 Vinod Sundaramoorthy
Vinod Sundaramoorthy Jessica M. Sultana
Jessica M. Sultana Julie D. Atkin
Julie D. Atkin- 1Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, Macquarie University Sydney, Sydney, NSW, Australia
- 2Department of Biochemistry and Genetics, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, VIC, Australia
Amyotrophic Lateral Sclerosis (ALS) is an invariably fatal neurodegenerative disorder, which specifically targets motor neurons in the brain, brain stem and spinal cord. Whilst the etiology of ALS remains unknown, fragmentation of the Golgi apparatus is detected in ALS patient motor neurons and in animal/cellular disease models. The Golgi is a highly dynamic organelle that acts as a dispatching station for the vesicular transport of secretory/transmembrane proteins. It also mediates autophagy and maintains endoplasmic reticulum (ER) and axonal homeostasis. Both the trigger for Golgi fragmentation and the functional consequences of a fragmented Golgi apparatus in ALS remain unclear. However, recent evidence has highlighted defects in vesicular trafficking as a pathogenic mechanism in ALS. This review summarizes the evidence describing Golgi fragmentation in ALS, with possible links to other disease processes including cellular trafficking, ER stress, defective autophagy, and axonal degeneration.
Introduction
The Golgi apparatus (referred to as “Golgi” hereafter) acts as a dispatching station whereby proteins and lipids newly synthesized in the ER are transported to the endosomal system, secretory granules, or plasma membrane. In spite of being a highly dynamic organelle (Griffiths et al., 1989), the Golgi normally maintains a characteristic morphology, consisting of flattened membrane stacks known as cisternae, and associated vesicles. The stacks of Golgi cisternae are interconnected laterally by tubules, forming a ribbon-like network (Rambourg and Clermont, 1990; Polishchuk and Mironov, 2004), usually in the perinuclear region of the cell, adjacent to the centrosome (Linstedt, 2004). The Golgi comprises of three functional compartments: the cis-Golgi, which being the nearest compartment to the ER, forms the entry face to the Golgi, the medial-Golgi, which is responsible for the modification, sorting and packaging of proteins for transportation, and finally the trans Golgi network, which forms the exit face of the Golgi (Rothman and Wieland, 1996; Glick and Nakano, 2009). Specific types of intracellular vesicles are associated with the Golgi. Secretory protein cargo buds from the ER via coat protein complex II (COPII) coated vesicles, to form tubulovesicular structures known as the ER-Golgi intermediate compartment (ERGIC), which eventually fuse with the cis-Golgi (Appenzeller-Herzog and Hauri, 2006). In contrast, the reticular trans-Golgi network (TGN) produces clathrin-coated vesicles which are targeted to endosomes and secretory vesicles in specialized cell types such as neurons (De Matteis and Luini, 2008). In addition to secretory trafficking, the Golgi is also responsible for the post-translational modification of proteins and lipids, including glycosylation (Stanley, 2011), sulfation (Baeuerle and Huttner, 1987), and proteolytic cleavage (Xu and Shields, 1993).
The Golgi in Neurons
Neurons are highly specialized cells with unique functional and morphological characteristics. Interestingly, in neurons the Golgi forms specialized “Golgi outposts” localized in axons and dendrites, which are discrete structures that are discontinuous from the somatic Golgi (Figure 1). These Golgi outposts are not fully characterized, but are thought to facilitate local secretory trafficking within neurites (Horton and Ehlers, 2003; Merianda et al., 2009). Axonal transport is an important property in neurons which involves trafficking of cellular proteins and vesicles within the axon, towards or away from the cell body. The relationship between axonal transport and transport within the soma is not fully understood, but these processes are clearly linked and involve the Golgi (Hirokawa and Takemura, 2005; Schwarz, 2013).
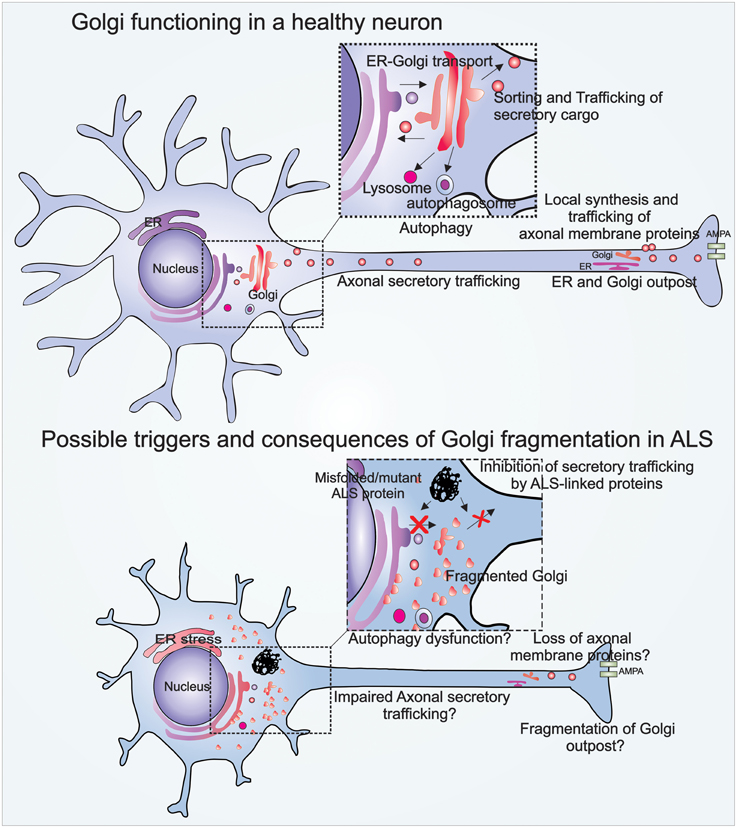
Figure 1. Illustration of Golgi functions in a healthy neuron, and Golgi fragmentation in an ALS-affected neuron. The Golgi in a healthy neuron regulates vesicular trafficking from the ER to the plasma membrane. The Golgi is also involved in the biogenesis of autophagosomes and lysosomes. Golgi outposts in healthy axons are involved in local synthesis and trafficking of axonal membrane proteins. Golgi fragmentation in ALS may be triggered by pathogenic mutant proteins that inhibit vesicular trafficking between the ER-Golgi, and Golgi to plasma membrane. Possible consequences of Golgi fragmentation in ALS include autophagy dysfunction, impaired axonal secretory trafficking, and loss of axonal homeostasis.
Fragmentation of the Golgi Apparatus
The Golgi is capable of undergoing profound morphological changes during normal cellular processes such as mitosis, as well as in pathological conditions. These morphological changes result in disruption of its characteristic ribbon-like network, forming a fragmented Golgi. Golgi fragmentation during mitosis facilitates equal distribution of the Golgi into the resulting daughter cells (Sütterlin et al., 2002). However, irreversible Golgi fragmentation occurs in pathological situations, when apoptosis is activated. Under these conditions, structural proteins within the Golgi are cleaved by the action of caspases (Lane et al., 2002). The Golgi also fragments when vesicular secretory trafficking is perturbed (Dascher and Balch, 1994; Wilson et al., 1994), which may also occur in pathological conditions. The morphological changes evident during fragmentation of the Golgi are attributed to two possibilities, either the Golgi membranes break into smaller dispersed vesicular structures (Figure 1), or the Golgi fuses with the ER upon fragmentation, which is then recycled, and it remerges at ER exit sites, dispersed throughout the cytoplasm (Cole et al., 1996; Storrie et al., 1998; Pelletier et al., 2000; Glick, 2002).
Golgi pathology is a feature of neurodegenerative diseases including Alzheimer's disease (Sun et al., 2008), Parkinson's disease (Fujita et al., 2006), Creutzfeldt-Jakob disease (Sakurai et al., 2000), multiple system atrophy (Sakurai et al., 2002), and ALS. Interestingly, Golgi fragmentation is often detected as an early event in these conditions, prior to apoptosis (Gosavi et al., 2002; Liazoghli et al., 2005; Atkin et al., 2014; van Dis et al., 2014), suggesting that Golgi fragmentation could be a trigger for neurodegeneration rather than a simple consequence of neuronal death. We review here the evidence describing Golgi fragmentation in ALS, and discuss recent studies implicating impairment of ER-Golgi mediated vesicular trafficking as a possible trigger. We also predict possible downstream consequences of Golgi fragmentation in ALS, and we examine links to other pathologies, including ER stress, autophagy dysfunction, and axonal degeneration.
Amyotrophic Lateral Sclerosis
Whilst 90% of ALS cases are sporadic, 10% of cases are familial, caused by mutations in genes encoding ubiquitously expressed proteins, including transactive response DNA binding protein (TDP-43), fused in sarcoma (FUS), optineurin, superoxide dismutase 1 (SOD1), and Chromosome 9 open reading frame 72 (C9orf72) (Renton et al., 2014) (Table 1). Although, the etiology of ALS remains unknown, RNA dysfunction and disruption to proteostasis are widely implicated as pathogenic mechanisms (Ling et al., 2013). Dysfunction to proteostasis includes protein misfolding and aggregation, ER stress, Golgi fragmentation, autophagy dysfunction, inhibition of cellular trafficking, and axonal degeneration. Like other neurodegenerative disorders, a pathological hallmark of ALS is the accumulation of intracellular inclusions containing misfolded protein aggregates (Wood et al., 2003; Blokhuis et al., 2013). Interestingly, wildtype (WT) forms of TDP-43, FUS, optineurin, and SOD1 may be recruited into ubiquitinated protein inclusions in sporadic ALS patients (Neumann et al., 2006; Deng et al., 2010; Blokhuis et al., 2013). Cytoplasmic accumulation, hyperphosphorylation and/or aggregation of TDP-43 is present in almost all cases of ALS (approximately 97%) (Ling et al., 2013). Transgenic mice overexpressing mutant SOD1G93A are the most widely used animal models of disease, which recapitulate many clinical and pathological features of ALS (Gurney et al., 1994). Increasing evidence now links ALS to frontotemporal dementia (FTD), with recent studies suggesting that ALS and FTD represent opposite ends of the disease spectrum (Ling et al., 2013).
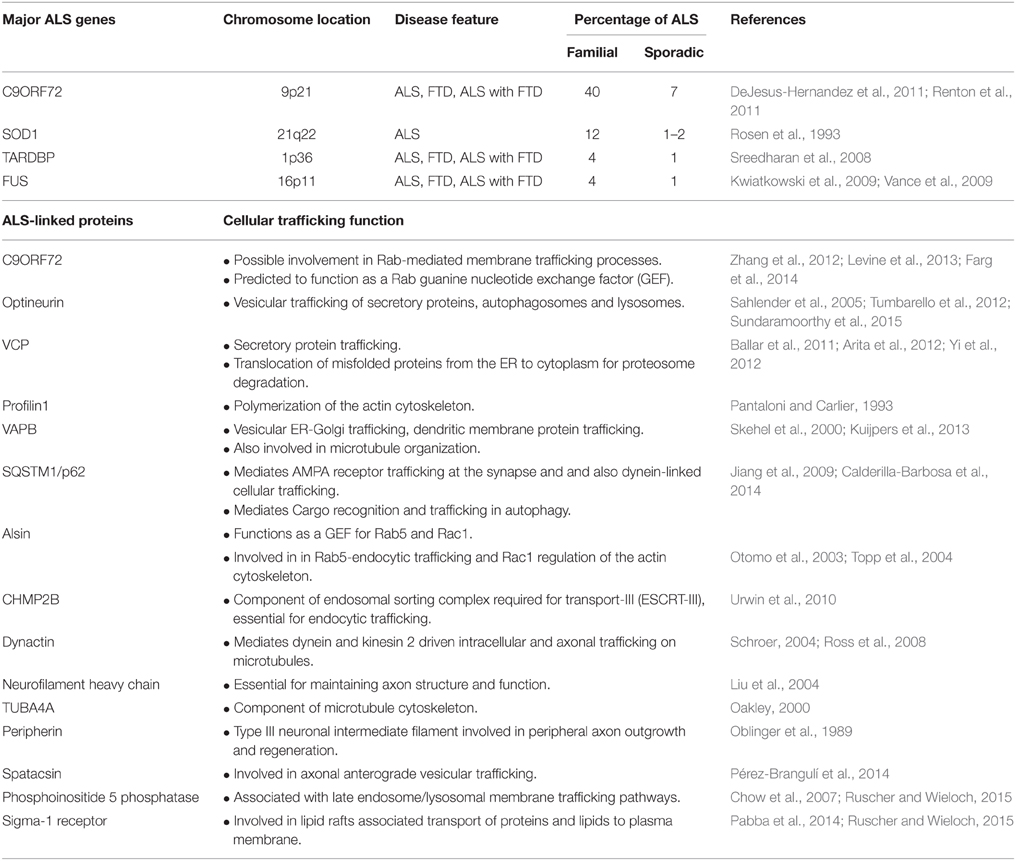
Table 1. List of major ALS genes and ALS-linked proteins with established intracellular and axonal trafficking functions.
Defects in intracellular trafficking, particularly within the axon, are implicated in ALS (Bilsland et al., 2010; Ikenaka et al., 2012; Alami et al., 2014). The “dying back” or slow degeneration of distal to proximal axons is associated with loss of motor neurons in ALS (Fischer et al., 2004; Dadon-Nachum et al., 2011; Moloney et al., 2014). In SOD1 mice, distal axonopathy and denervation of neuromuscular junctions are observed prior to the onset of clinical manifestations (Fischer et al., 2004). Fast-fatigable motor neurons with the longest axons and highest metabolic demands, are the most susceptible to axonal degeneration (Frey et al., 2000; Fischer et al., 2004). It has been suggested that lack of supply of essential proteins and lipids to distal axons is associated with axonal degeneration (Perlson et al., 2010).
Autophagy is an important proteostatic mechanism to degrade misfolded proteins in post-mitotic neurons (Thomas et al., 2013). It is therefore not surprising that defects in autophagy are present in ALS, however the nature of autophagy defects in ALS remains unclear. Autophagosomes accumulate in ALS patient brain tissues (Sasaki, 2011), implying that both induction of autophagy and inhibition of clearance of autophagsomes exist in ALS. However, more recent studies have demonstrated that formation of the autophagosome is impaired in cells expressing ALS mutant FU (Soo et al., 2015b) and in cells with reduced C9orf72 expression (Farg et al., 2014). Furthermore, mutations in proteins involved in endosomal sorting and trafficking which are required for the formation of autophagosomes (VCP, p62, dynactin, and RAB7) are also associated with ALS (Otomo et al., 2012). Activation of the unfolded protein response (UPR) and ER stress are well-documented pathogenic features in human ALS patients (Ilieva et al., 2007; Atkin et al., 2008; Oyanagi et al., 2008; Walker et al., 2010) and in animal/cellular disease models associated with mutant FUS, TDP-43, C9orf72, optineurin, and SOD1 (Atkin et al., 2006; Oh et al., 2008; Walker and Atkin, 2011; Farg et al., 2012; Walker et al., 2013; Zhang et al., 2014; Sundaramoorthy et al., 2015). Interestingly ER stress develops first in the most vulnerable motor neurons in SOD1G93A mice, 60 days before disease onset (Saxena et al., 2009), thus implicating ER stress as an active mechanism inducing cell death in ALS. Similarly, Golgi fragmentation is a prominent pathological feature in human ALS, and appears at a similar time point in SOD1 mice models (Gonatas et al., 1992; Mourelatos et al., 1994; van Dis et al., 2014).
Golgi Fragmentation in ALS
Fragmentation of the Golgi was first identified in ALS patient motor neurons over 20 years ago (Gonatas et al., 1992). In contrast to control patients, the Golgi in ALS patients was reduced and fragmented, appearing as disconnected punctate structures, similar to its morphology in cells treated with microtubule depolymerisation agents (Mourelatos et al., 1990; Gonatas et al., 1992). Since then, other studies have confirmed Golgi fragmentation in 10–50% sporadic patients (Gonatas et al., 2006; van Dis et al., 2014) and up to 70% of familial ALS patient motor neurons, bearing SOD1, FUS or optineurin mutations (Fujita et al., 2008; Ito et al., 2011). Interestingly, Golgi fragmentation is more prominent in larger human motor neurons, such as those in the cerebral cortex (Fujita et al., 1999) and anterior horn (Fujita et al., 2000), suggesting they are specifically vulnerable to disturbances in Golgi function.
Golgi fragmentation is also present in spinal anterior horn cells in sporadic ALS patients with cytoplasmic mislocalization of WT TDP-43, implying that a link exists between TDP-43 and Golgi pathologies (Fujita et al., 2008). Similarly, Golgi fragmentation is present in transgenic rats expressing mutant TDP-43M337V (Tong et al., 2012), in mutant SOD1G93A transgenic mice and in neuronal cells expressing SOD1G93A, G85R mutants (Mourelatos et al., 1996; Stieber et al., 2004). Interestingly, Golgi fragmentation precedes SOD1 inclusion formation, neuromuscular denervation, and mitochondrial-mediated apoptosis in low-copy number SOD1G93A transgenic mice, implying it is upstream in pathogenesis (van Dis et al., 2014). Similarly, ALS patients with optineurin mutations (<1% familial cases) demonstrate Golgi fragmentation in ~70% of anterior horn cells (Ito et al., 2011). Furthermore, Golgi fragmentation is present in cells expressing ALS-linked mutant FUS, optineurin and vesicle-associated membrane protein B (VAPB) (Teuling et al., 2007; Farg et al., 2013; Sundaramoorthy et al., 2015). However, despite being widely associated with ALS, the cellular events triggering Golgi fragmentation and the resulting consequences are not established. Increasing evidence implicates inhibition of vesicular trafficking between the ER-Golgi in ALS, which may explain the previous observations of Golgi fragmentation.
Impairment of Cellular Trafficking is A Trigger for Golgi Fragmentation in ALS
The ER-Golgi compartments form the first part of the cellular secretory pathway, hence they are sensitive to alterations in the rate of trafficking (Pelletier et al., 2000; Lee et al., 2004), and trafficking inhibition leads to dysfunction in both compartments. In the ER, accumulation of unfolded nascent secretory proteins in the lumen triggers the UPR. The UPR initially aims to reduce protein synthesis and increase protein folding (Graves et al., 2001; Preston et al., 2009). However, when impairment of trafficking persists, prolonged UPR results in activation of apoptosis (Szegezdi et al., 2006; Hetz, 2012). Similarly the organization of the Golgi depends on efficient bidirectional vesicular transport with the ER (Nassif et al., 2010). The formation of Golgi stacks requires continuous recycling of Golgi proteins to/from the ER (Lippincott-Schwartz et al., 2000). Inhibition of protein export from the ER disrupts Golgi organization (Storrie et al., 1998), resulting in the formation of tubulovesicular Golgi clusters, some of which fuse with the ER (Puri and Linstedt, 2003), which can further increase ER stress. Similarly, inhibition of vesicular trafficking from the Golgi to plasma membrane leads to protein accumulation within the Golgi. If prolonged, this can fragment the Golgi (Persson et al., 1992; Zolov and Lupashin, 2005; Zhou et al., 2013). Approximately one-third of the human proteome transverses through the Golgi destined for transmembrane, synaptic, axonal, or extracellular locations (Braakman and Bulleid, 2011). Hence disruption to intracellular trafficking involving the Golgi could severely compromise neuronal function and viability.
We recently demonstrated that Golgi-associated vesicular trafficking is inhibited in cells expressing ALS-mutant proteins: SOD1, FUS, TDP-43, and optineurin, providing an intriguing mechanism explaining Golgi fragmentation in patient tissues (Sundaramoorthy et al., 2013, 2015; Atkin et al., 2014; Soo et al., 2015a). Furthermore, inhibition of ER-Golgi transport by mutant SOD1 preceded all other cellular pathologies examined in neuronal cells, including ER stress, Golgi fragmentation, protein aggregation, inclusion formation, and apoptosis (Atkin et al., 2014). This implies that ER-Golgi trafficking defects may trigger ER-Golgi pathology in SOD1-ALS cases. More recently we demonstrated that mutant forms of both FUS and TDP-43 impair the incorporation of secretory cargo into COPII vesicles budding off from the ER, impeding protein export from the ER, while mutant SOD1 was shown to inhibit ERGIC-Golgi trafficking by destabilizing microtubules (Soo et al., 2015a). Furthermore, we have also demonstrated that misfolded WT SOD1 also impairs ER-Golgi trafficking similar to mutant SOD1, resulting in ER stress and Golgi fragmentation (Sundaramoorthy et al., 2015), although it remains controversial whether misfolded WT SOD1 is present in sporadic ALS tissues (Liu et al., 2009; Forsberg et al., 2010; Grad et al., 2014). However, it is tempting to speculate that impairment of ER-Golgi trafficking is a common trigger for Golgi fragmentation in sporadic and familial ALS. Similarly, we have also shown that expression of ALS-optineurin mutants impair myosin VI-mediated protein trafficking from the Golgi to plasma membrane, also inducing Golgi fragmentation (Sundaramoorthy et al., 2015). Hence these results imply that impairment of distinct protein trafficking pathways by different ALS-linked proteins are specific triggers for Golgi fragmentation in ALS (Figure 1).
Consistent with this notion, mutations in genes encoding proteins directly involved in intracellular trafficking are present in familial ALS (Table 1). Firstly, mutations in the gene encoding the p150Glued subunit of the dynein/dynactin complex were reported in sporadic and familial ALS (Münch et al., 2004). The ALS causing mutation impedes binding of p150Glued to microtubules, resulting in dysfunctional dynein/dynactin-mediated transport (Levy et al., 2006). Similarly, mutations in proteins directly involved in the ER-Golgi secretory pathway, including VAPB and VCP, are present in ALS (Nishimura et al., 2004; Johnson et al., 2010; Yi et al., 2012; Kuijpers et al., 2013). Recent findings of mutations in ALS-associated genes that encode cytoskeletal associated proteins provide additional evidence for trafficking disruption in ALS. Mutations in profilin 1, which mediates the conversion of soluble G-actin to functional F-actin, (Wu et al., 2012), and in tubulin alpha 4A (TUBA4A), a component of microtubules, were recently reported in familial ALS (Smith et al., 2014).
The identification of hexanucleotide (GGGGCC) repeat expansion mutations in C9ORF72 as the major cause of familial ALS and FTD (40%), further links cellular trafficking to ALS. Whilst the normal cellular function of C9orf72 was initially unknown, bioinformatics studies first predicted that C9orf72 functions in Rab-mediated trafficking (Zhang et al., 2012; Levine et al., 2013). Rab proteins form a large family of small guanosine triphosphate (GTP)ases that regulate vesicular trafficking at distinct cellular membranes (Stenmark and Olkkonen, 2001). Rab proteins are activated by conversion from an inactive guanosine diphosphate (GDP)-bound state to an active GTP-bound form, which is catalyzed by guanine nucleotide exchange factors (GEFs) (Stenmark and Olkkonen, 2001; Cherfils and Zeghouf, 2013). Bioinformatics predicted that C9orf72 functions as a RabGEF, because of the strong sequence and structural similarity to other evolutionary conserved differentially expressed in normal and neoplastic cells (DENN) domain-containing RabGEFs (Zhang et al., 2012; Levine et al., 2013). Consistent with these predictions, we demonstrated that C9orf72 associates with multiple Rabs including Rab1, which mediates ER-Golgi transport (Farg et al., 2014). Furthermore, we also found that depletion of C9orf72 using siRNA impaired autophagy and endocytic trafficking from the plasma membrane to Golgi (Farg et al., 2014). Whilst the hexanucleotide repeat expansion is present within an intronic region of C9ORF72, expression of C9orf72 protein is reduced in ALS patients causing haploinsufficiency (DeJesus-Hernandez et al., 2011; Haeusler et al., 2014). Hence this would disrupt the normal trafficking function of C9orf72 in ALS. However, recent studies have argued against this mechanism of pathogenesis (Koppers et al., 2015). Nevertheless, we also demonstrated increased association of C9orf72 with Rab7 and Rab11 in C9orf72-ALS patients, implying that intracellular trafficking is dysregulated in C9orf72-ALS, although the mechanism remains unclear (Farg et al., 2014). However, further studies are required to examine the relationship between C9orf72 and trafficking defects, including whether the Golgi is fragmented in C9orf72-ALS patients.
Golgi Fragmentation and Autophagy Dysfunction
The initial step in autophagy is the formation of a double-membraned phagophore, which then expands in size, engulfing defective proteins, damaged cellular organelles, or pathogens, forming the autophagosome (Reggiori and Klionsky, 2005). Although the membranes forming the autophagosome originate from multiple cellular organelles, the ER is implicated as the primary source of membrane because the omegasome, the autophagosome precursor, originates from ER cisternae (Hayashi-Nishino et al., 2009). However, the Golgi is necessary for subsequent autophagosome elongation, and Golgi-mediated trafficking provides membrane components for autophagosome biogenesis. Beclin1, which is located in the trans-Golgi network, recruits other autophagy-related (Atg) proteins for assembly into the autophagosome, (Kihara et al., 2001) and Atg9-positive vesicles cycle from the Golgi to deliver membranes to the developing autophagosome (Young et al., 2006; Webber et al., 2007). Blocking ER to cis-Golgi transport, or transport from the trans-Golgi to plasma membrane/endosomes, reduces autophagosome formation in mammalian cells (Zoppino et al., 2010; Guo et al., 2012). Furthermore, the Golgi recognizes and sorts lysosomal enzymes, which are then packaged into vesicles that bud from the trans-Golgi, forming lysosomes (Griffiths et al., 1988; Kornfeld and Mellman, 1989; Riederer et al., 1994). Hence these observations imply that disruption of Golgi-associated trafficking may impair autophagosome formation.
In contrast, fragmentation of the Golgi has also been shown to increase autophagosome biogenesis by feeding Atg9-positive fragmented Golgi membranes during starvation-induced autophagy (Takahashi et al., 2011). Pharmacological induction of Golgi fragmentation with Brefeldin A or Golgicide increases autophagosome biogenesis and induces accumulation of autophagosomes (Naydenov et al., 2012), but it can also block autophagosome formation in some cases (Nishida et al., 2009). Observations of Golgi fragmentation and autophagy dysfunction imply a possible link between these two pathologies in ALS. Hence examination of the pathological relationship between Golgi fragmentation and autophagy in ALS is warranted.
Golgi Fragmentation and Axonal Homeostasis
Motor neurons differ from other neurons in that they are exceptionally large, with long axons, up to 1 m in length in an adult human. These distal axons require membrane and cytoskeletal proteins, neurotransmitter receptors, and lipids to maintain synaptic plasticity, synaptogenesis, excitability, dendritic, and neurite outgrowth (Horton and Ehlers, 2004; Tuck and Cavalli, 2010; Ori-McKenney et al., 2012). These components must be transported from the ER/Golgi in the cell body over long distances along the axon. In addition to this traditional route, proteins are also synthesized via axonal ribosomes (Koenig et al., 2000; Kun et al., 2007) and mRNA (Taylor et al., 2009; Jung et al., 2012), thus facilitating local protein synthesis. Proteins synthesized in neurites are processed and secreted via Golgi outposts (Horton and Ehlers, 2003; Merianda et al., 2009) (Figure 1). These Golgi outposts share similar molecular markers to the somatic Golgi (Gardiol et al., 1999; Horton and Ehlers, 2003) and they handle secretion of essential axonal/dendritic membrane proteins (Lu et al., 2001; Passafaro et al., 2001). Similarly, cytoskeletal proteins processed via Golgi outposts are essential for axonal regeneration and plasticity of dendritic spines (Matus, 2000; Gu et al., 2008; Tuck and Cavalli, 2010; Shirao and González−Billault, 2013). Fragmentation of the neuronal Golgi would therefore be expected to impair normal axonal functions. In support of this notion, induction of Golgi fragmentation with Brefeldin A reduces synaptic potentiation and AMPA receptor expression on the postsynaptic membrane (Broutman and Baudry, 2001), and reduces axonal outgrowth (Jareb and Banker, 1997). Interestingly, fragmentation of somatic and dendritic Golgi in motor neurons accompanied by trafficking defects, preceded axonal retraction and muscle denervation in mice models of ALS (van Dis et al., 2014). Therefore, Golgi fragmentation may be an important trigger for loss of axonal homeostasis and degeneration of motor neurons in ALS.
Conclusion
An emerging concept in ALS is that the diverse mechanisms implicated in pathology are inter-linked, and that disturbances in one pathway induce other pathogenic mechanisms, resulting in neurodegeneration. Increasing evidence links Golgi fragmentation to recent pathological mechanisms implicated in ALS, including disruption of intracellular trafficking and ER stress. This warrants future studies examining the relationship between Golgi fragmentation to other somatic Golgi functions, including autophagy, and specific neuronal functions of the Golgi, such as axonal homeostasis (Figure 1). The unique characteristics of neurons and the existence of Golgi outposts may confer additional, more specialized functions of the Golgi in these cells which may render these cells more vulnerable to neurodegeneration in ALS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia (NHMRC) Project grants (1006141, 1030513, and 1086887), Bethlehem Griffiths Research Foundation, and Motor Neurone Disease Research Institute of Australia Angie Cunningham Laugh to Cure MND Grant, Zo-ee Research Grant and Grant-in-Aid.
References
Alami, N. H., Smith, R. B., Carrasco, M. A., Williams, L. A., Winborn, C. S., Han, S. S., et al. (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. doi: 10.1016/j.neuron.2013.12.018
Appenzeller-Herzog, C., and Hauri, H.-P. (2006). The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J. Cell Sci. 119, 2173–2183. doi: 10.1242/jcs.03019
Arita, M., Wakita, T., and Shimizu, H. (2012). Valosin-containing protein (VCP/p97) is required for poliovirus replication and is involved in cellular protein secretion pathway in poliovirus infection. J. Virol. 86, 5541–5553. doi: 10.1128/JVI.00114-12
Atkin, J. D., Farg, M. A., Soo, K. Y., Walker, A. K., Halloran, M., Turner, B. J., et al. (2014). Mutant SOD1 inhibits ER−Golgi transport in amyotrophic lateral sclerosis. J. Neurochem. 129, 190–204. doi: 10.1111/jnc.12493
Atkin, J. D., Farg, M. A., Turner, B. J., Tomas, D., Lysaght, J. A., Nunan, J., et al. (2006). Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 281, 30152–30165. doi: 10.1074/jbc.M603393200
Atkin, J. D., Farg, M. A., Walker, A. K., McLean, C., Tomas, D., and Horne, M. K. (2008). Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 30, 400–407. doi: 10.1016/j.nbd.2008.02.009
Baeuerle, P. A., and Huttner, W. B. (1987). Tyrosine sulfation is a trans-Golgi-specific protein modification. J. Cell Biol. 105, 2655–2664. doi: 10.1083/jcb.105.6.2655
Ballar, P., Pabuccuoglu, A., and Kose, F. A. (2011). Different p97/VCP complexes function in retrotranslocation step of mammalian ER-associated degradation (ERAD). Int. J. Biochem. Cell Biol. 43, 613–621. doi: 10.1016/j.biocel.2010.12.021
Bilsland, L. G., Sahai, E., Kelly, G., Golding, M., Greensmith, L., and Schiavo, G. (2010). Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 20523–20528. doi: 10.1073/pnas.1006869107
Blokhuis, A. M., Groen, E. J., Koppers, M., van den Berg, L. H., and Pasterkamp, R. J. (2013). Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 777–794. doi: 10.1007/s00401-013-1125-6
Braakman, I., and Bulleid, N. J. (2011). Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 80, 71–99. doi: 10.1146/annurev-biochem-062209-093836
Broutman, G., and Baudry, M. (2001). Involvement of the secretory pathway for AMPA receptors in NMDA-induced potentiation in hippocampus. J. Neurosci. 21, 27–34.
Calderilla-Barbosa, L., Seibenhener, M. L., Du, Y., Diaz-Meco, M.-T., Moscat, J., Yan, J., et al. (2014). Interaction of SQSTM1 with the motor protein dynein–SQSTM1 is required for normal dynein function and trafficking. J. Cell Sci. 127, 4052–4063. doi: 10.1242/jcs.152363
Cherfils, J., and Zeghouf, M. (2013). Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 93, 269–309. doi: 10.1152/physrev.00003.2012
Chow, C. Y., Zhang, Y., Dowling, J. J., Jin, N., Adamska, M., Shiga, K., et al. (2007). Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448, 68–72. doi: 10.1038/nature05876
Cole, N. B., Sciaky, N., Marotta, A., Song, J., and Lippincott-Schwartz, J. (1996). Golgi dispersal during microtubule disruption: regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol. Biol. Cell 7, 631–650. doi: 10.1091/mbc.7.4.631
Dadon-Nachum, M., Melamed, E., and Offen, D. (2011). The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 43, 470–477. doi: 10.1007/s12031-010-9467-1
Dascher, C., and Balch, W. E. (1994). Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J. Biol. Chem. 269, 1437–1448.
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes Chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
De Matteis, M. A., and Luini, A. (2008). Exiting the Golgi complex. Nat. Rev. Mol. Cell Biol. 9, 273–284. doi: 10.1038/nrm2378
Deng, H. X., Zhai, H., Bigio, E. H., Yan, J., Fecto, F., Ajroud, K., et al. (2010). FUS−immunoreactive inclusions are a common feature in sporadic and non−SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 67, 739–748. doi: 10.1002/ana.22051
Farg, M. A., Soo, K. Y., Walker, A. K., Pham, H., Orian, J., Horne, M. K., et al. (2012). Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 33, 2855–2868. doi: 10.1016/j.neurobiolaging.2012.02.009
Farg, M. A., Soo, K. Y., Warraich, S. T., Sundaramoorthy, V., Blair, I. P., and Atkin, J. D. (2013). Ataxin-2 interacts with FUS and intermediate-length polyglutamine expansions enhance FUS-related pathology in amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 717–728. doi: 10.1093/hmg/dds479
Farg, M. A., Sundaramoorthy, V., Sultana, J. M., Yang, S., Atkinson, R. A., Levina, V., et al. (2014). C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 23, 3579–3595. doi: 10.1093/hmg/ddu068
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. doi: 10.1016/j.expneurol.2003.10.004
Forsberg, K., Jonsson, P. A., Andersen, P. M., Bergemalm, D., Graffmo, K. S., Hultdin, M., et al. (2010). Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS ONE 5:e11552. doi: 10.1371/journal.pone.0011552
Frey, D., Schneider, C., Xu, L., Borg, J., Spooren, W., and Caroni, P. (2000). Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J. Neurosci. 20, 2534–2542.
Fujita, Y., Mizuno, Y., Takatama, M., and Okamoto, K. (2008). Anterior horn cells with abnormal TDP-43 immunoreactivities show fragmentation of the Golgi apparatus in ALS. J. Neurol. Sci. 269, 30–34. doi: 10.1016/j.jns.2007.12.016
Fujita, Y., Ohama, E., Takatama, M., Al-Sarraj, S., and Okamoto, K. (2006). Fragmentation of Golgi apparatus of nigral neurons with α-synuclein-positive inclusions in patients with Parkinson's disease. Acta Neuropathol. 112, 261–265. doi: 10.1007/s00401-006-0114-4
Fujita, Y., Okamoto, K., Sakurai, A., Amari, M., Nakazato, Y., and Gonatas, N. K. (1999). Fragmentation of the Golgi apparatus of Betz cells in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 163, 81–85. doi: 10.1016/S0022-510X(99)00014-3
Fujita, Y., Okamoto, K., Sakurai, A., Gonatas, N. K., and Hirano, A. (2000). Fragmentation of the Golgi apparatus of the anterior horn cells in patients with familial amyotrophic lateral sclerosis with SOD1 mutations and posterior column involvement. J. Neurol. Sci. 174, 137–140. doi: 10.1016/S0022-510X(00)00265-3
Gardiol, A., Racca, C., and Triller, A. (1999). Dendritic and postsynaptic protein synthetic machinery. J. Neurosci. 19, 168–179.
Glick, B. S. (2002). Can the Golgi form de novo? Nat. Rev. Mol. Cell Biol. 3, 615–619. doi: 10.1038/nrm877
Glick, B. S., and Nakano, A. (2009). Membrane traffic within the Golgi apparatus. Annu. Rev. Cell Dev. Biol. 25, 113. doi: 10.1146/annurev.cellbio.24.110707.175421
Gonatas, N. K., Stieber, A., and Gonatas, J. O. (2006). Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. J. Neurol. Sci. 246, 21–30. doi: 10.1016/j.jns.2006.01.019
Gonatas, N. K., Stieber, A., Mourelatos, Z., Chen, Y., Gonatas, J. O., Appel, S. H., et al. (1992). Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis. Am. J. Pathol. 140, 731.
Gosavi, N., Lee, H.-J., Lee, J. S., Patel, S., and Lee, S.-J. (2002). Golgi fragmentation occurs in the cells with prefibrillar α-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 277, 48984–48992. doi: 10.1074/jbc.M208194200
Grad, L. I., Yerbury, J. J., Turner, B. J., Guest, W. C., Pokrishevsky, E., O'Neill, M. A., et al. (2014). Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and-independent mechanisms. Proc. Natl. Acad. Sci. U.S.A. 111, 3620–3625. doi: 10.1073/pnas.1312245111
Graves, T. K., Patel, S., Dannies, P. S., and Hinkle, P. M. (2001). Misfolded growth hormone causes fragmentation of the Golgi apparatus and disrupts endoplasmic reticulum-to-Golgi traffic. J. Cell Sci. 114, 3685–3694.
Griffiths, G., Fuller, S. D., Back, R., Hollinshead, M., Pfeiffer, S., and Simons, K. (1989). The dynamic nature of the Golgi complex. J. Cell Biol. 108, 277–297. doi: 10.1083/jcb.108.2.277
Griffiths, G., Hoflack, B., Simons, K., Mellman, I., and Kornfeld, S. (1988). The mannose 6-phosphate receptor and the biogenesis of lysosomes. Cell 52, 329–341. doi: 10.1016/S0092-8674(88)80026-6
Gu, J., Firestein, B. L., and Zheng, J. Q. (2008). Microtubules in dendritic spine development. J. Neurosci. 28, 12120–12124. doi: 10.1523/JNEUROSCI.2509-08.2008
Guo, Y., Chang, C., Huang, R., Liu, B., Bao, L., and Liu, W. (2012). AP1 is essential for generation of autophagosomes from the trans-Golgi network. J. Cell Sci. 125, 1706–1715. doi: 10.1242/jcs.093203
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775. doi: 10.1126/science.8209258
Haeusler, A. R., Donnelly, C. J., Periz, G., Simko, E. A., Shaw, P. G., Kim, M.-S., et al. (2014). C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200. doi: 10.1038/nature13124
Hayashi-Nishino, M., Fujita, N., Noda, T., Yamaguchi, A., Yoshimori, T., and Yamamoto, A. (2009). A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 11, 1433–1437. doi: 10.1038/ncb1991
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Hirokawa, N., and Takemura, R. (2005). Molecular motors and mechanisms of directional transport in neurons. Nat. Rev. Neurosci. 6, 201–214. doi: 10.1038/nrn1624
Horton, A. C., and Ehlers, M. D. (2003). Dual modes of endoplasmic reticulum-to-Golgi transport in dendrites revealed by live-cell imaging. J. Neurosci. 23, 6188–6199.
Horton, A. C., and Ehlers, M. D. (2004). Secretory trafficking in neuronal dendrites. Nat. Cell Biol. 6, 585–591. doi: 10.1038/ncb0704-585
Ikenaka, K., Katsuno, M., Kawai, K., Ishigaki, S., Tanaka, F., and Sobue, G. (2012). Disruption of axonal transport in motor neuron diseases. Int. J. Mol. Sci. 13, 1225–1238. doi: 10.3390/ijms13011225
Ilieva, E. V., Ayala, V., Jové, M., Dalfó, E., Cacabelos, D., Povedano, M., et al. (2007). Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 130, 3111–3123. doi: 10.1093/brain/awm190
Ito, H., Nakamura, M., Komure, O., Ayaki, T., Wate, R., Maruyama, H., et al. (2011). Clinicopathologic study on an ALS family with a heterozygous E478G optineurin mutation. Acta Neuropathol. 122, 223–229. doi: 10.1007/s00401-011-0842-y
Jareb, M., and Banker, G. (1997). Inhibition of axonal growth by brefeldin A in hippocampal neurons in culture. J. Neurosci. 17, 8955–8963.
Jiang, J., Parameshwaran, K., Seibenhener, M. L., Kang, M. G., Suppiramaniam, V., Huganir, R. L., et al. (2009). AMPA receptor trafficking and synaptic plasticity require SQSTM1/p62. Hippocampus 19, 392–406. doi: 10.1002/hipo.20528
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi: 10.1016/j.neuron.2010.11.036
Jung, H., Yoon, B. C., and Holt, C. E. (2012). Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat. Rev. Neurosci. 13, 308–324. doi: 10.1038/nrn3274
Kihara, A., Kabeya, Y., Ohsumi, Y., and Yoshimori, T. (2001). Beclin–phosphatidylinositol 3−kinase complex functions at the trans−Golgi network. EMBO Rep. 2, 330–335. doi: 10.1093/embo-reports/kve061
Koenig, E., Martin, R., Titmus, M., and Sotelo-Silveira, J. R. (2000). Cryptic peripheral ribosomal domains distributed intermittently along mammalian myelinated axons. J. Neurosci. 20, 8390–8400.
Koppers, M., Blokhuis, A. M., Westeneng, H. J., Terpstra, M. L., Zundel, C. A., Vieira de Sá, R., et al. (2015). C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 78, 426–438. doi: 10.1002/ana.24453
Kornfeld, S., and Mellman, I. (1989). The biogenesis of lysosomes. Annu. Rev. Cell Biol. 5, 483–525. doi: 10.1146/annurev.cb.05.110189.002411
Kuijpers, M., Yu, K. L., Teuling, E., Akhmanova, A., Jaarsma, D., and Hoogenraad, C. C. (2013). The ALS8 protein VAPB interacts with the ER–Golgi recycling protein YIF1A and regulates membrane delivery into dendrites. EMBO J. 32, 2056–2072. doi: 10.1038/emboj.2013.131
Kun, A., Otero, L., Sotelo-Silveira, J. R., and Sotelo, J. R. (2007). Ribosomal distributions in axons of mammalian myelinated fibers. J. Neurosci. Res. 85, 2087–2098. doi: 10.1002/jnr.21340
Kwiatkowski, T. J. Jr. Bosco, D., Leclerc, A., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Lane, J. D., Lucocq, J., Pryde, J., Barr, F. A., Woodman, P. G., Allan, V. J., et al. (2002). Caspase-mediated cleavage of the stacking protein GRASP65 is required for Golgi fragmentation during apoptosis. J. Cell Biol. 156, 495–509. doi: 10.1083/jcb.200110007
Lee, M. C., Miller, E. A., Goldberg, J., Orci, L., and Schekman, R. (2004). Bi-directional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 20, 87–123. doi: 10.1146/annurev.cellbio.20.010403.105307
Levine, T. P., Daniels, R. D., Gatta, A. T., Wong, L. H., and Hayes, M. J. (2013). The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29, 499–503. doi: 10.1093/bioinformatics/bts725
Levy, J. R., Sumner, C. J., Caviston, J. P., Tokito, M. K., Ranganathan, S., Ligon, L. A., et al. (2006). A motor neuron disease–associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J. Cell Biol. 172, 733–745. doi: 10.1083/jcb.200511068
Liazoghli, D., Perreault, S., Micheva, K. D., Desjardins, M., and Leclerc, N. (2005). Fragmentation of the Golgi apparatus induced by the overexpression of wild-type and mutant human tau forms in neurons. Am. J. Pathol. 166, 1499–1514. doi: 10.1016/S0002-9440(10)62366-8
Ling, S.-C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Linstedt, A. D. (2004). Positioning the Golgi apparatus. Cell 118, 271–272. doi: 10.1016/j.cell.2004.07.015
Lippincott-Schwartz, J., Roberts, T. H., and Hirschberg, K. (2000). Secretory protein trafficking and organelle dynamics in living cells 1. Annu. Rev. Cell Dev. Biol. 16, 557–589. doi: 10.1146/annurev.cellbio.16.1.557
Liu, H. N., Sanelli, T., Horne, P., Pioro, E. P., Strong, M. J., Rogaeva, E., et al. (2009). Lack of evidence of monomer/misfolded superoxide dismutase−1 in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 66, 75–80. doi: 10.1002/ana.21704
Liu, Q., Xie, F., Siedlak, S. L., Nunomura, A., Honda, K., Moreira, P. I., et al. (2004). Neurofilament proteins in neurodegenerative diseases. Cell. Mol. Life Sci. 61, 3057–3075. doi: 10.1007/s00018-004-4268-8
Lu, W.-Y., Man, H.-Y., Ju, W., Trimble, W. S., MacDonald, J. F., and Wang, Y. T. (2001). Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29, 243–254. doi: 10.1016/S0896-6273(01)00194-5
Matus, A. (2000). Actin-based plasticity in dendritic spines. Science 290, 754–758. doi: 10.1126/science.290.5492.754
Merianda, T. T., Lin, A. C., Lam, J. S., Vuppalanchi, D., Willis, D. E., Karin, N., et al. (2009). A functional equivalent of endoplasmic reticulum and Golgi in axons for secretion of locally synthesized proteins. Mol. Cell. Neurosci. 40, 128–142. doi: 10.1016/j.mcn.2008.09.008
Moloney, E. B., de Winter, F., and Verhaagen, J. (2014). ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 8:252. doi: 10.3389/fnins.2014.00252
Mourelatos, Z., Adler, H., Hirano, A., Donnenfeld, H., Gonatas, J. O., and Gonatas, N. K. (1990). Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis revealed by organelle-specific antibodies. Proc. Natl. Acad. Sci. U.S.A. 87, 4393–4395. doi: 10.1073/pnas.87.11.4393
Mourelatos, Z., Gonatas, N. K., Stieber, A., Gurney, M. E., and Dal Canto, M. C. (1996). The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu, Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc. Natl. Acad. Sci. U.S.A. 93, 5472–5477. doi: 10.1073/pnas.93.11.5472
Mourelatos, Z., Hirano, A., Rosenquist, A. C., and Gonatas, N. K. (1994). Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis (ALS). Clinical studies in ALS of Guam and experimental studies in deafferented neurons and in beta, beta'-iminodipropionitrile axonopathy. Am. J. Pathol. 144, 1288.
Münch, C., Sedlmeier, R., Meyer, T., Homberg, V., Sperfeld, A. D., Kurt, A., et al. (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724–726. doi: 10.1212/01.WNL.0000134608.83927.B1
Nassif, M., Matus, S., Castillo, K., and Hetz, C. (2010). Amyotrophic lateral sclerosis pathogenesis: a journey through the secretory pathway. Antioxid. Redox Signal. 13, 1955–1989. doi: 10.1089/ars.2009.2991
Naydenov, N. G., Harris, G., Morales, V., and Ivanov, A. I. (2012). Loss of a membrane trafficking protein αSNAP induces non-canonical autophagy in human epithelia. Cell Cycle 11, 4613–4625. doi: 10.4161/cc.22885
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nishida, Y., Arakawa, S., Fujitani, K., Yamaguchi, H., Mizuta, T., Kanaseki, T., et al. (2009). Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461, 654–658. doi: 10.1038/nature08455
Nishimura, A. L., Mitne-Neto, M., Silva, H. C., Richieri-Costa, A., Middleton, S., Cascio, D., et al. (2004). A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 822–831. doi: 10.1086/425287
Oakley, B. R. (2000). An abundance of tubulins. Trends Cell Biol. 10, 537–542. doi: 10.1016/S0962-8924(00)01857-2
Oblinger, M. M., Wong, J., and Parysek, L. M. (1989). Axotomy-induced changes in the expression of a type III neuronal intermediate filament gene. J. Neurosci. 9, 3766–3775.
Oh, Y. K., Shin, K. S., Yuan, J., and Kang, S. J. (2008). Superoxide dismutase 1 mutants related to amyotrophic lateral sclerosis induce endoplasmic stress in neuro2a cells. J. Neurochem. 104, 993–1005. doi: 10.1111/j.1471-4159.2007.05053.x
Ori-McKenney, K. M., Jan, L. Y., and Jan, Y.-N. (2012). Golgi outposts shape dendrite morphology by functioning as sites of acentrosomal microtubule nucleation in neurons. Neuron 76, 921–930. doi: 10.1016/j.neuron.2012.10.008
Otomo, A., Hadano, S., Okada, T., Mizumura, H., Kunita, R., Nishijima, H., et al. (2003). ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum. Mol. Genet. 12, 1671–1687. doi: 10.1093/hmg/ddg184
Otomo, A., Pan, L., and Hadano, S. (2012). Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurol. Res. Int. 2012:498428. doi: 10.1155/2012/498428
Oyanagi, K., Yamazaki, M., Takahashi, H., Watabe, K., Wada, M., Komori, T., et al. (2008). Spinal anterior horn cells in sporadic amyotrophic lateral sclerosis show ribosomal detachment from, and cisternal distention of the rough endoplasmic reticulum. Neuropathol. Appl. Neurobiol. 34, 650–658. doi: 10.1111/j.1365-2990.2008.00941.x
Pabba, M., Wong, A. Y., Ahlskog, N., Hristova, E., Biscaro, D., Nassrallah, W., et al. (2014). NMDA receptors are upregulated and trafficked to the plasma membrane after Sigma-1 receptor activation in the rat hippocampus. J. Neurosci. 34, 11325–11338. doi: 10.1523/JNEUROSCI.0458-14.2014
Pantaloni, D., and Carlier, M.-F. (1993). How profilin promotes actin filament assembly in the presence of thymosin β4. Cell 75, 1007–1014. doi: 10.1016/0092-8674(93)90544-Z
Passafaro, M., Piëch, V., and Sheng, M. (2001). Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat. Neurosci. 4, 917–926. doi: 10.1038/nn0901-917
Pelletier, L., Jokitalo, E., and Warren, G. (2000). The effect of Golgi depletion on exocytic transport. Nat. Cell Biol. 2, 840–846. doi: 10.1038/35041089
Pérez-Brangulí, F., Mishra, H. K., Prots, I., Havlicek, S., Kohl, Z., Saul, D., et al. (2014). Dysfunction of spatacsin leads to axonal pathology in SPG11 linked hereditary spastic paraplegia. Hum. Mol. Genet. 23, 4859–4874. doi: 10.1093/hmg/ddu200
Perlson, E., Maday, S., Fu, M.-M., Moughamian, A. J., and Holzbaur, E. L. (2010). Retrograde axonal transport: pathways to cell death? Trends Neurosci. 33, 335–344. doi: 10.1016/j.tins.2010.03.006
Persson, R., Schnell, C. R., Borg, L. A., and Fries, E. (1992). Accumulation of Golgi-processed secretory proteins in an organelle of high density upon reduction of ATP concentration in rat hepatocytes. J. Biol. Chem. 267, 2760–2766.
Polishchuk, R. S., and Mironov, A. (2004). Structural aspects of Golgi function. Cell. Mol. Life Sci. 61, 146–158. doi: 10.1007/s00018-003-3353-8
Preston, A. M., Gurisik, E., Bartley, C., Laybutt, D. R., and Biden, T. J. (2009). Reduced endoplasmic reticulum (ER)-to-Golgi protein trafficking contributes to ER stress in lipotoxic mouse beta cells by promoting protein overload. Diabetologia 52, 2369–2373. doi: 10.1007/s00125-009-1506-5
Puri, S., and Linstedt, A. D. (2003). Capacity of the Golgi apparatus for biogenesis from the endoplasmic reticulum. Mol. Biol. Cell 14, 5011–5018. doi: 10.1091/mbc.E03-06-0437
Rambourg, A., and Clermont, Y. (1990). Three-dimensional electron microscopy: structure of the Golgi apparatus. Eur. J. Cell Biol. 51, 189.
Reggiori, F., and Klionsky, D. J. (2005). Autophagosomes: biogenesis from scratch? Curr. Opin. Cell Biol. 17, 415–422. doi: 10.1016/j.ceb.2005.06.007
Renton, A. E., Chiò, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23. doi: 10.1038/nn.3584
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Riederer, M. A., Soldati, T., Shapiro, A. D., Lin, J., and Pfeffer, S. R. (1994). Lysosome biogenesis requires Rab9 function and receptor recycling from endosomes to the trans-Golgi network. J. Cell Biol. 125, 573–582. doi: 10.1083/jcb.125.3.573
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Ross, J. L., Shuman, H., Holzbaur, E. L., and Goldman, Y. E. (2008). Kinesin and dynein-dynactin at intersecting microtubules: motor density affects dynein function. Biophys. J. 94, 3115–3125. doi: 10.1529/biophysj.107.120014
Rothman, J. E., and Wieland, F. T. (1996). Protein sorting by transport vesicles. Science 272, 227–234. doi: 10.1126/science.272.5259.227
Ruscher, K., and Wieloch, T. (2015). The involvement of the sigma-1 receptor in neurodegeneration and neurorestoration. J. Pharmacol. Sci. 127, 30–35. doi: 10.1016/j.jphs.2014.11.011
Sahlender, D. A., Roberts, R. C., Arden, S. D., Spudich, G., Taylor, M. J., Luzio, J. P., et al. (2005). Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J. Cell Biol. 169, 285–295. doi: 10.1083/jcb.200501162
Sakurai, A., Okamoto, K., Fujita, Y., Nakazato, Y., Wakabayashi, K., Takahashi, H., et al. (2000). Fragmentation of the Golgi apparatus of the ballooned neurons in patients with corticobasal degeneration and Creutzfeldt-Jakob disease. Acta Neuropathol. 100, 270–274. doi: 10.1007/s004010000182
Sakurai, A., Okamoto, K., Yaguchi, M., Fujita, Y., Mizuno, Y., Nakazato, Y., et al. (2002). Pathology of the inferior olivary nucleus in patients with multiple system atrophy. Acta Neuropathol. 103, 550–554. doi: 10.1007/s00401-001-0500-x
Sasaki, S. (2011). Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 70, 349–359. doi: 10.1097/NEN.0b013e3182160690
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype–selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Schroer, T. A. (2004). Dynactin. Annu. Rev. Cell Dev. Biol. 20, 759–779. doi: 10.1146/annurev.cellbio.20.012103.094623
Schwarz, T. L. (2013). Mitochondrial trafficking in neurons. Cold Spring Harb. Perspect. Biol. 5:a011304. doi: 10.1101/cshperspect.a011304
Shirao, T., and González-Billault, C. (2013). Actin filaments and microtubules in dendritic spines. J. Neurochem. 126, 155–164. doi: 10.1111/jnc.12313
Skehel, P. A., Fabian-Fine, R., and Kandel, E. R. (2000). Mouse VAP33 is associated with the endoplasmic reticulum and microtubules. Proc. Natl. Acad. Sci. U.S.A. 97, 1101–1106. doi: 10.1073/pnas.97.3.1101
Smith, B. N., Ticozzi, N., Fallini, C., Gkazi, A. S., Topp, S., Kenna, K. P., et al. (2014). Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84, 324–331. doi: 10.1016/j.neuron.2014.09.027
Soo, K. Y., Halloran, M., Sundaramoorthy, V., Parakh, S., Toth, R. P., Southam, K. A., et al. (2015a). Rab1-dependent ER–Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP-43 and FUS-associated ALS. Acta Neuropathol. 130, 679–697. doi: 10.1007/s00401-015-1468-2
Soo, K. Y., Sultana, J., King, A. E., Atkinson, R. A. K., Warraich, S. T., et al. (2015b). ALS-associated mutant FUS inhibits macroautophagy which is restored by overexpression of Rab1. Cell Death Discovery 1, 15030. doi: 10.1038/cddiscovery.2015.30
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Stanley, P. (2011). Golgi glycosylation. Cold Spring Harb. Perspect. Biol. 3:a005199. doi: 10.1101/cshperspect.a005199
Stenmark, H., and Olkkonen, V. M. (2001). The rab gtpase family. Genome Biol. 2:S3007. doi: 10.1186/gb-2001-2-5-reviews3007
Stieber, A., Gonatas, J. O., Moore, J. S., Bantly, A., Yim, H.-S., Yim, M. B., et al. (2004). Disruption of the structure of the Golgi apparatus and the function of the secretory pathway by mutants G93A and G85R of Cu, Zn superoxide dismutase (SOD1) of familial amyotrophic lateral sclerosis. J. Neurol. Sci. 219, 45–53. doi: 10.1016/j.jns.2003.12.004
Storrie, B., White, J., Röttger, S., Stelzer, E. H., Suganuma, T., and Nilsson, T. (1998). Recycling of Golgi-resident glycosyltransferases through the ER reveals a novel pathway and provides an explanation for nocodazole-induced Golgi scattering. J. Cell Biol. 143, 1505–1521. doi: 10.1083/jcb.143.6.1505
Sun, K.-H., de Pablo, Y., Vincent, F., Johnson, E. O., Chavers, A. K., and Shah, K. (2008). Novel genetic tools reveal Cdk5's major role in Golgi fragmentation in Alzheimer's disease. Mol. Biol. Cell 19, 3052–3069. doi: 10.1091/mbc.E07-11-1106
Sundaramoorthy, V., Walker, A. K., Tan, V., Fifita, J. A., Mccann, E. P., Williams, K. L., et al. (2015). Defects in optineurin- and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum. Mol. Genet. 24, 3830–3846. doi: 10.1093/hmg/ddv126
Sundaramoorthy, V., Walker, A. K., Yerbury, J., Soo, K. Y., Farg, M. A., Hoang, V., et al. (2013). Extracellular wildtype and mutant SOD1 induces ER-Golgi pathology characteristic of amyotrophic lateral sclerosis in neuronal cells. Cell. Mol. Life Sci. 70, 4181–4195. doi: 10.1007/s00018-013-1385-2
Sütterlin, C., Hsu, P., Mallabiabarrena, A., and Malhotra, V. (2002). Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell 109, 359–369. doi: 10.1016/S0092-8674(02)00720-1
Szegezdi, E., Logue, S. E., Gorman, A. M., and Samali, A. (2006). Mediators of endoplasmic reticulum stress−induced apoptosis. EMBO Rep. 7, 880–885. doi: 10.1038/sj.embor.7400779
Takahashi, Y., Meyerkord, C. L., Hori, T., Runkle, K., Fox, T. E., Kester, M., et al. (2011). Bif-1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy 7, 61–73. doi: 10.4161/auto.7.1.14015
Taylor, A. M., Berchtold, N. C., Perreau, V. M., Tu, C. H., Jeon, N. L., and Cotman, C. W. (2009). Axonal mRNA in uninjured and regenerating cortical mammalian axons. J. Neurosci. 29, 4697–4707. doi: 10.1523/JNEUROSCI.6130-08.2009
Teuling, E., Ahmed, S., Haasdijk, E., Demmers, J., Steinmetz, M. O., Akhmanova, A., et al. (2007). Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. J. Neurosci. 27, 9801–9815. doi: 10.1523/JNEUROSCI.2661-07.2007
Thomas, M., Alegre-Abarrategui, J., and Wade-Martins, R. (2013). RNA dysfunction and aggrephagy at the centre of an amyotrophic lateral sclerosis/frontotemporal dementia disease continuum. Brain 136, 1345–1360. doi: 10.1093/brain/awt030
Tong, J., Huang, C., Bi, F., Wu, Q., Huang, B., and Zhou, H. (2012). XBP1 depletion precedes ubiquitin aggregation and Golgi fragmentation in TDP−43 transgenic rats. J. Neurochem. 123, 406–416. doi: 10.1111/jnc.12014
Topp, J. D., Gray, N. W., Gerard, R. D., and Horazdovsky, B. F. (2004). Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J. Biol. Chem. 279, 24612–24623. doi: 10.1074/jbc.M313504200
Tuck, E., and Cavalli, V. (2010). Roles of membrane trafficking in nerve repair and regeneration. Commun. Integr. Biol. 3, 209–214. doi: 10.4161/cib.3.3.11555
Tumbarello, D. A., Waxse, B. J., Arden, S. D., Bright, N. A., Kendrick-Jones, J., and Buss, F. (2012). Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat. Cell Biol. 14, 1024–1035. doi: 10.1038/ncb2589
Urwin, H., Authier, A., Nielsen, J. E., Metcalf, D., Powell, C., Froud, K., et al. (2010). Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum. Mol. Genet. 19, 2228–2238. doi: 10.1093/hmg/ddq100
van Dis, V., Kuijpers, M., Haasdijk, E. D., Teuling, E., Oakes, S. A., Hoogenraad, C. C., et al. (2014). Golgi fragmentation precedes neuromuscular denervation and is associated with endosome abnormalities in SOD1-ALS mouse motor neurons. Acta Neuropathol. Commun. 2:38. doi: 10.1186/2051-5960-2-38
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Walker, A. K., and Atkin, J. D. (2011). Stress signaling from the endoplasmic reticulum: a central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB Life 63, 754–763. doi: 10.1002/iub.520
Walker, A. K., Farg, M. A., Bye, C. R., McLean, C. A., Horne, M. K., and Atkin, J. D. (2010). Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain 133, 105–116. doi: 10.1093/brain/awp267
Walker, A. K., Soo, K. Y., Sundaramoorthy, V., Parakh, S., Ma, Y., Farg, M. A., et al. (2013). ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS ONE 8:e81170. doi: 10.1371/journal.pone.0081170
Webber, J. L., Young, A. R., and Tooze, S. A. (2007). Atg9 trafficking in mammalian cells. Autophagy 3, 54–56. doi: 10.4161/auto.3419
Wilson, B. S., Nuoffer, C., Meinkoth, J. L., McCaffery, M., Feramisco, J. R., Balch, W. E., et al. (1994). A Rab1 mutant affecting guanine nucleotide exchange promotes disassembly of the Golgi apparatus. J. Cell Biol. 125, 557–571. doi: 10.1083/jcb.125.3.557
Wood, J. D., Beaujeux, T. P., and Shaw, P. J. (2003). Protein aggregation in motor neurone disorders. Neuropathol. Appl. Neurobiol. 29, 529–545. doi: 10.1046/j.0305-1846.2003.00518.x
Wu, C.-H., Fallini, C., Ticozzi, N., Keagle, P. J., Sapp, P. C., Piotrowska, K., et al. (2012). Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. doi: 10.1038/nature11280
Xu, H., and Shields, D. (1993). Prohormone processing in the trans-Golgi network: endoproteolytic cleavage of prosomatostatin and formation of nascent secretory vesicles in permeabilized cells. J. Cell Biol. 122, 1169–1184. doi: 10.1083/jcb.122.6.1169
Yi, P., Higa, A., Taouji, S., Bexiga, M. G., Marza, E., Arma, D., et al. (2012). Sorafenib-mediated targeting of the AAA+ ATPase p97/VCP leads to disruption of the secretory pathway, endoplasmic reticulum stress, and hepatocellular cancer cell death. Mol. Cancer Ther. 11, 2610–2620. doi: 10.1158/1535-7163.MCT-12-0516
Young, A. R., Chan, E. Y., Hu, X. W., Köchl, R., Crawshaw, S. G., High, S., et al. (2006). Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 119, 3888–3900. doi: 10.1242/jcs.03172
Zhang, D., Iyer, L. M., He, F., and Aravind, L. (2012). Discovery of novel DENN proteins: implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front. Genet. 3:283. doi: 10.3389/fgene.2012.00283
Zhang, Y.-J., Jansen-West, K., Xu, Y.-F., Gendron, T. F., Bieniek, K. F., Lin, W.-L., et al. (2014). Aggregation-prone c9FTD/ALS poly (GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 128, 505–524. doi: 10.1007/s00401-014-1336-5
Zhou, Z., Mogensen, M. M., Powell, P. P., Curry, S., and Wileman, T. (2013). Foot-and-mouth disease virus 3C protease induces fragmentation of the Golgi compartment and blocks intra-Golgi transport. J. Virol. 87, 11721–11729. doi: 10.1128/JVI.01355-13
Zolov, S. N., and Lupashin, V. V. (2005). Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. J. Cell Biol. 168, 747–759. doi: 10.1083/jcb.200412003
Keywords: amyotrophic lateral sclerosis, Golgi fragmentation, ER stress, axonal degeneration, secretory trafficking inhibition, autophagy dysfunction
Citation: Sundaramoorthy V, Sultana JM and Atkin JD (2015) Golgi fragmentation in amyotrophic lateral sclerosis, an overview of possible triggers and consequences. Front. Neurosci. 9:400. doi: 10.3389/fnins.2015.00400
Received: 28 August 2015; Accepted: 09 October 2015;
Published: 27 October 2015.
Edited by:
Georg Haase, Centre National de la Recherche Scientifique, FranceReviewed by:
Roland Brandt, University of Osnabrück, GermanyBruno Goud, Centre National de la Recherche Scientifique, France
Copyright © 2015 Sundaramoorthy, Sultana and Atkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie D. Atkin, anVsaWUuYXRraW5AbXEuZWR1LmF1