
- 1 Department of Pharmacology, Biocenter, University of Frankfurt, Frankfurt, Germany
- 2 Audiological and Vestibular Center of Azienda AUSL of Modena, Modena, Italy
- 3 Department of Neurology, University Hospital, Bordeaux, France
Piracetam, the prototype of the so-called nootropic drugs’ is used since many years in different countries to treat cognitive impairment in aging and dementia. Findings that piracetam enhances fluidity of brain mitochondrial membranes led to the hypothesis that piracetam might improve mitochondrial function, e.g., might enhance ATP synthesis. This assumption has recently been supported by a number of observations showing enhanced mitochondrial membrane potential, enhanced ATP production, and reduced sensitivity for apoptosis in a variety of cell and animal models for aging and Alzheimer disease. As a specific consequence, substantial evidence for elevated neuronal plasticity as a specific effect of piracetam has emerged. Taken together, this new findings can explain many of the therapeutic effects of piracetam on cognition in aging and dementia as well as different situations of brain dysfunctions.
Introduction
Piracetam, the prototype of the so-called “nootropic” drugs is used in many countries to treat cognitive impairment in aging, brain injuries, as well as dementia (Muller et al., 1999; Winblad, 2005). A recent meta-analysis of all available (published and not published) clinical studies provided compelling evidence for the global efficacy of piracetam in a diverse group of older subjects with cognitive impairment (Waegemans et al., 2002).
Similar to the situation in man, piracetam has also been shown to improve cognitive function in animals, but its mode of action is not yet finally known (Winblad, 2005). Findings that piracetam’s efficacy is usually associated with conditions of disturbed brain function like aging (young healthy animals usually benefit little or nothing from piracetam treatment) (Valzelli et al., 1980; Muller et al., 1997), has led to the speculation that piracetam’s mechanism of action is associated with biochemical deficits of the aged brain (Müller et al., 1994, 1999; Scheuer et al., 1999). This assumption was later supported by observations that piracetam specifically enhances membrane fluidity in aged brain material, showing no effect at all in membranes from young brains (Muller et al., 1997). This unique mechanism of action could be explained by the specific binding to the polar head-structures of membrane phospholipids leading to a more flexible arrangement of the fatty acid side chain structure, which got more rigid due to lipid peroxidation in aging or other situations associated with enhanced oxidative stress (Peuvot et al., 1995). At the subcellular level, piracetam’s effects on membrane fluidity could be demonstrated for synaptosomal plasma membranes as well as for mitochondrial membranes of aged mouse brain and for brain membranes of Alzheimer patients (Eckert et al., 1999; Muller et al., 1999). Since these effects were observed at concentrations also needed in pharmacological experiments to improve cognition (Saletu et al., 1995), we proposed that by restoring age-related membrane alterations piracetam improves brain function and finally cognition.
Piracetam’s improving effects on the fluidity of aged synaptosomal membranes could explain the beneficial effects of piracetam on age-related deficits on several mechanisms of signal transduction such as receptor density and function, and transmitter release, since these mechanisms are disturbed in the aging brain probably due to a decrease of membrane fluidity (Stoll et al., 1992; Viana et al., 1992; Cohen and Muller, 1993; Scheuer et al., 1999). On the other hand, initial evidence that piracetam’s beneficial effects on the fluidity of aged mitochondrial membranes could contribute to its therapeutic efficacy originated from observations that piracetam could improve glucose uptake and utilization as well as ATP production (Domanska-Janik and Zaleska, 1977; Heiss et al., 1988; Dormehl et al., 1999; Keil et al., 2006). These effects led to the term “metabolic enhancer,” sometimes used to characterize piracetam and related nootropics (Malik et al., 2007). However, the mechanism of these effects and its possible relationship to mitochondrial function remained obscure.
As a specific feature of piracetam and other similar compounds, beneficial effects on cognition are usually associated with impaired brain function such as aging, hypoxia, glucose deprivation, injuries, or even neurodegeneration. Thus, young healthy animals or men usually benefit less from piracetam indicating that this compound is not a “cognition booster” or a drug for neuro-enhancement. As all the pathological conditions mentioned above are typically associated with the vicious cycle between enhanced oxidative stress – elevated reactive oxygen species (ROS) production – mitochondrial damage – reduced energy supply – enhanced ROS production (Leuner et al., 2007; Mattson et al., 2008; Querfurth and LaFerla, 2010), we speculated that piracetam might enhance mitochondrial function or at least protect against mitochondrial damage under such conditions but mainly in brain aging, or dementia. In both situations, similar but also variant alterations of mitochondrial function are present (Leuner et al., 2007; Müller et al., 2010), representing possible targets and also plausible for therapeutic intervention by piracetam (Müller et al., 2010).
Mitochondrial Dysfunction and Oxidative Stress in Brain Aging and Alzheimer Disease
Like other differentiated tissues, the central nervous system is profoundly affected by aging and reacts to aging by a decline of several physiological abilities including sensory, motor, emotional, or cognitive functions (Sastre et al., 2000; Floyd and Hensley, 2002; Balaban et al., 2005; Mattson and Magnus, 2006; Onyango et al., 2010). Aging brain cells experience increasing amounts of oxidative stress, perturbed energy homeostasis, accumulation of damaged proteins, lesions in their nucleic acids and are characterized by impaired function of signaling mechanisms and altered gene expression. These changes were significantly exacerbated in neurodegenerative disorders and amplified in vulnerable neuronal populations be disease related processes such as accumulation of damaged proteins, e.g., Amyloid-beta (Aß) levels in Alzheimer’s disease (AD) (Cleary et al., 2005; Haass and Selkoe, 2007; Selkoe, 2008; Querfurth and LaFerla, 2010). Beside the described dysfunctions, mitochondrial perturbations are strongly associated with aging. Mitochondria play a central role in producing ATP as the central source of cellular energy and are critical regulators of programmed cell death during aging (Sastre et al., 2000; Atamna, 2004; Balaban et al., 2005; Schuessel et al., 2006). Mitochondrial function becomes less efficient during brain aging including decreased activities of complex I and to a lesser content of complex IV of the respiratory chain, which in turn leads to enhanced ROS production, reduced Ca2+ buffering capacity, and accumulation of mitochondrial DNA (mtDNA) mutations (reviews for this topic (Mattson, 2007; Mattson et al., 2008).
Abnormalities in mitochondrial function and oxidative stress are also relevant for the pathogenesis of Alzheimer’s disease (AD) (Hauptmann et al., 2006; Anandatheerthavarada and Devi, 2007; Leuner et al., 2007; Moreira et al., 2007; Reddy and Beal, 2008). In the brain of AD patients, defective energy metabolism and early defects in glucose utilization were observed already many years before (Munch et al., 1998). Moreover, the AD brain is specifically marked by accumulation of the misfolded proteins Aß and hyperphosphorylated tau, both contributing together with aging-related deficits to severe neurodegenerative alterations, such as the loss of synapses and neurons, atrophy, and the selective depletion of neurotransmitter systems (e.g., acetylcholine) in the hippocampus and cerebral cortex (Haass and Selkoe, 2007; Querfurth and LaFerla, 2010). At the mitochondrial level, complex I and complex IV seem to be specifically affected, where tau pathology mainly impairs complex I activity, and Aß complex IV activity (Eckert et al., 2008; Hauptmann et al., 2009; Rhein et al., 2009). One of the earliest changes observed in AD is synaptic failure which already starts in patients with mild cognitive impairment (MCI) and manifests during the disease process (Selkoe, 2002). Mitochondria are key players in synaptic plasticity providing the necessary energy (Mattson et al., 2008). The observed mitochondrial deficits result in enhanced oxidative stress. Importantly, mitochondrial dysfunction and reduced bioenergetics occur early in pathogenesis and precede the development of plaque formation (Hauptmann et al., 2009).
Pharmacological Strategies to Improve Mitochondrial Function in Aging or AD
While the concept of Aß-induced mitochondrial dysfunction as a major functionally relevant pathomechanism in AD has received substantial support over the last decade, improving mitochondrial function as a target for new drug development has rather not, as most interest has been directed to drugs leading to reduced Aß load (Lemere et al., 2004). However, as several compounds out of those disease-modifying drug classes have recently failed to show clinical effectiveness in AD trials (Gura, 2008), a report about substantial therapeutic effects of dimebon in a 1-year clinical trial (Doody et al., 2008) received large attention. Quite interestingly, although originally used as an antihistaminic drug, dimebon was later characterized as a mitochondrial stabilizer with rather similar properties as reported for piracetam in the present communication (Bachurin et al., 2003; Bernales et al., 2008). The concept of using mitochondrial protection as treatment strategy for dementia has recently been further supported by a preliminary report about again substantial clinical improvement in AD patients treated with methylene blue (Gura, 2008). Importantly, this drug not only has been shown to enhance cognitive functions in several animal studies associated with elevated oxygen consumption, but also seems to enhance mitochondrial function by activating complex I and IV activities at the cellular level (Callaway et al., 2002; Callaway et al., 2004; Atamna et al., 2008).
The close association and common basis of oxidative stress and mitochondrial dysfunction in brain aging and age-related neurodegenerative disorders like AD (see the previous chapters) explains that several antioxidants have a long history as possible treatments for AD and even have been and are used in this context. Initially mainly Vitamin E or Vitamin C or the combination of both was investigated (Bastianetto and Quirion, 2004; Farlow et al., 2008; Lee et al., 2009). While both at high concentrations definitively show antioxidant properties in vitro and in vivo, their therapeutical benefit to improve or even prevent cognitive impairment in the elderly is at present seen rather critically.
Another important class of naturally accruing antioxidant are flavonoids or other polyphenols, which also are fairly good antioxidants which reduced oxidative stress in vitro and in vivo. Flavonoids also improve mitochondrial dysfunction and seem to have therapeutical benefit for long-term treatment of age-related cognitive impairment animals and men (Schmitt-Schillig et al., 2005; Schaffer et al., 2006). The significant reduction of the risk in getting AD by Mediterranean diet is very likely explained to an impotent part by the high daily intake of flavonoids (Scarmeas et al., 2006, 2007). In general, even if the effectiveness of those natural occurring antioxidants to protect against AD seems to be limited, they seem to be the major players of diet in reducing oxidative stress and acting as a second however considerably weaker defense system.
Another case of a herbal drug is the standardized Ginkgo biloba extract (EGb 761), which has been used for many years as a prescription or OTC drug in many countries to treat aging-related cognitive disorders including AD (Christen and Maixent, 2002). EGb 761 contains 24% of flavonoids and 6% of terpenens (Sastre et al., 2002; Abdel-Kader et al., 2007). While the flavonoid fraction seems to be mainly responsible for the free radical scavenging properties, several terpene lactones (Ginkgolides, Bilobalide) show substantial mitochondria-protecting properties (Abdel-Kader et al., 2007).
Piracetam
Oxidative stress usually accumulates in aged or diseased brain tissue over years or even decades due to the vicious cycle between increased ROS, and enhanced mitochondrial dysfunction (Floyd and Hensley, 2002). Within this system, mitochondria play a crucial role as their impaired function not only leads to reduced ATP production and finally enhanced apoptosis (Mattson, 2000; Mattson et al., 2008) (Figure 1), but also since mitochondria are the relevant targets for their own production of free radicals (Figure 1). Accordingly, due to our initial findings of piracetam enhancing mitochondrial membrane fluidity (Eckert et al., 1999) and observations that membrane fluidity regulates mitochondrial function probably by enhancing the mobility and function of the complexes of the respiratory chain (Ricchelli et al., 1999; Muriel and Perez-Rojas, 2003; Colell et al., 2004; Aleardi et al., 2005), we speculated that piracetam might enhance mitochondrial function or at least might protect mitochondria in situations of enhanced damage. To investigate this assumption, we used different cell models in tissue culture and induced experimentally oxidative stress using different approaches to mirror within hours or a few days what is usually seen in aged or diseased brains at the end of the life span. Mitochondrial function was assessed by monitoring mitochondrial membrane potential (MMP) using specific membrane dyes, by measuring ATP production, and the release of proapoptotic factors (Keil et al., 2006; Kurz et al., 2010)
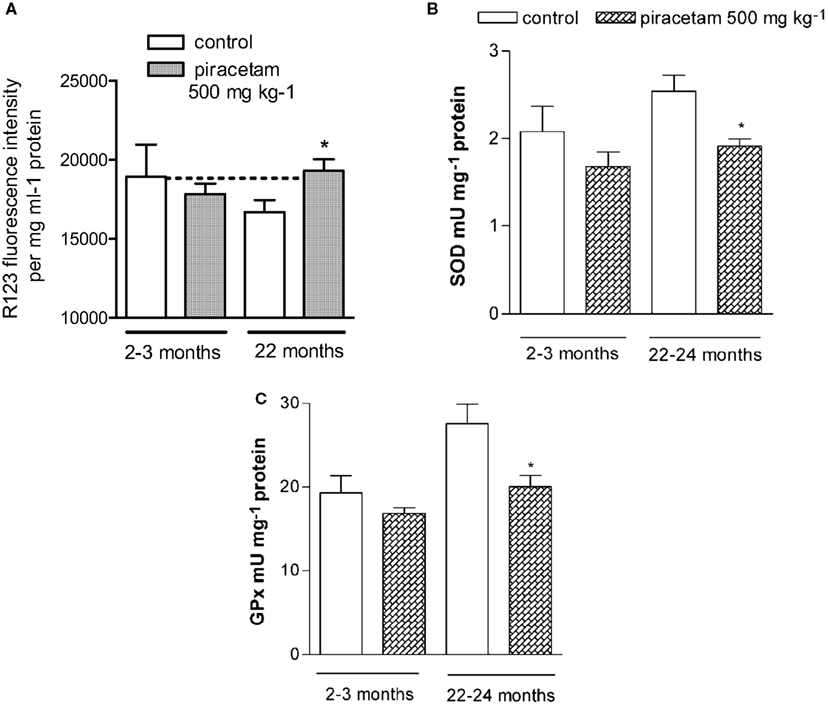
Figure 1. Piracetam reduces measures of mitochondrial dysfunction in NMRI mice – higher sensitivity in aged mice 2–3 months old and 22–24 months old NMRI mice were treated for 14 days with 0.5 g piracetam/kg in 0.9% NaCI solution p.o. once daily for 2 weeks. Control animals received 0.9% NaCI solution alone. All data are modified after Keil et al. (2006). (A) Dissociated brain cells were isolated and MMP was detected. The MMP was significantly reduced in old animals (22 months) and protected by Piracetam. Data are expressed as mean ± SEM (n = 6–8) *p < 0.05 piracetam treated old animals vs. aged controls. (B,C) Effects of aging and treatment with piracetam on SOD, and GPx in young and old animals. Activities of antioxidant enzymes are increased in aged mice. Treatment with piracetam reduces the enzyme activities significantly in old mice. Data are expressed as mean ± SEM (n = 7). *p < 0.05 piracetam treated old animals vs. aged controls.
Protection Against Oxidative Stress in Vitro
Beside the use of H2O2 in some initial experiments, we usually induced oxidative stress by the NO donor sodium nitroprusside (SNP), due to the important role of nitrosative stress in AD (Keil et al., 2004a,b). In PC12 cells, SNP, led to a reduction of MMP and ATP levels. Under basal conditions without additional SNP damage, piracetam did not affect both alterations even at rather high concentrations. However, piracetam was able to reduce both measures of mitochondrial dysfunction after pre- and post-incubation (Keil et al., 2006).
Another stressor, serum deprivation also leads to a decrease of mitochondrial membrane potential (MMP) and a decrease of ATP levels in neurons. Additionally, serum like glucose deprivation in PC12 cells causes peroxidation of their cell membrane lipids, decreases intracellular SOD activity, and enhances apoptosis in PC12 cells (Keil et al., 2004b, 2006). Interestingly, piracetam was able to protect MMP against cellular stress following serum deprivation. Under conditions of mild serum deprivation, when serum concentrations not lower than 2% were used, piracetam (500 μM) induced a nearly complete recovery of MMP. Furthermore, reduced ATP levels were already seen at 10% serum. Under this condition, piracetam completely restored ATP levels, while at lower serum concentrations only a partial restoration was seen (Keil et al., 2006).
As mentioned before, complex I and complex IV functions are impaired in aging and AD. Thus, the possible efficacy of piracetam to protect individual complexes of the mitochondrial respiratory chain after treatment with specific complex inhibitors was also investigated. Complexes I, II, and III were already protected at concentrations as low as 500 μM piracetam, while a significant protection of complexes IV and V was observed at a concentration of 1000 μM piracetam (Keil et al., 2006). This broad activity of piracetam is in line with the assumptions that improvement of complex activity might be due rather to its fluidity enhancing properties at mitochondrial membranes rather than by specific effects at the individual complexes. This is also supported by the data of Zhang et al. (2010) indicating enhanced activity of complexes I–IV in mice after induction of mitochondrial dysfunction with D-galactose. It is very important to note that the beneficial effects of piracetam have not only been seen at the level of MMP but also in using several other measures of mitochondrial function since MMP alterations are not always directly connected with changes of mitochondrial function (Cao et al., 2007; Kahlert et al., 2008).
As already mentioned, a major link between the mitochondrial defects of our brain accumulating during decades of aging and the specific Aß related toxicity in AD seems to be oxidative stress induced by Aß as well as Aß induced impairment of mitochondrial function, e.g., reduced activity of the complexes I and IV of the respiratory chain. This very slow process can experimentally be investigated by inducing mitochondrial dysfunction in cells in tissue culture following incubation with extracellularly applied Aß (Kurz et al., 2010). Since Aß1–42 is presently considered as the main toxic Aß species, we used this peptide and several experimental cell models (PC12 cells, HEK cells, dissociated mouse brain cells) to study possible protective effects of piracetam on mitochondrial deficits. When PC12 cells were treated with fibrillar Aß1–42 10 nM for 24 h a reduction of MMP was observed as described previously by our group (Keil et al., 2004a). The addition of piracetam 30 min after Aß1–42 substantially protected MMP at concentrations already beginning with 0.1 mM. Comparable protective effects of piracetam were observed for dissociated brain cells of Naval Medical Research Institute (NMRI) mice following incubation with fibrillar Aß1–42. In both cell types, piracetam alone has no effect on MMP.
Protection Against Oxidative Stress in Dissociated Brain Cells from Subchronically Treated Mice
After demonstrating piracetam’s efficacy in reducing mitochondrial dysfunction induced by several stressors in vitro, we also investigated its protective effects following treatment of aged mice or mice transgenic for the Swedish human Amyloid Precursor Protein (APP) gene which express a substantial amount of Aß in the brains (tgAPP mice) (Keil et al., 2006; Kurz et al., 2010). The rational for using aged animals mainly originates from a large number of findings that piracetam’s efficacy to improve cognitive functions usually is less pronounced in young healthy animals but gets much more prominent in aged animals (see Chapter 1.0). We used aged NMRI mice which are bread in our animal facilities not only showing clear cognitive impairment but also several measures of oxidative stress, and mitochondrial dysfunction at an age around 20 months including a decreased MMP compared to young mice (Müller et al., 2010). Piracetam treatment for 14 days normalized MMP in aged mice, while similar treatment of young animals had no effect (Figure 1).
On the other hand, a similar piracetam treatment not only protected brain cells of aged but also young mice against oxidative stress induced in vitro by addition of H2O2, SNP, or Aß1–42. Even if brain cells of young mice treated with piracetam also showed some benefit from piracetam treatment, aged animals usually responded most (Figure 1).
Antioxidative enzymes are the primary defense mechanism to protect biological macromolecules from oxidative damage, and are upregulated in aged mouse brain as an adaptive response to oxidative stress. Therefore, we investigated the effect of piracetam treatment on the activities of superoxide dismutase (SOD), glutathione peroxidase (GPx), and glutathione reductase (GR) in young mice (2–3 months old) and old mice (22–24 months old). We confirmed a significant increase in GPx and GR activity in aged mice compared to young mice. The activity of SOD had also a tendency to increase with age. Piracetam treatment decreased the activities of all three enzymes in aged mice nearly to the level of young animals. In young mice, a only small and not significant decrease of antioxidative enzymes could be observed (Figure 1). Quite similar mitochondria protecting effects have recently been observed in mice following experimentally induced mitochondrial dysfunction (g-galactose) after treatment with piracetam (300 mg/kg, 14 days) at the levels of MMP, activities of complexes I–IV, and ROS generation (Zhang et al., 2010). Unfortunately, both studies did not report glutathione levels, which also has a relevant role in regulating mitochondrial function (Jha et al., 2000).
Isolated brain cells of mice overexpressing mutated human amyloid precursor protein (tgAPP) show significant reductions of MMP and ATP synthesis relative to non-transgenic littermate controls confirming previous observation from our group (Hauptmann et al., 2009). Similar piracetam treatment (0.5 g/kg/day orally) already described for NMRI mice above again showed substantial improvement of MMP and ATP production (Figure 2). As reported earlier (Blanchard et al., 2003), these mice express substantial level of soluble Aß in the brain while littermates do not. Quite interestingly, piracetam treatment led to an about 25% reduction of soluble Aß (Kurz et al., 2010) (Figure 3). A related observation showing reduced Aß levels in the plasma of geriatric patients treated with piracetam was published by Blasko et al. (2005). In order to investigate if this effect of piracetam on Aß levels might also be associated with improved mitochondrial function, we used APPwt HEK 293 cells stably overexpressing human APP showing moderately enhanced Aß levels (Keil et al., 2004a). Piracetam lowered Aß levels under basal conditions (Kurz et al., 2010). In agreement with other findings (Guglielmotto et al., 2009) mitochondrial dysfunction induced with SNP elevates Aß1–40 levels substantially. Again, treatment with piracetam lowered Aß significantly by 15–20% (Figure 3). In addition, piracetam improves mitochondrial function under the same conditions in APPwt HEK 293 when Aß generation is decreased (Figure 3).
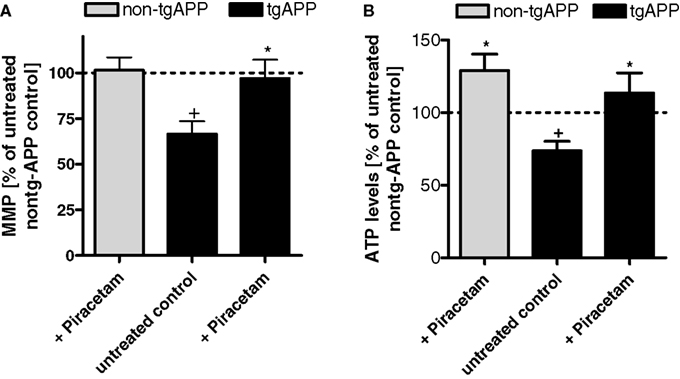
Figure 2. Piracetam improves measures of mitochondrial function in tgAPP mice. (A) Animals were treated for 14 days with 0.5 g piracetam/kg in 0.9% NaCI solution p.o. once daily for 2 weeks. Control animals received 0.9% NaCI solution alone. All data are modified after Kurz et al. (2010). The MMP was significantly reduced in tgAPP mice. Piracetam treatment normalizes the MMP to non-tgAPP levels. Data are expressed as mean ± SEM (n = 7–8). +p < 0.05 control non-tgAPP vs. control tgAPP, *p < 0.01 piracetam treated tgAPP vs. tgAPP control; Student’s unpaired t-test. (B) ATP levels were also significantly impaired in tgAPP mice. In contrast, piracetam treatment increases ATP levels not only in tgAPP animals but also in control animals. Data are expressed as mean ± SEM (n = 7-8). *p < 0.05 control non-tgAPP vs. piracetam treated non-tgAPP; +p < 0.05 control non-tgAPP vs. control tgAPP, *p < 0.01 piracetam treated tgAPP vs. tgAPP control; Student’s unpaired t-test.
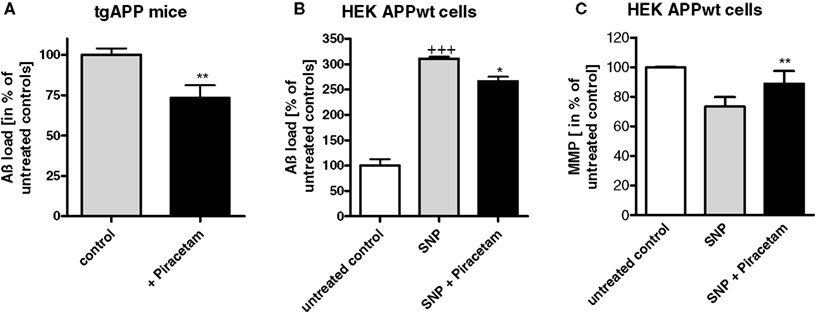
Figure 3. Piracetam ameliorates elevated Aβ production following mitochondrial dysfunction. (A) Normalized Aβ levels were quantified in brain homogenates from non-tg littermate and tgAPP mice (3 months old). **p < 0.01 piracetam treated tgAPP vs. control tgAPP; student’s unpaired t-test. (B) Piracetam reduces nitrosative stress induced elevation of Aβ in APPwt HEK293 cells. Cells were preincubated for 24 h with piracetam (1 mM) and stressed for additional 24 h with SNP (0.5 mM). Data are expressed as mean ± SEM (n = 3–4). +++p < 0.001 APPwt HEK293 control cells vs. cells treated with SNP, *p < 0.01 APPwt HEK293 cells stressed with SNP 0.5 mM vs. stressed cells preincubated with piracetam; Student’s unpaired t-test. (C) Piracetam ameliorates nitrosative stress induced reduction of mitochondrial membrane potential. Cells were preincubated for 24 h with piracetam (1 mM) and stressed for additional 24 h with SNP (0.5 mM). Data are expressed as mean ± SEM (n = 6). All data are modified after Kurz et al. (2010). **p < 0.01 APPwt HEK293 cells stressed with SNP 0.5 mM vs. stressed cells preincubated with piracetam, Student’s unpaired t-test.
Piracetam Ameliorates Aß Induced Impairment of Neuritic Outgrowth
In agreement with the pronounced loss of neurites and synapses in AD brain as one of the functionally most relevant histopathological lesions (Selkoe, 2002; Lacor et al., 2007), Aß peptides have been repeatedly demonstrated to reduce neuritic outgrowth in different neuronal cell lines in vitro including PC12 cells (Hirata et al., 2005; Hu et al., 2007; Evans et al., 2008). Oligomeric Aß seems to be more active than fibrillar Aß (Lacor et al., 2007; Evans et al., 2008). In agreement with these observations, the addition of oligomeric Aß1–42 (1 μM) to nerve growth factor (NGF) treated PC12 cells reduces neuritic length significantly. When the same experiment was carried out in the presence of piracetam (1 mM), the negative effect of oligomeric Aß1–42 was completely inhibited. In agreement with the assumption that enhanced oxidative stress might explain the Aß induced reduction of neuritic outgrowth (Guglielmotto et al., 2009) treating PC12 cells with SNP reduced neuritic outgrowth even stronger. Again, piracetam ameliorates this negative effect significantly under conditions of optimal NGF stimulation (Figure 4). A reduction of neuritic outgrowth depending on Aß load was also observed in our PC12 cells transgenic for human APP, where we observed a reduction of neuritic length, which could be substantially ameliorated by piracetam (Figure 4). The enhancing effect of piracetam was observed over the whole NGF concentration range (1–50 ng/ml) still leading to increased neuritic length under maximum NGF stimulation (Figure 4).
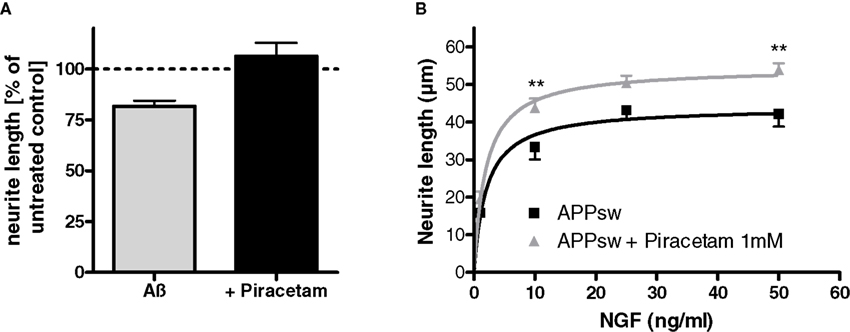
Figure 4. Piracetam improves neuritogenesis following Aß exposure. (A) PC12 cells were treated over 6 days with NGF 50 ng/ml in the presence of oligomeric Aβ 1 μM. Piracetam 1 mM was added and the effects on neurite outgrowth were investigated. (B) APPsw PC12 cells were treated over 6 days with NGF 50 ng/ml in the presence or absence of piracetam 1 mM and neurite outgrowth was measured. In the presence of different NGF concentrations (1–50 ng/ml) piracetam improved the neurotrophic effect of NGF in APPsw cells. All data are modified after Kurz et al. (2010). Data are expressed as mean ± SEM (n = 6–7) **p < 0.01, ***p < 0.001 vs. the respective control, two-way anova with bonferroni post test.
Summing Up
The data presented clearly show that piracetam protects mitochondria against different conditions associated with oxidative stress including aging. Piracetam’s protecting effects on mitochondrial damage induced in vitro are small, but reproducible and highly significant. This is not surprising, since the conditions used to induce oxidative stress in vitro are not pathophysiological but rather aggressive, in contrast to the small and slowly occurring changes induced by aging, which however were sometimes completely reversed by piracetam treatment. This was also the case for the adaptive elevation of antioxidant enzyme activities. When mild conditions were used in vitro (e.g. partial serum deprivation), a complete protection of mitochondrial function was seen by piracetam treatment in PC12 cells. Moreover, piracetam was highly effective in vivo in pathophysiologically relevant situations of brain dysfunction.
Piracetam does not possess radical scavenging properties (Keil et al., 2006). Thus, it seems quite likely that piracetam acts directly at the mitochondrial level, presumably by improving mitochondrial membrane properties. This is also supported by our observation that in several experiments protective effects were also seen in the recovery phase, when the oxidative stressor was already removed. Moreover, experiments from our lab also indicate comparable effects of piracetam on isolated mouse brain mitochondria of animals treated with piracetam. The concentrations of piracetam effective in vitro (100–1000 μM) and the doses used in the in vivo experiments (100–500 mg/kg) are quite well within the plasma concentrations seen in patients treated with the standard dose of about 5 g daily, which range between 200 and 2000 μM (Saletu et al., 1995; Wang et al., 2010). Thus, it is quite likely that similar effects are also taking place in the brain of piracetam treated patients. Moreover, a recent pharmacokinetic study in rats indicates plasma concentrations around 500–1000 μmol/l at oral doses of 500 mg/kg BW (Wang et al., 2010).
While the interaction of piracetam with neuronal membranes shows little changes of membrane properties under normal conditions, it significantly enhances reduced membrane fluidity, for example, in the aging or even Alzheimer brain. All the conditions associated with positive effects of piracetam on mitochondrial function in the experiments reviewed above also seem to be associated with decreased membrane fluidity mainly due to enhanced lipids peroxidation. Thus, it seems quite plausible that piracetam improves mitochondrial function by enhancing fluidity of mitochondrial membranes, which seems to be a critical factor regulating mitochondrial function. However, direct proof for this mechanism still needs to be shown.
Translation Into Effects on Cognition in Animals and Geriatric Patients
Piracetam was originally developed as a cyclic derivative of gamma-aminobutyric acid (GABA) to treat anxiety. While it failed as an anxiolytic, it showed considerable efficacy in tests of “central nystagmus” as a model of vertigo, which at that time was only seen for antihistaminic and anticholinergic compounds. Piracetam not only did not have antihistaminic or anticholinergic properties, but even did not show any central activity like sedation, stimulation, or influence on autonomic function. However, as a rather new spectrum of pharmacological properties piracetam did facilitate interhemispheric transfer, enhanced the cerebral resistance to noxious stimuli like hypoxia and, most importantly, it enhanced or facilitated learning and other cognitive functions (Giurgea, 1982). These cognition improving properties still represent the basis of the present therapeutical use of piracetam in many countries all over the world in the whole spectrum of geriatric memory disorders, in cases of impaired cognitive functions after head injuries, and also in vertigo. Even if piracetam can facilitate learning and other cognitive functions under normal conditions (e.g., young healthy volunteers), it has been a most consistent observation over nearly three decades of piracetam’s clinical use that the cognition improving properties are much more pronounced when brain function is impaired such as aging, hypoxia, cerebral injuries, or Aß load in AD, conditions which all have mitochondrial dysfunction as common final pathway. Since these preclinical and clinical data form the link between mitochondrial protection as major preclinical mechanism of action and the clinical efficacy of piracetam in a large variety of diverse groups of older subjects with cognitive impairment, some of these data will be reviewed shortly.
Two older studies have investigated the cognition improving effects of piracetam in young volunteers under mild hypoxia as a pharmacological model to mirror impaired brain function as seen in aging or dementia (Demay and Bande, 1980; Saletu et al., 1995). The data from Demay and Bande (1980) are given in Figure 5 showing considerably less errors in a memory task under hypoxia following 5 days of piracetam treatment with 4.8 g per day, the average therapeutic dose even used today (Figure 5). Hypoxia induced cognitive but also biochemical and physiological deficits mirroring the situation in dementia has also been used in a very recent study in rats (He et al., 2008). In this study, chronic cerebral hyperfusion was induced by ligature of bilateral common carotid arteries leading to acute but also subacute structural and functional brain deficits. Most importantly, spatial memory performance (Morris Water maze) was substantially impaired and showed pronounced improvement by piracetam treatment (600 mg) per day starting after surgery (Figure 6). Besides reducing histopathologic markers of neuronal damage and reducing the expression of proapoptotic proteins, piracetam also improved long term potentiation in the CA3 region of the hippocampus of lesioned animals (Figure 7). Together with data that piracetam reduces neuronal loss and improves synaptic reorganization after chronic alcohol consumption (Brandao et al., 1995; Brandao et al., 1996) with findings of enhanced restitution and reorganization of cortical neuronal circuits after brain damage (Coq and Xerri, 1999; Xerri and Zennou-Azogui, 2003), and with findings that piracetam improves neuritic outgrowth after oxidative stress and/or Aß mediated stress (Kurz et al., 2010), the findings of He et al. (2008) strongly support the concept that piracetam given chronically might improve many parameters of synaptic plasticity from LTP at the synaptic level, to enhanced neurite- as well as synaptogenesis at the structural level, and may even partially protect neurons from apoptosis.
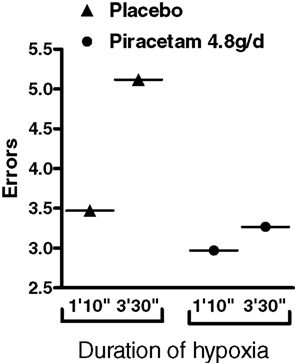
Figure 5. Piracetam improves cognitive function in men after hypoxia. The ability of 12 healthy volunteers to concentrate in a low oxygen pressure tank was assessed by measuring errors in a visual attention test in a placebo-controlled crossover trial in which volunteers were given 4.8 g piracetam daily for 4 days and 7.2 g on the fifth day. Depending on the time of hypoxia, errors were observed. Error number was significantly reduced by piracetam treatment, especially for the longer hypoxic period. Data are modified from Demay and Bande (1980).
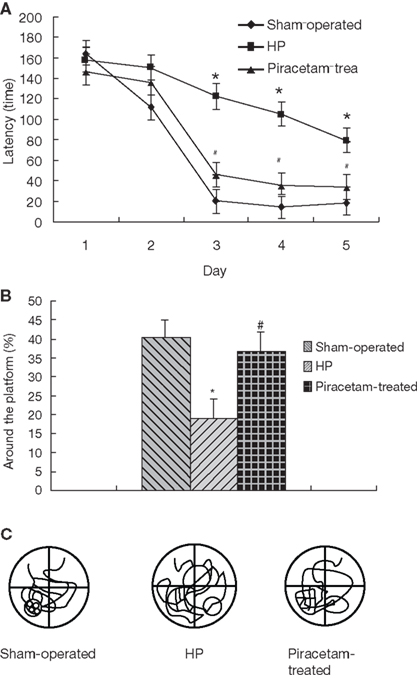
Figure 6. Piracetam improves cerebral hypoperfusion-induced deficits in learning and memory. Rats were treated for 30 days with piracetam 600mg/kg per day after ligature of bilateral carotid arteries. The Sham-operated group was only treated with saline without induction of hypoperfusion. The Morris water maze was performed with four trials per day during 5 days for the acquisition test (A), and performed with four trials on day 6 without the platform retention test (n = 10 ± SEM) (B). Representative examples of typical swim-tracking pathways in probe trial on training day 6 no platform (C). (n = 10 ± SEM). Data are reproduced from He et al. (2008) with permission. *p < 0,01 vs. Sham-operated, and #p < 0.05 vs. control hypoperfusion. Tukey’s test following a repeated ANOVA.
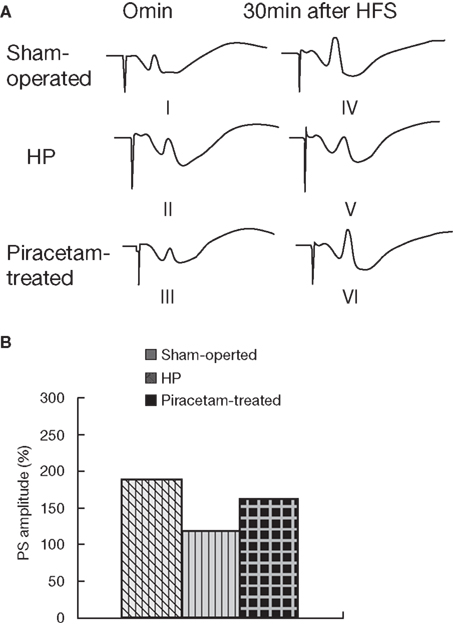
Figure 7. Piracetam improves LTP after cerebral hypoperfusion in CA3 synapses. Changes in LTP after ligature of carotoids induced as described by He et al. (2008). The animals were treated in the Sham-operated group (I, IV) and in the HP group (II, V) for 30 days with saline, and in the piracetam group (III, VI) with 600 mg/kg per day. (A) LTP was blocked in the HP group and improved in the piracetam treated group. (B) Improvement of the population spike (PS) by piracetam (n = 7–8 ± SEM). Data are reproduced from He et al. (2008) with permission. *p < 0,01 vs. Sham-operated, and #p < 0.05 vs. control hypoperfusion. One-way ANOVA followed by Fisher LSD test.
The new concept that by improving mitochondrial function piracetam stimulates synaptic function and plasticity in situations of impaired brain function easily integrates many previous preclinical findings see above and also data about clinical efficacy of piracetam in several not clearly related disease states.
• Cognitive deficits after coronary bypass surgery probably caused by periods of mild hypoxia are well known and represent a serious impairment of those patients. Treatment with piracetam has been shown in recent studies to reduce the severity of post surgery cognitive impairments (Uebelhack et al., 2003; Szalma et al., 2006; Holinski et al., 2008).
• Mild cognitive impairment (MCI) is a common prodromal state for late dementia. Piracetam treatment has been shown to improve several aspects of cognition in such patients, which had been selected at the time of the study on the basis of the previous and somewhat different concept of AAMI (age associated memory impairment) (Israel et al., 1994).
• Only few studies have been conducted in specific groups of patients with either Alzheimer type or vascular dementia. In most studies, mixed groups of demented patients were investigated. Nevertheless, cognitive improvement was seen under the treatment with piracetam regardless of the type of dementia (Waegemans et al., 2002). Most of these studies however were rather short. In the only one 1-year trial in AD patients, some evidence for a reduction of the cognitive decline over time was detected (Croisile et al., 1993), were the MMSE (minimental state evaluation did not change significantly in the Piracetam group but showed the expected 3 point decline in the placebo group over the year.
• Structural improvement might explain the therapeutical benefit in neurodevelopment disorders like dyslexia which is supported by several well controlled clinical trials (Wilsher, 1986; Wilsher et al., 1987; Ackerman et al., 1991).
• The effectiveness of piracetam in experimental models of central nystagmus was one of the first observations about pharmacological properties of piracetam (Giurgea, 1982). Even if this can be easily linked to its therapeutic use in vertigo, a plausible explanation of the improvement of vertigo by piracetam was not possible, especially since its effectiveness seems to be rather similar regardless the origin of vertigo. However, with the increasing awareness of higher brain regions for maintaining balance and preventing vertigo by coordinating all sensory inputs (Dieterich and Brandt, 2010), improvement of neuronal function, enhancement of cerebral communication, and improvement of neuronal plasticity could be an important aspect for piracetams effects on vertigo, e.g., enhancing the “cognitive aspects” of vertigo (Rosenhall et al., 1996; Oosterveld, 1999; Toupet, 2001).
• Even in myoclonus as in movement disorders (Hanna and Bhatia, 1997; Waldbaum and Patel, 2010), mitochondrial dysfunction seems to be part of the pathophysiological basis and might be the target of piracetam’s therapeutical benefit (Ikeda et al., 1996; Karacostas et al., 1999; Wolters and Benecke, 2009).
In conclusion, more than 30 years after the introduction of piracetam the new findings of mitochondrial protection will allow a new perspective for this old nootropic and might explain its consistent use in many countries around the world.
Conflict of Interest Statement
Kristina Leuner received honorarium for scientific lectures from UCB. Walter E. Müller is a paid consultant of several pharmaceutical companies, including UCB (Belgium), but receives no rewards from sales of any products.
Acknowledgment
The studies presented in this report and the publication costs were supported by an unrestricted research grant from UCB, Brussles (Belgium).
References
Abdel-Kader, R., Hauptmann, S., Keil, U., Scherping, I., Leuner, K., Eckert, A., and Muller, W. E. (2007). Stabilization of mitochondrial function by Ginkgo biloba extract (EGb 761). Pharmacol. Res. 56, 493–502.
Ackerman, P. T., Dykman, R. A., Holloway, C., Paal, N. P., and Gocio, M. Y. (1991). A trial of piracetam in 2 subgroups of students with dyslexia enrolled in summer tutoring. J. Learn Disabil. 24, 542–549.
Aleardi, A. M., Benard, G., Augereau, O., Malgat, M., Talbot, J. C., Mazat, J. P., Letellier, T., Dachary-Prigent, J., Solaini, G. C., and Rossignol, R. (2005). Gradual alteration of mitochondrial structure and function by beta-amyloids: importance of membrane viscosity changes, energy deprivation, reactive oxygen species production, and cytochrome c release. J. Bioenerg. Biomembr. 37, 207–225.
Anandatheerthavarada, H. K., and Devi, L. (2007). Amyloid precursor protein and mitochondrial dysfunction in Alzheimer’s disease. Neuroscientist 13, 626–638.
Atamna, H., Nguyen, A., Schultz, C., Boyle, K., Newberry, J., Kato, H., and Ames, B. N. (2008). Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 22, 703–712.
Bachurin, S. O., Shevtsova, E. P., Kireeva, E. G., Oxenkrug, G. F., and Sablin, S. O. (2003). Mitochondria as a target for neurotoxins and neuroprotective agents. Neuroprotective Agents 993, 334–344.
Balaban, R. S., Nemoto, S., and Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell 120, 483–495.
Bastianetto, S., and Quirion, R. (2004). Natural antioxidants and neurodegenerative diseases. Front. Biosci. 9, 3447–3452.
Bernales, S., Wagner, S., Protter, A. A., and Hung, D. T. (2008). Dimebon induces neuritic outgrowth and mitochondrial stabilization. Annual Meeting of the Society for Neuroscience, Washington D.C. [543.29/S12].
Blanchard, V., Moussaoui, S., Czech, C., Touchet, N., Bonici, B., Planche, M., Canton, T., Jedidi, I., Gohin, M., Wirths, O., Bayer, T. A., Langui, D., Duyckaerts, C., Tremp, G., and Pradier, L. (2003). Time sequence of maturation of dystrophic neurites associated with A beta deposits in APP/PS1 transgenic mice. Exp. Neurol. 184, 247–263.
Blasko, I., Kemmler, G., Krampla, W., Jungwirth, S., Wichart, I., Jellinger, K., Tragl, K. H., and Fischer, P. (2005). Plasma amyloid beta protein 42 in non-demented persons aged 75 years: Effects of concomitant medication and medial temporal lobe atrophy. Neurobiol. Aging 26, 1135–1143.
Brandao, F., CadeteLeite, A., Andrade, J. P., Madeira, M. D., and PaulaBarbosa, M. M. (1996). Piracetam promotes mossy fiber synaptic reorganization in rats withdrawn from alcohol. Alcohol 13, 239–249.
Brandao, F., PaulaBarbosa, M. M., and CadeteLeite, A. (1995). Piracetam impedes hippocampal neuronal loss during withdrawal after chronic alcohol intake. Alcohol 12, 279–288.
Callaway, N. L., Riha, P. D., Bruchey, A. K., Munshi, Z., and Gonzalez-Lima, F. (2004). Methylene blue improves brain oxidative metabolism and memory retention in rats. Pharmacol. Biochem. Behav. 77, 175–181.
Callaway, N. L., Riha, P. D., Wrubel, K. M., McCollum, D., and Gonzalez-Lima, F. (2002). Methylene blue restores spatial memory retention impaired by an inhibitor of cytochrome oxidase in rats. Neurosci. Lett. 332, 83–86.
Cao, J., Liu, Y., Jia, L., Zhou, H. M., Kong, Y., Yang, G., Jiang, L. P., Li, Q. J., and Zhong, L. F. (2007). Curcurnin induces apoptosis through mitochondrial hyperpolarization and mtDNA damage in human hepatoma G2 cells. Free Radic. Biol. Med. 43, 968–975.
Christen, Y., and Maixent, J. M. (2002). What is Ginkgo biloba extract EGb 761? An overview – From molecular biology to clinical medicine. Cell. Mol. Biol. 48, 601–611.
Cleary, J. P., Walsh, D. M., Hofmeister, J. J., Shankar, G. M., Kuskowski, M. A., Selkoe, D. J., and Ashe, K. H. (2005). Natural oligomers of the amyloid-protein specifically disrupt cognitive function. Nat. Neurosci. 8, 79–84.
Cohen, S. A., and Muller, W. E. (1993). Effects of piracetam on N-methyl-D-aspartate receptor properties in the aged mouse-brain. Pharmacology 47, 217–222.
Colell, A., Garcia-Ruiz, C., Mari, M., and Fernandez-Checa, J. C. (2004). Mitochondrial permeability transition induced by reactive oxygen species is independent of cholesterol-regulated membrane fluidity. FEBS Lett. 560, 63–68.
Coq, J. O., and Xerri, C. (1999). Acute reorganization of the forepaw representation in the rat SI cortex after focal cortical injury: neuroprotective effects of piracetam treatment. Eur. J. Neurosci. 11, 2597–2608.
Croisile, B., Trillet, M., Fondarai, J., Laurent, B., Mauguiere, F., and Billardon, M. (1993). Long-term and high-dose piracetam treatment of Alzheimers-disease. Neurology 43, 301–305.
Demay, F., and Bande, J. (1980). Effect of piracetam on volunteers in a low-pressure tank. J. Int. Med. Res. 8, 90–94.
Dieterich, M., and Brandt, T. (2010). Imaging cortical activity after vestibular lesions. Restor. Neurol. Neurosci. 28, 47–56.
Domanska-Janik, K., and Zaleska, M. (1977). The action of piracetam on 14C-glucose metabolism in normal, and posthypoxic rat cerebral coretx slices. Pol. J. Pharmacol. Pharm. 29, 111–116.
Doody, R. S., Gavrilova, S. I., Sano, M., Thomas, R. G., Aisen, P. S., Bachurin, S. O., Seely, L., and Hung, D. (2008). Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: a randomised, double-blind, placebo-controlled study. Lancet 372, 207–215.
Dormehl, I. C., Jordaan, B., Oliver, D. W., and Croft, S. (1999). SPECT monitoring of improved cerebral blood flow during long-term treatment of elderly patients with nootropic drugs. Clin. Nucl. Med. 24, 29–34.
Eckert, A., Hauptmann, S., Scherping, I., Meinhardt, J., Rhein, V., Drose, S., Brandt, U., Fandrich, M., Muller, W. E., and Gotz, J. (2008). Oligomeric and fibrillar species of beta-amyloid (A beta 42) both impair mitochondrial function in P301L tau transgenic mice. J. Mol. Med. 86, 1255–1267.
Eckert, G. P., Cairns, N. J., and Muller, W. E. (1999). Piracetam reverses hippocampal membrane alterations in Alzheimer’s disease – short communication. J. Neural Transm. 106, 757–761.
Evans, N. A., Facci, L., Owen, D. E., Soden, P. E., Burbidge, S. A., Prinjha, R. K., Richardson, J. C., and Skaper, S. D. (2008). A beta(1-42) reduces synapse number and inhibits neurite outgrowth in primary cortical and hippocampal neurons: a quantitative analysis. J. Neurosci. Methods 175, 96–103.
Farlow, M. R., Miller, M. L., and Pejovic, V. (2008). Treatment options in Alzheimer’s disease: Maximizing benefit, managing expectations. Dement. Geriatr. Cogn. Disord. 25, 408–422.
Floyd, R. A., and Hensley, K. (2002). Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging 23, 795–807.
Giurgea, C. E. (1982). The nootropic concept and its prospective implications. Drug Dev. Res. 2, 441–446.
Guglielmotto, M., Aragno, M., Autelli, R., Giliberto, L., Novo, E., Colombatto, S., Danni, O., Parola, M., Smith, M. A., Perry, G., Tamagno, E., and Tabaton, M. (2009). The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1 alpha. J. Neurochem. 108, 1045–1056.
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112.
Hanna, M. G., and Bhatia, K. P. (1997). Movement disorders and mitochondrial dysfunction. Curr. Opin. Neurol. 10, 351–356.
Hauptmann, S., Keil, U., Scherping, I., Bonert, A., Eckert, A., and Muller, W. E. (2006). Mitochondrial dysfunction in sporadic and genetic Alzheimer’s disease. Exp. Gerontol. 41, 668–673.
Hauptmann, S., Scherping, I., Drose, S., Brandt, U., Schulz, K. L., Jendrach, M., Leuner, K., Eckert, A., and Muller, W. E. (2009). Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 30, 1574–1586.
He, Z., Liao, Y., Zheng, M., Zeng, F. D., and Guo, L. J. (2008). Piracetam improves cognitive deficits caused by chronic cerebral hypoperfusion in rats. Cell. Mol. Neurobiol. 28, 613–627.
Heiss, W. D., Hebold, I., Klinkhammer, P., Ziffling, P., Szelies, B., Pawlik, G., and Herholz, K. (1988). Effect of piracetam on cerebral glucose-metabolism in Alzheimers-disease as measured by positron emission tomography. J. Cereb. Blood Flow Metab. 8, 613–617.
Hirata, K., Yamaguchi, H., Takamura, Y., Takagi, A., Fukushima, T., Iwakami, N., Saitoh, A., Nakagawa, M., and Yamada, T. (2005). A novel neurotrophic agent, T-817MA [1-{3-[2-(1-benzothiophen-5-yl) ethoxy] propyl}-3-azetidinol maleate], attenuates amyloid-beta-induced neurotoxicity and promotes neurite outgrowth in rat cultured central nervous system neurons. J. Pharmacol. Exp. Ther. 314, 252–259.
Holinski, S., Claus, B., Alaaraj, N., Dohmen, P. M., Kirilova, K., Neumann, K., Uebelhack, R., and Konertz, W. (2008). Cerebroprotective effect of piracetam in patients undergoing coronary bypass surgery. Med. Sci. Monitor 14, I53–I57.
Hu, M., Schurdak, M. E., Puttfarcken, P. S., El Kouhen, R., Gopalakrishnan, M., and Li, J. H. (2007). High content screen microscopy analysis of Ap(1-42)-induced neurite outgrowth reduction in rat primary cortical neurons: neuroprotective effects of alpha 7 neuronal nicotinic acetylcholine receptor ligands. Brain Res. 1151, 227–235.
Ikeda, A., Shibasaki, H., Tashiro, K., Mizuno, Y., Kimura, J., Kohara, N., Kashiwagi, W., Ichikawa, H., Sakuragi, A., and Urano, Y. (1996). Clinical trial of piracetam in patients with myoclonus: nationwide multiinstitution study in Japan. Mov. Disord. 11, 691–700.
Israel, L., Melac, M., Milinkevitch, D., and Dubos, G. (1994). Drug therapy and memory training programs: a double-blind randomized trial of general practice patients with age-associated memory impairment. Int. Psychogeriatr. 6, 155–170.
Jha, N., Jurma, O., Lalli, G., Liu, Y., Pettus, E. H., Greenamyre, J. T., Liu, R. M., Forman, H. J., and Andersen, J. K. (2000). Glutathione depletion in PC12 results in selective inhibition of mitochondrial complex I activity – iImplications for Parkinson’s disease. J. Biol. Chem. 275, 26096–26101.
Kahlert, S., Zundorf, G., and Reiser, G. (2008). Detection of de- and hyperpolarization of mitochondria of cultured astrocytes and neurons by the cationic fluorescent dye rhodamine 123. J. Neurosci. Methods 171, 87–92.
Karacostas, D., Doskas, T., Artemis, N., Vadicolias, K., and Milonas, I. (1999). Beneficial effect of piracetam monotherapy on post-ischaemic palatal myoclonus. J. Int. Med. Res. 27, 201–205.
Keil, U., Bonert, A., Marques, C. A., Scherping, I., Weyermann, J., Strosznajder, J. B., Muller-Spahn, F., Haass, C., Czech, C., Pradier, L., Muller, W. E., and Eckert, A. (2004a). Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J. Biol. Chem. 279, 50310–50320.
Keil, U., Bonert, A., Marques, C. A., Strosznajder, J. B., Muller-Spahn, F., Muller, W. E., and Eckert, A. (2004b). Elevated nitric oxide production mediates beta-amyloid-induced mitochondria failure. Pol. J. Pharmacol. 56, 631–634.
Keil, U., Scherping, I., Hauptmann, S., Schuessel, K., Eckert, A., and Muller, W. E. (2006). Piracetam improves mitochondrial dysfunction following oxidative stress. Br. J. Pharmacol. 147, 199–208.
Kurz, C., Ungerer, I., Lipka, U., Kirr, S., Schütt, T., Eckert, A., Leuner, K., and Müller, W. E. (2010). The metabolic enhancer piracetam ameliorates ß-amyloid peptide induced impairment of mitochondrial function and neuritic outgrowth. Br. J. Pharmacol. 160, 246–257.
Lacor, P. N., Buniel, M. C., Furlow, P. W., Clemente, A. S., Velasco, P. T., Wood, M., Viola, K. L., and Klein, W. L. (2007). Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 27, 796–807.
Lee, D. W., Huh, Y. S., and Kim, K. W. (2009). Evidence-based treatment of Alzheimer’s disease. J. Korean Am. Med. Assoc. 52, 417–425.
Lemere, C. A., Beierschmitt, A., Iglesias, M., Spooner, E. T., Bloom, J. K., Leverone J. F., Zheng J. B., Seabrook T. J., Louard D., Li D., Selkoe D. J., Palmour R. M., and Ervin F. R. (2004). Alzheimer’s disease A beta vaccine reduces central nervous system A beta levels in a non-human primate, the Caribbean Vervet. Am. J. Pathol. 165, 283–297.
Leuner, K., Hauptmann, S., Abdel-Kader, R., Scherping, I., Keil, U., Strosznajder, J. B., Eckert, A., and Muller, W. E. (2007). Mitochondrial dysfunction: the first domino in brain aging and Alzheimer’s disease? Antioxid. Redox Signal 9, 1659–1675.
Malik, R., Sangwan, A., Saihgal, R., Jindal, D. P., and Piplani, P. (2007). Towards better brain management: nootropics. Curr. Med. Chem. 14, 123–131.
Mattson, M. P. (2000). Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 1, 120–129.
Mattson, M. P. (2007). Mitochondrial regulation of neuronal plasticity. Neurochem. Res. 32, 707–715.
Mattson, M. P., Gleichmann, M., and Cheng, A. (2008). Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766.
Mattson, M. P., and Magnus, T. (2006). Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 7, 278–294.
Moreira, P. I., Santos, M. S., and Oliveira, C. R. (2007). Alzheimer’s disease: a lesson from mitochondrial dysfunction. Antioxid. Redox Signal 9, 1621–1630.
Müller, W. E., Eckert, A., Kurz, C., Eckert, G. P., and Leuner, K. (2010). Mitochondrial dysfunction: common final pathway in brain aging and Alzheimer’s disease – therapeutic aspects. Mol. Neurobiol. 41, 159–171.
Muller, W. E., Eckert, G. P., and Eckert, A. (1999). Piracetam: novelty in a unique mode of action. Pharmacopsychiatry 32, 2–9.
Müller, W. E., Hartmann, H., Koch, S., Scheuer, K., and Stoll, S. (1994). Neurotransmission in aging – therapeutic aspects. Int. Acad. Biomed. Drug Res. 7, 166–173.
Muller, W. E., Koch, S., Scheuer, K., Rostock, A., and Bartsch, R. (1997). Effects of piracetam on membrane fluidity in the aged mouse, rat, and human brain. Biochem. Pharmacol. 53, 135–140.
Munch, G., Schinzel, R., Loske, C., Wong, A., Durany, N., Li, J. J., Vlassara, H., Smith, M. A., Perry, G., and Riederer, P. (1998). Alzheimer’s disease – synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts. J. Neural Transm. 105, 439–461.
Muriel, P., and Perez-Rojas, J. M. (2003). Nitric oxide inhibits mitochondrial monoamine oxidase activity and decreases outer mitochondria membrane fluidity. Comp. Biochem. Physiol. C Comp. Pharmacol. Toxicol. 136, 191–197.
Onyango, I. G., Lu, J. H., Rodova, M., Lezi, E., Crafter, A. B., and Swerdlow, R. H. (2010). Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim. Biophys. Acta 1802, 228–234.
Peuvot, J., Schanck, A., Deleers, M., and Brasseur, R. (1995). Piracetam-induced changes to membrane physical-properties – a combined approach by P-31 nuclear-magnetic-resonance and conformational-analysis. Biochem. Pharmacol. 50, 1129–1134.
Reddy, P. H., and Beal, M. F. (2008). Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends. Mol. Med. 14, 45–53.
Rhein, V., Song, X. M., Wiesner, A., Ittner, L. M., Baysang, G., Meier, F., Ozmen, L., Bluethmann, H., Drose, S., Brandt, U., Savaskan, E., Czech, C., Gotz, J., and Eckert, A. (2009). Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. U.S.A. 106, 20057–20062.
Ricchelli, F., Gobbo, S., Moreno, G., and Salet, C. (1999). Changes of the fluidity of mitochondrial membranes induced by the permeability transition. Biochemistry 38, 9295–9300.
Rosenhall, U., Deberdt, W., Friberg, U., Kerr, A., and Oosterveld, W. (1996). Piracetam in patients with chronic vertigo – results of a double-blind, placebo-controlled study. Clin. Drug Investig. 11, 251–260.
Saletu, B., Hitzenberger, G., Grunberger, J., Anderer, P., Zyhlarz, G., Linzmayer, L., and Rameis, H. (1995). Double-blind, placebo-controlled, pharmacokinetic and pharmacodynamic studies with 2 new formulations of piracetam (infusion and syrup) under hypoxia in man. Int. J. Clin. Pharmacol. Ther. 33, 249–262.
Sastre, J., Lloret, A., Borras, C., Pereda, J., Garcia-Sala, D., Droy-Lefaix, M. T., Pallardo, F. V., and Vina, J. (2002). Ginkgo biloba extract EGb 761 protects against mitochondrial aging in the brain and in the liver. Cell. Mol. Biol. 48, 685–692.
Sastre, J., Pallardo, F. V., de la Asuncion, J. G., and Vina, J. (2000). Mitochondria, oxidative stress and aging. Free Radic. Res. 32, 189–198.
Scarmeas, N., Luchsinger, J. A., Mayeux, R., and Stern, Y. (2007). Mediterranean diet and Alzheimer disease mortality. Neurology 69, 1084–1093.
Scarmeas, N., Stern, Y., Mayeux, R., and Luchsinger, J. A. (2006). Mediterranean diet, Alzheimer disease, and vascular mediation. Arch. Neurol. 63, E1–E9.
Schaffer, S., Eckert, G. P., Schmitt-Schillig, S., and Muller, W. E. (2006). Plant foods and brain aging: a critical appraisal. Forum Nutr. 59, 86–115.
Scheuer, K., Rostock, A., Bartsch, R., and Muller, W. E. (1999). Piracetam improves cognitive performance by restoring neurochemical deficits of the aged rat brain. Pharmacopsychiatry 32, 10–16.
Schmitt-Schillig, S., Schaffer, S., Weber, C. C., Eckert, G. P., and Muller, W. E. (2005). Flavonoids and the aging brain. J. Physiol. Pharmacol. 56(Suppl. 1), 23–36.
Schuessel, K., Frey, C., Jourdan, C., Keil, U., Weber, C. C., Muller-Spahn, F., Muller, W. E., and Eckert, A. (2006). Aging sensitizes toward ROS formation and lipid peroxidation in PS1M146L transgenic mice. Free Radic. Biol. Med. 40, 850–862.
Selkoe, D. J. (2008). Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav. Brain Res. 192, 106–113.
Stoll, L., Schubert, T., and Muller, W. E. (1992). Age-related deficits of central muscarinic cholinergic receptor function in the mouse – partial restoration by chronic piracetam treatment. Neurobiol. Aging 13, 39–44.
Szalma, I., Kiss, A., Kardos, L., Horvath, G., Nyitrai, E., Tordai, Z., and Csiba, L. (2006). Piracetam prevents cognitive decline in coronary artery bypass: A randomized trial versus placebo. Ann. Thorac. Surg. 82, 1430–1435.
Toupet, M. (2001). Management of vertigo and impaired-balance syndromes in the ederly. Presse Med. 30, 1273–1274.
Uebelhack, R., Vohs, K., Zytowski, M., Schewe, H. J., Koch, C., and Konertz, W. (2003). Effect of piracetam on cognitive performance in patients undergoing bypass surgery. Pharmacopsychiatry 36, 89–93.
Valzelli, L., Bernasconi, S., and Sala, A. (1980). Piracetam activity may differ according to the age of the recipient mouse. Int. Pharmacopsychiatry 15, 150–156.
Viana, G. S. B., Marinho, M. M. F., and Sousa, F. C. F. (1992). Effect of piracetam administration on 3H-N-methylscopolamine binding in cerebral-cortex of young and old rats. Life Sci. 50, 971–977.
Waegemans, T., Wilsher, C. R., Danniau, A., Ferris, S. H., Kurz, A., and Winblad, B. (2002). Clinical efficacy of piracetam in cognitive impairment: a meta-analysis. Dement. Geriatr. Cogn. Disord. 13, 217–224.
Waldbaum, S., and Patel, M. (2010). Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 88, 23–45.
Wang, X., Zhu, J., Xu, R., Yang, X., Wu, H., Lin, D., Ye, F., and Hu, L. (2010). Determination of piracetam in rat plasma by LC-MS/MS and its application to pharmacokinetics. Biomed. Chromatogr. doi: 10.1002/bmc.1414 [Epub ahead of print].
Wilsher, C. R. (1986). Effects of piracetam on developmental dyslexia. Int. J. Psychophysiol. 4, 29–39.
Wilsher, C. R., Bennett, D., Chase, C. H., Conners, C. K., Diianni, M., Feagans, L., Hanvik, L. J., Helfgott, E., Koplewicz, H., Overby, P., Reader, M. J., Rudel, R. G., and Tallal, P. (1987). Piracetam and dyslexia - effects on reading tests. J. Clin. Psychopharmacol. 7, 230–237.
Winblad, B. (2005). Piracetam: a review of pharmacological properties and clinical uses. CNS Drug Rev. 11, 169–182.
Xerri, C., and Zennou-Azogui, Y. (2003). Influence of the postlesion environment and chronic piracetam treatment on the organization of the somatotopic map in the rat primary somatosensory cortex after focal cortical injury. Neuroscience 118, 161–177.
Keywords: mitochondrial dysfunction, alzheimer’s disease, aging, oxidative stress, piracetam
Citation: Leuner K, Kurz C, Guidetti G, Orgogozo J-M and Müller WE (2010) Improved mitochondrial function in brain aging and Alzheimer disease – the new mechanism of action of the old metabolic enhancer piracetam. Front. Neuropharma. 4:44. doi: 10.3389/fnins.2010.00044
Received: 10 May 2010;
Paper pending published: 29 May 2010;
Accepted: 08 June 2010;
Published online: 07 September 2010.
Edited by:
Xiao-Ming Ou, University of Mississippi Medical Center, USAReviewed by:
Lian-Jun Guo, Huazhong University of Science and Technology, ChinaGerald Muench, University of Western Sydney, Australia;
James Cook University, Australia
Copyright: © 2010 Leuner, Kurz, Guidetti, Orgogozo and Müller. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Walter E. Müller, Department of Pharmacology, Max-von-Laue-Strasse 9, 60438 Frankfurt, Germany. e-mail: pharmacolnat@em.uni-frankfurt.de