 Javaria Ashraf
Javaria Ashraf Jamil Ahmad
Jamil Ahmad Amjad Ali
Amjad Ali Zaheer Ul-Haq
Zaheer Ul-Haq
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neuroinform., 23 May 2018
Volume 12 - 2018 | https://doi.org/10.3389/fninf.2018.00026
This article is part of the Research TopicThe Alzheimer's Disease ChallengeView all 40 articles
Alzheimer's Disease (AD) is the most common neuro-degenerative disorder in the elderly that leads to dementia. The hallmark of AD is senile lesions made by abnormal aggregation of amyloid beta in extracellular space of brain. One of the challenges in AD treatment is to better understand the mechanism of action of key proteins and their related pathways involved in neuronal cell death in order to identify adequate therapeutic targets. This study focuses on the phenomenon of aggregation of amyloid beta into plaques by considering the signal transduction pathways of Calpain-Calpastatin (CAST) regulation system and Amyloid Precursor Protein (APP) processing pathways along with Ca2+ channels. These pathways are modeled and analyzed individually as well as collectively through Stochastic Petri Nets for comprehensive analysis and thorough understating of AD. The model predicts that the deregulation of Calpain activity, disruption of Calcium homeostasis, inhibition of CAST and elevation of abnormal APP processing are key cytotoxic events resulting in an early AD onset and progression. Interestingly, the model also reveals that plaques accumulation start early (at the age of 40) in life but symptoms appear late. These results suggest that the process of neuro-degeneration can be slowed down or paused by slowing down the degradation rate of Calpain-CAST Complex. In the light of this study, the suggestive therapeutic strategy might be the prevention of the degradation of Calpain-CAST complexes and the inhibition of Calpain for the treatment of neurodegenerative diseases such as AD.
Alzheimer's disease (AD) is a neurodegenerative disorder which has impacted nearly 44 million1 people around the world and this number is still increasing. AD is the leading cause of dementia in the old age (Ashford, 2004). Unfortunately, it is diagnosed only in one out of four people living with the disease1. Clinical characterization of AD includes memory loss and cognitive impairment which further lead to damaged behavioral activities and render a person completely dependent on an external aid (Budson and Price, 2005). AD establishes over time with the appearance of pathological emblems which are senile plaques and neurofibrillary tangles. These lesions comprise of extracellular deposits of Amyloid beta (Aβ) (Selkoe, 2000; Golde, 2005; Tam and Pasternak, 2012) and intracellular self-gathered clumps of tau proteins (Lee et al., 2001), respectively. Aβ is a 40–42 amino-acids long peptide which is formed after the proteolytic cleavage of Amyloid Precursor Protein (APP) (Selkoe, 2000; Golde, 2005; Tam and Pasternak, 2012). Previous studies have shown that Aβ monomers are initially non-toxic but their conversion to oligomers makes them toxic (Volles and Lansbury, 2002; Walsh and Selkoe, 2004). Eventually, the abnormal accumulation of oligomers form plaques (Walsh et al., 2002) that deposit into neuronal Endoplasmic Reticulum (ER) (Cuello, 2005) and in extracellular space (Trojanowski and Lee, 2000; Walsh et al., 2000). Aggregation of senile plaques and neurofibrillary tangles cause neuronal cell death and synaptic failure (Tiraboschi et al., 2000; Selkoe, 2002). During the last two decades, several lines of studies have pointed toward the imbalance between Aβ production and its clearance plays a central role in pathogenesis of AD. Since 1992, this hypothesis has earned acquiescence (Hardy and Higgins, 1992) and is known as “Amyloid cascade hypothesis (ACH)”. It suggests that Aβ and processing of APP are crucial in neuro-degeneration. In AD, aggregation of Aβ is the first step leading toward the formation of senile plaques (Hardy and Selkoe, 2002; Vassar, 2005). APP is a type1 trans-membrane protein produced in ER (Greenfield et al., 1999; Roussel et al., 2013). In neurons, production and metabolism of APP occurs rapidly which makes it a crucial element in neuro-pathogenesis (Lee et al., 2008). The main APP proteolytic processing steps occur at the cell surface and Trans-Golgi networks (TGNs). Proteolysis of APP can occur through the so-called non-amyloidogenic and amyloidogenic Pathways (Figure 1). The first step of non-amyloidogenic pathway is carried out by the enzyme alpha (α)-secretase that breaks down APP into soluble Amyloid precursor protein alpha (sAPPα) and alpha C-terminal fragment (αCTF / CTF83). The catalysis by α-secretase is imperative as it cuts APP within Aβ domain which blocks Aβ formation (Lichtenthaler, 2011). This initial step can also be driven by the beta (β)-secretase / β-site APP-cleaving enzyme (BACE), a transmembrane aspartyl protease (Vassar et al., 1999; Haass, 2004) (Figure 1), which constitute amyloidogenic pathway. BACE is a crucial enzyme, that acts as a rate limiting protein in Aβ generation. It breaks down the APP into soluble Amyloid precursor protein beta (sAPPβ) and beta C-terminal fragments (βCTF / CTF99) (Cai et al., 2001). The CTFs are intermediate products of the first step in both pathways which remain attached to the membrane and they are further cleaved by gamma (γ)-secretase (Zhang et al., 2011). In non-amyliodogenic pathway, the fragment α-CTF is cut down by γ-secretase into p38 and the Amyloid Precursor Protein Intracellular Cytoplasmic / C-terminal Domain (AICD). While in amyloidogenic pathway, γ-secretase degrades the βCTF into Aβ and AICD (O'Brien and Wong, 2011) (Figure 1).
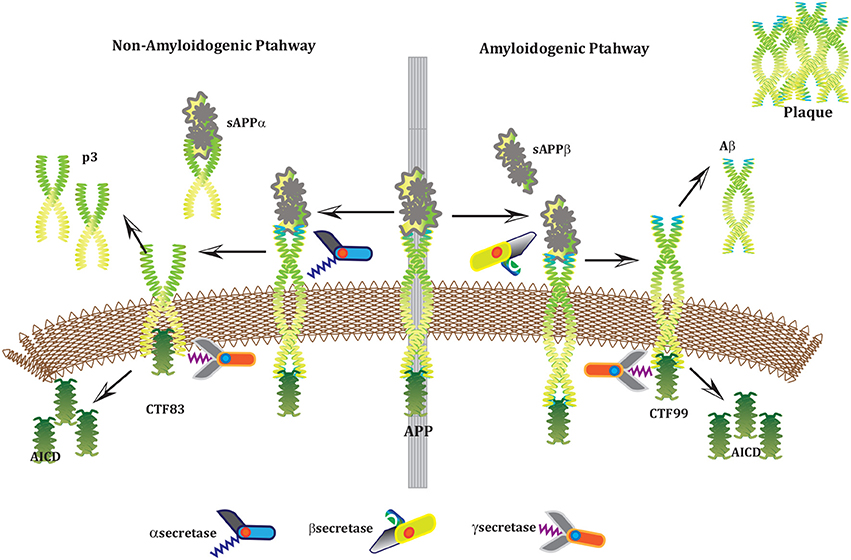
Figure 1. APP and processing products: APP is synthesized in the ER and then transported to the trans-Golgi-network (TGN) where it is cleaved by secretases. In non-amyloidogenic pathway (left), cleavage of APP by α-secretase results in the generation of sAPPα and C-terminal fragments CTF83 which is further cleaved by γ-secretase into p3 and AICD. Proteolysis by α-secretase prevents Aβ production as the cleavage site in APP is within the Aβ domain. In amyloidogenic pathway (right), APP is cleaved into sAPPβ and CTF99 by β−secretase / BACE activity. Furthermore, CTF99 breaks down into AICD and Aβ by γ-secretase activity. Aβ fragments oligomerize and fibrillize into plaques.
The Biological Regulatory Networks (BRN) of APP processing, depicted in Figure 2, is also built from Figure 1. APP processing depends on sequential cleavage by three secretases (α/β-secretase and γ-secretase). In normal conditions, α-secretase residing at the plasma membrane is constitutively active for APP coming to the cell surface and thus favoring non-amyloidogenic pathway (De Strooper and Annaert, 2000). Though there is an interesting fact about APP proteolysis that none of the secretases show special substrate specificity toward APP. There are several transmembrane proteins such as cell surface receptors and ligand, growth factors and cytokines besides APP which undergo ectodomain shedding by enzymes with α-secretase activity (Annaert and Saftig, 2009). In the same manner, BACE shows low affinity toward APP and it is not its exclusive physiological substrate (DeStrooper et al., 2006; Hu et al., 2006). Many observations highlight that in healthy cells APP is frequently processed through non-amyloidogenic pathway to resist amyloid generation while it is altered in pathological conditions (De Strooper and Annaert, 2000). Abnormal processing of APP is stated to be the first and fundamental step in plaques formation in AD pathogenesis (Jonsson et al., 2012). In neuropathological conditions, BACE affinity toward APP increases two folds which leads to enhanced Aβ production (Yang et al., 2003; Li and Südhof, 2004). Recent studies on transgenic mice model have shown that BACE activity is modulated by Calpain activation in AD pathology (Liang et al., 2010). Calpain-Calpastatin system also plays a key role in neurodegeneration. Transgenic mice models have shown that over expression of APP, increased production of Aβ, inhibition of Calpastatin (CAST) and activation of Calpain increase neuronal degeneration in AD (Higuchi et al., 2012).
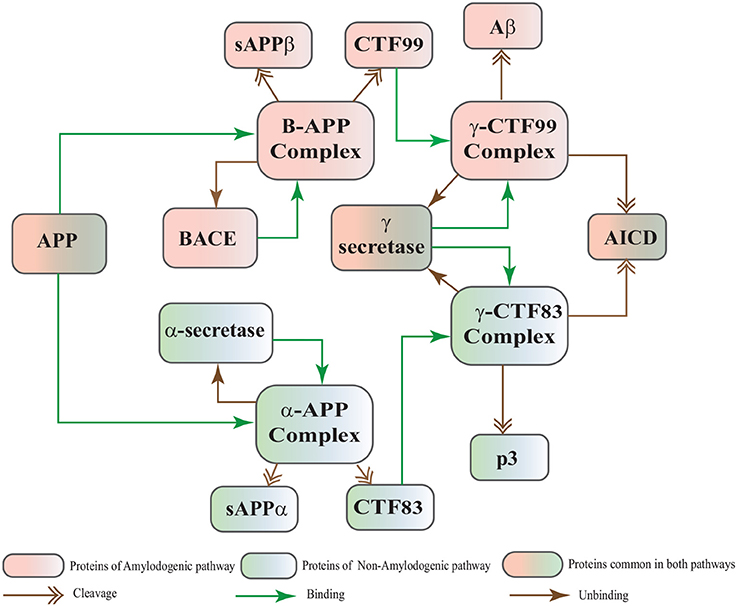
Figure 2. APP processing pathways BRN derived from Figure 1.
Calpains are protein clan of cysteine/ thiol proteases and their activity depends on Ca2+ concentration (Ferreira, 2012). The most studied Calpains, mu(μ)-Calpain (Calpain1) and m-Calpain (Calpain2) are present abundantly in neurons, central nervous system (CNS) and glial cells. Though their distribution differs, Calpain1 is ubiquitous and expressed more in neurons while Calpain2 is present in glial cells (Ono and Sorimachi, 2012; Santos et al., 2012). Calpain1 requires micro-molar concentration of Ca2+ (10–50μM), while Calpain2 is activated by mili-molar concentration of Ca2+ (250–350μM) in vitro (Goll et al., 2003; Ryu and Nakazawa, 2014). Ca2+ plays important role in ensuring the cell's vital functions. In addition to calcium, Calpain is tightly regulated in the cell by CAST which is also ubiquitous and solely a specific endogenous inhibitor for both Calpains (Melloni et al., 2006).
CAST is reported as an explicit suicide substrate for Calpain (Yang et al., 2013). The proportion of CAST in a cell is normally larger than Calpain, its ratio with location is crucial in controlling the extent of activation of Calpain within a cell (Todd et al., 2003). CAST interacts with Calpain at different stages i.e., first it constrains Calpain at the membrane where pro-Calpain is attached then it interacts with active Calpain inside cytosol (Hanna et al., 2008). CAST forms a reversible complex with Calpain at both the sites. At membrane, the reversible complex breaks down when Ca2+ influx increases to release Calpain. Inside cytosol, Calpain undergoes autolysis to attain active conformation. In response, CAST changes its cellular distribution to make itself widely available in the cytoplasm to counter active Calpain (Todd et al., 2003). Both active Calpain and CAST rejoin in a reversible complex to resist persistent activity of Calpain (De Tullio et al., 1999). Active Calpain modulates CAST by slowly digesting it into small inactive fragments which results in plethora of Calpain in cell leading to pathological condition (Averna et al., 2001b; Tompa et al., 2002) (Figure 3). It has been reported that in AD CAST becomes depleted from different regions of the brain as compared to healthy aged brain (Rao et al., 2008). It has also been observed that by controlling Calpain, CAST is indirectly preventing cell membrane damages induced by high Ca2+ and Aβ peptide (Vaisid et al., 2008).
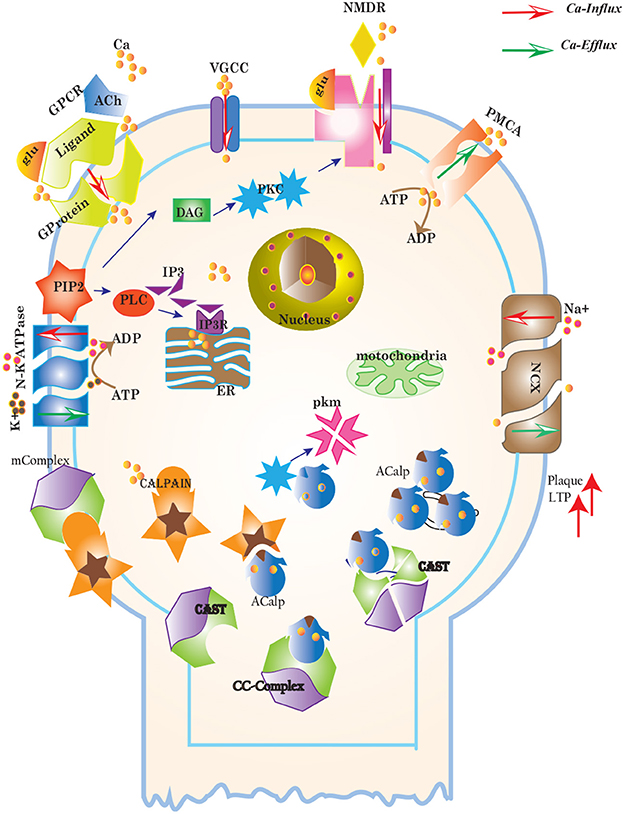
Figure 3. Different calcium channels work in harmony to establish homeostasis in neurons. Calcium influx is controlled by voltage gated (VGCCs) or receptor-ligand based (NMDAR, GPCR) channels. ER also release Ca2+ into the cytoplasm through inositol-1,4,5-trisphosphate (IP3R) and ryanodine receptors. Calcium efflux is carried out by energy (ATP) dependent channels such as plasma membrane calcium ATPase (PMCA), sodium-potassium ATPase (NKA) and sodium-calcium exchanger (NCX) channels. Calcium homeostasis influences Calpain-CAST system. At membrane, Calpain is bound to CAST to form mComplex at low Ca2+ level. At high Ca2+ concentration, Calpain is released into cytoplasm and autolysed to active form ACalp that again forms complex with CAST (cComplex). Gradually the complex breaks down and releases ACalp which enhances Plaque accumulation and LTP events.
CAST pool is regulated by reversible phosphorylation via PKC, which is a Ca2+-activated phospholipid dependent kinase. Moreover, it is de-phosphorylated by protein phosphatases (ppase) (Melloni et al., 2006). Phosphorylation control CAST inhibitory efficiency in brain (Averna et al., 2001a) to regulate its availability for calpain inhibition. Reversible protein phosphorylation regulates many neuronal functions and is important for neuronal signal transduction (Wu and Lynch, 2006). Inactive PKC is converted to Ca2+-bound activated form in the presence of diacylglycerol (DAG) which in turn is activated by receptor based hydrolysis of phosphoinositides 3 (IP3) (Courjaret et al., 2003). The N-terminal region of CAST which is responsible for the function of the protein has a site for phosphorylation by PKC. CAST is phosphorylated by PKC to decrease its inhibitory efficiency toward calpain (Averna et al., 2001a) (Figure 3). It has been observed that PKC also regulates APP processing by activating α-secretase (Rossner et al., 2001; Racchi et al., 2003), it promotes non-amylodogenic pathway over β-secretase (Lanni et al., 2004). In vivo studies show that in the presence of PKC, secretion of sAPPα increases and Aβ secretion declines (Chen and Fernandez, 2004). Other studies about AD found that PKC has substantial role in AD pathology (Etcheberrigaray et al., 2004; Alkon et al., 2007). Active Calpain also interacts with PKC and converts it into constitutive active enzyme (Yamakawa et al., 2001; Goll et al., 2003). Calpain1 directly starts depletion of PKC from cell by converting it into protein kinase M (PKM) (Yamakawa et al., 2001; Liu et al., 2008). The whole mechanism is also depicted in the form of Calpain-CAST system BRN in Figure 4.
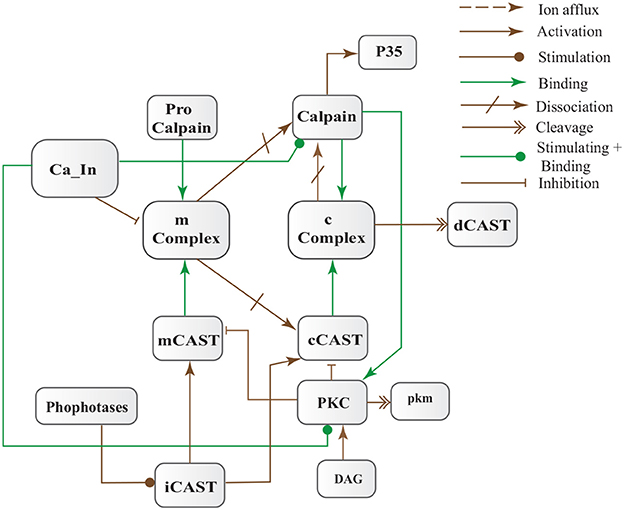
Figure 4. Calpain-CAST system BRN derived from Figure 3.
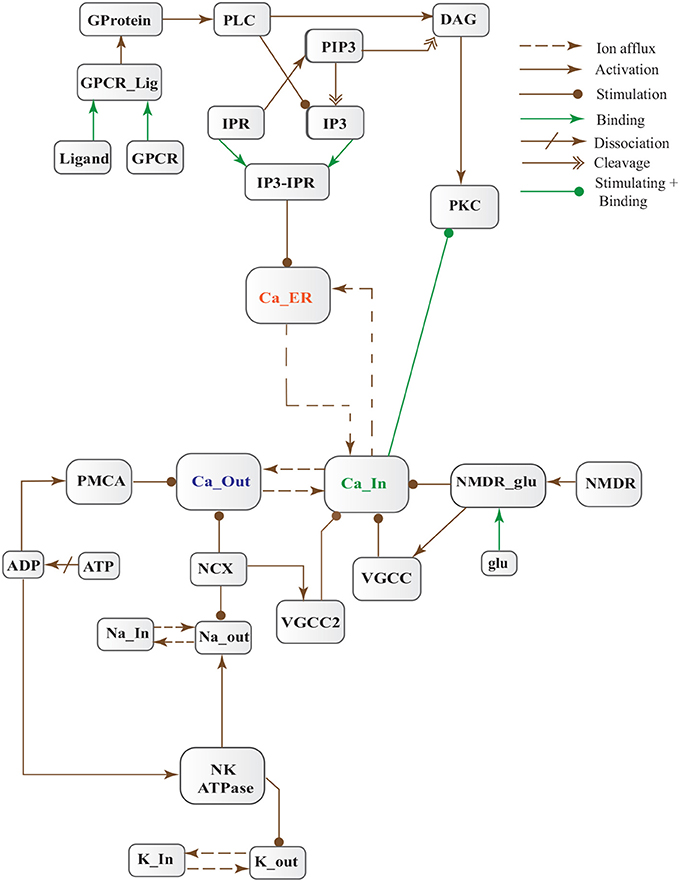
Figure 5. Calcium Influx Efflux BRN derived from Figure 3.
The dysregulation of Calcium homeostasis contributes in aging and neurodegeneration (Mattson, 2004; Smith et al., 2005; Stutzmann, 2005). A tremendous deal of work by calcium is tightly regulated in time, space and intensity by intracellular stores, influx and efflux channels (Stutzmann, 2005). At resting stage, extracellular Ca2+ concentration ranges from 1.5 to 2.0 mM (Orrenius et al., 2003). While magnitude of Ca2+ inside a cell is very low (between 50–100/ 50–300 nM) (LaFerla, 2002; Orrenius et al., 2003) and after activation it can rise to several micromoles. On contrary, inside ER, the level of Ca2+ is in the range 100-500μM (LaFerla, 2002) which is approximately 1000 times higher than cytosol concentration at the resting phase. Persistent alteration of Ca2+ homeostasis affects production and digestion of pathological proteins such as Calpain, Aβ and tau protein. Dysregulation of cellular Ca2+ level is an early and main feature of AD (Mattson et al., 2000; LaFerla, 2002; Small, 2009).
Cytosolic Ca2+ is maintained at very low level as compared to extracellular space through several homeostatic mechanisms, working both temporally and spatially (Figure 3). These equilibrating apparatuses include voltage-operated channels (VOCs) and receptor operated channels (ROCs) for Ca2+ inclusion, Ca2+ storage in organelles e.g., ER (Wojda et al., 2008) and Ca2+ extrusion to extracellular space. Different ATP-dependent membrane pumps such as plasma membrane calcium ATPase channel (PMCA) and sodium-calcium exchanger (NCX) which are dependent on sodium-potassium ATPase (NKA) (Wojda et al., 2008; Brittain et al., 2012) are used for Ca2+ efflux. In different physiological processes, elevation of Ca2+ is necessary to switch-on respective proteins. Ca2+ inclusion is administered by several routes such as N-methyl-D-aspartate receptor (NMDAR), an imperative type of ROCs, which switch into open conformation after binding of endogenous glutamate (glu) as ligand. Another important influx gateway is voltage gated Ca2+ channel (VGCC) which is in closed conformation when neuronal membrane is polarized (Schmolesky et al., 2002; Cain and Snutch, 2011). The VGCC adopts open conformation as plasma membrane depolarizes due to Ca2+/ sodium (Na+) influx through ROCs or ion channels (Weber, 2012). Ca2+ influx also increases from intracellular stores in ER through store-operated channels. There are two calcium channels in ER which are IP3-sensitive and ryanodine (RyRs)-sensitive Ca2+ stores (Berridge, 2009). IP3 driven release of Ca2+ starts by binding of G-protein coupled receptor (GPCR) on plasma membrane which induces Phospholipase C (PLC) mediated cleavage of phosphatidylinositol-4,5-bisphosphate (PIP2) on cell membrane into DAG and IP3. IP3 binds to its receptor on ER membrane and stimulate Ca2+ release into the cytoplasm (Berridge, 2009; Krebs et al., 2015). Furthermore, depletion of ER stores mediate influx of extracellular Ca2+ through store-operated channels (SOCs) (Emptage et al., 2001; Weber, 2012). The mechanism for lowering Ca2+ from cell is controlled by PMCA and NCX. Both PMCA and NCX are energy dependent while, NCX is also Na+ gradient dependent (Wojda et al., 2008). The BRN of Calcium channels, Figure 4, is also helpful in understanding the mechanism underlying the Ca2+ homeostasis.
To comprehend the above mentioned neuronal pathways, models are constructed to understand their dynamics. Stochastic approaches describe the randomness of biological system accurately as compared to ordinary differential equations. In BRNs, the activation or inhibition processes take place with random time delays, therefore, stochastic modeling frameworks are more suitable for their modeling. Petri nets provide complementary approach for both qualitative and quantitative modeling and simulation of the dynamical behavior of large systems in an intuitive way (Mounts and Liebman, 1997; Tsavachidou and Liebman, 2002; Tareen and Ahmad, 2015). The study (Tsavachidou and Liebman, 2002) shows that the Petri net models predict the experimental findings which support the soundness of these models. Stochastic petri nets (SPNs) have emerged as a promising tool for modeling and analyzing BRNs in the field of molecular biology (Goss and Peccoud, 1998). The dynamic behaviors of a variety of BRNs have been studied using stochastic simulations (Mura and Csikász-Nagy, 2008; Lamprecht et al., 2011; Castaldi et al., 2012; Marwan et al., 2012).
In this study, we have modeled and analyzed the neuronal physiological system constituting Ca2+ channels maintaining homeostasis, CAST regulating Calpain system and APP processing pathways separately and collectively at molecular level using SPNs to understand the AD progression mechanism. Particularly, we have analyzed neuronal patho-physiological dynamic behaviors causing the development of hallmark lesions in brain to answer many question e.g., how dysregulation of Ca2+ triggers AD? When CAST, the sole inhibitor of Calpain, depletes from the brain cells? how Aβ production increases? and when the accumulation of plaques start? The answers to these questions lie in the modeling of the combined BRN Figure 6. The model predicts that Calpain is the main cause of dysregulation, which start with the rise in Ca2+ levels in the cytosol. Calpain activates different pathways through which Aβ production and accumulation increases. Plaques start building with time at the age of forty and older. Plaques first enter lag phase and then into rapid growth phase. Calpain slowly degrades CAST which depletes from the cell and eventually neuronal degradation progresses. These results suggest that patho-physiological events such as dysregulation of Ca2+ homeostasis, Calpain hyper-activation, CAST degradation and abnormal digestion of APP, all are inter-connected and a cumulative study of these processes through SPN was needed.
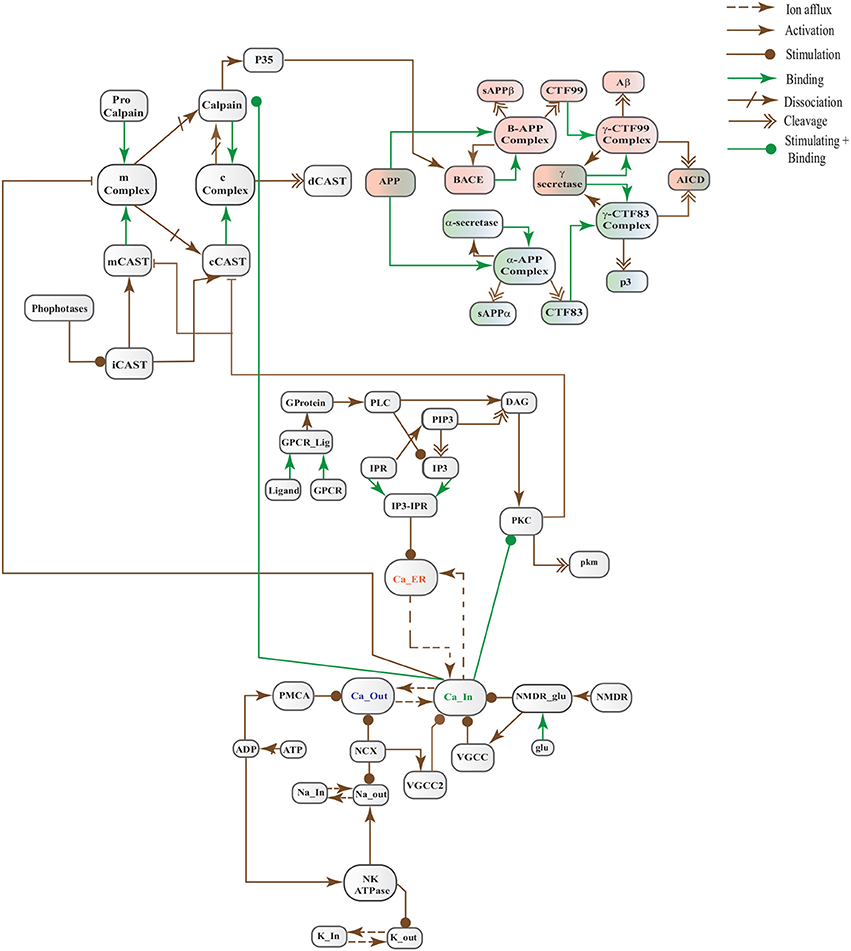
Figure 6. A crosstalk network of three pathways: APP processing pathways, Calcium Channels and Calpain-CAST system.
Petri nets (PNs) have three components namely places, transitions and edges. Place and transition are collectively known as nodes/ vertices of a PN. An arc or edge joins nodes such as a place (pre-place) to a transition or a transition to a place (post-place) to make a bipartite graph (Petri, 1966). Edges are directed and have weights associated with them. In biological systems, such as BRNs, places (○) represent biological entities e.g., protein or their complexes, gene, mRNA, ions, metabolites and cell or cellular components while transitions (□) represent biochemical reactions e.g., association, activation, decomposition, inhibition, phosphorylation, dephosphorylation and translocation (Tareen and Ahmad, 2015). The weights of edges represent the stoichiometry of reactions. The weight can be one (1) or greater than one (Blätke et al., 2011; Liu et al., 2016). Following are the different types of edges.
• Standard edge(→) is use to represents a simple biochemical reaction such as synthesis, decomposition, replacement and activation reactions. It is enabled when pre-places have adequate tokens. When the transition is fired, tokens from pre-places are removed and then deposited in post-places according to arc weights.
• Inhibitor edge(—◦) connects only a pre-place to a transition. If the corresponding pre-place is not adequately marked i.e., it has less tokens than weight of arc, then, the transition is enabled otherwise, it is un-firable. Tokens are not removed from pre-place in the inhibitor edge.
• Read edge (—•) connects only a pre-place to a transition. It enables the transition when the corresponding place is adequately marked. The tokens of a place are not altered when the transition is fired.
• Equal edge (–•−•) connects pre-place to transition. It may fire a transition when number of tokens in a pre-place is equal to corresponding arc weight. After firing of transition, tokens are not removed from the respective pre-place.
The following definitions are originally given in David and Alla (2010).
Definition 1 (Unmarked Petri Net). An unmarked Petri Net (PN) is a five-tuple ℙ = where:
• is a finite set of places i.e., = {, , …, },
• is a finite set of transitions i.e., = {, , , …, },
• = ∅, both and are non-empty sets,
• , is a set of input and output edges of transitions.
• 𝔭𝔯𝔢 : ℕ , is a weight function that assigns non-negative integers to input edges.
• 𝔭𝔬𝔰𝔱 : ℕ , is a weight function that assigns non-negative integers to output edges.
A simple unmarked PN is given in Figure 7A, to model a receptor-ligand association. A receptor is in closed conformation and ligand is in in-active form. The receptor must undergoes open conformation and then the ligand activates to form an association (RL_Complex) with receptor. The places in a PN may have tokens (black dots or positive real numbers) which represent marking of the places. In a marked PN, places are initially assigned tokens.
Definition 2 (Marked Petri Net). A Marked Petri Net (MPN) is a tuple 𝕄ℙ = where:
• ℙ = is an unmarked PN and
• : ℤ≥0, is an initial marking of the PN.
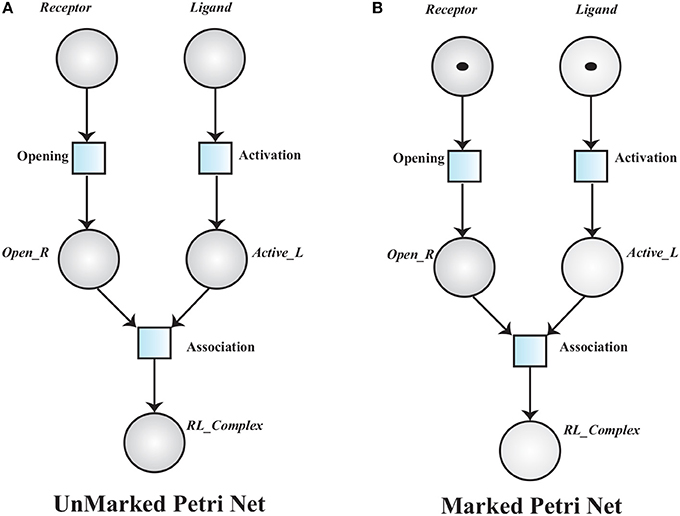
Figure 7. (A) An Unmarked Petri net model of Receptor Ligand binding. RL_Complex is formed when Receptor is in open conformation (Open_R) and Ligand is activated into Active_L. (B), A marked PN model has tokens which represent initial marking of the Receptor Ligand binding model.
The transition associated to a marked place is said to be enabled and it will be fired when the corresponding pre-places have tokens equal or greater than the weight of the associated arc. Receptor opening requires a Receptor and ligand activation depends on the presence of an inactive Ligand. An active ligand and an opened receptor form a Receptor-Ligand Complex (Figure 7B).
Definition 3 (Timed Petri Net). A Timed Petri Net (TPN) is a tuple 𝕋ℙ = where:
• 𝕄ℙ = is a MPN.
• : ℝ+ is a function that associates a positive real value i.e., time delay to each transition.
A TPN associates a time delay di to a transition . The transition is said to be enabled when it has sufficient tokens in its pre-places and it is fired when its deterministic delay time is elapsed (Figure 8). Stochastic Petri Net (SPN) are used when time delays are random variables. SPN (Figure 8C) (Marsan et al., 1994; Heiner et al., 2008) are explicitly derived from TPN (Figure 8A).
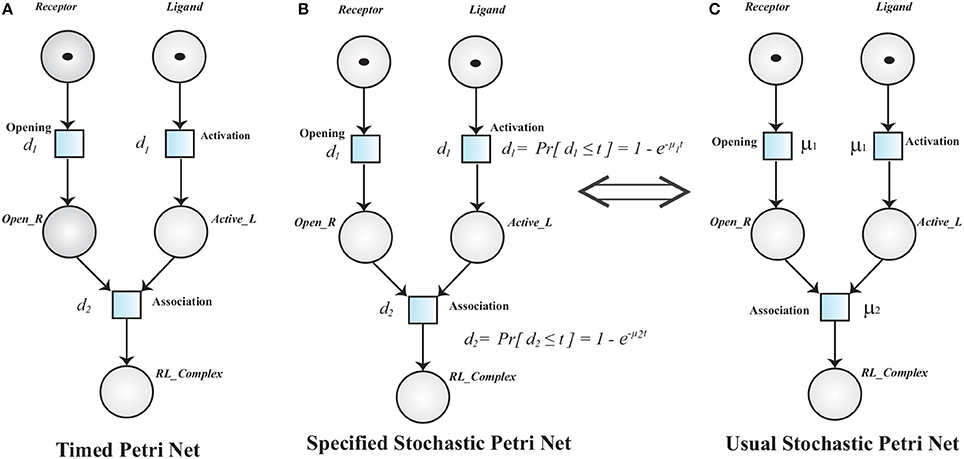
Figure 8. (A) A Timed Petri net model of Receptor Ligand Binding. Initially, the Receptor is in close conformation and the Ligand is inactive. After enabling of respective transition (Opening and Activation), Receptor adopts open conformation (Open_R) and Ligand is activated into Active_L respectively. The transition Opening and Activation have an associated time delay d1. The transitions are fired when the associated time delay is elapsed. RL_Complex is formed when Open_R, Active_L are present and time d2 is elapsed. (B,C) are Specified and Usual Stochastic Petri Nets respectively. The TPN can be converted into SPN when the deterministic firing function d changes into a random variable.
Definition 4 (Stochastic Petri Net). A marked stochastic petri net is a pair 𝕊ℙℕ = 〈𝕄ℙ, ℜ𝔞𝔱𝔢〉 where:
• 𝕄ℙ = is a MPN.
• ℜ𝔞𝔱𝔢 : ℝ+, is a function from the set of transition to the set of finite positive real numbers. ℜ𝔞𝔱𝔢() = μi, is the firing rate associated with transition .
The random variables di have assigned negative exponential probability distribution function (PDF) (t),
The marking of of SPN is a homogenous Markovanian Chain (HMC) process, which is a class of stochastic processes simply built from the reachability graph of a qualitative PN by assigning transition rates to edges between all the states (Heiner et al., 2008). Thus an HMC can be associated with every SPN. Gillespie was the first one who designed special case of Petri nets for reaction networks and called them SPNs (Gillespie, 1976). Snoopy (Marwan et al., 2012) tool is used to design, animate and simulate SPNs of neurodegeneration related BRNs. This tool has been extensively used to model a variety of systems such as software systems, biological systems and production systems. The supplementary file S1 contains a general Enzyme-Substrate BRN that is modeled and simulated through the Petri net modeling tool Snoopy. This file helps to explain the working of the tool (Snoopy).
In this study, stochastic modeling and analysis of the neuronal pathways involved in neuro-degradation in AD was carried out. The SPN models of the three main neuronal physiological pathways: Calcium Influx Efflux channels (Figure 5), Calpain-CAST regulatory system (Figure 4) and APP digesting pathways (Figure 2). The SPN model of the crosstalk of these three pathways is also presented.
The BRN of Calcium influx efflux channels in Figure 5 is translated into a SPN model as shown in Figure 9. The influx channels comprise of receptor based and voltage gated channels (NMDR, VGCC1 / VGCC2). In addition, the GPCR channel regulates intracellular trafficking of Ca2+ ions and stimulation of PKC signaling pathway. The energy dependent efflux channels (e.g., PMCA and Na-K ATPase) work efficiently in a sync with influx channels to maintain equilibrium between in-out flow of Ca2+ ions in neurons. Ca2+ ions move from Ca_In place (green) to Ca_Out place (blue) and vice versa. The places in green colors and transitions (yellow) constitute influx channels. The places in light blue colors and transitions in grey colors make efflux channels. Initially, NMDR_0 place represents that receptor is in close conformation that binds with glu to form the complex represented by NMDR_glu_0 place. This complex adapts open conformation represented by the place NMDR_glu_1 and then further triggers the opening of VGCC channel represented by the place VGCC_c1 to facilitate Ca2+ ions influx. Excess Ca2+ ions are deposited into the place Ca_ER through store-operated channel. The place GProtein_i represents inactive GPCR, that change into place GProtein_act by a complex of GPCR and ligand represented by places GPCR_Lig (complex), GPCR (receptor) and ligand respectively. GProtein_act activates PLC represented by place PLC_act, which mediate cleavage of PIP2 place into DAG and IP3. The place DAG activates PKC by adding Ca2+ ion in it. At the surface of ER, IP3 binds with its receptor IP3R, represented by places IP3 and IP3R, which induces Ca2+ ions flow into the place Ca_ER. At the same time, Ca outflow is maintained by PMCA, NCX and Na-K ATPase. PMCA channel represented by place PMCA_1 requires ATP (place ATP) to extrude one Ca2+ ion into extracellular space. The place NCX representing NCX pump ensures extrusion of one Ca2+ ion by adding three Na ions into the cytoplasm represented by place Na_In. The place Na_K_1 represents active (i.e., depends on ATP) Na-K ATPase pumping that pumps Na out of the neuron (place Na_out) and K into the neuron (place K_In). The place VGCC_c2 representing Ca inflow is also dependent on place NCX, which depolarizes the membrane. The SPN model is simulated after applying and adjusting the rates of transitions in order to reproduce physiological working of the pathway. The transitions are labeled by t, transition from t1–t22 are associated with influx channels. t1–t10 are linked to GPCR channel which help in storage of excess Ca2+ ions into place Ca_ER. t11 takes Ca2+ ions from Ca_ER into cytoplasm (place Ca_In). Remaining t12–t22 make NMDR and VGCC channels functional. The efflux channels, consisting of NCX, NKA channels and PMCA are connected with transitions t23–t29. The transitions t30 and t31 are involved in activation of PKC which is dependent on calcium. The rates of transitions are shown in parameter in Table 1. The simulation results have shown that there is an equilibrium behavior in all channels, which is required to maintain homeostasis in calcium flow. In Figure 10A, a stable oscillating behavior can be observed among the places Ca_out, Ca_ER and Ca_In. Concentration of Ca2+ ions in Ca_out place is higher while it is lower in place Ca_In. Na and K ions, (Figures 10B,C) are also oscillating properly, contributing to Ca2+ homeostasis and generating nerve impulses through polarization and depolarization. Ca2+ homeostasis has pivotal role in cell physiological working. Dysregulation of Ca2+ homeostasis will affect the regulation of most of the proteins, enzymes and genes which will have deleterious effects on the physiological processes.
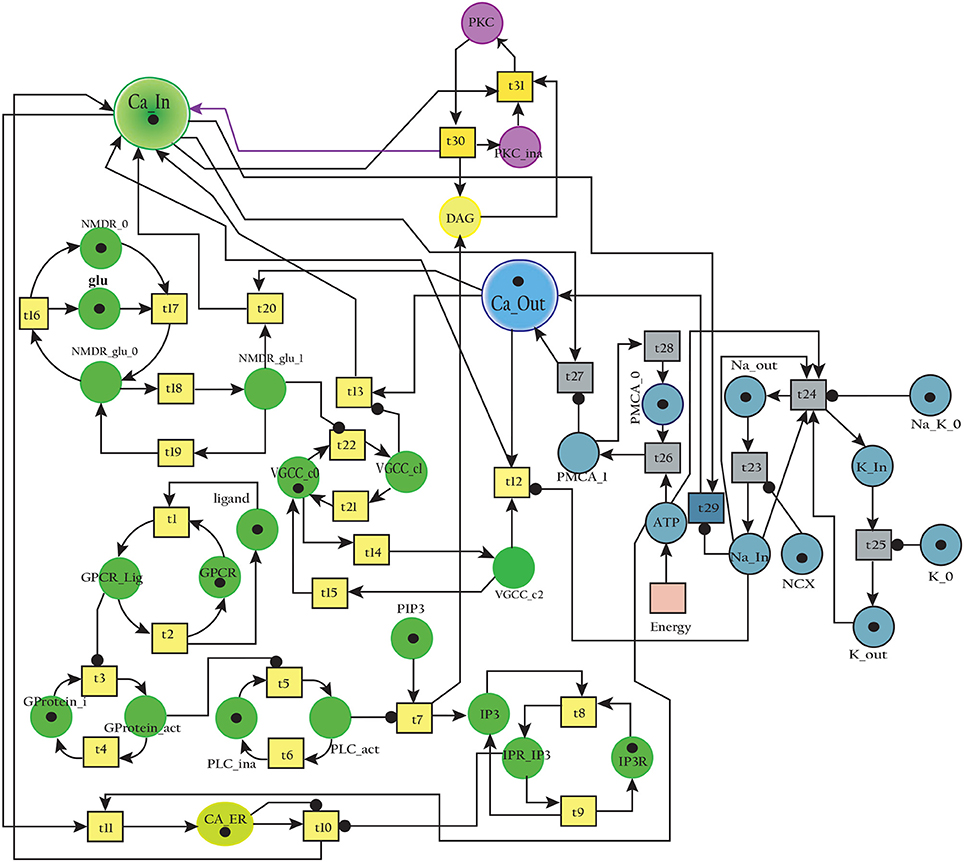
Figure 9. SPN model of the Calcium Channel.
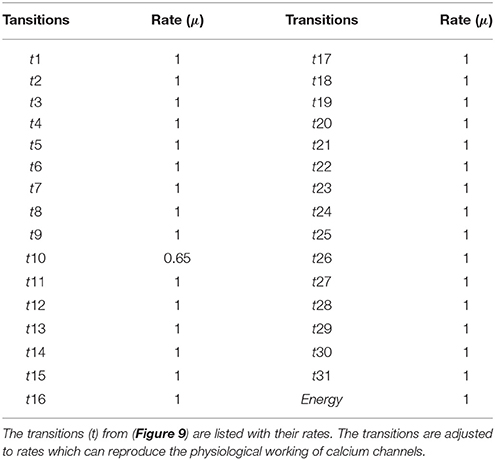
Table 1. Transition rates of the SPN model of Calcium Channel.
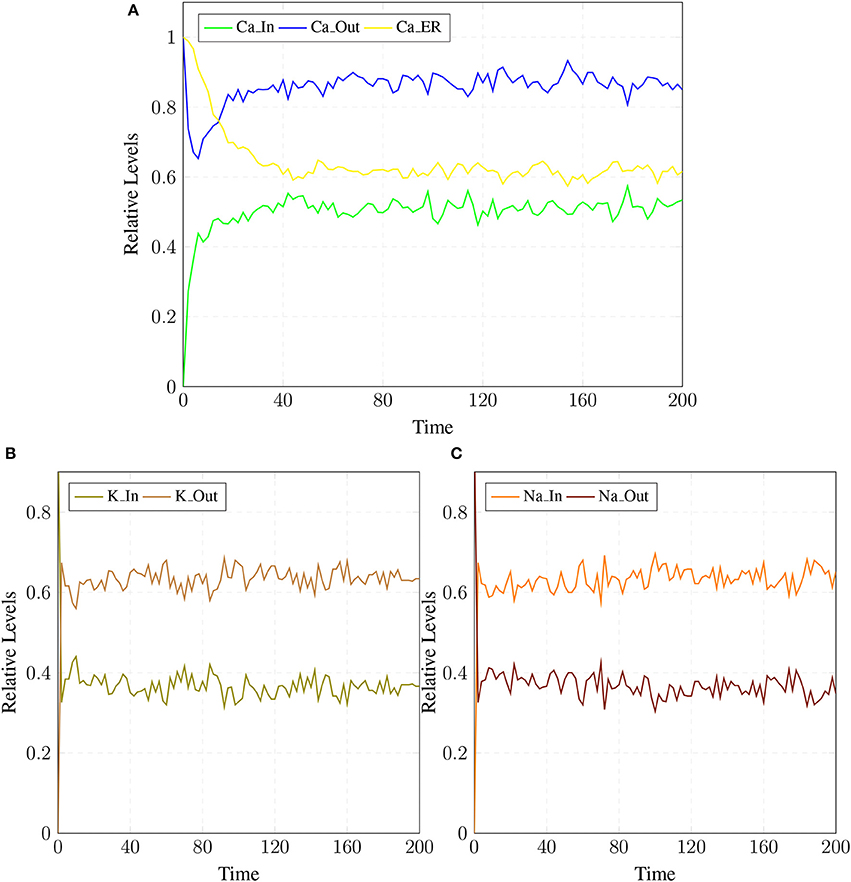
Figure 10. (A) shows Calcium homeostasis which is maintained by influx (Ca_In), efflux (Ca_Out) and intracellular storage (Ca_ER). Calcium efflux is further maintained by coordinated and controlled in and out flow of Potassium (K_In, K_Out) in (B) as well as of the Sodium (Na_In, Na_out) ions channels (C).
The model in Figure 11 represents Calpain-CAST regulatory system as given in Figure 4. In SPN model, due to lower Ca2+ level in cytosol, Calpain is in dormant form which is represented by place pCalp. It then attaches to CAST located on membrane (place mCT) to form a reversible complex of CAST and Calpain (place mC). Calpain is activated when cytosolic Ca2+ rises to specific concentration (Pal et al., 2001; Ryu and Nakazawa, 2014). Lower level of cytosolic Ca2+ represented by place Ca_In facilitates (place mC), membrane bound inactive reversible Calpain-CAST complex. After the concentration of Ca_In rises or crosses its threshold, then the short-lived mC is disintegrated into places CLP and cCT. The place CLP shows that Calpain moves to transmembrane and then converts into active form in the presence of Ca2+ ions and its autolysis. Now place CLP shows that Calpain is activated that is free to translocate to cytosol. The place cCT represents that CAST can again hinder over-activation of CLP by forming a reversible complex, represented by place cC. The place cC, splits into places CLP and dCT. In nature, active Calpain breaks down the bounded CAST into small subunits (Rao et al., 2008). The place CLP shows that it is active and which can be involved in various cellular processes. The place CLP can cleave place P35 into p25 which is involved in hyper-activation of cyclin dependent kinase 5 (cdk5) (Kusakawa et al., 2000; Lee et al., 2000). The transitions of this system are labeled with c. The transition c1 forms complex, place mC from pCalp and mCT at low Ca2+ concentration. The c2 breaks complex and activates CLP when level of Ca2+ rises into cytoplasm. The transition c4 forms complex place cC by binding CT and CLP in cytoplasm and c5, at a very low rate (Table 2) slowly degrades the CAST into dCT and makes the CLP free. Transitions c6, c12 and c13 are linked to CLP mediated P35 pathway. CLP also degrades PKC into pkm through c7. Remaining transitions c3, c8, c9, c10, c11 and c14 are associated to CAST regulation through PKC and phosphotases pp. The Table 2 lists all reaction rates of associated transitions which are obtained after fine tuning. In Figure 12A, the simulation shows that when Ca influx, Ca_In, is lower than its threshold then pCalp is bounded into complex of Calpain-CAST (mC) at membrane. When Ca_In crosses its threshold, the peak of mC runs down and CLP rises representing release and activation of Calpain, (Figure 12B). These results are according to experimental findings (De Tullio et al., 1999; Todd et al., 2003) (Figures 12A,B). To regulate CLP activation, cC appears but gradually the complex peak falls down showing that the cC is broken down into active CLP and dCT. The results in Figures 12C,D show that as CLP is rising, both the complexes (mC and cC) are gradually degrading causing depletion of CAST from the cytosol (Averna et al., 2001b; Tompa et al., 2002). In Figure 13, regulation of PKC, CAST and phophatases (represented as pp) can be observed. iPKC converted into PKC in the presence of Ca2+, which further converts into pkm by the action of CLP. As shown in the graph of Figure 13, CT is regulating through means of reversible phosphorylation and dephosphorylation expedited by PKC and pp, respectively.
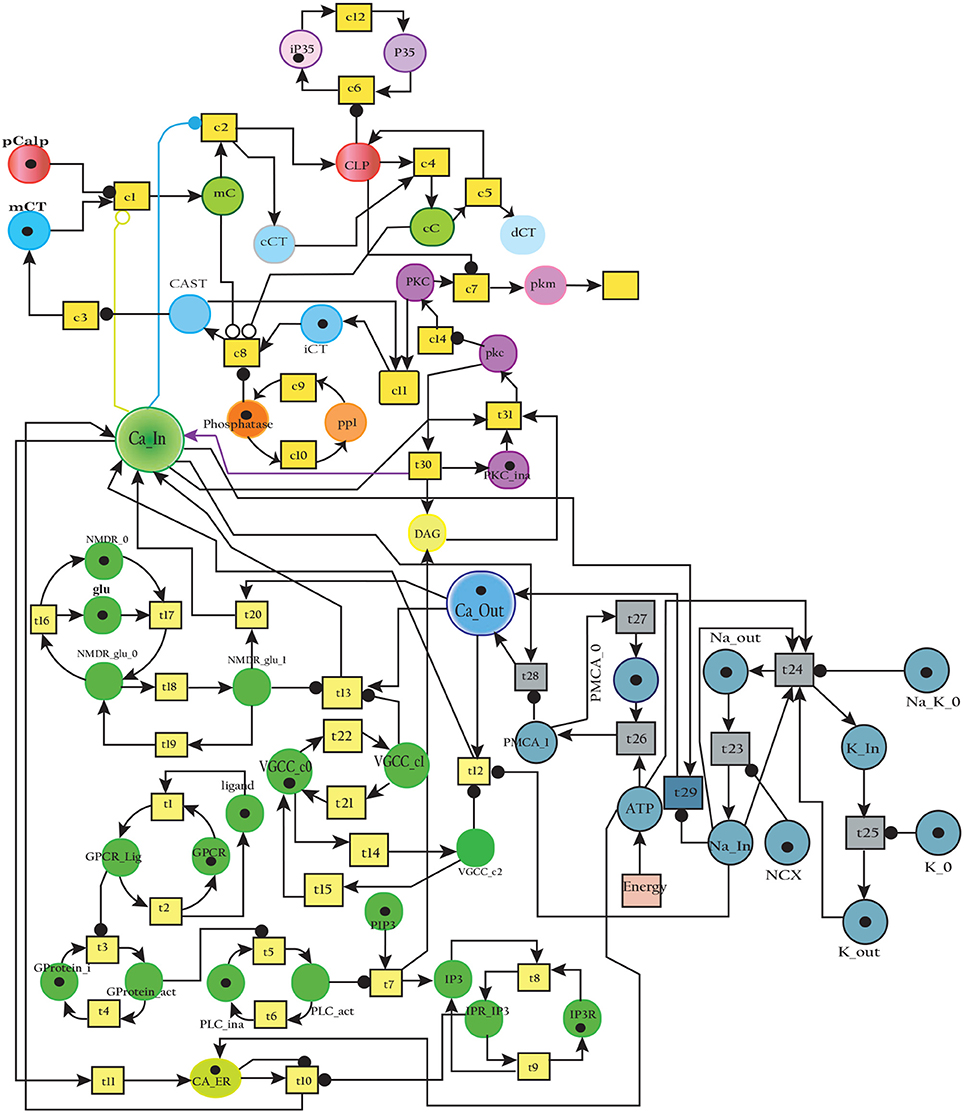
Figure 11. The SPN model of Calpain-CAST system.
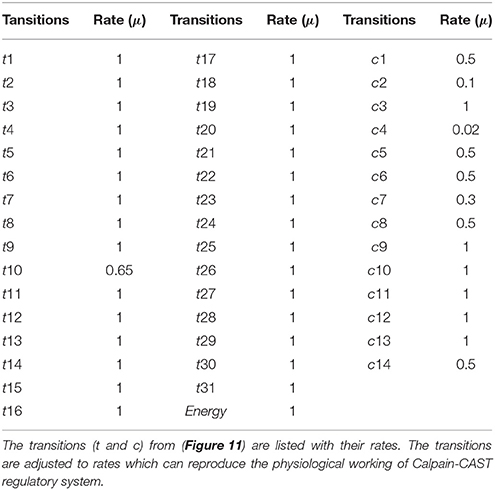
Table 2. The transition rates of the SPN model of Calpain-CAST regulatory system.
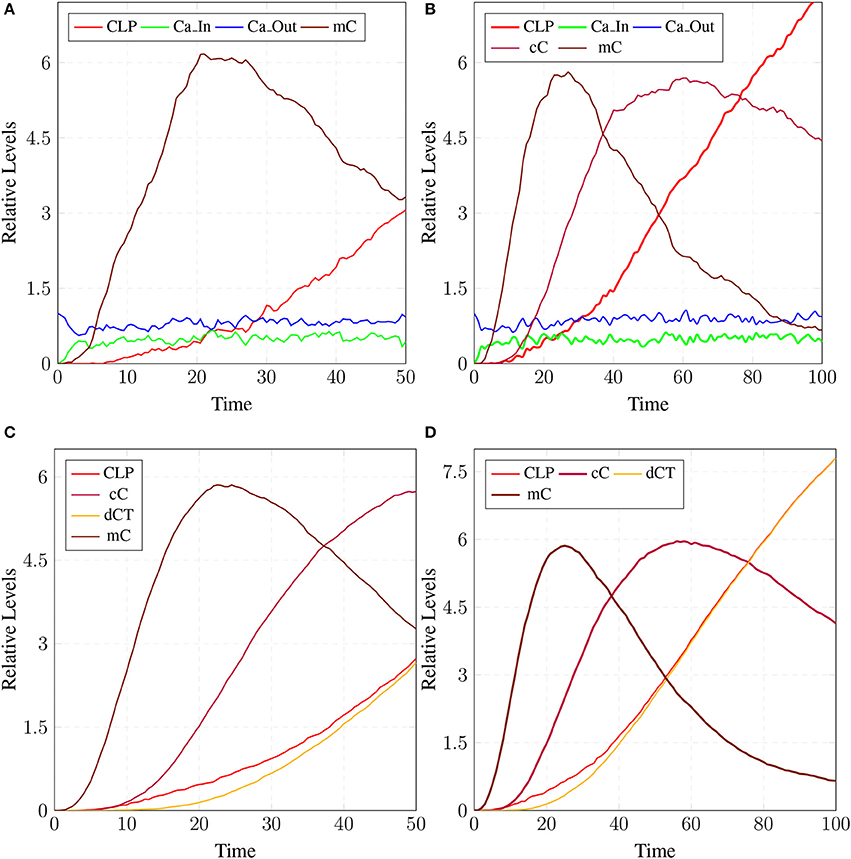
Figure 12. Calpain activation and regulation: In (A), CLP is bound to CAST in the form of mC at membrane. As the Ca_In increases and Ca_Out decreases the complex mC breaks down. In an elaborate view (B), CLP is produced by the break down of the mC which is again controlled by the formation of cC. In (C), mC is formed, maintained and eventually degraded into CAST and Calpain. Both Calpain and CAST form complex cC which decreases the availability of free CLP. CLP binding to cC slowly degrades the CAST which is represented as dCT. (D) Both complexes mC and cC are formed while cC is more stable than mC, as rate of degradation of mC is higher than (C).
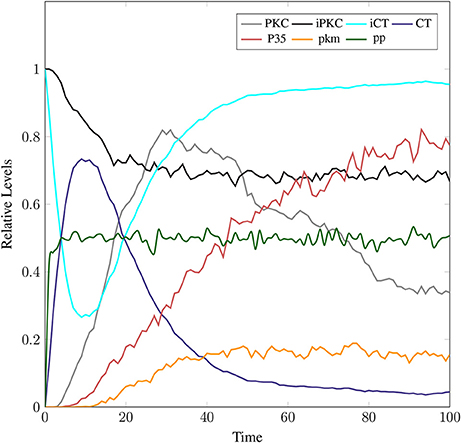
Figure 13. In physiological condition, the helping enzymes (PKC and phosphotases) are periodically activating and deactivating. iPKC is the dormant form of the cytosol; it activates into PKC to convert mCT and cCT into iCT. Phophotases (pp) convert the iCT into CT. PKC in the presence of CLP is degraded into pkm.
The SPN model of APP processing as shown in Figure 14 is built by combining both amyliodogenic and non-amyliodogenic pathways (Figure 2). APP (place aAPP) is catalyzed by α-secretase (place ALPHA) which produces CTF83 and sAPPα (places CTF83 and sAPPa), whereas digestion of APP (bAPP) by β-secretase (place BACE) yields CTF99 and sAPPβ, (represented by places CTF99 and sAPPb respectively). Further processing is carried out by enzyme γ-secretase (place GAMA) which converts CTF83 into p3 and AICD, while it converts CTF99 into Aβ and AICD. Aβ accumulate into Oligomer and finally into Plaq. The transitions a, a1 and g_a constitute non-amyliodogenic pathway, while transitions b, b1 and g_b are linked to amyloidogenic pathway. The parameters are set according to Table 3, in which the rate of transition of α-secretase (place ALPHA) mediated APP processing (aAPP) is higher than BACE (BACE) driven APP digestion (bAPP) (μ= 0.5, 0.3 respectively). The rates of plaques formation and accumulation (transitions: p1 and p2) are also adjusted according to the observations in literature which predict low Aβ burden in normal healthy brain (Mawuenyega et al., 2010; Rodrigue et al., 2012).
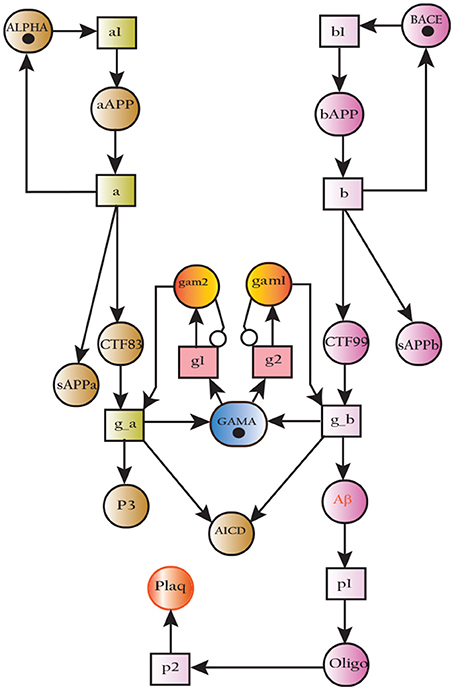
Figure 14. SPN model of non-amyliodogenic (left) and amyliodogenic (right) of APP processing pathways.

Table 3. The transition rates of the SPN model of APP processing pathways.
The simulations in Figure 15A shows that all the three enzymes (ALPHA, BACE, and GAMA) are available for the proteolysis of APP (in the cell to digest the around-the-clock production of) APP. The Figure 15B shows that the product sAPPa of non-amyloidogenic processing pathway is in higher concentration than the product sAPPb of amyloidogenic pathway. Production rate of sAPPa is higher than sAPPb. The graph in Figure 16A shows that in the healthy brain plaques are produced and accumulated in a linear fashion. Aβ burdens in the form of plaques at elder age i.e., approximately after 60 unit to 100 unit time. In Figure 16B, the linear behavior of plaques change to an exponential growth due to the fast accumulation rate with passage of time which depicts the lag phase (no plaques appearance) upto 80 unit time and after it evolves into growth phase (evolution).
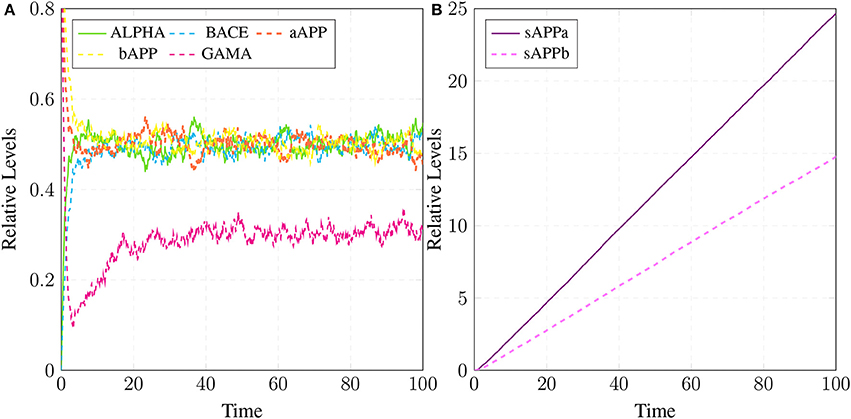
Figure 15. APP processing in physiological conditions: In (A), substrate aAPP binds with alpha secretase (ALPHA) and bAPP binds with BACE. All the three secretases ALPHA,BACE, and GAMA are oscillating. In (B), concentration of sAPPa is higher than the concentration of sAPPb which depicts healthy cell physiology.
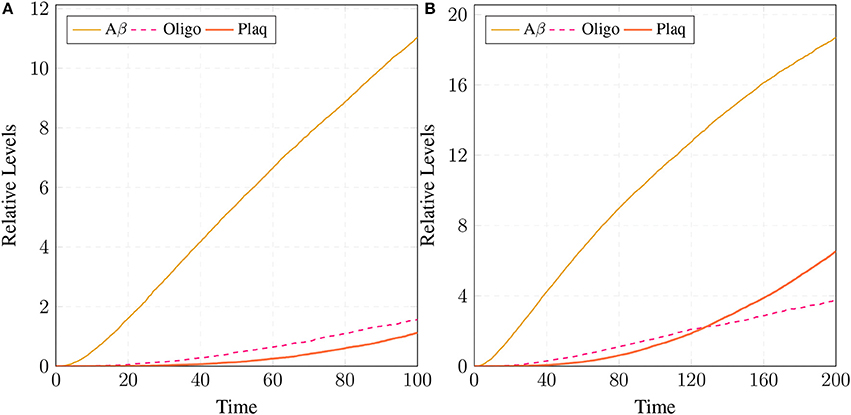
Figure 16. Plaques formation in healthy brain: In (A,B) production of plaque is shown over the time period of 100 and 200 unit time, respectively. Plaq accumulation starts after sixty to eighty unit time. Aβ and Oligo show fluctuating behaviors as they start to build-up but degrade with time. First, Aβ gathers into large quantity to form Oligo which gradually accumulates into Plaq.
The Figure 17 represents the SPN model of the crosstalk network in Figure 6. The connection that joins the pathways is established through places CLP, P35 and BACE. Active Calpain enhances β-secretase mediated APP cleavage twice than normal (Kusakawa et al., 2000; Liang et al., 2010) which initiates early production and accumulation of plaques (Jack et al., 2010; Braak et al., 2011). As a counter action, PKC PKC plays its role and enhances the activity of enzyme α-secretase (ALPHA) (Skovronsky et al., 2000). In the next connection, Calpain (CLP) hinders the functioning of PKC by degrading it into pkm (place pkm). Aβ oligomers represented by place Oligomer forms pores into the membrane which instantaneously increases influx of Ca2+ ions. The place Amyloidbeta inhibits the transition Energy which indirectly hinders Ca2+ extrusion into extracellular space. The new transitions in the crosstalk network are labeled with k. The transition k1 connects the place PKC with ALPHA while P35 is connected to Plaq through transitions c13, k2, k4, k5, and k6. The transition k3 joins Oligomer with Ca_In. The reaction rates of all the transitions are listed in Table 4.
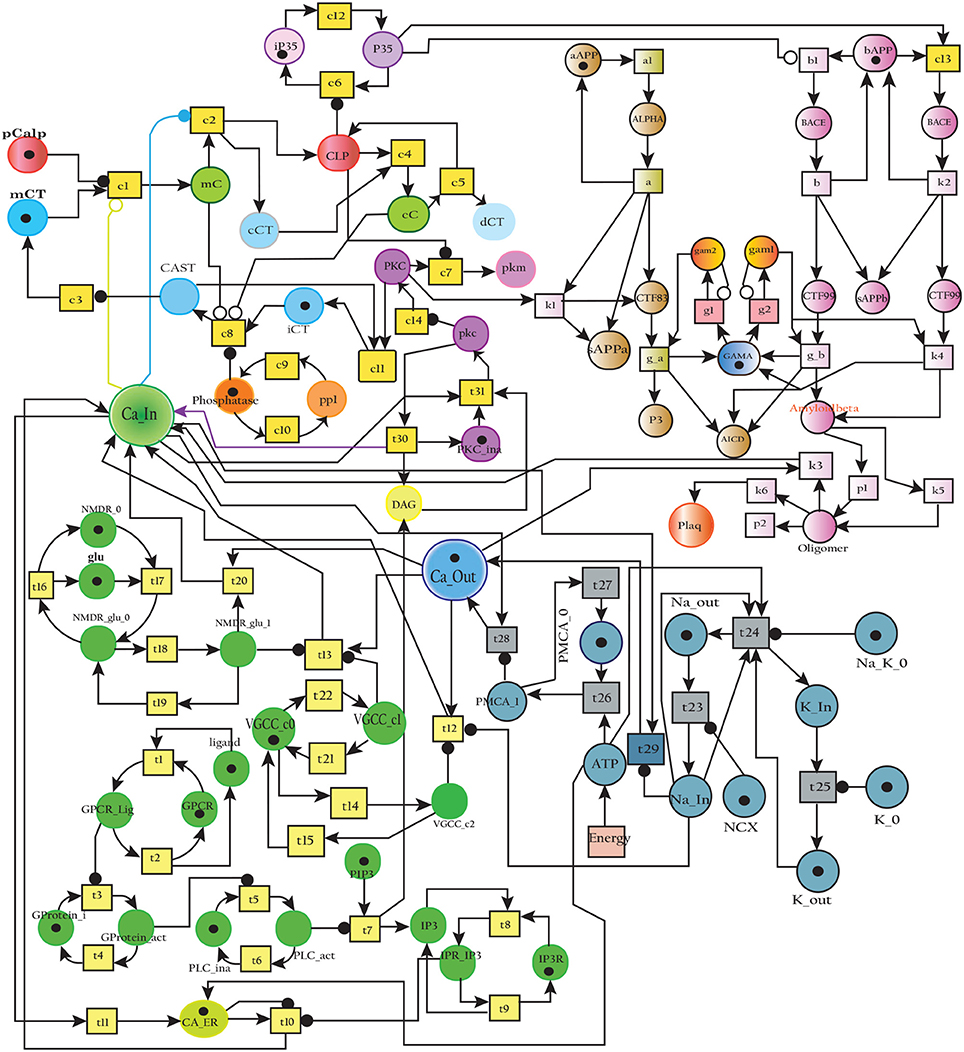
Figure 17. SPN model of the cross talk network of calcium homeostasis, APP processing and Calpain-CAST system Pathways.
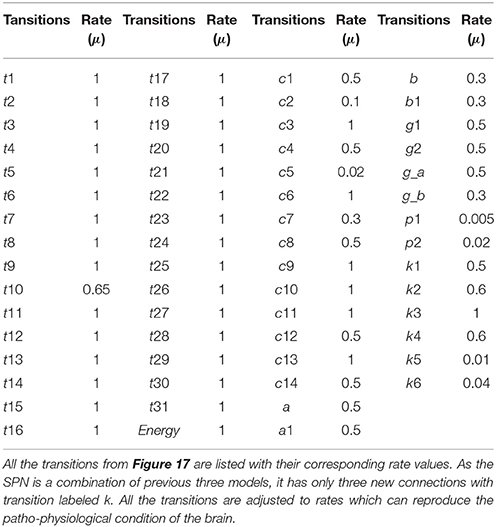
Table 4. The transitions rates of the SPN model of the crosstalk network.
The results in Figure 18 show that CLP and P35 are accumulating which can directly affect the plaques accumulation process by increase in their deposition rate, as compared to results in section 3.3 where accumulation of Plaq is minimal (Figure 16A). It can also be observed that mC and cC start declining at later stages which indicates depletion of CAST (Figure 18A).
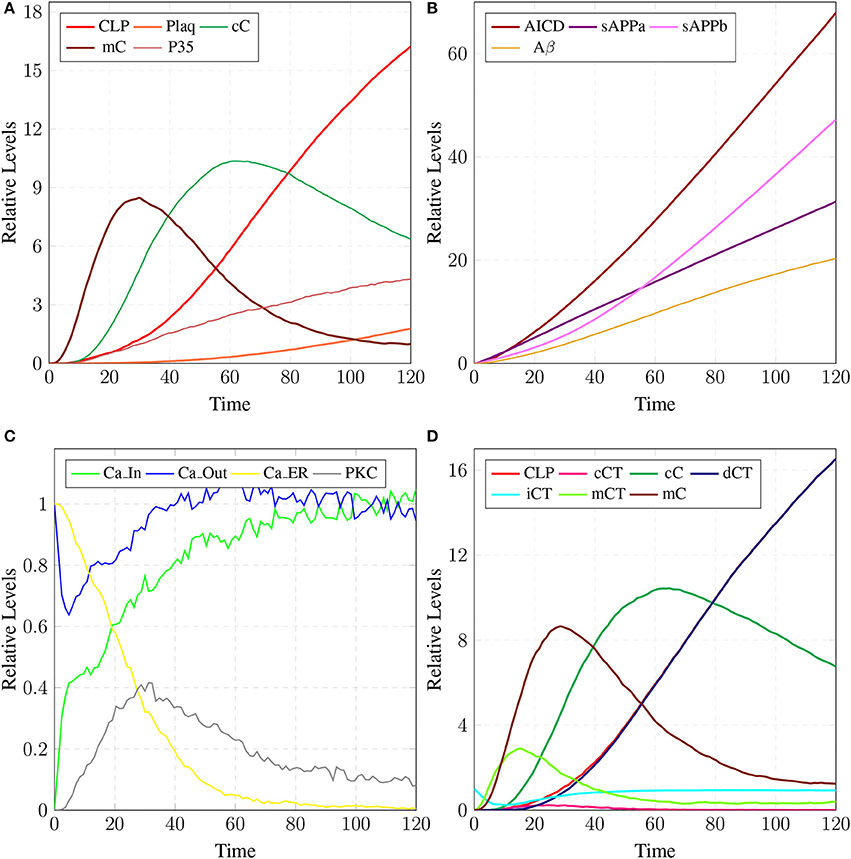
Figure 18. In (A), high concentration of CLP causes gradual degradation of cC and further increases the activation of P35 which then activates rapid production of Plaq leading to the onset of AD. In (B), the production of sAPPa is lower than the production sAPPb and AICD production is higher than both sAPPa and sAPPb. (C) shows slow dysregulation of calcium homeostasis by increase of concentration of Ca_In from Ca_Out and Ca_ER. (D) shows conversion of iCT into active form then translocation to membrane mCT and cytosol cCT. Furthermore, cCT binds with CLP to form cC. CLP gradually degrades CAST into dCT.
In Figure 18B, the concentration of sAPPb is higher than sAPPa protein, which is contrary to the Figure 15B. Hyper activity of CLP influences more than one processes in the pathological network by enhancing BACE activity which increases the growth of sAPPb. The decrease in PKC concentration and calcium dysregulation (Ca_In rises gradually, Ca_Out and Ca_ER both lower down) are also due to over-activation of CLP (Figure 18C). Instability in Calcium equilibrium in neurons appear after 60 unit time which also act as a signal indicating the development of AD. Additionally, degradation of PKC into pkm by the action of CLP also have bidirectional effect on the system. First the availability of active CT in the cell raises and then ALPHA secretase driven APP processing slows down (Figure 18B). Figure 18D represents the translocation and regulation of CAST (cC andcC) in the brain cell.
The parameter Table 5 shows that by changing the rate of reaction of two crucial transitions i.e., c2 and c5 (representing degradation rate of Calpain-CAST complex at membrane and cytosol, respectively) would provide effective strategy to control or stop the development of AD and other related neurological disorders. After applying in silico intervention, it can be observed that as the rate of transition c2 is decreased the neurological network move toward stability due to basal or low activity of CLP (Figure 19A). Further, the rate of c2 is set to zero which indicates that mC is made unbreakable or stable which implies that there would be no production of CLP (Figure 19B). In Figure 19A, both the complexes (mC and cC) are maintained for longer time duration which result in low production of CLP, Aβ and Plaq. In Figure 19B, there is neither the production of CLP nor the formation of cC due to long life of mC complex occur and there is negligible production of Aβ. The complex (mC) also ensures low level of sAPPb and high concentration of sAPPa and as a result there is minor accumulation of Plaq in the neurons.
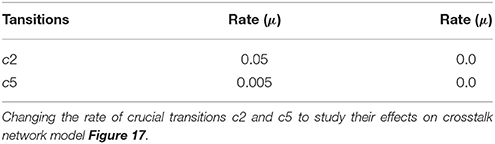
Table 5. Transition rate table.
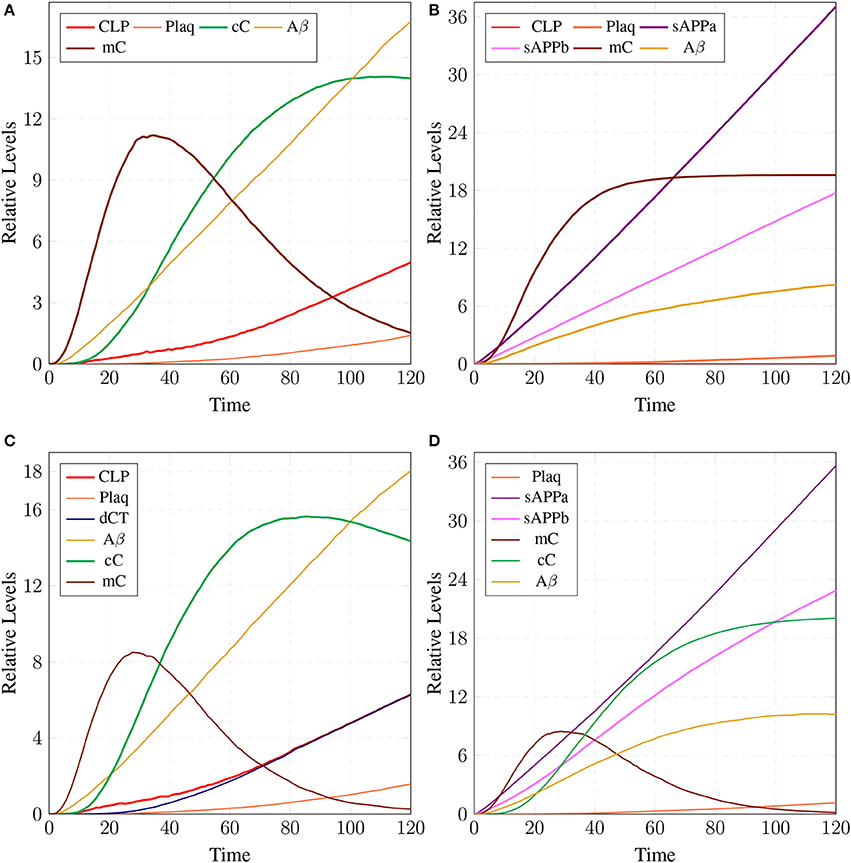
Figure 19. A mild cognitive impairment behavior in (A,C). CLP is in loosely controlled by mC and cC (c2 = 0.05 and c5 = 0.005). There is low level of Plaq and significant level of Aβ. By changing the rate of c2 = 0 (B) there is no conversion of pro-Calpain into CLP due to long life of mC. Production of sAPPb and Aβ are also decreased sufficiently and it lingers on the process of accumulation of Plaq. In (D), c5 is set to zero which controls level of CLP after its activation and saves CAST from depletion into dCT. The production of sAPPa is higher than production of sAPPb in (B,D).
The stability of mC have positive effects in maintaining the calcium homeostasis which regains its equilibrium accompanied by substantial concentration of PKC and no production of P35 Figure 20A. The smaller rate of c5 also favors stable condition of system and can delay the production of plaques. Figures 19C,D show that as the rate of transition of c5 decreases the system maintains a stable state due to prolonged life of cC which causes basal level production of CLP and eventually guard against rapid Plaq accumulation in brains. As shown in Figure 19D, cC helps in maintaining a balanced level of CAST, sAPPa and sAPPb and it lowers down the concentration and accumulation of Aβ and Plaq. Also it aids in maintaining homeostasis of other proteins and channels into the cell such as PKC, pkm, P35 and calcium homeostasis (Figure 20B). Therefore, these complexes are very critical targets since they regulate homeostasis of many crucial proteins.
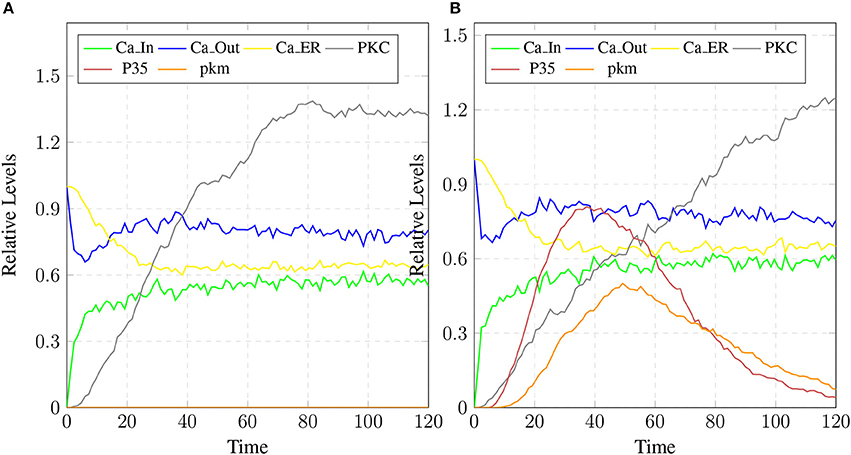
Figure 20. The restoration of Calcium homeostasis. In (A), PKC is present for longer time and there is no conversion into pkm also there is no production of P35. In (B), there is less production of P35 and pkm.
This study contributes in achieving long haul goal of AD research i.e., understanding and manipulating pathological conditions which include BACE and Calpain over-activation, Calcium dysregulation, CAST depletion and abnormal production of Aβ. In this study, neuronal pathways comprising Calpain-CAST system, APP processing pathways and Calcium channels were first modeled individually and then their cross-talk was also modeled. The simulation results of these models predicted a more clearer picture of AD pathogenisis also, on the basis of observations interventions were introduced to linger on the AD pathogenesis.
In a healthy physiological model, rate of Ca2+ ions inflow should be lower than rate of Ca2+ ions outflow with uniform oscillation within the safest level. Moreover, the concentration of Ca2+ ions in intracellular store (Ca_ER) is greater than (Ca_In) (Figures 10B,C) (Korol et al., 2008; Gutierrez-Merino et al., 2014). An equilibrated flow is also observed in other important ions channels such as NCX and NKA, which mediate influx and efflux of Na and K ions. Balance in their flow is obligatory for the adequate efflux of Ca2+ ions for maintaining homeostasis (Gutierrez-Merino et al., 2014). Another important physiological pathway, Calpain-CAST regulatory system also depends on Ca2+. The calcium influx and efflux channels are required for the activation and regulation of Calpain.
The simulation results in section 3.2, show that when concentartion of Ca2+ is lower inside the cytosol then Calpain is bounded in the complex of Calpain-CAST at membrane. As concentartion of Ca2+ rises, the complex degrades and active Calpain is produced (De Tullio et al., 1999; Todd et al., 2003). To regulate Calpain's activation, CAST appears to form Calpian-CAST complex in cytosol, but gradually its peak falls down which shows release of active Calpain and degradation of CAST. The results in Figures 12C,D show that as CLP rises, both the complexes (mC and cC) gradually degrade which causes depletion of CAST (Averna et al., 2001b; Tompa et al., 2002). PKC also has important role in building a feedback mechanism for Calpain by phosphorylating CAST into inactive form (Averna et al., 2001a). Calpain is also regulating PKC by cleaving it into pkm (Goll et al., 2003) which is persistently active catalytic fragment and quickly disappears (Yamakawa et al., 2001; Liu et al., 2008). PKC also has important roles in the system, it is favoring α-secretase mediated APP cleavage (Rossner et al., 2001; Racchi et al., 2003) and is also involved in inactivation of CAST through phosphorylation (Olariu et al., 2002). The inactive CAST regains its active conformation in the presence of phosphatases (Averna et al., 2001a).
Simulation results of the model APP processing pathways show that in a healthy brain all three enzymes of this pathway are available in the cell to digest the around-the-clock production of APP with α-secretase having high affinity as compared to BACE (De Strooper and Annaert, 2000). Consequently, production rate of sAPPα is higher than sAPPβ (Figure 15B) which is a depiction of normal physiological behavior (De Strooper and Annaert, 2000; Lichtenthaler, 2011). Studies showed that in healthy brain, plaques produce and accumulate in a linear fashion which is also observed in our studies. Moreover, Aβ burdens in the form of plaques at elder age. This linear behavior of plaques accumulation can adopt exponential growth due to the high accumulation rate with passage of time. In our results the formation of plaques first enter the lag phase (no plaques appearance) upto 80 unit time and then evolve into growth phase (evolution), which is also experimentally reported (Friedrich et al., 2010; Jack et al., 2010; Sperling et al., 2011).
In neuro-pathological model, simulation revealed that the intermediate product sAPPβ of amyloidogenic pathway rises in the AD brain. Higher concentration of sAPPβ can lead to early occurrence of AD (at age of 40 and onward) due to rapid deposition of the plaques in brain of AD patients (Freer et al., 2016). Higher level of sAPPβ also indicate the enhanced activity of BACE which is triggered by over-activation of Calpain. Enhanced activation of Calpain disturbs more than one processes in the pathological network i.e., increased BACE activity, decreased PKC functioning and calcium dysregulation. Initially CAST and its complex with pro-Calpain are in high quantity to control Calpain but with the elevation of calcium influx, the complex starts to degrade. The active Calpain accumulates in cytosol which is then controlled by cytosolic CAST by forming complex with it. After sometime Calpain degrades the CAST in the complex to release itself into the cytosol and eventually CAST and complex deplete from the cell (Higuchi et al., 2012). Depletion of CAST destroys the cell normal functioning and then nervous system deteriorates (Rao et al., 2008, 2014; Kurbatskaya et al., 2016).
After analyzing the neuropathological network, the vague picture of AD development becomes more clear. It provide useful observations such as loss of calcium homeostasis occur after 60 unit time due to intra-neuronal ATP depletion caused by elevation of Calpain (Lipton, 1999; Kurbatskaya et al., 2016) and formation of extracellular ion pores formed by Aβ oligomers (Small et al., 2009). Ca2+ disruption increases Calpain concentration in neurons which further elevates the BACE activity toward APP (Liang et al., 2010; Chami and Checler, 2012; Song et al., 2015) via cdk5 dependent pathway as a result of Aβ and sAPPβ levels in neurons built up (Sennvik et al., 2004). In normal healthy brain, the mean burden in the form of plaques are low and it increase in linear fashion from 60 to 90 unit time (Rodrigue et al., 2012; Freer et al., 2016; Kurbatskaya et al., 2016). It is noteworthy that both amyloidogenic and non-amyloidogenic pathways are enabled in an healthy individual but plaques in the brain of AD patient grow rapidly due to increased Calpain mediated cleavage of APP through BACE (Mawuenyega et al., 2010). It can be inferred that all the neuro-pathological events are inter-related where Calpain and its complexes are playing crucial role. Calpain can be called as bone of contention and the complexes are the defenders. This neuropathological network provides valuable knowledge about interventions and offers new therapeutic targets. To stop the Calpain destructive effects, it may be effective strategy to slightly modify the natural process. In the work of Emmaneul and coworkers on cardiovascular remodeling, the Calpain-CAST system has emerged as new effective strategy to prevent angiotension (Letavernier et al., 2008). In our study, Calpain-CAST complexes have also proved to be effective therapeutic targets for delaying the process of neuronal degradation which can save the brains from AD.
In AD, Aβ has central role as they accumulate into hallmark lesions i.e., plaques. Aβ generates from APP cleavage through amyloidogenic pathway. Progression of the corresponding processing pathway increases due to over-activation of Caplain and depletion of its sole inhibitor CAST. Calpain hyper activation depends on high Calcium concentration in the cytosol. Calpain triggers the production of P35 and the degradation of CAST and PKC. In-Addition, it also triggers the imbalance of calcium homeostasis. All these observations were incorporated into a Stochastic PN models. We gained insight into the mechanism of the AD progression and were able to derive some useful inferences. The first important inference is that under hyper-activation of Calpain, calcium homeostasis dysregulates and CAST starts to degrade gradually. APP processing enzyme (BACE) increases two folds which starts producing Aβ that slowly accumulates into plaques in AD brains at early age and lesions appear later. The second important inference is about the most crucial protein Calpain that influences many important proteins of neuronal network such as CAST, PKC, P35, and BACE. Furthermore, these proteins together dysregulate calcium homeostasis. Calpain regulation through CAST plays important role in keeping cells safe from neurodegradation. CAST encounters Calpain at two locations in the cell. Initially at membrane, CAST forms complex with inactive Calpain which is short lived and dissociates as the calcium influx increases. In the cytoplasm, active Calpain-CAST complex is long-standing which keeps the Calpain concentration in control but it also degraded slowly by the action of Calpain. From our study, Calpain and Calpain-CAST complexes have emerged as the potential therapeutic targets for the treatment of neurodegenerative pathologies. The pathway modeling of these networks have predicted that we can introduce delays in the production and accumulation of plaques by targeting the Calpain-CAST complexes. The production of Calpain should be kept in safe levels to avoid its hyper activity. It can be achieved implicitly by enhancing activity of Calpain-CAST complexes. The more durable are the complexes, the lesser would be the accumulation of plaques. Another useful strategy can be the designing of inhibitors against the active Calpain using in silico methods and in vitro experiments. By considering these effective interventions we can increase the chances of healthy life expectancy and can save many lives and families from the adversity of AD.
JAs and JAh conceived research idea, designed and performed the experiments, analyzed the data, wrote the paper, prepared figures and/or tables, reviewed drafts of the paper. AA and ZU-H analyzed the data, prepared tables and/or figures, wrote the paper, reviewed drafts of the paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fninf.2018.00026/full#supplementary-material
Alkon, D. L., Sun, M.-K., and Nelson, T. J. (2007). Pkc signaling deficits: a mechanistic hypothesis for the origins of alzheimer's disease. Trends Pharmacol. Sci. 28, 51–60. doi: 10.1016/j.tips.2006.12.002
Annaert, W. G., and Saftig, P. (2009). Regulated intramembrane proteolysis—a story about sheddases and I-CliPs. Semin. Cell Dev. Biol. 20:125. doi: 10.1016/j.semcdb.2009.03.005
Ashford, J. W. (2004). Apoe genotype effects on Alzheimer's disease onset and epidemiology. J. Mol. Neurosci. 23, 157–165. doi: 10.1385/JMN:23:3:157
Averna, M., De Tullio, R., Passalacqua, M., Salamino, F., Pontremoli, S., and Melloni, E. (2001a). Changes in intracellular calpastatin localization are mediated by reversible phosphorylation. Biochem. J. 354, 25–30. doi: 10.1042/bj3540025
Averna, M., De Tullio, R., Salamino, F., Minafra, R., Pontremoli, S., and Melloni, E. (2001b). Age-dependent degradation of calpastatin in kidney of hypertensive rats. J. Biol. Chem. 276, 38426–38432. doi: 10.1074/jbc.M101936200
Berridge, M. J. (2009). Inositol trisphosphate and calcium signalling mechanisms. Mol. Cell Res. 1793, 933–940. doi: 10.1016/j.bbamcr.2008.10.005
Blätke, M. A., Heiner, M., and Marwan, W. (2011). Petri Nets in Systems Biology. Technical report, Technical Report, Otto-von-Guericke University Magdeburg.
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathologic process in alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi: 10.1097/NEN.0b013e318232a379
Brittain, M. K., Brustovetsky, T., Sheets, P. L., Brittain, J. M., Khanna, R., Cummins, T. R., et al. (2012). Delayed calcium dysregulation in neurons requires both the nmda receptor and the reverse Na+/Ca2+ exchanger. Neurobiol. Dis. 46, 109–117. doi: 10.1016/j.nbd.2011.12.051
Budson, A. E., and Price, B. H. (2005). Memory dysfunction. New Engl. J. Med. 352, 692–699. doi: 10.1056/NEJMra041071
Cai, H., Wang, Y., McCarthy, D., Wen, H., Borchelt, D. R., Price, D. L., et al. (2001). Bace1 is the major β-secretase for generation of aβ peptides by neurons. Nat. Neurosci. 4, 233–234. doi: 10.1038/85064
Cain, S. M., and Snutch, T. P. (2011). Voltage-gated calcium channels and disease. Biofactors 37, 197–205. doi: 10.1002/biof.158
Castaldi, D., Maccagnola, D., Mari, D., and Archetti, F. (2012). “Stochastic simulation of the coagulation cascade: a petri net based approach,” in European Conference on Parallel Processing (Rhodos: Springer).
Chami, L., and Checler, F. (2012). Bace1 is at the crossroad of a toxic vicious cycle involving cellular stress and β-amyloid production in Alzheimer's disease. Mol. Neurodegenerat. 7:52. doi: 10.1186/1750-1326-7-52
Chen, M., and Fernandez, H. L. (2004). Stimulation of β-amyloid precursor protein α-processing by phorbol ester involves calcium and calpain activation. Biochem. Biophys. Res. Commun. 316, 332–340. doi: 10.1016/j.bbrc.2004.02.052
Courjaret, R., Grolleau, F., and Lapied, B. (2003). Two distinct calcium-sensitive and-insensitive pkc up-and down-regulate an α-bungarotoxin-resistant nachr1 in insect neurosecretory cells (dum neurons). Eur. J. Neurosci. 17, 2023–2034. doi: 10.1046/j.1460-9568.2003.02644.x
Cuello, A. C. (2005). Intracellular and extracellular aβ, a tale of two neuropathologies. Brain Pathol. 15, 66–71. doi: 10.1111/j.1750-3639.2005.tb00101.x
David, R., and Alla, H. (2010). Discrete, Continuous, and Hybrid Petri Nets. Grenoble: Springer Science & Business Media.
De Strooper, B., and Annaert, W. (2000). Proteolytic processing and cell biological functions of the amyloid precursor protein. J. Cell. Sci. 113, 1857–1870.
De Tullio, R., Passalacqua, M., Averna, M., Salamino, F., Melloni, E., and Pontremoli, S. (1999). Changes in intracellular localization of calpastatin during calpain activation. Biochem. J. 343, 467–472. doi: 10.1042/bj3430467
DeStrooper, B., Saftig, P., Birchmeier, C., and Haass, C. (2006). Control of peripheral nerve myelination by the beta-secretase bace1. Science 314, 664–666. doi: 10.1126/science.1132341
Emptage, N. J., Reid, C. A., and Fine, A. (2001). Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated ca 2+ entry, and spontaneous transmitter release. Neuron 29, 197–208. doi: 10.1016/S0896-6273(01)00190-8
Etcheberrigaray, R., Tan, M., Dewachter, I., Kuipéri, C., Van der Auwera, I., Wera, S., et al. (2004). Therapeutic effects of pkc activators in alzheimer's disease transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 101, 11141–11146. doi: 10.1073/pnas.0403921101
Ferreira, A. (2012). Calpain dysregulation in Alzheimer's disease. ISRN Biochem. 2012:728571. doi: 10.5402/2012/728571
Freer, R., Sormanni, P., Vecchi, G., Ciryam, P., Dobson, C. M., and Vendruscolo, M. (2016). A protein homeostasis signature in healthy brains recapitulates tissue vulnerability to Alzheimer's disease. Sci. Adv. 2:e1600947. doi: 10.1126/sciadv.1600947
Friedrich, R. P., Tepper, K., Rönicke, R., Soom, M., Westermann, M., Reymann, K., et al. (2010). Mechanism of amyloid plaque formation suggests an intracellular basis of aβ pathogenicity. Proc. Natl. Acad. Sci. U.S.A. 107, 1942–1947. doi: 10.1073/pnas.0904532106
Gillespie, D. T. (1976). A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 22, 403–434. doi: 10.1016/0021-9991(76)90041-3
Golde, T. E. (2005). The aβ hypothesis: Leading us to rationally-designed therapeutic strategies for the treatment or prevention of alzheimer disease. Brain Pathol. 15, 84–87. doi: 10.1111/j.1750-3639.2005.tb00104.x
Goll, D. E., Thompson, V. F., Li, H., Wei, W., and Cong, J. (2003). The calpain system. Physiol. Rev. 83, 731–801. doi: 10.1152/physrev.00029.2002
Goss, P. J., and Peccoud, J. (1998). Quantitative modeling of stochastic systems in molecular biology by using stochastic petri nets. Proc. Natl. Acad. Sci. U.S.A. 95, 6750–6755. doi: 10.1073/pnas.95.12.6750
Greenfield, J. P., Tsai, J., Gouras, G. K., Hai, B., Thinakaran, G., Checler, F., et al. (1999). Endoplasmic reticulum and trans-golgi network generate distinct populations of Alzheimer β-amyloid peptides. Proc. Natl. Acad. Sci. U.S.A. 96, 742–747. doi: 10.1073/pnas.96.2.742
Gutierrez-Merino, C., Marques-da Silva, D., Fortalezas, S., and Samhan-Arias, A. K. (2014). “Cytosolic calcium homeostasis in neurons–control systems, modulation by reactive oxygen and nitrogen species, and space and time fluctuations,” in Neurochemistry, ed T. Heinbockel (Rijeka: InTech), 59–110.
Haass, C. (2004). Take five–bace and the γ-secretase quartet conduct alzheimer's amyloid β-peptide generation. EMBO J. 23, 483–488. doi: 10.1038/sj.emboj.7600061
Hanna, R. A., Campbell, R. L., and Davies, P. L. (2008). Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature 456, 409–412. doi: 10.1038/nature07451
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184. doi: 10.1126/science.1566067
Heiner, M., Gilbert, D., and Donaldson, R. (2008). “Petri nets for systems and synthetic biology,” in International School on Formal Methods for the Design of Computer, Communication and Software Systems, eds M. Bernardo, P. Degano, and G. Zavattaro (Bertinoro: Springer), 215–264.
Higuchi, M., Iwata, N., Matsuba, Y., Takano, J., Suemoto, T., Maeda, J., et al. (2012). Mechanistic involvement of the calpain-calpastatin system in alzheimer neuropathology. FASEB J. 26, 1204–1217. doi: 10.1096/fj.11-187740
Hu, X., Hicks, C. W., He, W., Wong, P., Macklin, W. B., Trapp, B. D., et al. (2006). Bace1 modulates myelination in the central and peripheral nervous system. Nat. Neurosci. 9, 1520–1525. doi: 10.1038/nn1797
Jack, C. R., Knopman, D. S., Jagust, W. J., Shaw, L. M., Aisen, P. S., Weiner, M. W., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 9, 119–128. doi: 10.1016/S1474-4422(09)70299-6
Jonsson, T., Atwal, J. K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjornsson, S., et al. (2012). A mutation in app protects against Alzheimer's disease and age-related cognitive decline. Nature 488:96. doi: 10.1038/nature11283
Korol, T. Y., Kostyuk, E., and Kostyuk, P. (2008). Disruption of calcium homeostasis in Alzheimer's disease. Neurophysiology 40, 385–392. doi: 10.1007/s11062-009-9064-5
Krebs, J., Agellon, L. B., and Michalak, M. (2015). Ca2+ homeostasis and endoplasmic reticulum (er) stress: an integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 460, 114–121. doi: 10.1016/j.bbrc.2015.02.004
Kurbatskaya, K., Phillips, E. C., Croft, C. L., Dentoni, G., Hughes, M. M., Wade, M. A., et al. (2016). Upregulation of calpain activity precedes tau phosphorylation and loss of synaptic proteins in Alzheimer's disease brain. Acta Neuropathol. Commun. 4:34. doi: 10.1186/s40478-016-0299-2
Kusakawa, G.-i., Saito, T., Onuki, R., Ishiguro, K., Kishimoto, T., and Hisanaga, S.-i. (2000). Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J. Biol. Chem. 275, 17166–17172. doi: 10.1074/jbc.M907757199
LaFerla, F. M. (2002). Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat. Rev. Neurosci. 3, 862–872. doi: 10.1038/nrn960
Lamprecht, R., Smith, G. D., and Kemper, P. (2011). Stochastic petri net models of ca2+ signaling complexes and their analysis. Nat. Comput. 10, 1045–1075. doi: 10.1007/s11047-009-9143-y
Lanni, C., Mazzucchelli, M., Porrello, E., Govoni, S., and Racchi, M. (2004). Differential involvement of protein kinase c alpha and epsilon in the regulated secretion of soluble amyloid precursor protein. Eur. J. Biochem. 271, 3068–3075. doi: 10.1111/j.1432-1033.2004.04240.x
Lee, J., Retamal, C., Cuitiño, L., Caruano-Yzermans, A., Shin, J.-E., Van Kerkhof, P., et al. (2008). Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J. Biol. Chem. 283, 11501–11508. doi: 10.1074/jbc.M800642200
Lee, M.-s., Kwon, Y. T., Li, M., Peng, J., Friedlander, R. M., and Tsai, L.-H. (2000). Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405, 360–364. doi: 10.1038/35012636
Lee, V. M., Goedert, M., and Trojanowski, J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159. doi: 10.1146/annurev.neuro.24.1.1121
Letavernier, E., Perez, J., Bellocq, A., Mesnard, L., de Castro Keller, A., Haymann, J.-P., et al. (2008). Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin ii–induced hypertension. Circul. Res. 102, 720–728. doi: 10.1161/CIRCRESAHA.107.160077
Li, Q., and Südhof, T. C. (2004). Cleavage of amyloid-β precursor protein and amyloid-β precursor-like protein by bace 1. J. Biol. Chem. 279, 10542–10550. doi: 10.1074/jbc.M310001200
Liang, B., Duan, B.-Y., Zhou, X.-P., Gong, J.-X., and Luo, Z.-G. (2010). Calpain activation promotes bace1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of alzheimer disease. J. Biol. Chem. 285, 27737–27744. doi: 10.1074/jbc.M110.117960
Lichtenthaler, S. F. (2011). Alpha-secretase in Alzheimer's disease: molecular identity, regulation and therapeutic potential. J. Neurochem. 116, 10–21. doi: 10.1111/j.1471-4159.2010.07081.x
Lipton, P. (1999). Ischemic cell death in brain neurons. Physiol. Rev. 79, 1431–1568. doi: 10.1152/physrev.1999.79.4.1431
Liu, F., Heiner, M., and Yang, M. (2016). Fuzzy stochastic petri nets for modeling biological systems with uncertain kinetic parameters. PLoS ONE 11:e0149674. doi: 10.1371/journal.pone.0149674
Liu, J., Liu, M. C., and Wang, K. K. (2008). Calpain in the cns: from synaptic function to neurotoxicity. Sci. Signal. 1, re1. doi: 10.1126/stke.114re1
Marsan, M. A., Balbo, G., Conte, G., Donatelli, S., and Franceschinis, G. (1994). Modelling With Generalized Stochastic Petri Nets. New York, NY: John Wiley & Sons, Inc.
Marwan, W., Rohr, C., and Heiner, M. (2012). Petri nets in snoopy: a unifying framework for the graphical display, computational modelling, and simulation of bacterial regulatory networks. Bact. Mol. Netw. Methods Protocols 804, 409–437. doi: 10.1007/978-1-61779-361-5_21
Mattson, M. P. (2004). Pathways towards and away from alzheimer's disease. Nature 430, 631–639. doi: 10.1038/nature02621
Mattson, M. P., LaFerla, F. M., Chan, S. L., Leissring, M. A., Shepel, P. N., and Geiger, J. D. (2000). Calcium signaling in the er: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 23, 222–229. doi: 10.1016/S0166-2236(00)01548-4
Mawuenyega, K. G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased clearance of cns β-amyloid in Alzheimer's disease. Science 330, 1774–1774. doi: 10.1126/science.1197623
Melloni, E., Averna, M., Stifanese, R., De Tullio, R., Defranchi, E., Salamino, F., et al. (2006). Association of calpastatin with inactive calpain a novel mechanism to control the activation of the protease? J. Biol. Chem. 281, 24945–24954. doi: 10.1074/jbc.M601449200
Mounts, W. M., and Liebman, M. N. (1997). Application of petri nets and stochastic activity nets to modeling biological pathways and processes. Int. J. Comput. Simul. 20, 265–281.
Mura, I., and Csikász-Nagy, A. (2008). Stochastic petri net extension of a yeast cell cycle model. J. Theor. Biol. 254, 850–860. doi: 10.1016/j.jtbi.2008.07.019
O'Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer's disease. Annu. Rev. Neurosci. 34, 185–204. doi: 10.1146/annurev-neuro-061010-113613
Olariu, A., Yamada, K., Mamiya, T., Hefco, V., and Nabeshima, T. (2002). Memory impairment induced by chronic intracerebroventricular infusion of beta-amyloid (1–40) involves downregulation of protein kinase c. Brain Res. 957, 278–286. doi: 10.1016/S0006-8993(02)03608-9
Ono, Y., and Sorimachi, H. (2012). Calpains–an elaborate proteolytic system. Biochimica et Biophysica Acta 1824, 224–236. doi: 10.1016/j.bbapap.2011.08.005
Orrenius, S., Zhivotovsky, B., and Nicotera, P. (2003). Calcium: Regulation of cell death: the calcium–apoptosis link. Nat. Rev. Mol. Cell Biol. 4, 552. doi: 10.1038/nrm1150
Pal, G. P., Elce, J. S., and Jia, Z. (2001). Dissociation and aggregation of calpain in the presence of calcium. J. Biol. Chem. 276, 47233–47238. doi: 10.1074/jbc.M105149200
Petri, C. (1966). Kommunikation mit Automaten. Bonn: Institut für Instrumentelle Mathematik, Schriften Des IIM, No. 3 (1962). English Translation: Communication with Automata, Griffiss Air Force Base. Tech. Rep. RADC-TR-65-377, 1.
Racchi, M., Mazzucchelli, M., Pascale, A., Sironi, M., and Govoni, S. (2003). Role of protein kinase cα in the regulated secretion of the amyloid precursor protein. Mol. Psychiatry 8, 209–216. doi: 10.1038/sj.mp.4001204
Rao, M. V., McBrayer, M. K., Campbell, J., Kumar, A., Hashim, A., Sershen, H., et al. (2014). Specific calpain inhibition by calpastatin prevents tauopathy and neurodegeneration and restores normal lifespan in tau p301l mice. J. Neurosci. 34, 9222–9234. doi: 10.1523/JNEUROSCI.1132-14.2014
Rao, M. V., Mohan, P. S., Peterhoff, C. M., Yang, D.-S., Schmidt, S. D., Stavrides, P. H., et al. (2008). Marked calpastatin (cast) depletion in Alzheimer's disease accelerates cytoskeleton disruption and neurodegeneration: neuroprotection by cast overexpression. J. Neurosci. 28, 12241–12254. doi: 10.1523/JNEUROSCI.4119-08.2008
Rodrigue, K., Kennedy, K., Devous, M., Rieck, J., Hebrank, A., Diaz-Arrastia, R., et al. (2012). β-amyloid burden in healthy aging regional distribution and cognitive consequences. Neurology 78, 387–395. doi: 10.1212/WNL.0b013e318245d295
Rossner, S., Mendla, K., Schliebs, R., and Bigl, V. (2001). Protein kinase cα and β1 isoforms are regulators of α-secretory proteolytic processing of amyloid precursor protein in vivo. Eur. J. Neurosci. 13, 1644–1648. doi: 10.1046/j.0953-816x.2001.01525.x
Roussel, B. D., Kruppa, A. J., Miranda, E., Crowther, D. C., Lomas, D. A., and Marciniak, S. J. (2013). Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 12, 105–118. doi: 10.1016/S1474-4422(12)70238-7
Ryu, M., and Nakazawa, T. (2014). “Calcium and calpain activation,” in Neuroprotection and Neuroregeneration for Retinal Diseases, eds T. Nakazawa, Y. Kitaoka, and T. Harada (Tokyo: Springer), 13–24.
Santos, D. M., Xavier, J. M., Morgado, A. L., Sola, S., and Rodrigues, C. M. (2012). Distinct regulatory functions of calpain 1 and 2 during neural stem cell self-renewal and differentiation. PLoS ONE 7:e33468. doi: 10.1371/journal.pone.0033468
Schmolesky, M. T., Weber, J. T., Zeeuw, C. I., and Hansel, C. (2002). The making of a complex spike: ionic composition and plasticity. Ann. N.Y. Acad. Sci. 978, 359–390. doi: 10.1111/j.1749-6632.2002.tb07581.x
Selkoe, D. J. (2000). Toward a comprehensive theory for alzheimer's disease. hypothesis: Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity of amyloid β-protein. Ann. N.Y. Acad. Sci. 924, 17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x
Selkoe, D. J. (2002). Alzheimer's disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Sennvik, K., Bogdanovic, N., Volkmann, I., Fastbom, J., and Benedikz, E. (2004). β-secretase-cleaved amyloid precursor protein in Alzheimer brain: a morphologic study. J. Cell. Mol. Med. 8, 127–134. doi: 10.1111/j.1582-4934.2004.tb00267.x
Skovronsky, D. M., Moore, D. B., Milla, M. E., Doms, R. W., and Lee, V. M.-Y. (2000). Protein kinase c-dependent α-secretase competes with β-secretase for cleavage of amyloid-β precursor protein in the trans-golgi network. J. Biol. Chem. 275, 2568–2575. doi: 10.1074/jbc.275.4.2568
Small, D. H. (2009). Dysregulation of calcium homeostasis in Alzheimer's disease. Neurochem. Res. 34, 1824–1829. doi: 10.1007/s11064-009-9960-5
Small, D. H., Gasperini, R., Vincent, A. J., Hung, A. C., and Foa, L. (2009). The role of aβ-induced calcium dysregulation in the pathogenesis of Alzheimer's disease. J. Alzheimer's Dis. 16, 225–233. doi: 10.3233/JAD-2009-0951
Smith, I. F., Green, K. N., and LaFerla, F. M. (2005). Calcium dysregulation in alzheimer's disease: recent advances gained from genetically modified animals. Cell Calc. 38, 427–437. doi: 10.1016/j.ceca.2005.06.021
Song, W.-J., Son, M.-Y., Lee, H.-W., Seo, H., Kim, J. H., and Chung, S.-H. (2015). Enhancement of bace1 activity by p25/cdk5-mediated phosphorylation in Alzheimer's disease. PLoS ONE 10:e0136950. doi: 10.1371/journal.pone.0136950
Sperling, R. A., Aisen, P. S., Beckett, L. A., Bennett, D. A., Craft, S., Fagan, A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer's disease: recommendations from the national institute on aging-alzheimer's association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dement. 7, 280–292. doi: 10.1016/j.jalz.2011.03.003
Stutzmann, G. E. (2005). Calcium dysregulation, ip3 signaling, and Alzheimer's disease. Neuroscientist 11, 110–115. doi: 10.1177/1073858404270899
Tam, J. H., and Pasternak, S. H. (2012). Amyloid and alzheimer's disease: inside and out. Can. J. Neurol. Sci. 39, 286–298. doi: 10.1017/S0317167100013408
Tareen, S. H. K., and Ahmad, J. (2015). Modelling and analysis of the feeding regimen induced entrainment of hepatocyte circadian oscillators using petri nets. PLoS ONE 10:e0117519. doi: 10.1371/journal.pone.0117519
Tiraboschi, P., Hansen, L., Alford, M., Masliah, E., Thal, L., and Corey-Bloom, J. (2000). The decline in synapses and cholinergic activity is asynchronous in Alzheimer's disease. Neurology 55, 1278–1283. doi: 10.1212/WNL.55.9.1278
Todd, B., Moore, D., Deivanayagam, C. C., Chattopadhyay, D., Maki, M., Wang, K. K., et al. (2003). A structural model for the inhibition of calpain by calpastatin: crystal structures of the native domain vi of calpain and its complexes with calpastatin peptide and a small molecule inhibitor. J. Mol. Biol. 328, 131–146. doi: 10.1016/S0022-2836(03)00274-2
Tompa, P., Mucsi, Z., Orosz, G., and Friedrich, P. (2002). Calpastatin subdomains a and c are activators of calpain. J. Biol. Chem. 277, 9022–9026. doi: 10.1074/jbc.C100700200
Trojanowski, J. Q., and Lee, V. M.-Y. (2000). “fatal attractions” of proteins: a comprehensive hypothetical mechanism underlying Alzheimer's disease and other neurodegenerative disorders. Ann. N.Y. Acad. Sci. 924, 62–67. doi: 10.1111/j.1749-6632.2000.tb05561.x
Tsavachidou, D., and Liebman, M. N. (2002). Modeling and simulation of pathways in menopause. J. Am. Med. Inf. Ass. 9, 461–471. doi: 10.1197/jamia.M1103
Vaisid, T., Barnoy, S., and Kosower, N. (2008). Calpastatin overexpression attenuates amyloid-β-peptide toxicity in differentiated pc12 cells. Neuroscience 156, 921–931. doi: 10.1016/j.neuroscience.2008.07.072
Vassar, R. (2005). beta-secretase, app and abeta in Alzheimer's disease. Subcell Biochem. 38, 79–103. doi: 10.1007/0-387-23226-5_4
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., et al. (1999). β-secretase cleavage of alzheimer's amyloid precursor protein by the transmembrane aspartic protease bace. Science 286, 735–741. doi: 10.1126/science.286.5440.735
Volles, M. J., and Lansbury, P. T. (2002). Vesicle permeabilization by protofibrillar α-synuclein is sensitive to parkinson's disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 41, 4595–4602. doi: 10.1021/bi0121353
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Rowan, M. J., and Selkoe, D. J. (2002). Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem. Soc. Trans. 30, 552–557. doi: 10.1042/bst0300552
Walsh, D. M., and Selkoe, D. J. (2004). Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept. Lett. 11, 213–228. doi: 10.2174/0929866043407174
Walsh, D. M., Tseng, B. P., Rydel, R. E., Podlisny, M. B., and Selkoe, D. J. (2000). The oligomerization of amyloid β-protein begins intracellularly in cells derived from human brain. Biochemistry 39, 10831–10839. doi: 10.1021/bi001048s
Weber, J. T. (2012). Altered calcium signaling following traumatic brain injury. Front. Pharmacol. 3:60. doi: 10.3389/fphar.2012.00060
Wojda, U., Salinska, E., and Kuznicki, J. (2008). Calcium ions in neuronal degeneration. IUBMB Life 60, 575–590. doi: 10.1002/iub.91
Wu, H.-Y., and Lynch, D. R. (2006). Calpain and synaptic function. Mol. Neurobiol. 33, 215–236. doi: 10.1385/MN:33:3:215
Yamakawa, H., Banno, Y., Nakashima, S., Yoshimura, S.-i., Sawada, M., Nishimura, Y., et al. (2001). Crucial role of calpain in hypoxic pc12 cell death: calpain, but not caspases, mediates degradation of cytoskeletal proteins and protein kinase c-α and-δ. Neurol. Res. 23, 522–530. doi: 10.1179/016164101101198776
Yang, J., Weimer, R. M., Kallop, D., Olsen, O., Wu, Z., Renier, N., et al. (2013). Regulation of axon degeneration after injury and in development by the endogenous calpain inhibitor calpastatin. Neuron 80, 1175–1189. doi: 10.1016/j.neuron.2013.08.034
Yang, L.-B., Lindholm, K., Yan, R., Citron, M., Xia, W., Yang, X.-L., et al. (2003). Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 9, 3–4. doi: 10.1038/nm0103-3
Keywords: Calpain, CAST, calcium, PKC, APP, Stochastic petri net, Alzheimer disease, Amyloid beta
Citation: Ashraf J, Ahmad J, Ali A and Ul-Haq Z (2018) Analyzing the Behavior of Neuronal Pathways in Alzheimer's Disease Using Petri Net Modeling Approach. Front. Neuroinform. 12:26. doi: 10.3389/fninf.2018.00026
Received: 11 December 2017; Accepted: 30 April 2018;
Published: 23 May 2018.
Edited by:
Athanasios Alexiou, Novel Global Community Educational Foundation (NGCEF), Hebersham, AustraliaReviewed by:
Faez Iqbal Khan, Rhodes University, South AfricaCopyright © 2018 Ashraf, Ahmad, Ali and Ul-Haq. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jamil Ahmad, amFtaWwuYWhtYWRAcmNtcy5udXN0LmVkdS5waw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.