid intraneuronal accumulation of Aβ, and induce new Aβ (especially Aβ42) biosynthesis, preventing mobilization of less soluble, and potentially more toxic Aβ species from the brain parenchyma.
The relation between synaptic/neuronal activity and AD pathology is complex and of high interest for AD research, since it affects the homeostasis of APP, Aβ and tau, and since functional alterations can be detected very early in subjects at risk for AD.
We hypothesize that physiologic synaptic activation (without induction of epileptic seizure) might be protective for neuronal preservation, and persistence of normal cognitive functions during aging (Figure 1). The higher activity observed in preclinical or early MCI patients might be the protraction of a compensatory/defensive response attempting to promote survival pathways, and preventing Aβ accumulation within neurons by maintaining its degradation and physiological secretion.
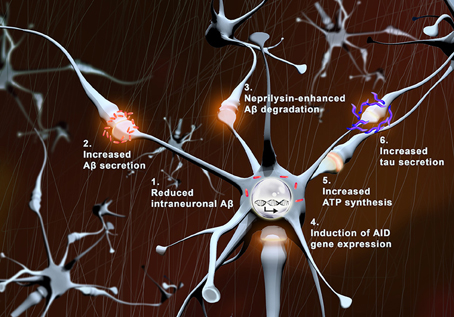
Figure 1. Protective effects of synaptic activity. With synaptic activation: (1) intraneuronal levels of Aβ (red) are reduced, (2) Aβ secretion is augmented, (3) neprilysin-induced Aβ degradation is enhanced, (4) transcription of pro-survival genes (AID) increases, (5) local ATP synthesis rises at synapses, and (6) tau secretion is augmented.
Unveiling new mechanisms linking synaptic activity with APP/Aβ and tau biology might provide new important findings on AD pathogenesis, and could lead to novel therapeutic approaches.
The writing of this review was possible by the support of Institut Professeur Baulieu to DT.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author thanks Prof. Gilbert Di Paolo (Columbia University, New York, US) for useful comments, Dr. Magali Dumont (ICM-Inserm U1127, Paris, France) for helpful discussions, and Mr. Matteo Tampellini (Tampelliniart, Varese, Italy) for the figure preparation.
Almeida, C. G., Tampellini, D., Takahashi, R. H., Greengard, P., Lin, M. T., Snyder, E. M., et al. (2005). Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 20, 187–198. doi: 10.1016/j.nbd.2005.02.008
Avila, J. (2010). Intracellular and extracellular tau. Front. Neurosci. 4:49. doi: 10.3389/fnins.2010.00049
Bakker, A., Krauss, G. L., Albert, M. S., Speck, C. L., Jones, L. R., Stark, C., et al. (2012). Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74, 467–474. doi: 10.1016/j.neuron.2012.03.023
Bas-Orth, C., and Bading, H. (2013). The divergence-convergence model of acquired neuroprotection. Mech. Dev. 130, 396–401. doi: 10.1016/j.mod.2012.09.008
Bayer, T. A., and Wirths, O. (2010). Intracellular accumulation of amyloid-beta – a predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease. Front. Aging Neurosci. 2:8. doi: 10.3389/fnagi.2010.00008
Bero, A. W., Yan, P., Roh, J. H., Cirrito, J. R., Stewart, F. R., Raichle, M. E., et al. (2011). Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat. Neurosci. 14, 750–756. doi: 10.1038/nn.2801
Billings, L. M., Oddo, S., Green, K. N., McGaugh, J. L., and LaFerla, F. M. (2005). Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron 45, 675–688. doi: 10.1016/j.neuron.2005.01.040
Binder, L. I., Frankfurter, A., and Rebhun, L. I. (1985). The distribution of tau in the mammalian central nervous system. J. Cell Biol. 101, 1371–1378. doi: 10.1083/jcb.101.4.1371
Blennow, K., Hampel, H., Weiner, M., and Zetterberg, H. (2010). Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 6, 131–144. doi: 10.1038/nrneurol.2010.4
Briones, T. L., Rogozinska, M., and Woods, J. (2009). Environmental experience modulates ischemia-induced amyloidogenesis and enhances functional recovery. J. Neurotrauma 26, 613–625. doi: 10.1089/neu.2008.0707
Brody, D. L., Magnoni, S., Schwetye, K. E., Spinner, M. L., Esparza, T. J., Stocchetti, N., et al. (2008). Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science 321, 1221–1224. doi: 10.1126/science.1161591
Buchhave, P., Minthon, L., Zetterberg, H., Wallin, A. K., Blennow, K., and Hansson, O. (2012). Cerebrospinal fluid levels of beta-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch. Gen. Psychiatry 69, 98–106. doi: 10.1001/archgenpsychiatry.2011.155
Busciglio, J., Pelsman, A., Wong, C., Pigino, G., Yuan, M., Mori, H., et al. (2002). Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down's syndrome. Neuron 33, 677–688. doi: 10.1016/S0896-6273(02)00604-9
Casas, C., Sergeant, N., Itier, J. M., Blanchard, V., Wirths, O., van der Kolk, N., et al. (2004). Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am. J. Pathol. 165, 1289–1300. doi: 10.1016/S0002-9440(10)63388-3
Cataldo, A. M., Petanceska, S., Terio, N. B., Peterhoff, C. M., Durham, R., Mercken, M., et al. (2004). Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol. Aging 25, 1263–1272. doi: 10.1016/j.neurobiolaging.2004.02.027
Chin, J., Palop, J. J., Puoliväli, J., Massaro, C., Bien-Ly, N., Gerstein, H., et al. (2005). Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J. Neurosci. 25, 9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005
Chin, J., Palop, J. J., Yu, G. Q., Kojima, N., Masliah, E., and Mucke, L. (2004). Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J. Neurosci. 24, 4692–4697. doi: 10.1523/JNEUROSCI.0277-04.2004
Cirrito, J. R., Kang, J. E., Lee, J., Stewart, F. R., Verges, D. K., Silverio, L. M., et al. (2008). Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron, 58, 42–51. doi: 10.1016/j.neuron.2008.02.003
Cirrito, J. R., May, P. C., O'Dell, M. A., Taylor, J. W., Parsadanian, M., Cramer, J. W., et al. (2003). In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J. Neurosci. 23, 8844–8853.
Cirrito, J. R., Yamada, K. A., Finn, M. B., Sloviter, R. S., Bales, K. R., May, P. C., et al. (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. doi: 10.1016/j.neuron.2005.10.028
Coleman, P. D., and Yao, P. J. (2003). Synaptic slaughter in Alzheimer's disease. Neurobiol. Aging 24, 1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001
Cruz, J. C., Kim, D., Moy, L. Y., Dobbin, M. M., Sun, X., Bronson, R. T., et al. (2006). p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J. Neurosci. 26, 10536–10541. doi: 10.1523/JNEUROSCI.3133-06.2006
D'Andrea, M. R., Nagele, R. G., Wang, H. Y., Peterson, P. A., and Lee, D. H. (2001). Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology 38, 120–134. doi: 10.1046/j.1365-2559.2001.01082.x
Drechsel, D. N., Hyman, A. A., Cobb, M. H., and Kirschner, M. W. (1992). Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 3, 1141–1154. doi: 10.1091/mbc.3.10.1141
Echeverria, V., Ducatenzeiler, A., Dowd, E., Jänne, J., Grant, S. M., Szyf, M., et al. (2004). Altered mitogen-activated protein kinase signaling, tau hyperphosphorylation and mild spatial learning dysfunction in transgenic rats expressing the beta-amyloid peptide intracellularly in hippocampal and cortical neurons. Neuroscience 129, 583–592. doi: 10.1016/j.neuroscience.2004.07.036
Eckert, M. J., and Abraham, W. C. (2013). Effects of environmental enrichment exposure on synaptic transmission and plasticity in the hippocampus. Curr. Top. Behav. Neurosci. 15, 165–187. doi: 10.1007/7854_2012_215
Elman, J. A., Oh, H., Madison, C. M., Baker, S. L., Vogel, J. W., Marks, S. M., et al. (2014). Neural compensation in older people with brain amyloid-beta deposition. Nat. Neurosci. 17, 1316–1318. doi: 10.1038/nn.3806
Frandemiche, M. L., De Seranno, S., Rush, T., Borel, E., Elie, A., Arnal, I., et al. (2014). Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J. Neurosci. 34, 6084–6097. doi: 10.1523/JNEUROSCI.4261-13.2014
Garcia-Osta, A., and Alberini, C. M. (2009). Amyloid beta mediates memory formation. Learn. Mem. 16, 267–272. doi: 10.1101/lm.1310209
Gerenu, G., Dobarro, M., Ramirez, M. J., and Gil-Bea, F. J. (2013). Early cognitive stimulation compensates for memory and pathological changes in Tg2576 mice. Biochim. Biophys. Acta 1832, 837–847. doi: 10.1016/j.bbadis.2013.02.018
Gómez-Ramos, A., Díaz-Hernández, M., Cuadros, R., Hernández, F., and Avila, J. (2006). Extracellular tau is toxic to neuronal cells. FEBS Lett. 580, 4842–4850. doi: 10.1016/j.febslet.2006.07.078
Gouras, G. K., Tampellini, D., Takahashi, R. H., and Capetillo-Zarate, E. (2010). Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol. 119, 523–541. doi: 10.1007/s00401-010-0679-9
Gouras, G. K., Tsai, J., Naslund, J., Vincent, B., Edgar, M., Checler, F., et al. (2000). Intraneuronal Abeta42 accumulation in human brain. Am. J. Pathol. 156, 15–20. doi: 10.1016/S0002-9440(10)64700-1
Hall, A. M., Throesch, B. T., Buckingham, S. C., Markwardt, S. J., Peng, Y., Wang, Q., et al. (2015). Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer's disease. J. Neurosci. 35, 6221–6230. doi: 10.1523/JNEUROSCI.2552-14.2015
Ho, G. J., Hashimoto, M., Adame, A., Izu, M., Alford, M. F., Thal, L. J., et al. (2005). Altered p59Fyn kinase expression accompanies disease progression in Alzheimer's disease: implications for its functional role. Neurobiol. Aging 26, 625–635. doi: 10.1016/j.neurobiolaging.2004.06.016
Hong, S., Quintero-Monzon, O., Ostaszewski, B. L., Podlisny, D. R., Cavanaugh, W. T., Yang, T., et al. (2011). Dynamic analysis of amyloid beta-protein in behaving mice reveals opposing changes in ISF versus parenchymal Abeta during age-related plaque formation. J. Neurosci. 31, 15861–15869. doi: 10.1523/JNEUROSCI.3272-11.2011
Hsieh, H., Boehm, J., Sato, C., Iwatsubo, T., Tomita, T., Sisodia, S., et al. (2006). AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843. doi: 10.1016/j.neuron.2006.10.035
Hu, N. W., Klyubin, I., Anwyl, R., and Rowan, M. J. (2009). GluN2B subunit-containing NMDA receptor antagonists prevent Abeta-mediated synaptic plasticity disruption in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 20504–20509. doi: 10.1073/pnas.0908083106
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Iwata, N., Tsubuki, S., Takaki, Y., Watanabe, K., Sekiguchi, M., Hosoki, E., et al. (2000). Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat. Med. 6, 143–150. doi: 10.1038/77399
Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., Iwatsubo, T., et al. (2003). APP processing and synaptic function. Neuron 37, 925–937. doi: 10.1016/S0896-6273(03)00124-7
LaFerla, F. M., Green, K. N., and Oddo, S. (2007). Intracellular amyloid-beta in Alzheimer's disease. Nat. Rev. Neurosci. 8, 499–509. doi: 10.1038/nrn2168
Lazarov, O., Robinson, J., Tang, Y. P., Hairston, I. S., Korade-Mirnics, Z., Lee, V. M., et al. (2005). Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell 120, 701–713. doi: 10.1016/j.cell.2005.01.015
Levenga, J., Krishnamurthy, P., Rajamohamedsait, H., Wong, H., Franke, T. F., Cain, P., et al. (2013). Tau pathology induces loss of GABAergic interneurons leading to altered synaptic plasticity and behavioral impairments. Acta Neuropathol. Commun. 1:34. doi: 10.1186/2051-5960-1-34
Lu, W., Man, H., Ju, W., Trimble, W. S., MacDonald, J. F., and Wang, Y. T. (2001). Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29, 243–254. doi: 10.1016/S0896-6273(01)00194-5
Mandelkow, E. M., and Mandelkow, E. (1998). Tau in Alzheimer's disease. Trends Cell Biol. 8, 425–427. doi: 10.1016/S0962-8924(98)01368-3
Marchetti, C., and Marie, H. (2011). Hippocampal synaptic plasticity in Alzheimer's disease: what have we learned so far from transgenic models? Rev. Neurosci. 22, 373–402. doi: 10.1515/rns.2011.035
Menkes-Caspi, N., Yamin, H. G., Kellner, V., Spires-Jones, T. L., Cohen, D., and Stern, E. A. (2015). Pathological tau disrupts ongoing network activity. Neuron 85, 959–966. doi: 10.1016/j.neuron.2015.01.025
Mori, C., Spooner, E. T., Wisniewsk, K. E., Wisniewski, T. M., Yamaguch, H., Saido, T. C., et al. (2002). Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid 9, 88–102.
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
O'Brien, J. L., O'Keefe, K. M., LaViolette, P. S., DeLuca, A. N., Blacker, D., Dickerson, B. C., et al. (2010). Longitudinal fMRI in elderly reveals loss of hippocampal activation with clinical decline. Neurology 74, 1969–1976. doi: 10.1212/WNL.0b013e3181e3966e
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., et al. (2003). Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. doi: 10.1016/S0896-6273(03)00434-3
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 55, 697–711. doi: 10.1016/j.neuron.2007.07.025
Pooler, A. M., Noble, W., and Hanger, D. P. (2014). A role for tau at the synapse in Alzheimer's disease pathogenesis. Neuropharmacology 76(Pt A), 1–8. doi: 10.1016/j.neuropharm.2013.09.018
Pooler, A. M., Phillips, E. C., Lau, D. H., Noble, W., and Hanger, D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394. doi: 10.1038/embor.2013.15
Potter, R., Patterson, B. W., Elbert, D. L., Ovod, V. Kasten, T., Sigurdson, W., et al. (2013). Increased in vivo amyloid-beta42 production, exchange, and loss in presenilin mutation carriers. Sci. Trans. Med. 5:189ra177. doi: 10.1126/scitranslmed.3005615
Puzzo, D., Privitera, L., Fa, M., Staniszewski, A., Hashimoto, G., Aziz, F., et al. (2011). Endogenous amyloid-beta is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 69, 819–830. doi: 10.1002/ana.22313
Puzzo, D., Privitera, L., Leznik, E., Fà, M., Staniszewski, A., Palmeri, A., et al. (2008). Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 28, 14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008
Rajendran, L., Honsho, M., Zahn, T. R., Keller, P., Geiger, K. D., Verkade, P., et al. (2006). Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 103, 11172–11177. doi: 10.1073/pnas.0603838103
Ramírez, A. E., Pacheco, C. R., Aguayo, L. G., and Opazo, C. M. (2014). Rapamycin protects against Abeta-induced synaptotoxicity by increasing presynaptic activity in hippocampal neurons. Biochim. Biophys. Acta 1842, 1495–1501. doi: 10.1016/j.bbadis.2014.04.019
Rangaraju, V., Calloway, N., and Ryan, T. A. (2014). Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. doi: 10.1016/j.cell.2013.12.042
Reiman, E. M., Chen, K., Alexander, G. E., Caselli, R. J., Bandy, D., Osborne, D., et al. (2004). Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc. Natl. Acad. Sci. U.S.A. 101, 284–289. doi: 10.1073/pnas.2635903100
Reiman, E. M., Quiroz, Y. T., Fleisher, A. S., Chen, K., Velez-Pardo, C., Jimenez-Del-Rio, M., et al. (2012). Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 11, 1048–1056. doi: 10.1016/S1474-4422(12)70228-4
Roy, M., Cardoso, C., Dorieux, O., Malgorn, C., Epelbaum, S., Petit, F., et al. (2014). Age-associated evolution of plasmatic amyloid in mouse lemur primates: relationship with intracellular amyloid deposition. Neurobiol. Aging 36, 149–156. doi: 10.1016/j.neurobiolaging.2014.07.017
Sanchez, P. E., Zhu, L., Verret, L., Vossel, K. A., Orr, A. G., Cirrito, J. R., et al. (2012). Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc. Natl. Acad. Sci. U.S.A. 109, E2895–E2903. doi: 10.1073/pnas.1121081109
Selkoe, D. J. (2002). Alzheimer's disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Smith, G. S., Laxton, A. W., Tang-Wai, D. F., McAndrews, M. P., Diaconescu, A. O., Workman, C. I., et al. (2012). Increased cerebral metabolism after 1 year of deep brain stimulation in Alzheimer disease. Arch. Neurol. 69, 1141–1148. doi: 10.1001/archneurol.2012.590
Sperling, R. A., Laviolette, P. S., O'Keefe, K., O'Brien, J., Rentz, D. M., Pihlajamaki, M., et al. (2009). Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63, 178–188. doi: 10.1016/j.neuron.2009.07.003
Stargardt, A., Swaab, D. F., and Bossers, K. (2015). The storm before the quiet: neuronal hyperactivity and Abeta in the presymptomatic stages of Alzheimer's disease. Neurobiol. Aging 36, 1–11. doi: 10.1016/j.neurobiolaging.2014.08.014
Stern, Y., Gurland, B., Tatemichi, T. K., Tang, M. X., Wilder, D., and Mayeux, R. (1994). Influence of education and occupation on the incidence of Alzheimer's disease. JAMA 271, 1004–1010. doi: 10.1001/jama.1994.03510370056032
Stern, Y. (2006). Cognitive reserve and Alzheimer disease. Alzheimer Dis. Assoc. Disord. 20, S69–S74. doi: 10.1097/00002093-200607001-00010
Swaab, D. F., and Bao, A. M. (2010). (Re-)activation of neurons in aging and dementia: lessons from the hypothalamus. Exp. Gerontol. 46, 178–184. doi: 10.1016/j.exger.2010.08.028
Tai, H. C., Serrano-Pozo, A., Hashimoto, T., Frosch, M. P., Spires-Jones, T. L., and Hyman, B. T. (2012). The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 181, 1426–1435. doi: 10.1016/j.ajpath.2012.06.033
Takahashi, R. H., Almeida, C. G., Kearney, P. F., Yu, F., Lin, M. T., Milner, T. A., et al. (2004). Oligomerization of Alzheimer's beta-amyloid within processes and synapses of cultured neurons and brain. J. Neurosci. 24, 3592–3599. doi: 10.1523/JNEUROSCI.5167-03.2004
Takahashi, R. H., Milner, T. A., Li, F., Nam, E. E., Edgar, M. A., Yamaguchi, H., et al. (2002). Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 161, 1869–1879. doi: 10.1016/S0002-9440(10)64463-X
Tampellini, D., Capetillo-Zarate, E., Dumont, M., Huang, Z., Yu, F., Lin, M. T., et al. (2010). Effects of synaptic modulation on {beta}-Amyloid, synaptophysin, and memory performance in Alzheimer's Disease transgenic mice. J. Neurosci. 30, 14299–14304. doi: 10.1523/JNEUROSCI.3383-10.2010
Tampellini, D., and Gouras, G. K. (2010). Synapses, synaptic activity and intraneuronal Abeta in Alzheimer's disease. Front. Aging Neurosci. 2:13 doi: 10.3389/fnagi.2010.00013
Tampellini, D., Magrané, J., Takahashi, R. H., Li, F., Lin, M. T., Almeida, C. G., et al. (2007). Internalized antibodies to the Abeta domain of APP reduce neuronal Abeta and protect against synaptic alterations. J. Biol. Chem. 282, 18895–18906. doi: 10.1074/jbc.M700373200
Tampellini, D., Rahman, N., Gallo, E. F., Huang, Z., Dumont, M., Capetillo-Zarate, E., et al. (2009). Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J. Neurosci. 29, 9704–9713. doi: 10.1523/JNEUROSCI.2292-09.2009
Tampellini, D., Rahman, N., Lin, M. T., Capetillo-Zarate, E., and Gouras, G. K. (2011). Impaired beta-amyloid secretion in Alzheimer's disease pathogenesis. J. Neurosci. 31, 15384–15390. doi: 10.1523/JNEUROSCI.2986-11.2011
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. doi: 10.1002/ana.410300410
Trinchese, F., Liu, S., Battaglia, F., Walter, S., Mathews, P. M., and Arancio, O. (2004). Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann. Neurol. 55, 801–814. doi: 10.1002/ana.20101
Tu, S., Okamoto, S., Lipton, S. A., and Xu, H. (2014). Oligomeric Abeta-induced synaptic dysfunction in Alzheimer's disease. Mol. Neurodegener. 9:48. doi: 10.1186/1750-1326-9-48
Verret, L., Mann, E. O., Hang, G. B., Barth, A. M., Cobos, I., Ho, K., et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. doi: 10.1016/j.cell.2012.02.046
Warmus, B. A., Sekar, D. R., McCutchen, E., Schellenberg, G. D., Roberts, R. C., McMahon, L. L., et al. (2014). Tau-mediated NMDA receptor impairment underlies dysfunction of a selectively vulnerable network in a mouse model of frontotemporal dementia. J. Neurosci. 34, 16482–16495. doi: 10.1523/JNEUROSCI.3418-14.2014
Xia, D., Li, C., and Götz, J. (2015). Pseudophosphorylation of Tau at distinct epitopes or the presence of the P301L mutation targets the microtubule-associated protein Tau to dendritic spines. Biochim. Biophys. Acta 1852, 913–924. doi: 10.1016/j.bbadis.2014.12.017
Yamada, K., Holth, J. K., Liao, F., Stewart, F. R., Mahan, T. E., Jiang, H., et al. (2014). Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 211, 387–393. doi: 10.1084/jem.20131685
Yamamoto, K., Tanei, Z., Hashimoto, T., Wakabayashi, T., Okuno, H., Naka, Y., et al. (2015). Chronic optogenetic activation augments abeta pathology in a mouse model of Alzheimer disease. Cell Rep. 11, 859–865. doi: 10.1016/j.celrep.2015.04.017
Keywords: Alzheimer, synapses, synaptic activity, beta-amyloid, tau
Citation: Tampellini D (2015) Synaptic activity and Alzheimer's disease: a critical update. Front. Neurosci. 9:423. doi: 10.3389/fnins.2015.00423
Received: 23 July 2015; Accepted: 19 October 2015;
Published: 04 November 2015.
Edited by:
Marc Dhenain, Commissariat à l'Énergie Atomique, FranceReviewed by:
Serge Marty, Ecole Normale Supérieure, FranceCopyright © 2015 Tampellini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Davide Tampellini, ZGF2aWRlLnRhbXBlbGxpbmlAaW5zZXJtLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.