 Elise Drapé
Elise Drapé Typhaine Anquetil
Typhaine Anquetil Bruno Larrivée
Bruno Larrivée Alexandre Dubrac
Alexandre Dubrac
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Hum. Neurosci. , 24 November 2022
Sec. Brain Health and Clinical Neuroscience
Volume 16 - 2022 | https://doi.org/10.3389/fnhum.2022.1006115
This article is part of the Research Topic Brain Arteriovenous Malformations: Cerebrovasculature Behaving Badly View all 11 articles
Hereditary hemorrhagic telangiectasia (HHT) is a genetic disorder characterized by vessel dilatation, such as telangiectasia in skin and mucosa and arteriovenous malformations (AVM) in internal organs such as the gastrointestinal tract, lungs, and brain. AVMs are fragile and tortuous vascular anomalies that directly connect arteries and veins, bypassing healthy capillaries. Mutations in transforming growth factor β (TGFβ) signaling pathway components, such as ENG (ENDOGLIN), ACVRL1 (ALK1), and SMAD4 (SMAD4) genes, account for most of HHT cases. 10–20% of HHT patients develop brain AVMs (bAVMs), which can lead to vessel wall rupture and intracranial hemorrhages. Though the main mutations are known, mechanisms leading to AVM formation are unclear, partially due to lack of animal models. Recent mouse models allowed significant advances in our understanding of AVMs. Endothelial-specific deletion of either Acvrl1, Eng or Smad4 is sufficient to induce AVMs, identifying endothelial cells (ECs) as primary targets of BMP signaling to promote vascular integrity. Loss of ALK1/ENG/SMAD4 signaling is associated with NOTCH signaling defects and abnormal arteriovenous EC differentiation. Moreover, cumulative evidence suggests that AVMs originate from venous ECs with defective flow-migration coupling and excessive proliferation. Mutant ECs show an increase of PI3K/AKT signaling and inhibitors of this signaling pathway rescue AVMs in HHT mouse models, revealing new therapeutic avenues. In this review, we will summarize recent advances and current knowledge of mechanisms controlling the pathogenesis of bAVMs, and discuss unresolved questions.
Hereditary hemorrhagic telangiectasia (HHT), or Rendu-Osler disease, is an autosomal-dominant inherited syndrome with a prevalence of around 1:5,000–8,000 people (Bideau et al., 1989; Kjeldsen et al., 1999), characterized by vascular anomalies. The major lesions found are telangiectasia, widened small vessels located near the surface of the skin or mucous membranes such as lips, tongue, nasal, buccal, and gastrointestinal mucosa. These lesions are fragile and prone to bleeding mostly in the nasal mucosa and gastrointestinal tract (Shovlin, 2010). Most HHT patients also develop pulmonary, hepatic, spinal or brain arteriovenous malformations (AVMs), which are abnormal connections between arteries and veins. While 33% of patients develop pulmonary AVMs, 10–20% will develop brain AVMs (bAVMs) (Haitjema et al., 1995). Although bAVMs are less frequent, their consequences can be detrimental for the patient. Indeed, in addition to intracranial hemorrhages, bAVMs can lead to blood-brain-barrier (BBB) defects, promoting neuronal dysfunction and seizure such as epilepsy (Mohr et al., 2013; Rohn et al., 2014). However, bAVM pathogenesis is not well understood.
It is now well known that ALK1 signaling, one of the canonical pathways of the TGFβ superfamily, plays a critical role in vascular morphogenesis (Roman and Hinck, 2017). BMP9 and BMP10 are cytokines produced by the liver and the heart, respectively (Figure 1; Neuhaus et al., 1999; Bidart et al., 2012). BMP9 expression has also been reported in the lung and the brain septum, although at a significantly lower level than in the liver (López-Coviella et al., 2000; Bidart et al., 2012). Cardiac BMP10 expression is associated with the development of the trabeculated myocardium during embryonic development and then becomes mainly restricted to the right atria in post-natal life (Neuhaus et al., 1999; Somi et al., 2004). These cytokines can have an autocrine and paracrine action but can also enter blood circulation to act on endothelial cells (David et al., 2008; Chen H. et al., 2013). Indeed, BMP9/10 bind with high affinity to the TGFβ receptor 1 ALK1, a serine/threonine kinase receptor, and its coreceptor, ENDOGLIN (Figure 1), which is expressed predominantly on ECs (David et al., 2006; Alt et al., 2012; Townson et al., 2012). BMP9/10 ligands bind to ALK1 which will heterodimerize with BMP receptor II (BMPRII) to transduce downstream signaling. The ENDOGLIN receptor interacts with the receptor complex to promote the phosphorylation of the transcription factors SMAD1/5/8 (Chen and Massagué, 1999; Oh et al., 2000), leading to their association with the common regulator SMAD4 (Nakao et al., 1997). Then, the SMAD complex translocates and accumulates into the nucleus (Figure 1) in order to regulate the expression of specific target genes such as DNA-binding protein inhibitor 1 (ID1) and ID3 (Valdimarsdottir et al., 2002). For example, the BMP9/ALK1 signaling pathway regulates angiogenesis by modulating the expression of genes involved in vascular quiescence, cell junction, response to shear stress, and the recruitment of mural cells.

Figure 1. Endothelial BMP/ALK1 signaling pathway. Soluble BMP9 and BMP10, secreted, respectively, by the liver and the heart in the bloodstream, bind their receptor ALK1, a SER/THR kinase, and its coreceptors BMPRII and ENDOGLIN at the surface of ECs. The activation of the BMP receptor complex will trigger the downstream SMAD signaling. Phosphorylated SMAD1/5/8 will recruit SMAD4. This SMAD complex accumulates in nucleus to regulate transcription of target genes, including genes involved in angiogenic processes.
HHT patients can be subdivided into five groups. Mutations in the ENG gene (encoding for ENDOGLIN) are responsible for HHT1 (McAllister et al., 1994), while mutation of ACVRL1 (encoding for ALK1) leads to HHT2 (Johnson et al., 1996). HHT1 and HHT2 represent around 90% of HHT cases (Prigoda et al., 2006). Moreover, mutations of SMAD4 represent 2% of a type of HHT called juvenile polyposis (JP)-HHT (Howe et al., 1998; Gallione et al., 2004). Finally, loci on chromosomes 5 and 7, unlinked to ENG, ACVRL1, or SMAD4 mutations, have been associated with subsets of HHT patients and have been referred to as HHT3 and HHT4, respectively (Cole et al., 2005; Bayrak-Toydemir et al., 2006), while HHT features (HHT5) have also been observed in individuals with heterozygous mutations of growth differentiation factor 2 (GDF2), which encodes BMP9 (Wooderchak-Donahue et al., 2013). It should be noted that the vascular phenotype varies depending on the mutated HHT genes but also on the individual. Indeed, mutations in the same gene can trigger different vascular anomalies from one patient to another, suggesting context-dependent pathogenicity of vascular malformations (Kjeldsen et al., 2005; Bayrak-Toydemir et al., 2006).
This review will focus on HHT bAVMs and summarize recent advances in organotypic pathogenicity, animal models, cellular and molecular mechanisms, and therapeutics.
The blood supply of the human brain relies on only two pairs of large arteries: the internal carotids, which supply the blood to the cerebrum, and the vertebral arteries, which join distally to form the basilar artery, and finally vascularize the cerebellum and the brain stem. Proximally, the basilar artery and the internal carotid arteries join to form a ring at the brain’s base known as the circle of Willis.
The circle of Willis gives rise to three pairs of main arteries: the anterior, middle, and posterior cerebral arteries, which divide progressively into smaller arteries and pial arterioles within the brain’s subarachnoid space meninges. Then, penetrating arterioles dive into the cortex and divide into precapillary arterioles and capillaries, the most predominant brain vessels. Similarly, the pial venular network from the brain surface is connected to ascending venules diving into the parenchyma, followed by postcapillary venules, which branch to capillaries.
Even if human brain vascularization is well described, its development is based on studies done in animal models, such as rodents. Comprehensive studies on the mouse brain showed that vascularization starts at embryonic day (E) 7.5–8.5 by vasculogenesis at the ventral region of the neural tube, followed by the invasion of capillary sprouts into the neuroepithelium at E9.5 (Hogan et al., 2004). Branching and arborization of capillary sprouts from the pial surface subsequently lead to the formation of the perineural vascular plexus (PNVP) at the brain’s surface (Tata et al., 2015; Biswas et al., 2020). Then, the first sprouts of the PNVP invade the ventrolateral forebrain following an angiogenic gradient of Vascular Endothelial Growth Factor (VEGF) induced by the hypoxic tissue at postnatal day (P) 9.5 (Bautch and James, 2009; Tata et al., 2015; Peguera et al., 2021). The vascularization will then continue to invade the tissue in a ventrolateral to dorsomedial fashion by angiogenesis, the growth of pre-existing blood vessels. The cerebral vascular network is still expanding and remodeling for a few weeks after birth (Biswas et al., 2020). While canonical pro-angiogenic signaling pathways, such as VEGF, WNT, and TGFß, are essential, mechanisms controlling this postnatal angiogenesis wave remain unclear (Paredes et al., 2018). Neo-angiogenic sprouts and EC proliferation are observed until P14. Then, from P14 to P25, a progressive decrease in the angiogenic rate will favor the stabilization of the vasculature (Biswas et al., 2020; Coelho-Santos and Shih, 2020). Moreover, the capillary density varies depending on the brain region. Indeed, the angiogenesis rate is far higher in gray matter, enriched in neuronal cell bodies, than in white matter to supply the energy needed (Coelho-Santos and Shih, 2020).
The ECs lining vessels have developed a unique and highly selective BBB to maintain brain homeostasis and neural function (Daneman and Prat, 2015). This organotypic feature of the CNS capillaries depends on the interaction and communication between the neurovascular unit (NVU) cells, which includes ECs, pericytes (PCs), astrocyte endfeet as well as neurons (Siegenthaler et al., 2013; Kaplan et al., 2020). In the mouse, brain angiogenesis begins at E9.5-E11.5, and the initiation of BBB starts soon after (Biswas et al., 2020). ECs acquire both paracellular and transcellular barriers to prevent the movement of small molecules between ECs (Daneman and Prat, 2015; Biswas et al., 2020). The endothelial BBB is characterized by abundant tight junctions (paracellular barrier) and a low rate of endocytosis (transcellular barrier) to control passive exchanges and caveolae-dependent receptor-mediated transcytosis. Therefore, the BBB allows a high control of the nutrient exchange, low permeability, and immune privileges (Siegenthaler et al., 2013; Daneman and Prat, 2015; Haddad-Tóvolli et al., 2017).
A healthy blood vascular network has capillary plexuses between arteries, a high flow region, and veins, a low flow region. Capillaries are essential for the delivery of oxygen and nutrients. Therefore, neurovascular coupling, the blood flow regulation by neurons, is essential for neuronal function (Kaplan et al., 2020). The main characteristic of AVM is a direct connection between veins and arteries, bypassing the capillary bed. The loss of capillaries in bAVMs leads to cerebral hemodynamic changes with the increased blood flow in the arteries and veins. Thus, bAVMs are considered high-flow vascular malformations. bAVMs are also characterized by one or several feeding arteries and one draining vein. Moreover, bAVMs are associated with ECs proliferation and mural cell coverage defects.
There are different types of bAVMs defined by specific features (Matsubara et al., 2000): arteriovenous fistulas (AVF), nidal AVMs, and capillary vascular malformations (CVM). AVFs correspond to direct fistula connections between one artery and one vein, whereas nidal AVMs and CVMs are characterized by the presence of a nidus, defined as tangles of abnormal blood vessels. Nidal AVMs have a nidus between 1 and 3 cm with multiple feeding arteries and veins, whereas the nidus of CVM is less than 1 cm and has one feeding artery and vein (Matsubara et al., 2000; Griauzde et al., 2020). Patients with AVF and nidal AVM have more chance of rupture risk and may present symptoms such as seizure, hemorrhage, and headache, whereas most patients with CVM are asymptomatic (Matsubara et al., 2000; Brinjikji et al., 2017). The different types of bAVMs can also be discerned by the age of the patients. Indeed, AVFs are observed in young children, whereas nidus AVMs and CVMs develop later in life (Matsubara et al., 2000; Krings et al., 2005).
Even if the different AVMs have different characteristics, no differences in the localization in the brain have been reported (Krings et al., 2005). AVMs are located mostly in superficial regions of the cerebral cortex, with a majority in the supratentorial region compared to the infratentorial region. However, very few AVMs can be found in deeper areas such as the basal ganglia or brain stem (Matsubara et al., 2000; Saleh et al., 2013; Komiyama et al., 2015; Krings et al., 2015).
Most bAVMs are asymptomatic, and the diagnosis is often made by brain imaging after hospitalization or examination for another medical reason. However, some patients may develop symptoms such as headaches, seizures, and hemorrhages (Brown et al., 1996). In addition, it seems that there is a correlation between the hemodynamic changes, the patients’ age, and the AVM’s cerebral location (Gross and Du, 2013; Abecassis et al., 2014). The exact causes of seizures are not yet well understood. However, frontal or temporal lobe locations of AVMs are associated with a higher risk of seizure compared to a deep location (Hoh et al., 2002). In addition, it also appears that hemodynamic changes may influence the occurrence of seizures (Fierstra et al., 2011).
The risks of hemorrhage may be increased by the position of the AVM in the brain, with a deep location having a higher risk of rupture (Yamada et al., 2007). However, the bAVMs found in HHT patients predominantly have a cortical area and therefore have a relatively low risk of rupture (around 2% per year) (Willemse et al., 2000). Moreover, it has also been documented that the size of the AVM and its association with aneurysms may also influence the risk of hemorrhage (Gross and Du, 2013). The high blood pressure generated by bAVM may also increase the risk of rupture (Langer et al., 1998). Hemorrhage after bAVM rupture leads to mortality or morbidity development (Fleetwood and Steinberg, 2002). Indeed, it has been shown that patients with hemorrhagic bAVMs have a higher risk of subsequent epileptic seizure (Josephson et al., 2011).
However, mechanisms that lead to the development of those symptoms are not yet well understood, mainly because only a few patients develop cerebral symptoms, the distribution of these symptoms are heterogeneous among patients, and the lack of experimental models.
It is crucial to develop animal models that recapitulate HHT pathophysiology to improve our knowledge of AVMs’ pathogenesis and reveal new therapeutic avenues. Several studies have used new sophisticated models to decipher AVM mechanisms in the last two decades, as summarized below (Tual-Chalot et al., 2015).
Three transgenic mouse models with global knockout for Eng were first developed in order to recapitulate HHT1 (Bourdeau et al., 1999; Li et al., 1999; Arthur et al., 2000). These mice show marked defects in yolk sac angiogenesis and cardiac development, leading to lethality at E10.5. Similarly, global knockout mice for Acvrl1 have been developed to study HHT2 (Oh et al., 2000; Urness et al., 2000; Srinivasan et al., 2003). These mice also die around E10.5 with pronounced angiogenic defects. Indeed, mutant embryos exhibit hyperdilated vessels and the formation of arteriovenous shunts. The embryonic lethality of these mutant mice highlights the importance of the BMP/ALK1/ENG signaling pathway in vascular development and morphogenesis but makes it challenging to study the mechanisms underlying these angiogenic defects. Thus, the following studies were carried out on heterozygous mice. In addition, to be viable, these models are closer to HHT physiopathology, as HHT patients also carry heterozygous mutations (Srinivasan et al., 2003; Torsney et al., 2003). Moreover, heterozygous mutant mice display some features found in patients, such as mucocutaneous vascular lesions in internal organs, such as lungs, liver, spleen, and intestines. However, these mice develop very few cerebral AVMs at a lower frequency than that found in patients.
The creation of inducible and tissue-specific mutant mice was crucial for the investigation of the role of key genes in developmental and pathological processes. Reporter mice have shown that ALK1 and ENG are mainly expressed in ECs of blood vessels (Jonker and Arthur, 2002; Seki et al., 2003). In line with these findings, the endothelial-specific Acvrl1 deletion is sufficient to trigger blood vessels dilation, hemorrhages and AVMs in the brain, lungs and intestine (Park et al., 2009). However, endothelial mutant mice showed improved survival until postnatal day 5 compared to global KO which induces embryonic lethality. New inducible and endothelial-specific models allowed studying AVMs in postnatal mice. Interestingly, an early postnatal endothelial-specific deletion of Eng or Acvrl1 is sufficient to induce an HHT-like phenotype, including AVM (Table 1; Allinson et al., 2007; Park et al., 2008, 2009; Mahmoud et al., 2010). However, the deletion of Eng in perivascular smooth muscle cells does not promote arteriovenous shunt formation, in line with its expression profile (Garrido-Martin et al., 2014). Altogether, these findings revealed the essential role of ECs in the pathogenesis of HHT. These models also confirmed the hypothesis that a second hit is necessary to induce AVM in HHTs, also known as the three events hypothesis (Tual-Chalot et al., 2015). This refers to the fact that 3 stimuli are necessary for AVM formation: gene loss, protein loss, and then an angiogenic stimulus. While an early endothelial deletion of Eng and Acvrl1 promotes AVMs formation in the angiogenic context of developing tissues, the deletion in adults is not sufficient unless there is an angiogenic or inflammatory stimulus. Indeed, local injection of VEGF or LPS following an endothelial Acvrl1 or Eng deletion induces several vascular anomalies including bAVMs in adult mice (Walker et al., 2011; Choi et al., 2012, 2014; Han et al., 2014). In addition, AVMs also develop in the skin of adult Acvrl1 mutant mice when angiogenesis is induced with a wound, proving the necessity of an angiogenic hit for AVM formation (Park et al., 2009).

Table 1. Summary of the major HHT mouse models.
In patients, heterozygous mutations in Smad4 lead to JP-HHT, but the mechanisms are still poorly understood. Similar to Acvrl1 and Eng knockout mice, a global loss of Smad4 results in lethality at the E10.5 (Lan et al., 2007). Constitutive endothelial Smad4 deletion decreases blood vessel integrity and leads to cerebral bleeding, specifically during embryonic development (Li et al., 2011). In addition, postnatal loss of Smad4 induces the formation of AVMs in the retina, brain, and gastrointestinal tract, phenocopying vascular phenotypes observed in endothelial Acvrl1 and Eng mutant mice (Table 1; Ola et al., 2018).
Mutations in the GDF2 gene are responsible for less than 1% of HHTs (Wooderchak-Donahue et al., 2013; Hernandez et al., 2015). In mice, injection of antibodies blocking both BMP9 and BMP10 has been shown to phenocopy endothelial Eng, Acvrl1, and Smad4 mutant mice to some degree, with AVM formation in the retina and the gastrointestinal tract, suggesting that it could be a model of HHT (Table 1; Baeyens et al., 2016; Ola et al., 2016; Ruiz et al., 2016, 2020). Moreover, while deletion of either Gdf2 or Bmp10 does not promote vessel dilatation and AVM formation (Ricard et al., 2012), postnatal double Gdf2 and Bmp10 deletion induces vascular anomalies, including AVMs in the gastrointestinal tract (Bouvard et al., 2021).
Recently, new genetic tools have been developed to delete genes in specific EC subtypes, such as capillaries, veins, arteries, and tip cells, to study the onset and progression of AVMs. Therefore, it has been shown that deletion of Eng (Singh et al., 2020), Smad4 (Lee et al., 2021), and Acvrl1 (Park H. et al., 2021) in venous ECs is sufficient to induce AVMs, unlike deletion in other EC subpopulations (Table 1). The underlying mechanisms will be discussed in later sections.
Despite the development of new mouse models, the study of bAVMs remains complicated due to their 3D structure and unpredicted location in the brain. Therefore, many studies have been performed on the retina which has a vascular network growing in 2D that can be imaged in wholemount. Moreover, retina and brain blood vessels share numerous features including the NVU and Blood Retina Barrier (BRB). The retina is a part of the central nervous system and is a well-established model for studying angiogenic processes such as endothelial sprouting, arterio-venous specification, and vascular remodeling (Adams and Alitalo, 2007; Fonseca et al., 2020). In mice, the vascularization of the retina starts at birth, and blood vessels grow from the optic nerve to the periphery until P7 to form a superficial vascular plexus. Then, blood vessels dive into the neuroretina to form the deep vascular plexus until P12, followed by the formation of the intermediate plexus until P15, and the vascular network is completed and fully mature at P21 (Gariano and Gardner, 2005). Compared to the brain, the superficial retinal plexus is a stereotypic vascular network that can be imaged by wholemount, allowing the study of complex vascular structures such as AVMs. Therefore, we and others have shown that endothelial deletion of Eng, Acvrl1, or Smad4 induces AVMs in mouse retinas, which has greatly improved our understanding of the mechanisms involved in the pathogenesis (Mahmoud et al., 2010; Tual-Chalot et al., 2014; Baeyens et al., 2016; Ola et al., 2016, 2018; Crist et al., 2018).
All this work shows that endothelial deletion of Eng, Acvrl1 and Smad4 are appropriate models to study pathology. In the retina, in addition to the formation of AVMs, mutants show an increase in EC sprouting, proliferation, and vessel dilatation. Mural cells are also affected with a decrease in pericyte coverage on the one hand and arterialization of the veins on the other with the recruitment of vascular smooth muscle cells (vSMC). However, despite some degree of similarity in the vascular phenotype, these mutant mice also present distinct phenotypes. For example, 60% of Acvrl1 mutants develop AVMs in the retina (Tual-Chalot et al., 2014), whereas this percentage is around 70 and 82% in Eng and Smad4 mutants, respectively (Mahmoud et al., 2010; Crist et al., 2018). Moreover, both Eng and Smad4 mutants exhibit a delay in vessel outgrowth but not Acvrl1 mutants. Acvrl1 mutants show a hyperbranching more pronounced than Eng and Smad4 mutants, which is associated with a strong increase in the number of tip cells. However, it should be noted that the dose of tamoxifen used and the age of the mice at time of injection could also influence these differences. Further investigation is required to accurately compare vascular phenotypes of those models.
Overall, these new transgenic mouse models showed that the BMP/ALK1/ENG/SMAD4 signaling pathway is essential for both developmental and adult vascular morphogenesis. Indeed, in addition to AVMs, the postnatal endothelial deletion of Eng, Acvrl1, and Smad4 are lethal, and survival varies depending on the gene and age.
Zebrafish also represent an important model for studying bAVMs. First, because of the high degree of homology between zebrafish and vertebrates regarding vasculature and angiogenic mechanisms (Schuermann et al., 2014). Secondly, thanks to the transparency of the embryos it is easy to follow the vasculature and the development of vascular malformation using fluorescence imaging. Finally, the facility to perform genetic mutations makes the zebrafish an essential model for studying vascular development and disease. acvrl1 mutation in zebrafish induces cranial vessel defects and enlargement, ultimately leading to the formation of AVMs with direct connections between the basilar and basal communicating arteries with the primordial midbrain and hindbrain veins (Roman et al., 2002; Corti et al., 2011). Moreover, bmp10 mutants phenocopy acvrl1 mutants, with vessel enlargement, increase of ECs number and formation of direct connection between arteries and the midbrain and hindbrain veins (Laux et al., 2013). Indeed, bmp10 mutants exhibit blood vessel abnormalities in liver, heart dysmorphology, vascular defects correlating with increased cardiac output and embryonic lethal cranial AVMs indistinguishable from acvrl1 mutants (Capasso et al., 2020), highlighting that in zebrafish, bmp10 encodes the only required Alk1 ligand in the juvenile-to-adult period. By contrast, bmp9 mutants survive to adulthood and do not develop cranial AVMs (Capasso et al., 2020), although they do display transient remodeling defects of the caudal venous plexus (Wooderchak-Donahue et al., 2013). Recently, Sugden et al. (2017) have shown that zebrafish mutated for eng at the embryonic stage survive, whereas they develop AVM-like vascular defects. The authors also showed that loss of eng in adults leads to dilation of cerebral vessels, and that only induction of an angiogenic hit through fin amputation lead to bAVM formation (Sugden et al., 2017). Finally, the contribution of Smad signaling in AVM formation was also highlighted in zebrafish by a study showing that Smad9 morphants develop cranial AVMs with morphologic similarities to human bAVMs (Walcott et al., 2018).
AVMs develop in high blood flow regions and are often associated with vascular proliferation. The ECs lining blood vessels are exposed to blood flow which induces mechanical forces. The generated laminar shear stress (LSS), within the physiological range, triggers a number of flow responses in EC, including PI3K/AKT signaling pathway activation, alignment in the direction of flow (Tzima et al., 2003; Coon et al., 2015), migration against the blood flow (Parmar et al., 2006; Ouarné et al., 2021), inhibition of proliferation (Akimoto et al., 2000), and PCs recruitment (Van Gieson et al., 2003). Therefore, LSS is essential for blood vessel integrity, vascular remodeling, and morphogenesis. Recent studies have demonstrated an essential role for the BMP/ALK1/ENG signaling pathway in the LSS-induced PI3K/AKT activation and vascular quiescence (Laux et al., 2013; Ola et al., 2016, 2018; Jin et al., 2017). Indeed, PI3K/AKT signaling pathway is an important regulator of EC proliferation (Graupera and Potente, 2013), and it can be inhibited to promote vascular quiescence. Therefore, the understanding of the BMP/ALK1/ENG-LSS crosstalk could allow the identification of new therapeutic strategies.
Angiogenic ECs express high levels of Eng in development and disease, whereas its expression is attenuated in adult quiescent endothelium (Miller et al., 1999; Jonker and Arthur, 2002; Torsney et al., 2002). Moreover, Eng depletion inhibits TGFβ-induced proliferation arrest in vitro and in vivo, resulting in hyperproliferation and migration defects (Park et al., 2013).
In endothelial Eng mutant mice mimicking HHT1, 70% of retinas develop multiple AVMs and bleeding. These retinas also exhibit several other vascular anomalies, such as increased vascular branching, sprouting, vessel diameter, and endothelial proliferation (Mahmoud et al., 2010). Loss of Eng induces arterial and venous identity defects, and it can also be noted that AVMs are positive for venous markers such as EPHRIN B4 (EphB4) and APELIN receptor (AplnR). Blood flow was shown to promote the association of ALK1 and ENG at the EC membrane, thereby enhancing BMP signaling and vascular quiescence (Baeyens et al., 2016). A recent study showed that Eng mutant ECs fail to migrate against the flow and exhibit an increase in VEGF-induced PI3K/AKT activation (Jin et al., 2017). Inhibition of VEGFR2 or PI3K/AKT signaling decreased the number of AVMs in Eng mutant retinas. In addition, the authors also showed that Eng mutant ECs tend to proliferate mainly in arteries. One study in which Eng was specifically deleted only in capillaries and veins but not in arteries (Singh et al., 2020) showed that these mice still have AVMs in the retinas, suggesting that the loss of Eng in arteries is not involved in the development of AVMs. Similar observations have been made in zebrafish, where eng mutations lead to the formation of AVMs with an increase in vessel diameter (Sugden et al., 2017) caused by defects in flow response and cell polarization leading to vessel enlargement. The role of Eng in the flow response and the pathogenesis of AVMs, therefore, appears to be a conserved process. Altogether, these studies demonstrate that Eng is essential for EC specification, migration against the flow, and shear stress-induced quiescence to prevent AVMs formation.
In the HHT2 mouse model, the endothelial Acvrl1 deletion induces an increase in vascular density, endothelial sprouting as well as the formation of AVMs in the retina (Park et al., 2009; Larrivée et al., 2012; Ola et al., 2016). Similar to HHT1 models, endothelial Acvrl1 deletion decreases PC recruitment, arterial marker expression such as JAGGED1, and AVMs are positive for the venous marker EPHRIN B4. Interestingly, retinal shunts develop near the optic nerve in high flow regions, while hyperbranching and hypersprouting develop mainly at the migration front, a low flow region (Bernabeu et al., 2014; Baeyens et al., 2016). These data suggest that, like the HHT1 models, LSS is important in the development of HHT2’s AVMs. Moreover, several studies in zebrafish have shown that acvrl1 expression is induced by blood flow (Corti et al., 2011; Laux et al., 2013). acvrl1 deletion also decreased the expression of genes involved in the flow-response, such as Edn1 and Cxcr4. However, the expression of Klf2, a transcription factor essential for the endothelial flow-response (Parmar et al., 2006), is not altered. Although endothelial Acvrl1 inhibition promotes arterial enlargement associated with an increase in the number of ECs, a recent study showed AVMs formation requires mutant capillary and venous and not arterial ECs (Park H. et al., 2021). While arterial-specific Acvrl1 deletion did not induce obvious vascular phenotypes and lethality, deletion in venous ECs results in retinal AVMs and is lethal 5–6 days after gene deletion. Moreover, Acvrl1 inhibition in endothelial tip cells, cells leading vascular sprouts and arteries formation (Xu et al., 2014; Pitulescu et al., 2017), failed to induce retinal AVMs (Park H. et al., 2021). However, these mutant mice develop capillary malformations in the retina, intestinal villi, and brain, and they die 10–11 days after tamoxifen injection, suggesting that they might develop AVMs in other tissues. Therefore, Park H. et al. (2021) proposed that HHT2’s AVMs originate from defective venous ECs. Indeed, mutant ECs present a polarization defect and migrate randomly instead of being directed against the blood flow. This phenomenon has also been observed in the zebrafish model (Rochon et al., 2016).
Skin biopsies of HHT2 patients showed an increase in the PI3K/AKT pathway suggesting an involvement of this pathway in AVM formation (Alsina-Sanchís et al., 2018). In vitro, BMP9 stimulation blocks VEGF-induced EC proliferation, and this effect is lost with the ACVRL1 knockdown. Moreover, inhibition of ACVRL1 increased VEGF-induced VEGFR2 activation and AKT phosphorylation (Ola et al., 2016; Alsina-Sanchís et al., 2018). While VEGFR2 is the main VEGF receptor that mediates VEGF-induced EC proliferation and angiogenesis, VEGFR1 cannot transduce VEGF signaling and acts as a decoy receptor. Therefore, VEGFR1 traps VEGF ligand, and VEGFR1 inhibition increases VEGF bioavailability and signaling. Interestingly, patients and mouse models of HHT2 show decreased VEGFR1 expression. Thalgott et al. (2018) have recently shown that blocking VEGFR1 induces retinal AVMs in a VEGFR2-dependent manner. In addition, VEGFR2 inhibition, using DC101 blocking antibody, can also rescue the formation of AVMs, as well as hypersprouting and hyperbranching, in Acvrl1 mutant retinas highlighting that exacerbated VEGF signaling is associated with AVM formation in Acvrl1 mutant models (Ola et al., 2016).
Pharmacological or genetic inhibition of the PI3K/AKT signaling pathway also rescues EC proliferation and AVMs in Acvrl1 mutant mice (Ola et al., 2016; Alsina-Sanchís et al., 2018), demonstrating the contribution of this signaling pathway in the development of AVMs. PTEN is a phosphatase that negatively regulates the PI3K/AKT pathway by modulating the phosphatidylinositol 3,4,5-trisphosphate (PIP3) conversion into phosphatidylinositol 4,5-bisphosphate (PIP2) (Graupera and Potente, 2013). In vitro studies have shown that BMP9/ALK1 signaling increase PTEN activity in ECs, thereby decreasing PI3K/AKT signaling (Ola et al., 2016; Alsina-Sanchís et al., 2018). However, while the loss of endothelial PTEN in ECs increases EC proliferation and induces retinal vascular hyperplasia, it does not induce AVM formation (Serra et al., 2015).
LSS-induced EC polarity and migration are regulated by integrin binding to extracellular matrix proteins and signaling to CDC42 (cell division control protein 42 homolog) (Etienne-Manneville and Hall, 2001; Tzima et al., 2003). Integrins are transmembrane receptors that allow the adhesion of ECs to the extracellular matrix, which is an important process for angiogenesis (Serini et al., 2008). Some integrins interact with VEGFR2 at the EC membrane regulating its activity and the activation of the PI3K/AKT and YAP/TAZ signaling pathways. A recent study showed that Acvrl1 deletion increased the expression of integrin β1, α5, and αv in AVMs (Park H. et al., 2021). They also found an increase in the integrins-VEGFR2 interaction and the YAP/TAZ signaling pathway activation. In addition, pharmacological inhibition of integrins or YAP/TAZ signaling prevents the formation of AVMs as well as the endothelial polarization defects in Acvrl1 mutant mice.
All these studies demonstrate that BMP9/ALK1 signaling is essential to control LSS-induced EC quiescence and polarity via the VEGFR2-integrins complex and the activation of downstream signaling pathways, such as PI3K/AKT and YAP/TAZ.
In the mouse model of JP-HHT, postnatal endothelial Smad4 deletion induces retinal AVMs and hypersprouting, as observed in the Acvrl1 and Eng mutant mice (Crist et al., 2018; Ola et al., 2018). However, the authors found a vascular outgrowth defect that was also found in the HHT1 models but not in the HHT2 model (Crist et al., 2018). Similar to Acvrl1 and Eng deletion, AVMs developed near the optic nerve where blood flow is high, and Smad4 AVMs originate from defective venous ECs (Lee et al., 2021). In line with its upstream effectors, Smad4 deletion also increased EC proliferation and PI3K/AKT signaling and inhibited EC migration against the flow, suggesting a similar mechanism in SMAD4 AVMs. Indeed, the inhibition of the BMP9/ALK1/SMAD4 signaling pathway promotes excessive AKT signaling, which inhibits the transcription factor Forkhead box protein O1 (FOXO1) and increases the expression of the proto-oncogene c-MYC (Ola et al., 2018). Ola et al. (2018) proposed that SMAD4 activation regulates Casein Kinase 2 (CK2) expression, which is responsible for PTEN phosphorylation and inhibition. Moreover, CK2 inhibition prevents PI3K/AKT overactivation and the formation of AVMs in Smad4 mutant mice. These data suggest that the BMP9/ALK1/SMAD4 signaling pathway is essential in venous EC to prevent abnormal PI3K/AKT signaling, proliferation, and LSS-induced migration defect.
In line with these observations, knockdown of receptor-regulated SMADs (R-SMADs) SMAD1 and SMAD5 has also been shown to result in the formation of AVMs. Benn and colleagues reported that endothelial-specific simultaneous deletion of Smad1 and Smad5 resulted in the formation of retinal AVMs in areas with high blood flow, while reduced vessel regression and increased loop formation were observed in areas of lower blood flow (Benn et al., 2020). The formation of vascular shunts was also observed in yolk sacs of endothelial-specific Smad1/5 knockout embryos at E9.25, which occurred in part as a consequence of decreased Cx37 expression, leading to vessel enlargement and the formation of shunts in vessel segments of higher flow (Peacock et al., 2020). Altogether, these studies further highlight the crucial role of SMAD signaling in preventing AVM development.
Constitutive Smad4 deletion in brain ECs increased EC proliferation, leading to cerebral hemorrhages (Li et al., 2011). In addition, the authors observed a decrease in NOTCH expression, suggesting a connection between the NOTCH and ALK1/SMAD4 pathways in the maintenance of brain vascular integrity. Interestingly, mutant mouse embryos for NOTCH effectors, such as delta-like-4 (Dll4) and Recombination Signal Binding Protein For Immunoglobulin Kappa J Region (Rbpj), exhibit bAVMs (Krebs et al., 2004). The involvement of this pathway in bAVM formation is discussed in a later section.
BMP9 and BMP10 signaling have redundant functions in mice during postnatal life, and both ligands have to be inhibited to phenocopy Acvrl1 and Eng mutant mice. Indeed, the injection of BMP9 and BMP10 blocking antibodies in neonatal pups induces retinal AVMs near the optic nerve, as well as hypersprouting and hyperplasia (Baeyens et al., 2016; Ruiz et al., 2016). Moreover, BMP9 inhibition is sufficient to increase PI3K/AKT signaling and proliferation of EC under-flow in vitro. Similar to other HHT models, inhibition of the PI3K/AKT or VEGFR2 pathway prevents and rescues EC proliferation and AVMs formation induced by BMP9 and BMP10 blockade (Baeyens et al., 2016; Ola et al., 2016).
Interestingly, sirolimus, an inhibitor of AKT downstream effector mammalian target of rapamycin (mTOR) pathway, can rescue SMAD1/5/8 signaling and AVMs by activating ALK2 in BMP9/10 immunodepleted mice (Ruiz et al., 2020).
Despite the specificities of distinct HHT types, the different mouse models suggest that there is one common mechanism that controls AVM formation. The loss of the BMP/ALK1/SMAD4 signaling pathway in venous ECs leads to an abnormal response to blood flow, including the increase of PI3K/AKT signaling activation and EC proliferation and inhibition of LSS-induced EC polarity and migration against the flow. Then, mutant ECs accumulate in abnormal capillaries, which get enlarged, experience higher blood flow, and acquire venous markers. Whereas arterial ECs might be dispensable, the decrease of NOTCH signaling and arterial marker expression are associated with AVMs. Ultimately, the loss of capillary bed and distinct EC phenotypes forms fragile AVMs (Figure 2). Further studies are required to investigate whether this mechanism is conserved in other tissues and humans.
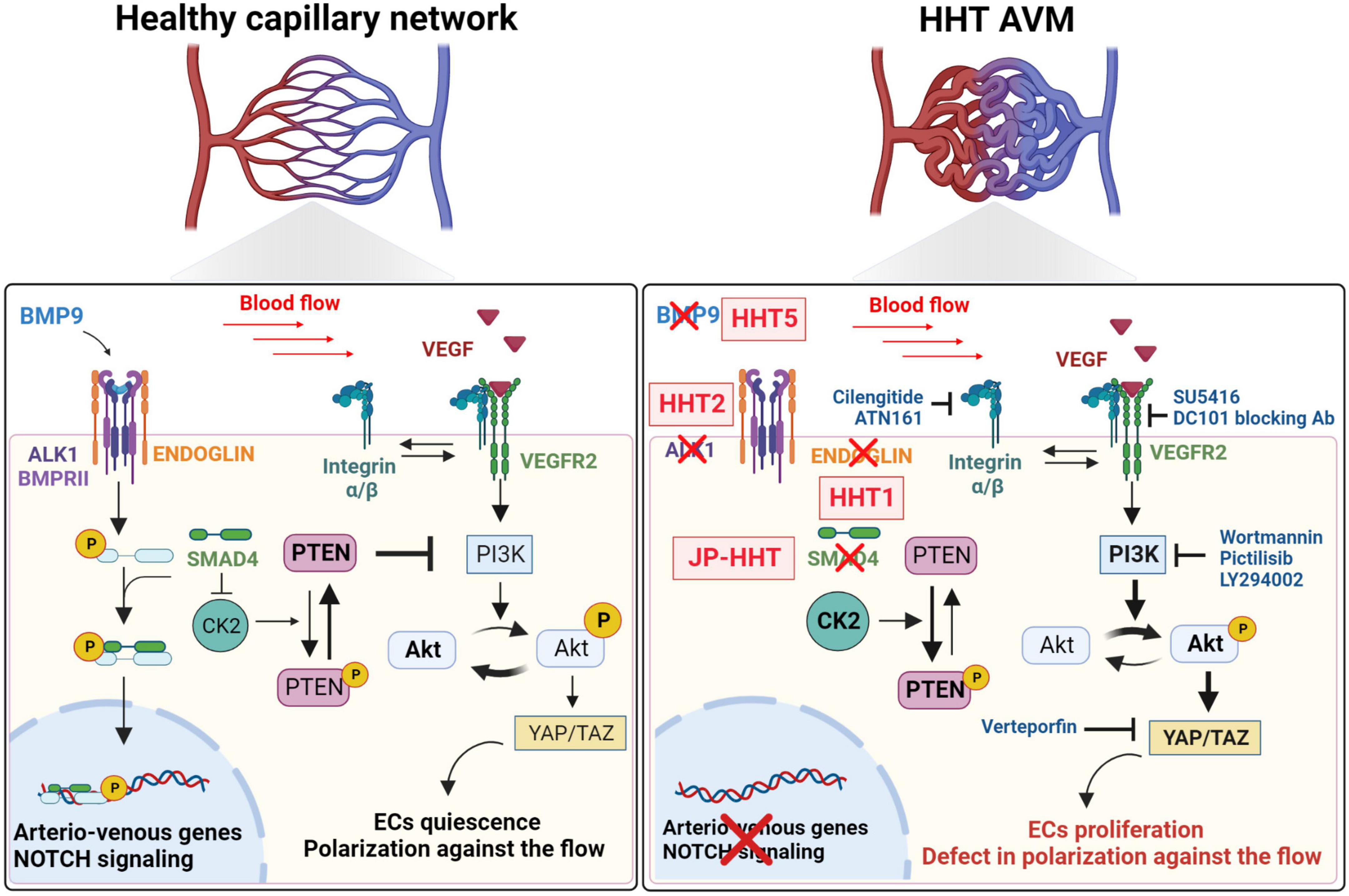
Figure 2. Signaling pathways known to be involved in the pathogenesis of AVMs. In healthy capillary networks, blood flow potentiates BMP/ALK1 signaling pathway in ECs to regulate the expression of genes involved in arterio-venous specification and NOTCH signaling pathway. BMP/ALK1/SMAD4 pathway will inhibit blood flow- and VEGFR2-induced PI3K/AKT signaling. Indeed SMAD4 inhibits the expression of CK2, which allows the phosphorylation, and inhibition of PTEN, a negative regulator of the PI3K/AKT signaling, which will promote ECs quiescence and polarization against the flow. HHT1, HHT2, JP-HHT, and HHT5 are induced by ENG, ACVRL1 (ALK1), SMAD4, and GDF2 (BMP9) heterozygous mutations, respectively. CK2 inhibition is removed and its expression increases leading to an enhanced phosphorylation of PTEN which will not be able to inhibit PI3K/AKT signaling induced by blood flow and VEGFR2. Increased AKT phosphorylation leads to higher YAP/TAZ expression and activity, hyperproliferation of ECs and migration defects against the flow.
The NOTCH pathway is a highly conserved cell signaling system involved in cell fate decisions during several developmental processes, including cardiovascular development and neurogenesis. Mammalian cells possess four specific transmembrane NOTCH receptors, NOTCH1, NOTCH2, NOTCH3, and NOTCH4 (Gordon et al., 2008). NOTCH ligands are also transmembrane proteins that are members of the Delta-like and Jagged families in mammals: delta-like-1 (DLL1), DLL3, and DLL4, JAGGED-1, and JAGGED-2 (D’Souza et al., 2008). Upon activation by their specific ligands, NOTCH receptors undergo proteolytic processing, releasing the NOTCH intracellular domain (NICD), which translocates to the nucleus, where it can form a complex with the DNA-binding protein RBP-J to initiate transcription of its downstream targets, such as members of the Hairy and enhancer-of-split (HES), and Hairy and enhancer-of-split-related (HEY, HESR, HRT, or CHF) gene families (Castel et al., 2013). NOTCH signaling plays a critical role during vascular morphogenesis, as demonstrated by several studies in which deletion of genes coding for NOTCH receptors, ligands, or downstream effectors results in severe angiogenic defects (Swiatek et al., 1994; Hamada et al., 1999; Xue et al., 1999; Krebs et al., 2000; Domenga et al., 2004; Duarte et al., 2004). Specifically loss of NOTCH signaling has been associated with impaired tip/stalk EC specification in several in vitro and animal models (Noguera-Troise et al., 2006; Hellström et al., 2007; Leslie et al., 2007; Suchting et al., 2007). In addition to its roles in sprouting angiogenesis, NOTCH signaling has also been implicated in the specification of artery/vein EC. Indeed, studies have shown that NOTCH signaling induces the expression of several arterial markers and can suppress the expression of venous markers in developing blood vessels (Lawson et al., 2001; Iso et al., 2006; Trindade et al., 2008). As such, both gain-of-function and loss-of-function in NOTCH have resulted in abnormal arterial and venous specification (Lawson et al., 2001; Duarte et al., 2004; Trindade et al., 2008).
The abnormal arteriovenous specification has in several cases been associated with the development of arteriovenous shunts, suggesting a role for NOTCH signaling in the pathophysiology of AVMs. Interestingly, elevated NOTCH signaling has been found in human patients with idiopathic AVMs (ZhuGe et al., 2009, 2013; Yao et al., 2013; Li et al., 2014). Moreover, AVMs have been found in both Notch gain- and loss-of-function animal models, indicating that NOTCH signaling must be tightly regulated in ECs to regulate proper vascular morphogenesis. In mice, the expression of a constitutively active NOTCH4 results in AVMs in the liver, uterus, skin, and brain (Murphy et al., 2008, 2014). Similarly, EC-specific, constitutively active NOTCH1 also results in AVM formation during embryonic development, suggesting that increased activity of either Notch receptor is sufficient to cause brain AVMs (Krebs et al., 2010). DLL4 overexpression models have also been shown to be prone to AVM formation (Trindade et al., 2008). Conversely, decreased NOTCH signaling has also been associated with the development of AVM lesions. In zebrafish embryos, reduction of NOTCH signaling results in the formation of AVMs (Lawson et al., 2001), while in mouse embryos, both Dll4 ± and Rbpj null embryos exhibit the presence of AVMs (Krebs et al., 2004). Endothelial-specific deletion of Rbpj from birth also results in abnormal AV shunting and tortuous vessels in the brain, intestine, and heart of P14 mice (Nielsen et al., 2014).
Several studies have documented the crosstalk between endothelial BMP/ALK1/SMAD and NOTCH signaling pathways during vascular development (Larrivée et al., 2012; Moya et al., 2012; Ricard et al., 2012). Functionally, it was observed that BMP9 stimulation could counteract the effects of NOTCH inhibition using in vitro angiogenesis assays as well as mouse models of vascular development (Larrivée et al., 2012). It was also demonstrated that NOTCH target genes are decreased in Eng-deficient cells, but that ALK1 overexpression could rescue their expression confirming the regulatory effects of ALK1 on NOTCH signaling (Hwan Kim et al., 2020). Interestingly, activated SMADs can co-immunoprecipitate with NICD to potentiate HEY1 expression, suggesting a direct interaction between components of these signaling pathways (Itoh et al., 2004). Finally, similarly to flow-dependent activation of ALK1 (Baeyens et al., 2016), LSS at an arterial magnitude also activates NOTCH signaling to control arterial specification and ECs quiescence (Fang et al., 2017). These insights suggest that mechanical forces may also contribute to the crosstalk between ALK1 and NOTCH signaling. Taken together, these studies demonstrate that ALK1/BMP/SMAD signaling is an important modulator of NOTCH signaling in ECs.
In contrast to several models of idiopathic AVMs, which display increased NOTCH signaling, AVMs in HHT models tend to display decreased NOTCH signaling. In Acvrl1 knockout mouse models, the development of AVMs has been associated with reduced Notch1 and Jag1 expression (Tual-Chalot et al., 2014). Other components of BMP signaling, which directly affect ALK1 signaling and lead to AVM formation, have also been shown to have decreased Notch signaling. For example, deletion of Mgp, which can act as a regulatory protein for BMPs, causes AVM formation in multiple organs partly by modulating Acvrl1 expression. Mgp depletion in ECs upregulated Acvrl1 expression and, in turn, increased NOTCH signaling in an ALK1-dependent manner (Yao et al., 2011, 2013). On the other hand, other studies did not report alterations in NOTCH signaling following impairment of ALK1 or ENG signaling. In zebrafish, Rochon et al. (2015) showed that AVMs in acvrl1 mutants arose independently of perturbations in NOTCH signaling, suggesting that it may not be critical for AVM formation in this context. Therefore, the role of NOTCH signaling in HHT pathophysiology still remains unclear, and further evaluations of NOTCH signaling components and downstream targets in AVMs from HHT patients will be needed to offer additional insights into its role in the pathogenesis of these lesions.
bAVMs found in HHT patients are a familial form with well identified mutations. However, most cases of bAVMs are sporadic, involving somatic mutations mainly associated with RAS signaling, and mechanisms are not well understood.
The MAPK/ERK signaling pathway is also involved in controlling EC proliferation and arteriovenous specification during development. Growth factor receptors, such as EGFR2 or VEGFR2, activate proto-oncogene Rat sarcoma virus proteins (RAS; HRAS, KRAS, and NRAS in humans), which triggers the downstream MAPK/ERK chain, including Rapidly Accelerated Fibrosarcoma (RAF) and Mitogen-activated protein kinase kinase (MEK). Finally, the activated ERK will translocate into the nucleus to regulate the expression of genes involved in cell proliferation (Karar and Maity, 2011; Tan et al., 2013; Simons et al., 2016).
Somatic mutations in the MAPK pathway lead to AVMs formation, such as activating mutations of KRAS, BRAF, or MAPK1/2. Moreover, loss-of-function mutations of RASA1, a negative regulator of RAS signaling, are associated with capillary-malformation arteriovenous malformation (CM-AVM1) and bAVMs in humans (Eerola et al., 2003; Revencu et al., 2008). These AVMs are all characterized by increased ERK phosphorylation and cell proliferation (Eerola et al., 2003; Boon et al., 2005; Couto et al., 2017; Hong et al., 2019).
In mice, overexpression of KRAS in brain ECs specifically leads to the formation of bAVMs, confirming the importance of ECs in the pathogenesis of AVMs (Fish et al., 2020; Park H. et al., 2021). Furthermore, overexpression of KRAS in zebrafish embryos leads to AVMs and increased activity of proteins involved in angiogenesis, such as DLL4 (Al-Olabi et al., 2018; Fish et al., 2020). Interestingly, overexpression of KRAS in mouse cerebral ECs results in overexpression of VEGFR2 expression (Park E. S. et al., 2021). Surprisingly, while Rasa1 loss of function mice exhibit vascular and lymphatic defects, they did not feature bAVMs (Henkemeyer et al., 1995; Lapinski et al., 2012, 2017; Lubeck et al., 2014; Chen et al., 2019). MEK inhibition decreases the number of shunts induced by KRAS overexpression in brain ECs. Interestingly, inhibition of the PI3K/AKT pathway has no effect (Fish et al., 2020). Therefore, while EC proliferation is a common feature with HHT AVM, mechanisms might differ regarding EC polarity and migration against the flow.
The various HHT models developed in recent years have demonstrated the importance of the endothelial BMP/ALK1 pathway in the pathogenesis of AVMs. However, PCs are also essential for vascular integrity and morphogenesis (Armulik et al., 2011). To control capillary coverage by PCs, ECs express soluble Platelet-derived growth factor B (PDGFB), which activates the PDGF receptor (PDGFR) at the PC membrane. PDGFB deletion induces PC loss and micro aneurysms (Lindahl et al., 1997; Hellström et al., 2001; Lindblom et al., 2003). While AVMs are associated with decreased PC coverage in mice and humans (Chen W. et al., 2013; Winkler et al., 2018), the PC contribution to HHT AVMs remains unclear.
It has been shown that following Acvrl1 deletion, there was a decrease in the expression of PDGFRB, but not PDGFB, which could explain the defect in PC recruitment (Chen W. et al., 2013). Thalidomide had already been shown to inhibit angiogenesis. The injection of thalidomide, a drug that reduced the severity and frequency of epistaxis in HHT patients, into mice depleted for Eng prevented AVMs-induced bleeding and excessive angiogenesis. Interestingly, it also restores PC coverage by increasing PDGFRB levels (Lebrin et al., 2010; Zhu et al., 2018).
These data suggest that PCs may be involved in the onset or progression of AVMs. Recently, it was shown that a PC-specific deletion of Rbpj was sufficient to induce the formation of retinal AVMs. Interestingly, the deletion of Rbpj in SMC leads to the formation of AVMs in the retina (Nadeem et al., 2020). It was also reported that Notch1 ±; Notch3-/- mice developed AVMs in the retina (Kofler et al., 2015). Notch3 deficiency in this model compromised PC function, which in turn exacerbated EC activation caused by Notch1 haploinsufficiency.
vSMCs are cells found in arteries and veins, and this coverage is also reduced in bAVMs. However, their involvement in the formation of AVMs is controversial. Indeed, in 2014 a study compared the effect of Acvrl1 and Eng deletion in ECs and SMCs using Scl-CreER and Myh11-CreER driver mice (Table 1). The authors showed that only endothelial deletion allowed AVMs formation in a model of injury-induced skin AVMs (Garrido-Martin et al., 2014). However, a recent study showed that deletion of Acvrl1 in SMCs using Tagln-Cre mice induced the formation of bAVMs associated with a better survival of the mice that allows the monitoring of bAVMs formation over time, and to have a pathological model more representative of the patients. However, Tagln-CRE can be expressed by some ECs which could be sufficient to induce AVMs. It is therefore possible that Acvrl1 mosaic deletion in ECs would be the primary cause of these bAVMs over time rather than the deletion in SMCs (Han et al., 2021). Further studies are needed to understand the exact role of SMCs and PCs in developing bAVMs.
Currently, treatment options for bAVMs are limited and only based on surgeries: surgical resection, endovascular embolization, and/or stereotactic radiosurgery. Knowing the risk of intracranial hemorrhage (ICH) (which varies from 0.9 to 34.3%), the treatment of unruptured bAVMs has become controversial, as untreated patients’ fate may be less morbid than those treated with invasive therapies. Thus, the demand for pharmacologic treatment options is high (Chen W. et al., 2014; Shaligram et al., 2019).
Because of the enhanced angiogenesis associated with bAVMs formation, the most common pharmacologic target is VEGF. The anti-VEGF drug bevacizumab is currently undergoing phase III clinical trial for HHT (NCT 03227263) after showing promising effects on other HHT symptoms such as epistaxis and gastrointestinal bleedings in phase II clinical trials and case reports (Oosting et al., 2009), without showing adverse effects. Altogether, these results suggest that anti-VEGF therapies could represent good alternatives or complements to surgical procedures (Chen W. et al., 2014; Epperla and Hocking, 2015; Snodgrass et al., 2021).
Elucidation of the signaling events associated with HHT has confirmed the contribution of inadequate BMP9/ALK1/SMAD signaling in the formation of bAVMs, and as such, some research was done using tacrolimus, an anti-inflammatory FDA-approved drug, to evaluate whether it could potentiate the activity of the impaired BMP9/ALK1/SMAD pathway present in HHT patients resulting from haploinsufficiency. Indeed, a preclinical study demonstrated that tacrolimus could reduce the development of retinal AVMs in BMP9/10 immunodepleted mice, and it is currently undergoing a clinical trial to evaluate its ability to reduce epistaxis (NCT 03152019) (Robert et al., 2020; Snodgrass et al., 2021).
A better understanding of the signaling pathways inducing bAVMs opens up opportunities for pharmacological treatments using other drugs such as tyrosine kinase inhibitors. Some case reports already showed an improvement of epistaxis and telangiectasia in mostly HHT1 and two patients with different tyrosine kinase inhibitors (Robert et al., 2020; Snodgrass et al., 2021) targeting the VEGFR2 and the ANGPT2 receptor TIE2, which are overactivated in HHT. Thus, two clinical trials are beginning to test the impact of pazopanib, a selective multi-targeted receptor tyrosine kinase inhibitor, on epistaxis and telangiectasia (NCT03850964/NCT03850964). However, preclinical studies in mouse models showed no improvement in wound-induced skin AVMs, leaving little hope for a potential rescue of bAVMs (Robert et al., 2020; Snodgrass et al., 2021).
Other potential treatments are based on inhibiting the PI3K/AKT/mTOR pathway, which is highly activated downstream of ANGPT2 and VEGF signaling. The most common mTOR inhibitor is sirolimus (rapamycin), which was shown to prevent bleeding and anemia in preclinical mouse models of HHT by controlling VEGFR2 and mTOR overactivation (Robert et al., 2020). One case report and a clinical study suggested that sirolimus could control and rescue HHT-induced bleedings and vascular malformations (Skaro et al., 2006; Dupuis-Girod et al., 2010). Apart from sirolimus, using PI3K inhibitors that are already FDA-approved for chemotherapy could also represent an excellent potential therapeutic avenue.
Altogether, although preclinical and clinical tests on pharmacological treatments are ongoing, the probability of reversing the bAVMs is still weak. Despite advances in research to identify the mechanisms, surgical treatments are still the best option to treat bAVMs. Thus, improving surgical conditions and tools currently remains the optimal avenue to improve the survival of HHT patients (Chen W. et al., 2014; Shaligram et al., 2019; Robert et al., 2020).
Identifying the molecular mechanisms associated with bAVM formation is an essential step toward elucidating the mechanisms of these malformations. Several additional questions, however, remain to be addressed. Are there modifying genes that may explain the clinical variability of bAVMs? How does inadequate ALK1/ENG signaling affect the response of ECs to mechanical stress, and how does this contribute to bAVM formation? Is PI3K/AKT signaling increased in HHT bAVMs, and could it be targeted to improve the malformations? What is the contribution of NOTCH signaling in the formation of bAVMs in HHT patients? In addition to these questions, one significant challenge remains to characterize and understand the common mechanisms underlying idiopathic bAVMs and those observed in HHT patients. The recent identification of several signaling pathways involved in bAVMs formation, as well as the analysis of several experimental animal models, have provided several clues to this goal and could pave the way for developing novel therapies.
ED, TA, BL, and AD wrote the manuscript, collected literature information, and edited the manuscript. All authors reviewed and commented on the manuscript.
This work was supported by the Canadian Institutes of Health Research (project grant PJT-165871 and PJT-183658) and the Vision Health Research Network (project grant RRSV_PP1920) to AD, from Fonds de Recherche en Ophtalmologie de l’Université de Montréal (FROUM) to ED and TA, and from the Canadian Institutes of Health Research (project grant FRN-148525) and Fonds de Recherche du Québec - Santé (FRQS) AMD Research Program (Funding reference #269835) to BL. AD and BL were FRQS Research Scholars.
We apologize to colleagues whose work could not be cited in this review due to space limitations. All figures were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ACVRL1, Activin A receptor like type 1; AVF, Arteriovenous fistulas; AVM, Arteriovenous malformations; bAVM, Brain AVM; BBB, Blood-brain-barrier; BMP, Bone Morphogenetic Protein; CK2, Casein Kinase 2; CNS, Central nervous system; CVM, Capillary vascular malformation; EC, Endothelial cell; ENG, Endoglin; HHT, Hereditary hemorrhagic telangiectasia; JP-HHT, Juvenile polyposis-HHT; LSS, Laminar Shear Stress; NVU, Neurovascular unit; PC, Pericyte; PDGF, Platelet-derived growth factor B; PDGFR, Platelet-derived growth factor B receptor; PNVP, Perineural vascular plexus; SMC, Smooth muscle cells; TGFβ, Transforming growth factor β; VEGF, Vascular endothelial growth factor; VEGFR, Vascular endothelial growth factor receptor.
Abecassis, I. J., Xu, D. S., Batjer, H. H., and Bendok, B. R. (2014). Natural history of brain arteriovenous malformations: A systematic review. Neurosurg. Focus 37:E7. doi: 10.3171/2014.6.FOCUS14250
Adams, R. H., and Alitalo, K. (2007). Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 8, 464–478. doi: 10.1038/nrm2183
Akimoto, S., Mitsumata, M., Sasaguri, T., and Yoshida, Y. (2000). Laminar shear stress inhibits vascular endothelial cell proliferation by inducing cyclin-dependent kinase inhibitor p21Sdi1/Cip1/Waf1. Circ. Res. 86, 185–90. doi: 10.1161/01.RES.86.2.185
Allinson, K. R., Carvalho, R. L., van den Brink, S., Mummery, C. L., and Arthur, H. M. (2007). Generation of a floxed allele of the mouse endoglin gene. Genesis 45, 391–95. doi: 10.1002/dvg.20284
Al-Olabi, L., Polubothu, S., Dowsett, K., Andrews, K. A., Stadnik, P., Joseph, A. P., et al. (2018). Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J. Clin. Investig. 128, 1496–1508. doi: 10.1172/JCI98589
Alsina-Sanchís, E., García-Ibáñez, Y., Figueiredo, A. M., Riera-Domingo, C., Figueras, A., Matias-Guiu, X., et al. (2018). ALK1 loss results in vascular hyperplasia in mice and humans through PI3K activation. Arterioscler. Thromb. Vasc. Biol. 38, 1216–29. doi: 10.1161/ATVBAHA.118.310760
Alt, A., Miguel-Romero, L., Donderis, J., Aristorena, M., Blanco, F. J., and Round, A. (2012). Structural and functional insights into endoglin ligand recognition and binding. PLoS One 7:12. doi: 10.1371/journal.pone.0029948
Armulik, A., Genové, G., and Betsholtz, C. (2011). Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 21, 193–215. doi: 10.1016/j.devcel.2011.07.001
Arthur, H. M., Ure, J., Smith, A. J., Renforth, G., Wilson, D. I., Torsney, E., et al. (2000). Endoglin, an ancillary TGFβ receptor, Is required for extraembryonic angiogenesis and plays a key role in heart development. Dev. Biol. 217, 42–53. doi: 10.1006/dbio.1999.9534
Baeyens, N., Larrivée, B., Ola, R., Hayward-Piatkowskyi, B., Dubrac, A., Huang, B., et al. (2016). Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J. Cell Biol. 214, 807–16. doi: 10.1083/jcb.201603106
Bautch, V. L., and James, J. M. (2009). Neurovascular development. Cell Adhes. Migr. 3, 199–204. doi: 10.4161/cam.3.2.8397
Bayrak-Toydemir, P., McDonald, J., Markewitz, B., Lewin, S., Miller, F., Chou, L. S., et al. (2006). Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: Mutations and manifestations. Am. J. Med. Genet. Part A 140, 463–70. doi: 10.1002/ajmg.a.31101
Benn, A., Alonso, F., Mangelschots, J., Génot, E., Lox, M., and Zwijsen, A. (2020). BMP-SMAD1/5 Signaling regulates retinal vascular development. Biomolecules 10:488. doi: 10.3390/biom10030488
Bernabeu, M. O., Jones, M. L., Nielsen, J. H., Krüger, T., Nash, R. W., Groen, D., et al. (2014). Computer simulations reveal complex distribution of haemodynamic forces in a mouse retina model of angiogenesis. J. R. Soc. Interface 11:20140543. doi: 10.1098/rsif.2014.0543
Bidart, M., Ricard, N., Levet, S., Samson, M., Mallet, C., David, L., et al. (2012). BMP9 Is produced by hepatocytes and circulates mainly in an active mature form complexed to its prodomain. Cell. Mol. Life Sci. 69, 313–24. doi: 10.1007/s00018-011-0751-1
Bideau, A., Plauchu, H., Brunet, G., and Robert, J. (1989). Epidemiological investigation of rendu-osler disease in France: Its geographical distribution and prevalence. Popul. Engl. Sel. 44, 3–22.
Biswas, S., Cottarelli, A., and Agalliu, D. (2020). Neuronal and glial regulation of CNS angiogenesis and barriergenesis. Development 147:dev182279. doi: 10.1242/dev.182279
Boon, L. M., Mulliken, J. B., and Vikkula, M. (2005). RASA1: Variable phenotype with capillary and arteriovenous malformations. Curr. Opin. Genet. Dev. Genet. Dis. 15, 265–69. doi: 10.1016/j.gde.2005.03.004
Bourdeau, A., Dumont, D. J., and Letarte, M. (1999). A murine model of hereditary hemorrhagic telangiectasia. J. Clin. Investig. 104, 1343–51.
Bouvard, C., Tu, L., Rossi, M., Desroches-Castan, A., Berrebeh, N., Helfer, E., et al. (2021). Different cardiovascular and pulmonary phenotypes for single- and double-knock-out mice deficient in BMP9 and BMP10. Cardiovasc. Res. 118, 1805–20. doi: 10.1093/cvr/cvab187
Brinjikji, W., Iyer, V. N., Lanzino, G., Thielen, K. R., and Wood, C. P. (2017). Natural history of brain capillary vascular malformations in hereditary hemorrhagic telangiectasia patients. J. Neurointerv. Surg. 9, 26–28. doi: 10.1136/neurintsurg-2015-012252
Brown, R. D., Wiebers, D. O., Torner, J. C., and O’Fallon, W. M. (1996). Frequency of intracranial hemorrhage as a presenting symptom and subtype analysis: A population-based study of intracranial vascular malformations in Olmsted County, Minnesota. J. Neurosurg. 85, 29–32. doi: 10.3171/jns.1996.85.1.0029
Capasso, T. L., Li, B., Volek, H. J., Khalid, W., Rochon, E. R., Anbalagan, A., et al. (2020). BMP10-mediated ALK1 signaling is continuously required for vascular development and maintenance. Angiogenesis 23, 203–20. doi: 10.1007/s10456-019-09701-0
Castel, D., Mourikis, P., Bartels, S. J., Brinkman, A. B., Tajbakhsh, S., and Stunnenberg, H. G. (2013). Dynamic binding of RBPJ is determined by notch signaling status. Genes Dev. 27, 1059–71. doi: 10.1101/gad.211912.112
Chen, D., Teng, J. M., North, P. E., Lapinski, P. E., and King, P. D. (2019). RASA1-Dependent cellular export of collagen IV controls blood and lymphatic vascular development. J. Clin. Investig. 129, 3545–61. doi: 10.1172/JCI124917
Chen, H., Brady Ridgway, J., Sai, T., Lai, J., Warming, S., Chen, H., et al. (2013). Context-dependent signaling defines roles of BMP9 and BMP10 in embryonic and postnatal development. Proc. Natl. Acad. Sci. U.S.A. 110, 11887–92. doi: 10.1073/pnas.1306074110
Chen, W., Choi, E. J., McDougall, C. M., and Su, H. (2014). Brain arteriovenous malformation modeling, pathogenesis, and novel therapeutic targets. Transl. Stroke Res. 5, 316–29. doi: 10.1007/s12975-014-0343-0
Chen, W., Guo, Y., Walker, E. J., Shen, F., Jun, K., Oh, S. P., et al. (2013). Reduced mural cell coverage and impaired vessel integrity after angiogenic stimulation in the Alk1-deficient brain. Arterioscler. Thromb. Vasc. Biol 33, 305–10. doi: 10.1161/ATVBAHA.112.300485
Chen, Y. G., and Massagué, J. (1999). Smad1 recognition and activation by the ALK1 group of transforming growth factor-β family receptors. J. Biol. Chem. 274, 3672–77. doi: 10.1074/jbc.274.6.3672
Choi, E. J., Chen, W., Jun, K., Arthur, H. M., Young, W. L., and Su, H. (2014). Novel brain arteriovenous malformation mouse models for type 1 hereditary hemorrhagic telangiectasia. PLoS One 9:e88511. doi: 10.1371/journal.pone.0088511
Choi, E. J., Walker, E. J., Shen, F., Oh, S. P., Arthur, H. M., and Young, W. L. (2012). Minimal homozygous endothelial deletion of eng with VEGF stimulation is sufficient to cause cerebrovascular dysplasia in the adult mouse. Cerebrovasc. Dis. 33, 540–47. doi: 10.1159/000337762
Coelho-Santos, V., and Shih, A. Y. (2020). Postnatal development of cerebrovascular structure and the neurogliovascular unit. Wiley Interdiscip. Rev. Dev. Biol. 9:e363. doi: 10.1002/wdev.363
Cole, S. G., Begbie, M. E., Wallace, G. M. F., and Shovlin, C. L. (2005). A new locus for Hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J. Med. Genet. 42, 577–82. doi: 10.1136/jmg.2004.028712
Coon, B. G., Baeyens, N., Han, J., Budatha, M., Ross, T. D., Fang, J. S., et al. (2015). Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J. Cell Biol. 208, 975–86. doi: 10.1083/jcb.201408103
Corti, P., Young, S., Chen, C. Y., Patrick, M. J., Rochon, E. R., and Pekkan, K. (2011). Interaction between Alk1 and blood flow in the development of arteriovenous malformations. Development 138, 1573–82. doi: 10.1242/dev.060467
Couto, J. A., Huang, A. Y., Konczyk, D. J., Goss, J. A., Fishman, S. J., and Mulliken, J. B. (2017). Somatic MAP2K1 mutations are associated with extracranial arteriovenous malformation. Am. J. Hum. Genet. 100, 546–54. doi: 10.1016/j.ajhg.2017.01.018
Crist, A. M., Lee, A. R., Patel, N. R., Westhoff, D. E., and Meadows, S. M. (2018). Vascular deficiency of Smad4 causes arteriovenous malformations: A mouse model of hereditary hemorrhagic telangiectasia. Angiogenesis 21, 363–80. doi: 10.1007/s10456-018-9602-0
Daneman, R., and Prat, A. (2015). The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 7:a020412. doi: 10.1101/cshperspect.a020412
David, L., Mallet, C., Keramidas, M., Lamandé, N., Gasc, J. M., and Dupuis-Girod, S. (2008). Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ. Res. 102, 914–22. doi: 10.1161/CIRCRESAHA.107.165530
David, L., Mallet, C., Mazerbourg, S., Feige, J. J., and Bailly, S. (2006). Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 109, 1953–61. doi: 10.1182/blood-2006-07-034124
Domenga, V., Fardoux, P., Lacombe, P., Monet, M., Maciazek, J., Krebs, L. T., et al. (2004). Notch3 Is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 18, 2730–35. doi: 10.1101/gad.308904
D’Souza, B., Miyamoto, A., and Weinmaster, G. (2008). The many facets of Notch ligands. Oncogene 27, 5148–67. doi: 10.1038/onc.2008.229
Duarte, A., Hirashima, M., Benedito, R., Trindade, A., Diniz, P., and Bekman, E. (2004). Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 18, 2474–78. doi: 10.1101/gad.1239004
Dupuis-Girod, S., Chesnais, A. L., Ginon, I., Dumortier, J., Saurin, J. C., and Finet, G. (2010). Long-term outcome of patients with hereditary hemorrhagic telangiectasia and severe hepatic involvement after orthotopic liver transplantation: A single-center study. Liver Transplant. 16, 340–47. doi: 10.1002/lt.21990
Eerola, I., Boon, L. M., Mulliken, J. B., Burrows, P. E., Dompmartin, A., Watanabe, S., et al. (2003). Capillary malformation–arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am. J. Hum. Genet. 73, 1240–49. doi: 10.1086/379793
Epperla, N., and Hocking, W. (2015). Blessing for the bleeder: Bevacizumab in hereditary hemorrhagic telangiectasia. Clin. Med. Res. 13, 32–35. doi: 10.3121/cmr.2013.1205
Etienne-Manneville, S., and Hall, A. (2001). Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCζ. Cell 106, 489–98. doi: 10.1016/S0092-8674(01)00471-8
Fang, J. S., Coon, B. G., Gillis, N., Chen, Z., Qiu, J., Chittenden, T. W., et al. (2017). Shear-induced notch-Cx37-P27 axis arrests endothelial cell cycle to enable arterial specification. Nat. Commun. 8:2149. doi: 10.1038/s41467-017-01742-7
Fierstra, J., Conklin, J., Krings, T., Slessarev, M., Han, J. S., Fisher, J. A., et al. (2011). Impaired Peri-Nidal Cerebrovascular Reserve in Seizure Patients with Brain Arteriovenous Malformations. Brain 134, 100–109. doi: 10.1093/brain/awq286
Fish, J. E., Flores Suarez, C. P., Boudreau, E., Herman, A. M., Gutierrez, M. C., Gustafson, D., et al. (2020). Somatic gain of KRAS function in the endothelium is sufficient to cause vascular malformations that require MEK but not PI3K signaling. Circ. Res. 127, 727–43. doi: 10.1161/CIRCRESAHA.119.316500
Fleetwood, I. G., and Steinberg, G. K. (2002). Arteriovenous malformations. Lancet 359, 863–73. doi: 10.1016/S0140-6736(02)07946-1
Fonseca, C. G., Barbacena, P., and Franco, C. A. (2020). Endothelial cells on the move: Dynamics in vascular morphogenesis and disease. Vasc. Biol. 2:H29–43. doi: 10.1530/VB-20-0007
Gallione, C. J., Repetto, G. M., Legius, E., Rustgi, A. K., Schelley, S. L., and Tejpar, S. (2004). A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 363, 852–59. doi: 10.1016/S0140-6736(04)15732-2
Gariano, R. F., and Gardner, T. W. (2005). Retinal angiogenesis in development and disease. Nature 438, 960–66. doi: 10.1038/nature04482
Garrido-Martin, E. M., Nguyen, H. L., Cunningham, T. A., Choe, S. W., Jiang, Z., and Arthur, H. M. (2014). Common and distinctive pathogenetic features of arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 and hereditary hemorrhagic telangiectasia 2 animal models—brief report. Arterioscler. Thromb. Vasc. Biol. 34, 2232–36. doi: 10.1161/ATVBAHA.114.303984
Gordon, W. R., Arnett, K. L., and Blacklow, S. C. (2008). The molecular logic of notch signaling – a structural and biochemical perspective. J. Cell Sci. 121, 3109–19. doi: 10.1242/jcs.035683
Graupera, M., and Potente, M. (2013). Regulation of angiogenesis by PI3K signaling networks. Exp. Cell Res. 319, 1348–55. doi: 10.1016/j.yexcr.2013.02.021
Griauzde, J., Wilseck, Z. M., Chaudhary, N., Pandey, A. S., Vercler, C. J., Kasten, S. J., et al. (2020). Endovascular treatment of arteriovenous malformations of the head and neck: Focus on the yakes classification and outcomes. J. Vasc. Int. Radiol. 31, 1810–16. doi: 10.1016/j.jvir.2020.01.036
Gross, B. A., and Du, R. (2013). Natural history of cerebral arteriovenous malformations: A meta-analysis: Clinical article. J. Neurosurg. 118, 437–43. doi: 10.3171/2012.10.JNS121280
Haddad-Tóvolli, R., Dragano, N. R. V., Ramalho, A. F. S., and Velloso, L. A. (2017). Development and function of the blood-brain barrier in the context of metabolic control. Front. Neurosci. 11:224. doi: 10.3389/fnins.2017.00224
Haitjema, T., Disch, F., Overtoom, T. T., Westermann, C. J., and Lammers, J. W. (1995). Screening family members of patients with hereditary hemorrhagic telangiectasia. Am. J. Med. 99, 519–24. doi: 10.1016/S0002-9343(99)80229-0
Hamada, Y., Kadokawa, Y., Okabe, M., Ikawa, M., Coleman, J. R., and Tsujimoto, Y. (1999). Mutation in ankyrin repeats of the mouse notch2 gene induces early embryonic lethality. Development 126, 3415–24. doi: 10.1242/dev.126.15.3415
Han, C., Choe, S.-W., Kim, Y. H., Acharya, A. P., Keselowsky, B. G., Sorg, B. S., et al. (2014). VEGF neutralization can prevent and normalize arteriovenous malformations in an animal model for hereditary hemorrhagic telangiectasia 2. Angiogenesis 17, 823–30. doi: 10.1007/s10456-014-9436-3
Han, C., Lang, M. J., Nguyen, C. L., Melendez, E. L., Mehta, S., Turner, G. H., et al. (2021). Novel experimental model of brain arteriovenous malformations using conditional Alk1 gene deletion in transgenic mice. J. Neurosurg. 137, 163–74. doi: 10.3171/2021.6.JNS21717
Hellström, M., Gerhardt, H., Kalén, M., Li, X., Eriksson, U., Wolburg, H., et al. (2001). Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol. 153, 543–54. doi: 10.1083/jcb.153.3.543
Hellström, M., Phng, L.-K., Hofmann, J. J., Wallgard, E., Coultas, L., Lindblom, P., et al. (2007). Dll4 signalling through notch1 regulates formation of tip cells during angiogenesis. Nature 445, 776–80. doi: 10.1038/nature05571
Henkemeyer, M., Rossi, D. J., Holmyard, D. P., Puri, M. C., Mbamalu, G., Harpal, K., et al. (1995). Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature 377, 695–701. doi: 10.1038/377695a0
Hernandez, F., Huether, R., Carter, L., Johnston, T., Thompson, J., Gossage, J. R., et al. (2015). Mutations in RASA1 and GDF2 identified in patients with clinical features of hereditary hemorrhagic telangiectasia. Hum. Genome Var. 2:15040. doi: 10.1038/hgv.2015.40
Hogan, K. A., Ambler, C. A., Chapman, D. L., and Bautch, V. L. (2004). The neural tube patterns vessels developmentally using the VEGF signaling pathway. Development 131, 1503–13. doi: 10.1242/dev.01039
Hoh, B. L., Chapman, P. H., Loeffler, J. S., Carter, B. S., and Ogilvy, C. S. (2002). Results of multimodality treatment for 141 patients with brain arteriovenous malformations and seizures: Factors associated with seizure incidence and seizure outcomes. Neurosurgery 51, 303–11.
Hong, T., Yan, Y., Li, J., Radovanovic, I., Ma, X., Shao, Y. W., et al. (2019). High prevalence of KRAS/BRAF somatic mutations in brain and spinal cord arteriovenous malformations. Brain 142, 23–34. doi: 10.1093/brain/awy307
Howe, J. R., Roth, S., Ringold, J. C., Summers, R. W., Järvinen, H. J., and Sistonen, P. I., et al. (1998). Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 280, 1086–88. doi: 10.1126/science.280.5366.1086
Hwan Kim, Y., Vu, P. N., Choe, S. W., Jeon, C. J., Arthur, H. M., Vary, C. P. H., et al. (2020). Overexpression of activin receptor-like kinase 1 in endothelial cells suppresses development of arteriovenous malformations in mouse models of hereditary hemorrhagic telangiectasia. Circ. Res. 127, 1122–37. doi: 10.1161/CIRCRESAHA.119.316267
Iso, T., Maeno, T., Oike, Y., Yamazaki, M., Doi, H., Arai, M., et al. (2006). Dll4-selective notch signaling induces EphrinB2 gene expression in endothelial cells. Biochem. Biophys. Res. Commun. 341, 708–14. doi: 10.1016/j.bbrc.2006.01.020
Itoh, F., Itoh, S., Goumans, M.-J., Valdimarsdottir, G., Iso, T., Dotto, G. P., et al. (2004). Synergy and antagonism between Notch and BMP receptor signaling pathways in endothelial cells. EMBO J. 23, 541–51. doi: 10.1038/sj.emboj.7600065
Jin, Y., Muhl, L., Burmakin, M., Wang, Y., Duchez, A. C., Betsholtz, C., et al. (2017). Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat. Cell Biol. 19, 639–52. doi: 10.1038/ncb3534
Johnson, D. W., Berg, J. N., Baldwin, M. A., Gallione, C. J., Marondel, I., Yoon, S.-J., et al. (1996). Mutations in the activin receptor–like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 13, 189–95. doi: 10.1038/ng0696-189
Jonker, L., and Arthur, H. M. (2002). Endoglin expression in early development is associated with vasculogenesis and angiogenesis. Mech. Dev. 110, 193–96. doi: 10.1016/S0925-4773(01)00562-7
Josephson, C. B., Leach, J.-P., Duncan, R., Roberts, R. C., Counsell, C. E., and Al-Shahi Salman, R. (2011). Seizure risk from cavernous or arteriovenous malformations. Neurology 76, 1548–54. doi: 10.1212/WNL.0b013e3182190f37
Kaplan, L., Chow, B. W., and Gu, C. (2020). Neuronal regulation of the blood–brain barrier and neurovascular coupling. Nat. Rev. Neurosci. 21, 416–32. doi: 10.1038/s41583-020-0322-2
Karar, J., and Maity, A. (2011). PI3K/AKT/mTOR pathway in angiogenesis. Front. Mol. Neurosci. 4:51. doi: 10.3389/fnmol.2011.00051
Kjeldsen, A. D., Møller, T. R., Brusgaard, K., Vase, P., and Andersen, P. E. (2005). Clinical symptoms according to genotype amongst patients with hereditary haemorrhagic telangiectasia. J. Intern. Med. 258, 349–55. doi: 10.1111/j.1365-2796.2005.01555.x
Kjeldsen, A. D., Vase, P., and Green, A. (1999). Hereditary haemorrhagic telangiectasia: A population-based study of prevalence and mortality in danish patients. J. Intern. Med. 245, 31–39. doi: 10.1046/j.1365-2796.1999.00398.x
Kofler, N. M., Cuervo, H., Uh, M. K., Murtomäki, A., and Kitajewski, J. (2015). Combined deficiency of Notch1 and Notch3 causes pericyte dysfunction, models CADASIL, and results in arteriovenous malformations. Sci. Rep. 5:16449. doi: 10.1038/srep16449
Komiyama, M., Terada, A., Ishiguro, T., Watanabe, Y., Nakajima, H., and Yamada, O. (2015). Neuroradiological manifestations of hereditary hemorrhagic telangiectasia in 139 Japanese patients. Neurol. Med. Chir. 55, 479–86. doi: 10.2176/nmc.oa.2015-0040
Krebs, L. T., Shutter, J. R., Tanigaki, K., Honjo, T., Stark, K. L., and Gridley, T. (2004). Haploinsufficient lethality and formation of arteriovenous malformations in notch pathway mutants. Genes Dev. 18, 2469–73. doi: 10.1101/gad.1239204
Krebs, L. T., Starling, C., Chervonsky, A. V., and Gridley, T. (2010). Notch1 activation in mice causes arteriovenous malformations phenocopied by ephrinB2 and EphB4 mutants. Genesis 48, 146–50. doi: 10.1002/dvg.20599
Krebs, L. T., Xue, Y., Norton, C. R., Shutter, J. R., Maguire, M., Sundberg, J. P., et al. (2000). Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 14, 1343–52. doi: 10.1101/gad.14.11.1343
Krings, T., Kim, H., Power, S., Nelson, J., Faughnan, M. E., and Young, W. L. (2015). Neurovascular manifestations in hereditary hemorrhagic telangiectasia: Imaging features and genotype-phenotype correlations. Am. J. Neuroradiol. 36, 863–70. doi: 10.3174/ajnr.A4210
Krings, T., Ozanne, A., Chng, S. M., Alvarez, H., Rodesch, G., and Lasjaunias, P. L. (2005). Neurovascular phenotypes in hereditary haemorrhagic telangiectasia patients according to age. Neuroradiology 47, 711–20. doi: 10.1007/s00234-005-1390-8
Lan, Y., Liu, B., Yao, H., Li, F., Weng, T., Yang, G., et al. (2007). Essential role of endothelial Smad4 in vascular remodeling and integrity. Mol. Cell. Biol. 27, 7683–92. doi: 10.1128/MCB.00577-07
Langer, D. J., Lasner, T. M., Hurst, R. W., Flamm, E. S., Zager, E. L., and King, J. T. (1998). Hypertension, small size, and deep venous drainage are associated with risk of hemorrhagic presentation of cerebral arteriovenous malformations. Neurosurgery 42, 481–86. doi: 10.1097/00006123-199803000-00008
Lapinski, P. E., Kwon, S., Lubeck, B. A., Wilkinson, J. E., Srinivasan, R. S., Sevick-Muraca, E., et al. (2012). RASA1 maintains the lymphatic vasculature in a quiescent functional state in mice. J. Clin. Investig. 122, 733–47. doi: 10.1172/JCI46116
Lapinski, P. E., Lubeck, B. A., Chen, D., Doosti, A., Zawieja, S. D., and Davis, M. J. (2017). RASA1 regulates the function of lymphatic vessel valves in mice. J. Clin. Investig. 127, 2569–85. doi: 10.1172/JCI89607
Larrivée, B., Prahst, C., Gordon, E., del Toro, R., Mathivet, T., Duarte, A., et al. (2012). ALK1 signaling inhibits angiogenesis by cooperating with the notch pathway. Dev. Cell 22, 489–500. doi: 10.1016/j.devcel.2012.02.005
Laux, D. W., Young, S., Donovan, J. P., Mansfield, C. J., Upton, P. D., and Roman, B. L. (2013). Circulating Bmp10 Acts through endothelial Alk1 to mediate flow-dependent arterial quiescence. Development 140, 3403–12. doi: 10.1242/dev.095307
Lawson, N. D., Scheer, N., Pham, V. N., Kim, C. H., Chitnis, A. B., and Campos-Ortega, J. A. (2001). Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 128, 3675–83. doi: 10.1242/dev.128.19.3675
Lebrin, F., Srun, S., Raymond, K., Martin, S., van den Brink, S., Freitas, C., et al. (2010). Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat. Med. 16, 420–28. doi: 10.1038/nm.2131
Lee, H.-W., Xu, Y., He, L., Choi, W., Gonzalez, D., Jin, S.-W., et al. (2021). Role of venous endothelial cells in developmental and pathologic angiogenesis. Circulation 144, 1308–22. doi: 10.1161/CIRCULATIONAHA.121.054071
Leslie, J. D., Ariza-McNaughton, L., Bermange, A. L., McAdow, R., Johnson, S. L., and Lewis, J. (2007). Endothelial signalling by the notch ligand delta-like 4 restricts angiogenesis. Development 134, 839–44. doi: 10.1242/dev.003244
Li, D. Y., Sorensen, L. K., Brooke, B. S., Urness, L. D., Davis, E. C., Taylor, D. G., et al. (1999). Defective angiogenesis in mice lacking endoglin. Science 284, 1534–37. doi: 10.1126/science.284.5419.1534
Li, F., Lan, Y., Wang, Y., Wang, J., Yang, G., Meng, F., et al. (2011). Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with notch. Dev. Cell 20, 291–302. doi: 10.1016/j.devcel.2011.01.011
Li, S., Wang, R., Wang, Y., Li, H., Zheng, J., Duan, R., et al. (2014). Receptors of the notch signaling pathway are associated with hemorrhage of brain arteriovenous malformations. Mol. Med. Rep. 9, 2233–38. doi: 10.3892/mmr.2014.2061
Lindahl, P., Johansson, B. R., Levéen, P., and Betsholtz, C. (1997). Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 277, 242–45. doi: 10.1126/science.277.5323.242
Lindblom, P., Gerhardt, H., Liebner, S., Abramsson, A., Enge, M., Hellstrom, M., et al. (2003). Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 17, 1835–40. doi: 10.1101/gad.266803
López-Coviella, I., Berse, B., Krauss, R., Thies, R. S., and Blusztajn, J. K. (2000). Induction and maintenance of the neuronal cholinergic phenotype in the central nervous system by BMP-9. Science 289, 313–16. doi: 10.1126/science.289.5477.313
Lubeck, B. A., Lapinski, P. E., Bauler, T. J., Oliver, J. A., Hughes, E. D., Saunders, T. L., et al. (2014). Blood vascular abnormalities in Rasa1R780Q knockin mice: Implications for the pathogenesis of capillary malformation–arteriovenous malformation. Am. J. Pathol. 184, 3163–69. doi: 10.1016/j.ajpath.2014.08.018
Mahmoud, M., Allinson, K. R., Zhai, Z., Oakenfull, R., Ghandi, P., Adams, R. H., et al. (2010). Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ. Res. 106, 1425–33. doi: 10.1161/CIRCRESAHA.109.211037
Matsubara, S., Mandzia, J. L., ter Brugge, K., Willinsky, R. A., and Faughnan, M. E. (2000). Angiographic and clinical characteristics of patients with cerebral arteriovenous malformations associated with hereditary hemorrhagic telangiectasia. AJNR 21, 1016–20.
McAllister, K. A., Grogg, K. M., Johnson, D. W., Gallione, C. J., Baldwin, M. A., Jackson, C. E., et al. (1994). Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 8, 345–51. doi: 10.1038/ng1294-345
Miller, D. W., Graulich, W., Karges, B., Stahl, S., Ernst, M., Ramaswamy, A., et al. (1999). Elevated expression of endoglin, a component of the TGF-β-receptor complex, correlates with proliferation of tumor endothelial cells. Int. J. Cancer 81, 568–72.
Mohr, J. P., Kejda-Scharler, J., and Spellman, J. P. - (2013). Diagnosis and treatment of arteriovenous malformations. Curr. Neurol. Neurosci. Rep. 13:324. doi: 10.1007/s11910-012-0324-1
Moya, I. M., Umans, L., Maas, E., Pereira, P. N. G., Beets, K., Francis, A., et al. (2012). Stalk cell phenotype depends on integration of notch and Smad1/5 signaling cascades. Dev. Cell 22, 501–14. doi: 10.1016/j.devcel.2012.01.007
Murphy, P. A., Kim, T. N., Huang, L., Nielsen, C. M., Lawton, M. T., Adams, R. H., et al. (2014). Constitutively active Notch4 receptor elicits brain arteriovenous malformations through enlargement of capillary-like vessels. Proc. Natl. Acad. Sci. U.S.A. 111, 18007–12. doi: 10.1073/pnas.1415316111
Murphy, P. A., Lam, M. T. Y., Wu, X., Kim, T. N., Vartanian, S. M., Bollen, A. W., et al. (2008). Endothelial Notch4 signaling induces hallmarks of brain arteriovenous malformations in mice. Proc. Natl. Acad. Sci. U.S.A. 105, 10901–6. doi: 10.1073/pnas.0802743105
Nadeem, T., Bogue, W., Bigit, B., and Cuervo, H. (2020). Deficiency of notch signaling in pericytes results in arteriovenous malformations. JCI Insight 5:e125940. doi: 10.1172/jci.insight.125940
Nakao, A., Imamura, T., Souchelnytskyi, S., Kawabata, M., Ishisaki, A., Oeda, E., et al. (1997). TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 16, 5353–62. doi: 10.1093/emboj/16.17.5353
Neuhaus, H., Rosen, V., and Thies, R. S. (1999). Heart specific expression of mouse BMP-10 a novel member of the TGF-beta superfamily. Mech. Dev. 80, 181–84. doi: 10.1016/s0925-4773(98)00221-4
Nielsen, C. M., Cuervo, H., Ding, V. W., Kong, Y., Huang, E. J., and Wang, R. A. (2014). Deletion of Rbpj from postnatal endothelium leads to abnormal arteriovenous shunting in mice. Development 141, 3782–92. doi: 10.1242/dev.108951
Noguera-Troise, I., Daly, C., Papadopoulos, N. J., Coetzee, S., Boland, P., Gale, N. W., et al. (2006). Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444, 1032–37. doi: 10.1038/nature05355
Oh, S. P., Seki, T., Goss, K. A., Imamura, T., Yi, Y., Donahoe, P. K., et al. (2000). Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 97, 2626–31. doi: 10.1073/pnas.97.6.2626
Ola, R., Dubrac, A., Han, J., Zhang, F., Fang, J. S., Larrivée, B., et al. (2016). PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat. Commun. 7:13650. doi: 10.1038/ncomms13650
Ola, R., Künzel, S. H., Zhang, F., Genet, G., Chakraborty, R., Pibouin-Fragner, L., et al. (2018). SMAD4 prevents flow induced arteriovenous malformations by inhibiting casein kinase 2. Circulation 138, 2379–94. doi: 10.1161/CIRCULATIONAHA.118.033842
Oosting, S., Nagengast, W., and de Vries, E. (2009). More on bevacizumab in hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 361:931. doi: 10.1056/NEJMc091271
Ouarné, M., Pena, A., and Franco, C. A. (2021). From remodeling to quiescence: The transformation of the vascular network. Cells Dev. Quan. Cell Dev. Biol. 168:203735. doi: 10.1016/j.cdev.2021.203735
Paredes, I., Himmels, P., and Ruiz de Almodóvar, C. (2018). Neurovascular communication during CNS development. Dev. Cell 45, 10–32. doi: 10.1016/j.devcel.2018.01.023
Park, E. S., Kim, S., Huang, S., Yoo, J. Y., Körbelin, J., Lee, T. J., et al. (2021). Selective endothelial hyperactivation of oncogenic KRAS induces brain arteriovenous malformations in mice. Ann. Neurol. 89, 926–41. doi: 10.1002/ana.26059
Park, H., Furtado, J., Poulet, M., Chung, M., Yun, S., Lee, S., et al. (2021). Defective flow-migration coupling causes arteriovenous malformations in hereditary hemorrhagic telangiectasia. Circulation 144, 805–22. doi: 10.1161/CIRCULATIONAHA.120.053047
Park, S., DiMaio, T. A., Liu, W., Wang, S., Sorenson, C. M., and Sheibani, N. (2013). Endoglin regulates the activation and quiescence of endothelium by participating in canonical and non-canonical TGF-β signaling pathways. J. Cell Sci. 126, 1392–1405. doi: 10.1242/jcs.117275
Park, S., Ok, M., Wankhede, Y. J., Lee, E.-J., Choi, N., Fliess, S.-W., et al. (2009). Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J. Clin. Investig. 119, 3487–96. doi: 10.1172/JCI39482
Park, S. O., Lee, Y. J., Seki, T., Hong, K. H., Fliess, N., Jiang, Z., et al. (2008). ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood 111, 633–42. doi: 10.1182/blood-2007-08-107359
Parmar, K. M., Larman, H. B., Dai, G., Zhang, Y., Wang, E. T., Moorthy, S. N., et al. (2006). Integration of flow-dependent endothelial phenotypes by kruppel-like factor 2. J. Clin. Investig. 116, 49–58. doi: 10.1172/JCI24787
Peacock, H. M., Tabibian, A., Criem, N., Caolo, V., Hamard, L., Deryckere, A., et al. (2020). Impaired SMAD1/5 mechanotransduction and Cx37 (Connexin37) expression enable pathological vessel enlargement and shunting. Arterioscler. Thromb. Vasc. Biol. 40:e87–104. doi: 10.1161/ATVBAHA.119.313122
Peguera, B., Segarra, M., and Acker-Palmer, A. (2021). Neurovascular crosstalk coordinates the central nervous system development. Curr. Opin. Neurobiol. Mol. Neurosci. 69, 202–13. doi: 10.1016/j.conb.2021.04.005
Pitulescu, M. E., Schmidt, I., Giaimo, B. D., Antoine, T., Berkenfeld, F., Ferrante, F., et al. (2017). Dll4 and notch signalling couples sprouting angiogenesis and artery formation. Nat. Cell Biol. 19, 915–27. doi: 10.1038/ncb3555
Prigoda, N. L., Savas, S., Abdalla, S. A., Piovesan, B., Rushlow, D., Vandezande, K., et al. (2006). Hereditary haemorrhagic telangiectasia: Mutation detection, test sensitivity and novel mutations. J. Med. Genet. 43, 722–28. doi: 10.1136/jmg.2006.042606
Revencu, N., Boon, L. M., Mulliken, J. B., Enjolras, O., Cordisco, M. R., Burrows, P. E., et al. (2008). Parkes weber syndrome, vein of galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum. Mutat. 29, 959–65. doi: 10.1002/humu.20746
Ricard, N., Ciais, D., Levet, S., Subileau, M., Mallet, C., Zimmers, T. A., et al. (2012). BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood 119, 6162–71. doi: 10.1182/blood-2012-01-407593
Robert, F., D-Castan, A., Bailly, S., Girod, S. D., and Feige, J.-J. (2020). Future treatments for hereditary hemorrhagic telangiectasia. Orphanet J. Rare Dis. 15:4. doi: 10.1186/s13023-019-1281-4
Rochon, E. R., Menon, P. G., and Roman, B. L. (2016). Alk1 controls arterial endothelial cell migration in lumenized vessels. Development 143, 2593–2602. doi: 10.1242/dev.135392
Rochon, E. R., Wright, D. S., Schubert, M. M., and Roman, B. L. (2015). Context-specific interactions between notch and ALK1 cannot explain ALK1-associated arteriovenous malformations. Cardiovasc. Res. 107, 143–52. doi: 10.1093/cvr/cvv148
Rohn, B., Haenggi, D., Etminan, N., Kunz, M., Turowski, B., and Steiger, H.-J. (2014). Epilepsy, headache, and quality of life after resection of cerebral arteriovenous malformations. J. Neurol. Surg. Part A 75, 282–88. doi: 10.1055/s-0033-1358611
Roman, B. L., and Hinck, A. P. (2017). ALK1 signaling in development and disease: New paradigms. Cell. Mol. Life Sci. 74, 4539–60. doi: 10.1007/s00018-017-2636-4
Roman, B. L., Pham, V. N., Lawson, N. D., Kulik, M., Childs, S., Lekven, A. C., et al. (2002). Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels. Development 129, 3009–19. doi: 10.1242/dev.129.12.3009
Ruiz, S., Zhao, H., Chandakkar, P., Chatterjee, P. K., Papoin, J., Blanc, L., et al. (2016). A mouse model of hereditary hemorrhagic telangiectasia generated by transmammary-delivered immunoblocking of BMP9 and BMP10. Sci. Rep. 5:37366. doi: 10.1038/srep37366
Ruiz, S., Zhao, H., Chandakkar, P., Papoin, J., Choi, H., Kitabayashi, A. N., et al. (2020). Correcting Smad1/5/8, MTOR, and VEGFR2 treats pathology in hereditary hemorrhagic telangiectasia models. J. Clin. Investig. 130, 942–57. doi: 10.1172/JCI127425
Saleh, M., Carter, M. T., Latino, G. A., Dirks, P., and Ratjen, F. (2013). Brain arteriovenous malformations in patients with hereditary hemorrhagic telangiectasia: Clinical presentation and anatomical distribution. Pediatr. Neurol. 49, 445–50. doi: 10.1016/j.pediatrneurol.2013.07.021
Schuermann, A., Helker, C. S. M., and Herzog, W. (2014). Angiogenesis in zebrafish. Semin. Cell Dev. Biol. 31, 106–14. doi: 10.1016/j.semcdb.2014.04.037
Seki, T., Yun, J., and Oh, S. P. (2003). Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circ. Res. 93, 682–89. doi: 10.1161/01.RES.0000095246.40391.3B
Serini, G., Napione, L., Arese, M., and Bussolino, F. (2008). Besides adhesion: New perspectives of integrin functions in angiogenesis. Cardiovasc. Res. 78, 213–22. doi: 10.1093/cvr/cvn045
Serra, H., Chivite, I., A-Urarte, A., Soler, A., Sutherland, J. D., A-Aristorena, A., et al. (2015). PTEN mediates notch-dependent stalk cell arrest in angiogenesis. Nat. Commun. 6:7935. doi: 10.1038/ncomms8935
Shaligram, S. S., Winkler, E., Cooke, D., and Su, H. (2019). Risk factors for hemorrhage of brain arteriovenous malformation. CNS Neurosci. Ther. 25, 1085–95. doi: 10.1111/cns.13200
Shovlin, C. L. (2010). Hereditary haemorrhagic telangiectasia: Pathophysiology, diagnosis and treatment. Blood Rev. 24, 203–19. doi: 10.1016/j.blre.2010.07.001
Siegenthaler, J. A., Sohet, F., and Daneman, R. (2013). ‘Sealing off the CNS’: Cellular and molecular regulation of blood–brain barriergenesis. Curr. Opin. Neurobiol. 23, 1057–64. doi: 10.1016/j.conb.2013.06.006
Simons, M., Gordon, E., and Welsh, L. C. (2016). Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 17, 611–25. doi: 10.1038/nrm.2016.87
Singh, E., Redgrave, R. E., Phillips, H. M., and Arthur, H. M. (2020). Arterial endoglin does not protect against arteriovenous malformations. Angiogenesis 23, 559–66. doi: 10.1007/s10456-020-09731-z
Skaro, A. I., Marotta, P. J., and McAlister, V. C. (2006). Regression of cutaneous and gastrointestinal telangiectasia with sirolimus and aspirin in a patient with hereditary hemorrhagic telangiectasia. Ann. Intern. Med. 144, 226–27. doi: 10.7326/0003-4819-144-3-200602070-00030
Snodgrass, R. O., Chico, T. J. A., and Arthur, H. M. (2021). Hereditary haemorrhagic telangiectasia, an inherited vascular disorder in need of improved evidence-based pharmaceutical interventions. Genes 12:174.
Somi, S., Buffing, A. A. M., Moorman, A. F. M., and Van Den Hoff, M. J. B. (2004). Expression of bone morphogenetic protein-10 MRNA during chicken heart development. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 279, 579–82. doi: 10.1002/ar.a.20052
Srinivasan, S., Hanes, M. A., Dickens, T., Porteous, M. E. M., Oh, S. P., Hale, L. P., et al. (2003). A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum. Mol. Genet. 12, 473–82. doi: 10.1093/hmg/ddg050
Suchting, S., Freitas, C., Noble, F. L., Benedito, R., Bréant, C., Duarte, A., et al. (2007). The notch ligand delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc. Natl. Acad. Sci. U.S.A. 104, 3225–30. doi: 10.1073/pnas.0611177104
Sugden, W. W., Meissner, R., Wilmsen, T. A., Tsaryk, R., Leonard, E. V., Bussmann, J., et al. (2017). Endoglin controls blood vessel diameter through endothelial cell shape changes in response to haemodynamic cues. Nat. Cell Biol. 19, 653–65. doi: 10.1038/ncb3528
Swiatek, P. J., Lindsell, C. E., del Amo, F. F., Weinmaster, G., and Gridley, T. (1994). Notch1 Is essential for postimplantation development in mice. Genes Dev. 8, 707–19. doi: 10.1101/gad.8.6.707
Tan, W. H., Popel, A. S., and Gabhann, F. M. (2013). Computational model of VEGFR2 pathway to ERK activation and modulation through receptor trafficking. Cell. Signal. 25, 2496–510. doi: 10.1016/j.cellsig.2013.08.015
Tata, M., Ruhrberg, C., and Fantin, A. (2015). Vascularisation of the central nervous system. Mech. Dev. 138, 26–36. doi: 10.1016/j.mod.2015.07.001
Thalgott, J. H., D-Santos-Luis, D., Hosman, A. E., Martin, S., Lamandé, N., Bracquart, D., et al. (2018). Decreased expression of vascular endothelial growth factor receptor 1 contributes to the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Circulation 138, 2698–2712. doi: 10.1161/CIRCULATIONAHA.117.033062
Torsney, E., Charlton, R., Diamond, A. G., Burn, J., Soames, J. V., and Arthur, H. M. (2003). Mouse model for hereditary hemorrhagic telangiectasia has a generalized vascular abnormality. Circulation 107, 1653–57. doi: 10.1161/01.CIR.0000058170.92267.00
Torsney, E., Parums, R. C. D., Collis, M., and Arthur, H. M. (2002). Inducible expression of human endoglin during inflammation and wound healing in vivo. Inflamm. Res. 51, 464–70. doi: 10.1007/PL00012413
Townson, S. A., M-Hackert, E., Greppi, C., Lowden, P., Sako, D., Liu, J., et al. (2012). Specificity and structure of a high affinity activin receptor-like kinase 1 (ALK1) signaling complex. J. Biol. Chem. 287, 27313–25. doi: 10.1074/jbc.M112.377960
Trindade, A., Kumar, S. R., Scehnet, J. S., Lopes-da-Costa, L., Becker, J., Jiang, W., et al. (2008). Overexpression of delta-like 4 induces arterialization and attenuates vessel formation in developing mouse embryos. Blood 112, 1720–29. doi: 10.1182/blood-2007-09-112748
Tual-Chalot, S., Mahmoud, M., Allinson, K. R., Redgrave, R. E., Zhai, Z., Oh, S. P., et al. (2014). Endothelial depletion of acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PLoS One 9:e98646. doi: 10.1371/journal.pone.0098646.
Tual-Chalot, S., Oh, P., and Arthur, H. (2015). Mouse models of hereditary haemorrhagic telangiectasia: Recent advances and future challenges. Front. Genet. 6:25. doi: 10.3389/fgene.2015.00025
Tzima, E., Kiosses, W. B., del Pozo, M. A., and Schwartz, M. A. (2003). Localized Cdc42 activation, detected using a novel assay, mediates microtubule organizing center positioning in endothelial cells in response to fluid shear stress. J. Biol. Chem. 278, 31020–3. doi: 10.1074/jbc.M301179200
Urness, L. D., Sorensen, L. K., and Li, D. Y. (2000). Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat. Genet. 26, 328–31. doi: 10.1038/81634
Valdimarsdottir, G., M-J, Goumans, Rosendahl, A., Brugman, M., Itoh, S., Lebrin, F., et al. (2002). Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation 106, 2263–70. doi: 10.1161/01.cir.0000033830.36431.46
Van Gieson, E. J., Murfee, W. L., Skalak, T. C., and Price, R. J. (2003). Enhanced smooth muscle cell coverage of microvessels exposed to increased hemodynamic stresses in vivo. Circ. Res. 92, 929–36. doi: 10.1161/01.RES.0000068377.01063.79
Walcott, B. P., Winkler, E. A., Zhou, S., Birk, H., Guo, D., Koch, M. J., et al. (2018). Identification of a rare BMP pathway mutation in a non-syndromic human brain arteriovenous malformation via exome sequencing. Hum. Genome Var. 5:18001. doi: 10.1038/hgv.2018.1
Walker, E. J., Su, H., Shen, F., Choi, E.-J., Oh, S. P., Chen, G., et al. (2011). Arteriovenous malformation in the adult mouse brain resembling the human disease. Ann. Neurol. 69, 954–62. doi: 10.1002/ana.22348
Willemse, R. B., Mager, J. J., Westermann, C. J., Overtoom, T. T., Mauser, H., and Wolbers, J. G. (2000). Bleeding risk of cerebrovascular malformations in hereditary hemorrhagic telangiectasia. J. Neurosurg. 92, 779–84. doi: 10.3171/jns.2000.92.5.0779
Winkler, E. A., Birk, H., Burkhardt, J.-K., Chen, X., Yue, J. K., Guo, D., et al. (2018). Reductions in brain pericytes are associated with arteriovenous malformation vascular instability. J. Neurosurg. 129, 1464–74.
Wooderchak-Donahue, W. L., McDonald, J., O’Fallon, B., Upton, P. D., Li, W., Roman, B. L., et al. (2013). BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am. J. Hum. Genet. 93, 530–37. doi: 10.1016/j.ajhg.2013.07.004
Xu, C., Hasan, S. S., Schmidt, I., Rocha, S. F., Pitulescu, M. E., Bussmann, J., et al. (2014). Arteries are formed by vein-derived endothelial tip cells. Nat. Commun. 5:5758. doi: 10.1038/ncomms6758
Xue, Y., Gao, X., Lindsell, C. E., Norton, C. R., Chang, B., Hicks, C., et al. (1999). Embryonic lethality and vascular defects in mice lacking the notch ligand jagged1. Hum. Mol. Genet. 8, 723–30. doi: 10.1093/hmg/8.5.723
Yamada, S., Takagi, Y., Nozaki, K., Kikuta, K.-I., and Hashimoto, N. (2007). Risk factors for subsequent hemorrhage in patients with cerebral arteriovenous malformations. J. Neurosurg. 107, 965–72. doi: 10.3171/JNS-07/11/0965
Yao, Y., Jumabay, M., Wang, A., and Boström, K. I. (2011). Matrix Gla protein deficiency causes arteriovenous malformations in mice. J. Clin. Investig. 121, 2993–3004. doi: 10.1172/JCI57567
Yao, Y., Yao, J., Radparvar, M., Blazquez-Medela, A. M., Guihard, P. J., Jumabay, M., et al. (2013). Reducing Jagged 1 and 2 levels prevents cerebral arteriovenous malformations in matrix Gla protein deficiency. Proc. Natl. Acad. Sci. U.S.A. 110, 19071–76. doi: 10.1073/pnas.1310905110
Zhu, W., Chen, W., Zou, D., Wang, L., Bao, C., Zhan, L., et al. (2018). Thalidomide reduces hemorrhage of brain arteriovenous malformations in a mouse model. Stroke 49, 1232–40. doi: 10.1161/STROKEAHA.117.020356
ZhuGe, Q., Wu, Z., Huang, L., Zhao, B., Zhong, M., Zheng, W., et al. (2013). Notch4 is activated in endothelial and smooth muscle cells in human brain arteriovenous malformations. J. Cell. Mol. Med. 17, 1458–64. doi: 10.1111/jcmm.12115
Keywords: HHT, AVM, BMP, ALK1, ENG, SMAD4, endothelial cells
Citation: Drapé E, Anquetil T, Larrivée B and Dubrac A (2022) Brain arteriovenous malformation in hereditary hemorrhagic telangiectasia: Recent advances in cellular and molecular mechanisms. Front. Hum. Neurosci. 16:1006115. doi: 10.3389/fnhum.2022.1006115
Received: 29 July 2022; Accepted: 27 October 2022;
Published: 24 November 2022.
Edited by:
Richard Daneman, University of California, San Diego, United StatesReviewed by:
Sabine Bailly, INSERM, FranceCopyright © 2022 Drapé, Anquetil, Larrivée and Dubrac. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruno Larrivée, YnJ1bm8ubGFycml2ZWVAdW1vbnRyZWFsLmNh; Alexandre Dubrac, YWxleGFuZHJlLmR1YnJhY0B1bW9udHJlYWwuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.