



94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Hum. Neurosci. , 02 November 2010
Sec. Brain Health and Clinical Neuroscience
Volume 4 - 2010 | https://doi.org/10.3389/fnhum.2010.00204
An important development in behavioral neuroscience in the past 20 years has been the demonstration that it is possible to stimulate functional recovery after cerebral injury in laboratory animals. Rodent models of cerebral injury provide an important tool for developing such rehabilitation programs. The models include analysis at different levels including detailed behavioral paradigms, electrophysiology, neuronal morphology, protein chemistry, and epigenetics. A significant challenge for the next 20 years will be the translation of this work to improve the outcome from brain injury and disease in humans. Our goal in the article will be to synthesize the multidisciplinary laboratory work on brain plasticity and behavior in the injured brain to inform the development of rehabilitation programs.
Clinical neurologists have long known that some recovery of function is possible after cerebral injury, but the rules that govern how and when recovery occurs are poorly understood. As a result, the prescription of specific treatments is based largely on hunch and habit rather than evidence-based randomized clinical trials (Teasell et al., 2006). A significant challenge for basic research is to identify the nature and mechanisms of the mediating processes underlying functional improvement so that rehabilitation strategies can be aimed at the appropriate targets. Over the past decade considerable progress has been made in the use of laboratory animal models to begin to understand some of the targets. This is the topic of the current review.
At least five obstacles can be identified to the development of evidence-based therapies. First, there is no generally accepted definition of what constitutes “recovery.” The word can be used to imply a complete return of function, a marked improvement in function, or indeed any degree of improvement. We like to call the problem of recovery the “problem of the three-legged cat.” If a cat sustains an accident that requires amputation of a hind leg, there are obviously significant difficulties in walking. But over the ensuing months the cat “recovers” and often can be almost as agile as before the accident. The cat obviously has only three legs so did not really “recover” but actually learned to compensate with new behavioral strategies for locomoting about the world. Functional improvement after most cerebral injury is really compensation. There is still an area of dead or dysfunctional tissue but the person has learned to navigate around the disability, either cognitively or physically.
Second, the organization of the brain is not static and the brain has the capacity to alter its structure and function in reaction to environmental diversity as well as to perturbations including injury. Although the term brain plasticity is now widely used, it is not easily defined and is used to refer to changes at many levels in the nervous system ranging from molecular events, such as changes in gene expression, to behavior (e.g., Shaw and McEachern, 2001). One problem is that by its very nature, the relationship between brain plasticity and behavior is correlational. This is a problem only insofar as one’s comfort level in the proof of causation is concerned. A single correlative study is not a reason to rush to the clinic but a corpus of data collected over dozens of studies in different laboratories does provide a rationale for moving toward evidence-based treatments.
Third, basic studies looking for the nature and mechanisms of processes mediating recovery usually must begin in the laboratory using animal models. This has proven problematic in translating to the clinic because of uncertainty over the appropriateness of animal models. Consider the history of the search for neuroprotective agents for stroke victims. A decade of animal studies using rats identified a series of compounds that appeared to be neuroprotective but none panned out in the clinic. The problem was not that the models were unsatisfactory but rather that the mechanisms of neuroprotection were not proven. In fact, in many studies the compounds had the effect of lowering body temperature in the lab animals so that the real mechanism was cryoprotection and not the action on some cellular channel. The compounds failed to work in people because humans are much larger and body temperature is not so easily lowered as it is in a small animal like a rat. This obviously speaks to the issue of correlation but there are other problems.
A fourth obstacle is the choice of human candidates for clinical trials. Most animal studies have well-defined injuries that are controlled by the investigators whereas there is far more variance in human conditions. A general rule of thumb in animal studies is that smaller injuries show much better response to therapies than larger injuries yet in clinical practice it is those people with large injuries that are most in need of treatments. There is thus a tendency to choose patients with larger injuries in clinical trials even though the proof of principle might be easier to demonstrate in people with less severe disabilities.
Finally, there is a significant issue regarding the gray to white matter ratio in human versus rodent brains. Humans have much more white matter and many injuries in humans are confined largely to white matter (e.g., strokes, traumatic brain injury). There are very few studies of localized white matter injuries in rodents, in part because they would be difficult to do given the reduced white matter volume. The question becomes one of whether treatments that are effective in stimulating enhanced compensation in laboratory animals with gray matter injury will generalize to people with white matter injury. Part of the answer is related to understanding what the mechanisms underlying the functional improvement might be. This is an empirical question and basic research can provide clues. It is our view that rejecting the results on the basis of concerns about the generality is misguided. On what other basis will we identify new treatments? To quote Harry Harlow, “If the competent fail to generalize, the incompetent will fill the field” (Harlow et al., 1972).
Before we address the treatments that can enhance plasticity in the injured brain we must briefly review several key principles of plasticity in the normal brain.
A change in behavior must certainly result from some change in the brain but there are many ways to investigate such changes. Changes may be inferred from global measures of brain activity, such as in the various forms of in vivo imaging, but such changes are far removed from the molecular processes that drive them. Global changes presumably reflect synaptic changes but synaptic changes result from more molecular changes such as modifications in channels, gene expression, and so on. The problem in studying brain plasticity is to choose a surrogate marker that best suits the question being asked. Changes in calcium channels may be perfect for studying synaptic changes at specific synapses that might be related to simple learning but are impractical for understanding sex differences in language processing. The latter might best be studied by in vivo imaging or postmortem analysis of cell morphology (e.g., Jacobs and Scheibel, 1993). The appropriate level must be targeted at the research question at hand. Studies investigating strategies for stimulating functional improvement after injury most commonly use anatomical (cell morphology and connectivity), physiological (cortical stimulation), and in vivo imaging. Each of these levels can be linked to behavioral outcomes in both human and non-human studies whereas more molecular levels have proven to be much more difficult to relate to behavior, and especially mental behavior.
The brain is altered by a surprisingly wide variety of experiences throughout the lifespan as summarized in Table 1. Many of these experiences are obvious such as sensorimotor training such as one might expect to see in rehabilitation programs. Many are far less intuitive, however. The brain can be changed by virtually any experience, and even a thought. If one can remember some idea a week after having it, the brain must have changed in order to store the thought. Although such changes are fascinating in themselves, it is really the magnitude of the change that is going to be important for rehabilitation. Some of the biggest changes in brain functioning come from drugs such as psychomotor stimulants or the administration of neurotrophic factors when used in combination with other experiences such as sensorimotor training.
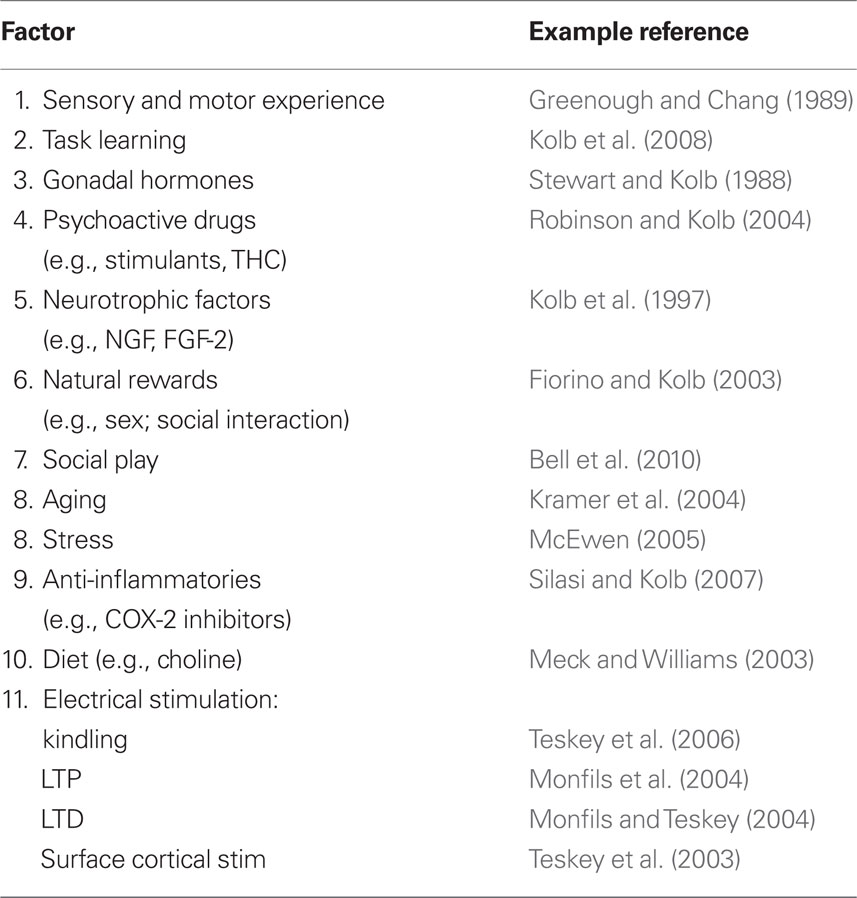
Table 1 Factors affecting the synaptic organization of the normal brain.
There has been a tendency in the literature to see different neuronal changes as surrogates for one another. One of the most common is to assume that changes in spine density reflect changes in dendritic length and vice versa. This turns out not to be the case as the two measures can vary independently and sometimes in opposite directions (e.g., Kolb et al., 2008; Comeau et al., 2010). Furthermore, cells in different cortical layers, but in the same presumptive columns, can show very different responses to the same experiences (e.g., Teskey et al., 2006).
All cortical regions show remarkable plasticity but the sensory and motor regions respond differently to most experiences than the prefrontal or hippocampal regions. For example, whereas psychoactive drugs or social/sexual behaviors produce large changes in dendritic organization in the prefrontal regions, these drugs do little to the rest of the neocortex (e.g., Robinson and Kolb, 2004). In contrast, whereas the sensory and motor regions show large chronic changes in response to complex housing (sometimes called enriched housing), the prefrontal regions show only transient changes that disappear after a few days (e.g., Kolb et al., 2003b; Comeau et al., 2010). This difference is important for designing therapies because it is clear that the location of injury is important in predicting which treatments will be most effective.
Humans have a lifetime of experiences, including drug experiences, prior to cerebral injury and this may influence not only spontaneous recovery but also the effectiveness of postinjury treatments. Little is known about these interactions, either in laboratory animals or humans but it will become an important topic in the coming years. Recently, we have shown in laboratory animals that if animals are exposed to psychomotor stimulants either as juveniles or in adulthood, later experiences have a much-attenuated (or sometimes absent) effect. For example, when rats are given methylphenidate as juveniles or amphetamine as adults and then sometime later are placed in complex environments or trained on learning tasks, the later experience-dependent changes are blocked (see Figure 1). What is surprising is that although the drugs do not show any obvious direct effect on sensory cortical regions, prior exposure prevents the expected changes in these regions (e.g., Kolb et al., 2003a). These drug–experience interactions are not unidirectional however. When infant rats are given tactile stimulation with a fine brush for 15 min 3 times per day from birth until weaning, they show a dramatic attenuation in the effects of amphetamine in adulthood (Muhammad et al., 2009). Such results imply that treatment for brain injuries will not be “one fits all” because people have different histories, including drug histories. Although our level of ignorance on plastic interactions is obviously large, the interactions loom as an important clinical concern in the future. Furthermore, it again may give pause to simple translations from the lab to the clinic.
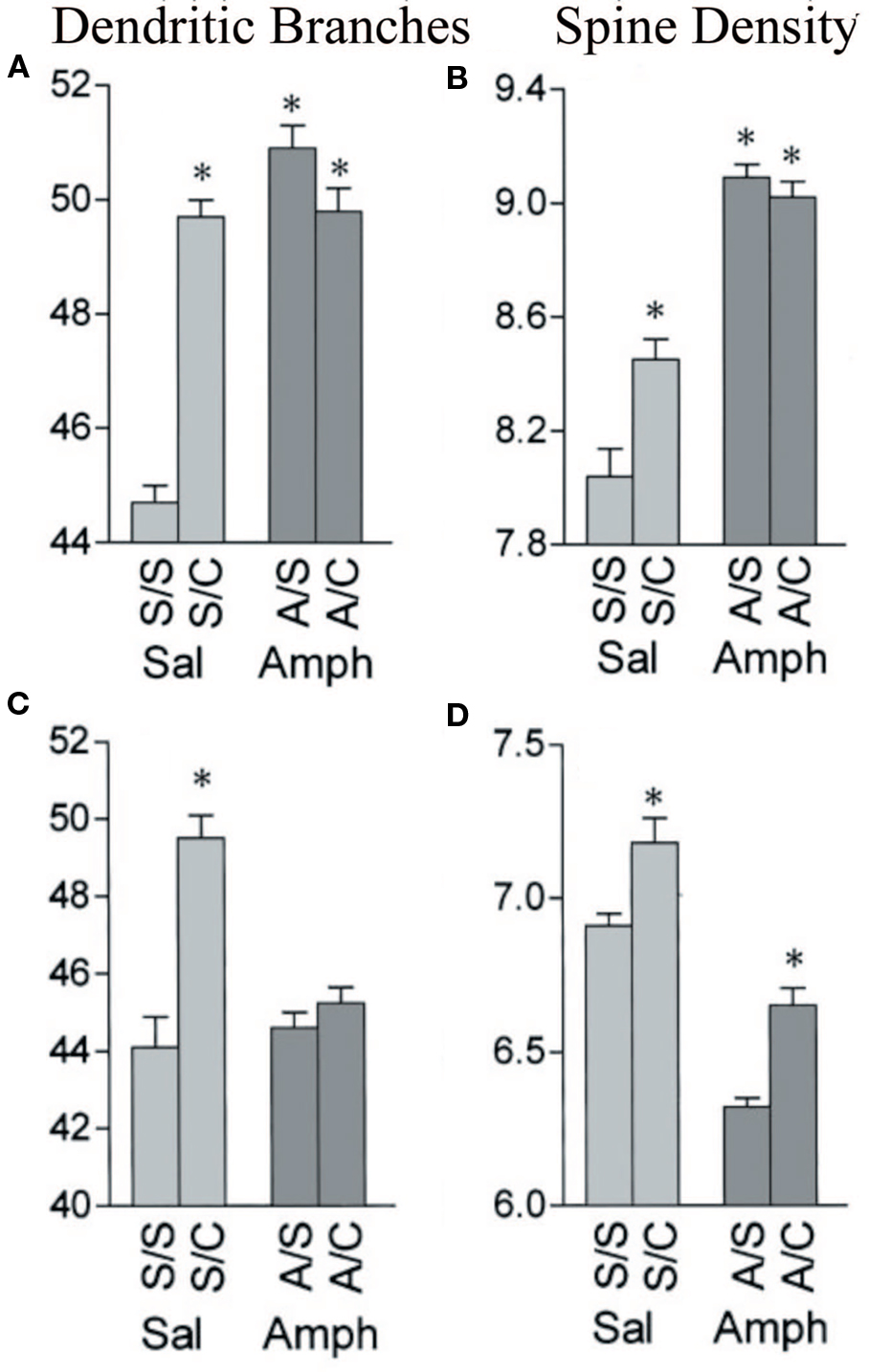
Figure 1 Effects of amphetamine and complex housing on dendritic branches (A,C) and spines (B,D) on both apical (A,B) and basilar (C,D) of layer III pyramidal cells in parietal cortex. Branching: complex housing (C) increased the number of both apical and basilar branches, but amphetamine (A) had no effect. Prior amphetamine completely blocked the effect of housing in the complex environment. Spines: complex housing increased the number of spines and amphetamine decreased density. Spine density remained lower in the amphetamine group (A/C) than in the control group (S/C) (after Kolb et al., 2003a).
Plastic changes need not be permanent and they may change dramatically over time. One of the clearest demonstrations of this comes from studies of brain plasticity in response to kindling. Kindling refers to the progressive intensification of electrographic and behavioral seizure activity with repeated stimulation and is thus a model of brain sensitization (see Teskey, 2001 for a review). In electrical kindling the stimulation is normally applied in brief trains, usually once daily, to a specific brain site (Racine, 1972). The development and expression of seizures is associated with dynamic changes in dendritic arborization and spine density that is dependent on the pyramidal cell layer. In layer III there was an initial reduction in dendritic length and spine density after the cessation of seizures followed by a rebound and increase 3 weeks later. Surprisingly the opposite changes occur in layer V with an initial lengthening of dendrites followed by a rebound and reduction at the 3-week time point (Teskey et al., 2006). The differential effect between layer V pyramidal neurons and layer III pyramidal neurons suggests that these areas play different roles in the expression of seizures and the adaptation of the brain to the persistent effect of kindling.
Time-dependent plasticity is not unique to electrical stimulation, however. For example, when rats are placed in complex environments there is a transient increase in dendritic length in the prefrontal cortex that can be seen after 4 days of complex housing but has disappeared after 14 days (Figure 2). In contrast, there are no obvious changes in sensory cortex after 4 days but clear, and seemingly permanent, changes after 14 days (Comeau et al., 2010).
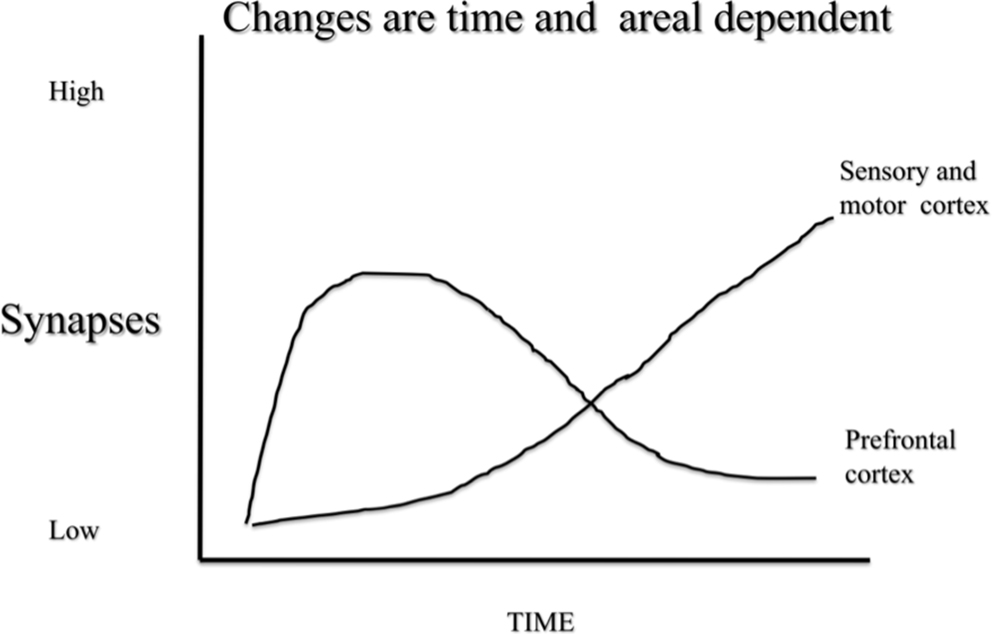
Figure 2 A schematic illustration of synaptic change in response to complex housing in rats. Pyramidal neurons in prefrontal cortex shows acute changes that revert by baseline in about 10 days. In contrast pyramidal neurons in sensory and motor regions show a slower change that remains months after treatment.
The possibility that there are different chronic and transient experience-dependent changes in cerebral neurons is consistent with genetic studies showing that there are different genes expressed acutely and chronically in response to complex environments (e.g., Rampon et al., 2000). The difference in how transient and persistent changes in neuronal networks relate to behavior is unknown.
One implication of time-dependent changes in brain plasticity relates to the question of how the brain changes in response to injury over time. It is known, for example, that there is a cascade of molecular and cellular changes that evolve after injury. Thus, during the days and weeks following an injury there are degenerative processes that can be seen in the death of neurons and the atrophy of synapses in regions related to the injury. As time progresses and the degenerative processes stabilize, there is a slow development of new neural extended neural networks throughout the forebrain (e.g., Gonzalez and Kolb, 2003). A key issue is to ask when it would be most beneficial to begin rehabilitative programs to take advantage of the regenerative plasticity and perhaps even influence the evolution of the degenerative changes. We are ignorant here and this is no doubt one reason why there is so little known about when treatments should begin after injury.
It is generally presumed that the developing brain will be more responsive to experiences than the adult or senescent brain. This is most certainly correct but there is another important wrinkle: there are qualitatively different changes in the brain in response to what appears to be the same experience at different ages. For example, when weanling, adult, or senescent rats were placed in a complex environment, all groups showed large synaptic changes but they were surprisingly different. Specifically, whereas we anticipated an increase in spine density in response to complex housing, this was only true in adult and senescent rats. Rats placed in the environments as juveniles showed a decrease in spine density (Kolb et al., 2003c). A similar drop in spine density was found in later studies in which newborn rats were given tactile stimulation with a soft brush for 15 min, three times daily over the first 10 days of life (Kolb and Gibb, 2010). The age-dependent nature of synaptic change is clearly important for understanding what treatments might be effective for pediatric versus adult neurological disorders.
Our studies of brain plasticity in animals without cerebral injury were designed to allow us to develop new strategies for designing evidence-based rehabilitation programs. The underlying assumption was that the normal and injured brain would respond the same way to the same experience. This is not always the case. For example, we used postinjury tactile stimulation to facilitate recovery from both adult and infant cerebral injuries (Gibb et al., 2010; Kolb and Gibb, 2010) and expected to find that the synaptic changes were at least qualitatively similar in the normal and injured brains. In fact, they were not. For example, whereas there was an increase in spine density in cortical pyramidal neurons in animals with perinatal prefrontal injuries, there was a decrease in the sham operates. Nonetheless, both brain-injured and sham animals showed significantly enhanced motor and cognitive performance relative to untreated animals. This was unexpected and once again illustrates our ignorance of the underlying mechanisms responsible for the beneficial effects of tactile stimulation.
Although the differential effects of the same experiences on the normal and injured brain give us pause for thought, they do not impugn the logic of looking for treatments to facilitate functional recovery by using experiences that change the normal brain. After all, the treatments worked. However, they do remind us that there is a lot that we do not know about how experience changes the brain.
Although the general gist of the literature is that plastic changes in the brain support improved motor and cognitive functions, plastic changes can interfere with behavior too. A good example is the drug-induced changes seen in response to psychomotor stimulants (e.g., Robinson and Kolb, 2004). It is reasonable to propose that some of the maladaptive behavior of drug addicts could result from drug-related changes in prefrontal neuronal morphology.
There are many other examples of pathological plasticity including pathological pain (Baranauskas, 2001), pathological response to sickness (Raison et al., 2006), epilepsy (Teskey, 2001), schizophrenia (Black et al., 2004), and dementia (Mattson et al., 2001). One important caveat in designing rehabilitation programs for brain injury is to ensure that the treatments do not induce plastic changes that could actually interfere with recovery. One case in point may be the routine use of SSRIs following stroke. These drugs, such as fluoxetine (Prozac) are often given to prevent or reduce poststroke depressive symptoms. Few studies have examined this in laboratory animals but a study by Keith et al. (2007) is worrying. These authors found that when rats with hippocampal lesions were given fluoxetine postinjury the animals showed impaired recovery on spatial memory tasks compared to untreated lesion rats. A similar study by Windle and Corbett (2005) looked at the effect of fluoxetine on motor recovery from an ischemic injury of cortex. These authors had hypothesized that because fluoxetine is known to produce various plastic changes in the brain, it might facilitate recovery. They found no beneficial effects although unlike the Keith et al. (2007) study there were no deleterious effects either. The differences in outcome could be related to type of injury, location or injury, and/or behavioral measures but they do illustrate the problem of potentially inadvertently stimulating pathological plasticity in the injured brain.
We have already alluded to one of the difficulties in using laboratory rodents to model human conditions, namely the different gray to white matter ratio in rodents and humans. Another problem is just as problematic, however. This problem relates to the question of what is the most suitable injury etiology to use in the laboratory. There are multiple ways of inducing strokes, traumatic brain injuries, dementias, and other conditions. But which one is most “natural.” This is an important question and there is a temptation among many clinicians to dismiss rodent models because they are different than human diseases and thus not relevant. This is a bit like saying that we cannot generalize about neurorehabilitation from one human condition to another because the conditions are so different. There is little doubt that dementia and ischemia are different but we are searching for ways to change the brain and, in principle, if a treatment is successful for one condition it ought to be a good bet to try in another.
It is our view that the same is true for animal models of human conditions. There is an important caveat here, however. It is essential in the laboratory to compare multiple models where possible to ensure that treatment effects are not model-dependent. A study by Gonzalez and Kolb (2003) is instructive here. These authors compared the effect of several types of cerebral injury and found that the pattern of synaptic changes was etiology-dependent but all were correlated with functional improvement over time. For example, ischemic strokes to the motor cortex led to increased synaptogenesis in the contralateral motor cortex and bilaterally in the medial prefrontal cortex. Excision of the same tissue led to synaptogenesis in the striatum bilaterally. Curiously, however, the functional improvement over time was similar in the two cases, suggesting that there are multiple routes to facilitating functional improvement. The authors did not compare the effect of a specific treatment in animals with different etiologies but given that the brain’s “natural” response to the injuries was different, there obviously would be different changes in response to rehabilitation experiences. The key question is whether the treatment would be equally effective with different etiologies and again, we are ignorant. The answer, however, is not to dismiss the findings but to do the research to find out what treatments are effective.
One final issue with models using laboratory animals is the question of what behavioral measures are appropriate to allow comparsions to humans. This is not a simple question because it is not possible to use any language-based measures in lab animals. The laboratory rat is the most extensively studied lab animal and there is a vast literature on the analysis of the behavior of the laboratory rat (see volume by Whishaw and Kolb, 2005). In general, studies of recovery from cerebral injuries have focused primarily on two types of measures – namely motor and cognitive behaviors.
Although the original studies of motor behavior tended to use a wide range of analyses (e.g., Kolb and Whishaw, 1983) most current studies use sophisticated measures of skilled forepaw use, such as reaching for pellets through a slot, or measures of hindlimb and forelimb placing (e.g., Metz and Whishaw, 2002). Most measures of cognitive function in brain-injured rats derive from the long history of studying rats in various forms of neuropsychological tests. The primary types of tests used in studies of recovery of function include various measures of object recognition and memory (e.g., Mumby, 2005) or measures of spatial navigation that can be used to make inferences about the type of cognitive representations that the animals have of their world (e.g., Sutherland, 2005). A key feature of these analyses is that there are multiple neural networks underlying different forms of cognitive processing in mammals and it requires different behavioral measures to assess different cerebral regions in rodents, just as it does in humans (e.g., McDonald et al., 2004).
We have identified a range of obstacles and issues that must be considered in our search for novel neurorehabilitation strategies. We have also made it clear that we are woefully ignorant about many of the details and complexities but nonetheless, a lot has been learned in the past two decades. The place to start modulating the effect of cortical injury is to consider the range of factors that induce neuroplasticity in the normal brain, which we saw in Table 1. The events that are most likely to be beneficial can be grouped as postinjury experience, pharmacotherapy, cell-based therapy, electrical stimulation, and diet. Note that all of these studies are from laboratory animals because such studies would be impractical in humans.
Studies of normal laboratory animals have consistently shown that the single most effective way to change both brain and behavior is to place groups of animals in complex environments for a month or longer (for a review, see Kolb and Whishaw, 1998). The complex-housed animals typically have a lot of novel and changing stimulation (such as toys, tunnels, runways, etc) and get a lot of exercise. Such experience profoundly changes the brain with the most obvious change being an increase of about 5% in overall brain weight. In addition, there are generalized increases in synapse number, astrocyte number and complexity, and angiogenesis. Animals typically have improved cognitive and motor functions on a wide range of neuropsychological tasks and the benefits far outlast the period of complex housing. Studies from literally dozens of laboratories have also shown this experience to be the most successful treatment strategy for optimizing functional recovery from a variety of forms of experimental brain damage including cortical ablation, cortical ischemia, and head trauma (e.g., Kolb and Elliott, 1987; Will and Kelche, 1992; Johansson, 1996; Biernaskie and Corbett, 2001). Although the mechanism of the beneficial effects of complex housing is not known, there are extensive changes in gene expression that are related to neuronal structure and function (Rampon et al., 2000). It has been hypothesized that changes in gene expression may increase the synthesis of neurotrophic factors, which in turn facilitate synaptic plasticity (e.g., Johansson, 2000).
One important question to consider is “what would be the equivalent treatment for humans?” It can be argued that the caged rats were actually deprived so the complex housing was simply making things more “normal.” This is likely partly true but we hasten to point out that people with brain injuries often find themselves in rather simple environments, either in hospital or at home, and often with little social contact. For active working people with a full life, this really is a form of deprivation. However, this begs the question of how an equivalent treatment could be designed for people. Most studies have kept the animals in the complex environments for 24 h/day, which would be impractical for human therapies. Our best guess is that an equivalent therapy would have to be intense, daily, and include a variety of different treatments including cognitive and physical therapy. If the mechanism(s) underlying the profound plastic changes in the brain were better understood we might be better able to design equivalent treatments, namely ones that act on the same mechanisms.
One of the effects of complex housing is that the animals receive a lot sensory stimulation, and especially tactile stimulation. With this in mind we reasoned that tactile stimulation might be effective in facilitating recovery. Our first studies looked at recovery from perinatal cortical injuries and we were able to show dramatic recovery in tests of spatial navigation and skilled forelimb movements after frontal or posterior parietal injuries (Kolb and Gibb, 2010). The tactile stimulation was given just three times daily (15 min each time) for 2–3 weeks after the injuries and in adulthood there were clear morphological changes in cortical pyramidal neurons. Specifically, whereas animals with injuries but no treatment showed extensive atrophy of cortical pyramidal neurons, those with the treatments showed a reversal of the atrophy correlated with functional recovery. Parallel findings have also now been shown in adult rats with either aspiration lesions of frontal cortex or ischemic lesions of motor cortex (Gibb et al., 2010). Once again, the recovery was correlated with a reversal of atrophy in cortical pyramidal neurons.
The results of the tactile stimulation studies are provocative, especially given that massage has long been used as a supportive treatment for brain-injured patients, albeit without a known mechanism of action. Although the effects of tactile stimulation are not as large as those from complex housing, they are not trivial and the intensity of the stimulation (45 min/day) is much reduced from the complex housing (24 h/day) and thus may be more practical clinically. But what is the mechanism of action? Three likely candidates are increased cholinergic activation of the cortex, increased neurotrophic factor expression, and increased cerebral activity. We have found increased acetylcholinesterase (AChE) levels in cortex and increased expression of at least one neurotrophic factor, fibroblast growth factor-2 (FGF-2), in both skin and brain (R. Gibb and B. Kolb, unpublished observations). We have also seen similar changes in AChE and FGF-2 expression in cortex of animals with 60 days of complex housing (Kolb et al., 1998). No doubt there will be many other changes observed in both paradigms, and especially other neurotrophic factors such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF).
There are studies that have utilized NGF directly in animals with cerebral injuries with promising results (e.g., Hart et al., 1978). A later study by Kolb et al. (1997) showed that rats with large cortical strokes showed about a 20% decrease in dendritic arborization in the remaining motor regions and that this was completely reversed by NGF. Although the results of this study were compelling, the difficulty with NGF as a potential treatment is that it is expensive and does not pass the blood–brain barrier and thus must be infused into cerebral ventricles.
Physiotherapists often treat motor deficits resulting from brain injury by using various forms of repetitive motor training. There is some evidence that repetitive motor training can be effective both in humans and non-human primates (e.g., Nudo et al., 1996). Such treatments have not always been found to be beneficial, however, and the differences may be related to the details of the training including intensity and the postinjury timing. These latter issues have been poorly studied in both animal laboratory studies as well as in clinical trials. It seems likely that a couple of hours a week will have only modest effects in both laboratory animals and human patients. Our best hunch from our animal work is that the best practice would include daily training that would include at least 2 h per day of actual therapy.
One likely combination of treatments that may prove effective is to use increased postinjury experience in combination with other treatments such as neurotrophic factors. Witt-Lajeunesse et al. (2010) combined repetitive reach training for 2 h/day or complex housing for 24/day with FGF-2 in animals with motor cortex injury. Although neither FGF-2 nor the reach training proved beneficial, the combined treatment was effective as illustrated in Figure 3. Notice that the combination of reach training and FGF-2 was as effective as complex housing alone or in combination with FGF-2. The combined effect of the two treatments is again encouraging because of the reduced time commitment compared to the complex housing.
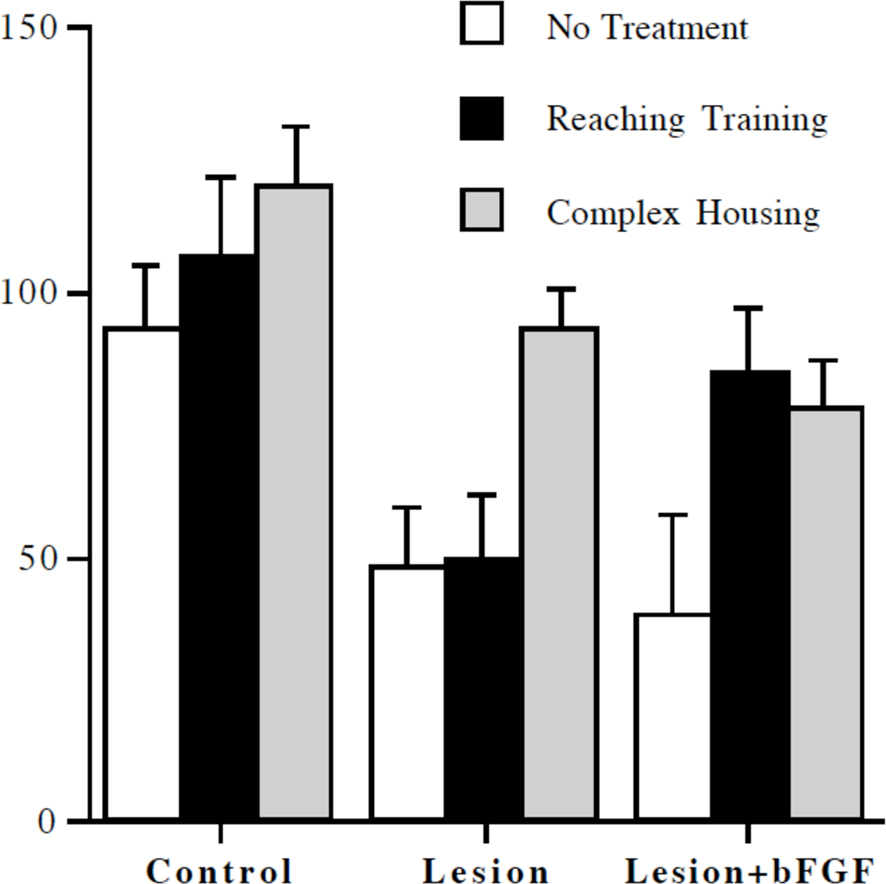
Figure 3 Reaching accuracy expressed as a percent of preoperative performance level. Complex housing improved performance in all groups. Reaching training was effective only in combination with infusion of basic fibroblast growth factor (bFGF) (from Witt-Lajeunesse et al., 2010).
Psychomotor stimulants (amphetamine, cocaine, nicotine) stimulate plastic changes in the dorsal striatum, nucleus accumbens, and prefrontal cortex (e.g., Lena and Changeux, 1999; Robinson and Kolb, 2004). In addition, nicotine (but not amphetamine or cocaine) stimulates synaptic growth in the motor cortex as well. It is reasonable, therefore, to hypothesize that psychomotor stimulants might facilitate recovery from cerebral injury – especially if given in combination with some sort of cognitive or motor training. Early studies using amphetamine as a postinjury treatment showed striking benefits in motor behaviors in rats (e.g., Feeney and Sutton, 1987; Goldstein, 2003) but later clinical studies have had mixed success. One reason appears to be lesion size. Moroz and Kolb (2005) compared the effects amphetamine on recovery from focal versus more extensive strokes. The main finding was that whereas amphetamine is effective in producing both synaptic change and behavioral improvement after focal cortical injuries, there was little benefit after large middle cerebral occlusions.
More recently, Papadopoulos et al. (2009) showed that a combination of amphetamine and complex housing produced a benefit in motor recovery after stroke in rats that was greater than either treatment alone, and was correlated with enhanced axonal sprouting from contralesional projection neurons into deafferentated subcortical areas.
Other studies have looked at the effect of nicotine on postinjury recovery and found significant beneficial effects, even after larger injuries in skilled forelimb movements (e.g., Gonzalez et al., 2006). The recovery is correlated with increased dendritic length and spine density that reverses the stroke-induced atrophy. Thus, nicotine may be a better choice for clinical trials and has the advantage that it can be administered in slow released patches that are already available. One difficulty with nicotine is that exposure to nicotine prior to cerebral injury appears to attenuate or block the effect of the drug on stimulating recovery. Presumably the brain already has been changed by the prior exposure to the drug and additional doses postinjury may thus be ineffective. It is quite possible that prior exposure to nicotine will also reduce the effectiveness of amphetamine as a treatment and this may have influenced the effectiveness of amphetamine in the clinical trials mentioned earlier.
Methylphenidate (Ritalin) also produces plastic changes in prefrontal cortex and striatum and thus may also be a candidate for clinical trials. We are unaware of any preclinical studies (i.e., lab animal) showing effects (or not) of methylphenidate on the injured brain but we are aware of rehabilitation physicians who prescribe it in low doses. Their logic is not based upon the plastic changes in the cortex so much as the use of the drug to enhance attention during cognitive rehabilitation. To our knowledge there are no randomized clinical trials examining the effects of methylphenidate. Both preclinical and clinical trials are obviously needed. They might prove especially valuable in the treatment of children.
Two other types of pharmacotherapies have taken advantage of plasticity-inducing properties of two compounds that enhance axonal sprouting after cerebral injury. The first is inosine, which is a purine nucleoside that can activate an intracellular signaling pathway that regulates the expression of multiple genes involved in axon outgrowth. Chen et al. (2002) showed that in adult rats with unilateral cortical strokes inosine stimulated neurons on the undamaged side of the brain to extend new projections to denervated areas of the midbrain and spinal cord. This growth was paralleled by improved performance on several measures of forelimb motor skills (Figure 4; see also Zai et al., 2009). More recently, inosine has also proved effective in stimulating recovery from traumatic brain injury (Smith et al., 2007). This latter study had an added wrinkle in that they compared the effectiveness of inosine and complex housing in stimulating recovery in motor function. Although animals given the complex housing recovered more quickly, after 28 days the inosine-treated animals were not functionally different than the complex-housed animals.
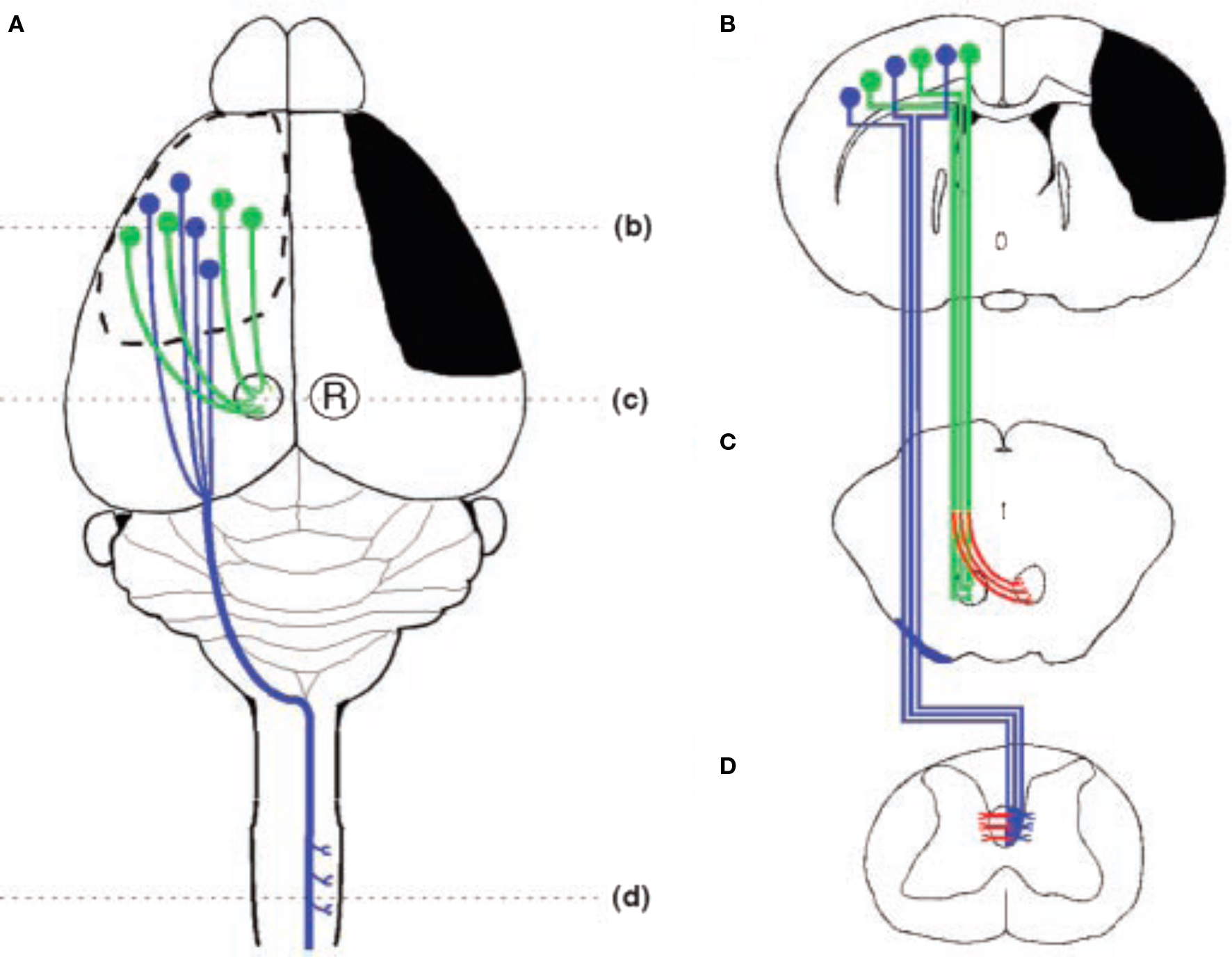
Figure 4 Schematic illustration of the stroke and major cortical efferent pathways. (A) The sensorimotor cortex and other right hemisphere structures were damaged unilaterally by occluding the right middle cerebral artery. The pathways originating from the intact left hemisphere that were traced include the corticorubral (green) and corticospinal (blue) tracts. (B) Coronal section through the forebrain showing the extent of injury (black) and the cells of origin of the intact corticospinal (blue) and corticorubral (green) tracts. (C) Projections from the sensorimotor cortex to the ipsilateral red nucleus. Compensatory growth to the denervated (right) red nucleus is shown in red. (D) Corticospinal tract projections from the intact hemisphere decussate in the caudal medulla, course in the contralateral dorsal funiculus, and synapse primarily on layer 4–6 interneruons in the cervical and lumbar enlargements of the spinal cord. Inosine-induced collateral projections to the denervated side are shown in red (after Chen et al., 2002).
The second compound is an antibody to NoGo-A, which is a molecule that blocks axon regeneration after both spinal cord and cerebral injury (for a review, see Schwab, 2004). Papadopoulos et al. (2006) showed that administration of an antibody to NoGo-A stimulated axon generation much as in the inosine studies, and in addition, increased synaptogenesis in cortical pyramidal neurons. These morphological changes were correlated with functional recovery on measures of skilled reaching.
The isolation and identification of multipotent neural stem cells from the adult forebrain (Reynolds and Weiss, 1992) and their localization to the subventricular zone (SVZ) of the lateral ventricles in both rodents and humans (Morshead et al., 1994; Doetsch et al., 1999; Sanai et al., 2004) has spurred significant interest in the therapeutic potential of rostral forebrain neurogenesis (Alvarez-Buylla and Lim, 2004; Lie et al., 2004; Lindvall et al., 2004). In rodents and non-human primates, the normal biologic function of SVZ NSCs is the generation of new olfactory interneurons (Doetsch and Hen, 2005). Recently, a series of studies have provided evidence that a middle cerebral artery occlusion model of stroke results in enhanced proliferation within the SVZ, neuronal migration toward the ischemic cortex and striatum, and subsequent differentiation into a few new neurons (Arvidsson et al., 2002; Parent et al., 2002; Jin et al., 2003; Zhang et al., 2004). Clearly, this phenomenon is insufficient to repair significant brain damage; however, a select combination of signals might enhance this process such that substantial functional tissue regeneration can occur in major structures, such as the cerebral cortex.
Epidermal growth factor (EGF) is known to be a powerful mitogen for adult forebrain SVZ neural stem cells, both in vitro and in vivo (Morshead et al., 1994; Craig et al., 1999) and thus may be particularly supportive of precursor cell re-direction through the generation of radial glial cells that support neuronal migration into the adult forebrain parenchyma (Gregg and Weiss, 2003). One difficulty with EGF infusions in vivo is that although there is a proliferation of precursor cells, these cells do not normally differentiate into neurons. One solution to this problem is to take advantage of the fact that erythropoietin (EPO) can promote the differentiation of neurons from neural precursor cells (Shingo et al., 2001).
Kolb et al. (2007) infused a combination of EGF and EPO into the lateral ventricle of rats with motor cortex strokes and were able to stimulate the growth of new cells in the subventricular zone. Cell tracking studies showed that cells migrated from the SVZ to the site of infarct to form a plug of tissue that contained glia and immature neurons (Figure 5). The formation of the plug was correlated with enhanced motor function on several measures of skilled forelimb movements. Removal of the regenerated cortical tissue reversed the functional recovery, although not immediately. Rather, the functions deteriorated over a 1–2 weeks, which suggests that although the new tissue played some role in the recovery of function, it was indirect, perhaps by influencing the function of the peri-infarct tissue. Subsequent studies found similar effects of EGF and EPO treatments on more posterior injuries. For example, the contralateral neglect induced by posterior parietal infarcts showed recovery with EGF and EPO treatments and again there was a tissue plug in the lesion cavity.
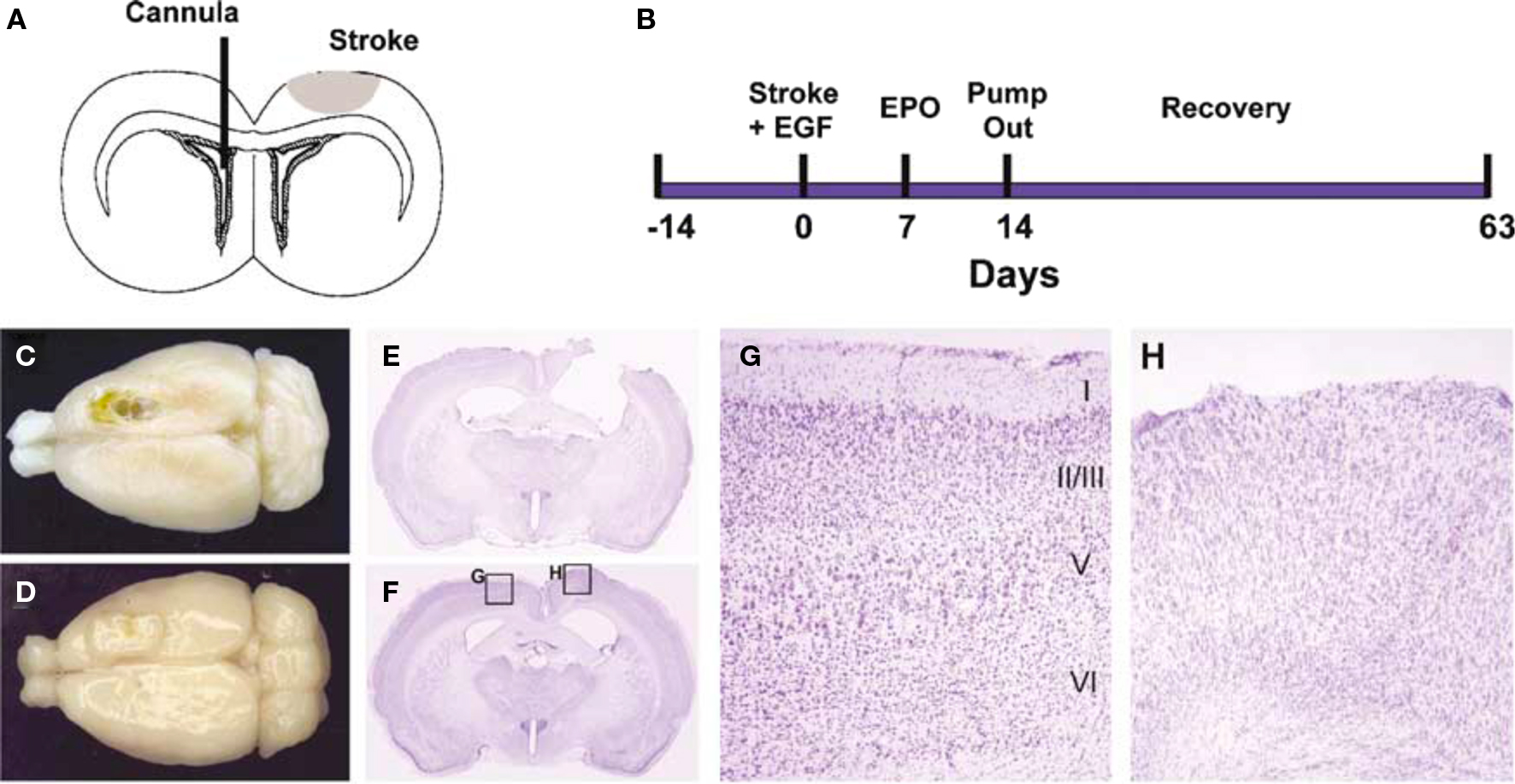
Figure 5 Epidermal growth factor + EPO infusions led to tissue regeneration in the motor cortex after focal stroke. (A,B) Experimental paradigm. Devascularizing lesion on day 0 was followed by EGF and/or EPO infusion via an intraventricular cannula in the contralateral hemisphere beginning on day 7 poststroke. EGF was infused for 7 days followed by EPO for 7 days. (C,D) Dorsal photographs of lesions brains (42 days after stroke), infused with either CSF + CSF (C) or EGF + EPO (D). The stroke produced a chronic cavity whereas treatment with EGF + EPO led to the development of newly generated cortical tissue. (E–H) Coronal cresyl violet-stained sections showing the lesion cavity in a CSF + CSF lesion brain (E) and an EGF + EPO lesion brain (F). The lesion cavity is filled with tissue in the latter brain. The intact hemisphere of the EGF + EPO brain has clear lamination characteristic of motor cortex (G) but there is no obvious organization in the newly generated cortical tissue and a complete absence of a layer I (H) (modified from Kolb et al., 2007).
These findings suggest that specific combinations of growth factors can mobilize endogeneous adult neural stem cells to promote functional recovery after stroke. One difficulty is that EGF does not pass the blood–brain barrier and must be infused directly into the brain. One solution is to use other compounds to induce cell genesis and a preliminary study by Strickland and Weiss used prolactin, which does cross the blood–brain barrier, in combination with EPO and replicated the EGF and EPO results (T. Strickland and S. Weiss, personal communication, 2009), suggesting that less invasive treatment regimes may prove beneficial.
Fibroblast growth factor-2 is also a potent mitogen for precursor cells in the SVZ. Sun et al. (2009) infused FGF-2 into the lateral ventricle of rats that had received traumatic brain injuries in adulthood. The FGF-2 treatment stimulated cell proliferation in the dentate gyrus and led to enhanced cognitive recovery in a measure of spatial navigation. In a parallel study of rats with unilateral ischemic injuries to motor cortex Leker et al. (2007) found a similar result. The FGF-2 stimulated recovery is encouraging although there has been a suggestion that it is effective only in young adult rats and not in aged rats (Won et al., 2006), which is obviously an important factor given that stroke is most likely to occur in older people.
One advantage of FGF-2 over EGF is that it passes the blood–brain barrier so it should be possible to influence recovery without an invasive mode of administration. A series of studies by Monfils et al. (2005, 2006, 2008) gave 10-day old rats motor cortex lesions followed by FGF-2 subcutaneously. The studies showed regeneration of the lost tissue, functional recovery, and evidence of functional cortico-spinal projections. Curiously, the FGF-2 induced regeneration was age and location-dependent. Thus, it was effective in stimulating regeneration after day 10 injuries but not day 3 injuries, although it did facilitate recovery at the younger age time (Comeau et al., 2007). Furthermore, although FGF-2 give after day 10 prefrontal and motor cortex injuries does stimulate regeneration, similar treatment was not effective in stimulating regeneration after more posterior injuries, possibly because of the longer distance the new cells would have to travel. Once again, however, although there was no regeneration after posterior lesions, the FGF-2 did facilitate functional recovery. These findings suggest that there are at least two separate FGF-2-related mechanisms at play here: mitogenesis and synaptogenesis.
Direct electrical stimulation of the brain to modulate local neuronal activity during motor performance holds promise for enhancing behavioral recovery following a stroke. The central idea is that stimulation of the peri-infarcted cortex facilitates the recruitment of neurons into a functional neural network that is responsible for behaviors like reaching with the forelimb (Brown et al., 2003). Thus cortical stimulation serves as an adjunctive treatment during behavioral therapy presumably through enhancing activity-dependent synaptic plasticity. As a proof of principle, studies in rats with infarcts of the sensorimotor cortex indicate that the efficacy of rehabilitation of motor functions is improved by coupling it with cortical stimulation through electrodes positioned over peri-infarct areas (Adkins et al., 2006). First the minimum amount of current to elicit a forelimb movement is first determined. This is important because it allows the investigator to know that the electrode has been placed in a viable location and is capable of inducing movement of a desired set of muscles. Parenthetically, it may be the case that a phase III clinical trial in stroke patients using this approached failed to show efficacy because the level of stimulation was insufficient (movements were never induced) and the electrodes were in the wrong location. One obvious conclusion is that it is important to closely parallel the human and non-human experimental manipulations. After recovery from electrode implantation and during daily training on a behavioral task, which requires skilled usage of the forelimb, animals receive current at an intensity that is subthreshold (usually 50%) to evoke movements. Several laboratories using a variety of ischemic models (endothelin-1, pial strip, and electrocautery) have shown efficacy with bipolar or monopolar stimulation between 40 to 70% of initial movement thresholds and between 50 and 100 Hz.
The details of the neural mechanisms underlying these functional effects are still not known. However, cortical stimulation-induced improvements in reaching success coincide with a variety of neuroplastic changes in the stimulated region of the sensorimotor cortex. One laboratory observed increased surface density of layer V dendritic processes, greater density of synapses with multisynaptic boutons and perforated postsynaptic densities that are presumed to be more efficacious (Adkins-Muir and Jones, 2003; Adkins et al., 2008). Expansion of motor maps in rats (Kleim et al., 2003) and monkeys (Plautz et al., 2003) also occurs. The polysynaptic component of a motor cortical evoked potential, that is indicative of propagating neural activity through a network, was observed to increase (Teskey et al., 2003). All of these enhancements were observed relative to a comparison group of animals receiving rehabilitation alone. Presumably understanding the specific neural mechanisms mediating these functional improvements should assist us in our endeavor to improve motor “re-learning” after brain damage.
It is generally presumed that the body heals better when it is given good nutrition so it is reasonable to predict that recovery from cortical injury might be facilitated by vitamin and/or mineral supplements. Although there is little study of this possibility after adult brain injury, dietary choline supplementation during the perinatal period produces a variety of changes both to behavior and brain of laboratory rats (Meck and Williams, 2003). For example, perinatal choline supplementation leads to enhanced spatial memory in various spatial navigation tests (e.g., Meck et al., 1988; Tees and Mohammadi, 1999) and increases the levels of NGF in hippocampus and neocortex (e.g., Sandstrom et al., 2002). Halliwell and Kolb, (2003) added choline to the drinking water of pregnant rats and continued the treatment until weaning. Rats with neonatal prefrontal lesions and the choline treatment showed significant recovery of function on various cognitive tasks. The functional recovery was correlated with dendritic growth in cortical pyramidal cells. In a follow-up study, dams and later their pups were given a diet enriched with vitamins and minerals using a formula that had been found to be beneficial in treating human bipolar patients (Kaplan et al., 2001, 2004). Like the choline treatment, there was a facilitation of recovery correlated with dendritic growth in cortical pyramidal cells (C. Halliwell and B. Kolb, unpublished observations).
The possibility that diet might be important for stimulating recovery after early cortical injury is further supported by a study by Dabydeen et al. (2008). Human neonates with perinatal brain damage were randomly allocated to receive either a high- (120% recommended average intake) or average (100% recommended average intake) energy and protein diet. The effect on recovery was so dramatic that the study was terminated before completion so that all infants could be placed on the higher energy diet. Although direct anatomical measurements of synaptic growth could not be done in such a study, non-invasive imaging showed that axonal diameters in the corticospinal tract, length, and weight were also significantly increased.
Animal models of cerebral plasticity and recovery from injury have allowed us to identify the nature and mechanisms of the mediating processes underlying functional improvement so that rehabilitation strategies can be aimed at the appropriate targets. Throughout the animal literature there is a consistent demonstration that placing rats in complex environments benefits functional recovery after cerebral injuries at different ages and of different etiologies. It is uncertain what the equivalent treatment would be in humans but complex environment treatment provides a useful “gold standard” against which to compare other treatments. Although few treatments are as effective, many are nearly as effective (e.g., cell-based therapies, pharmacotherapies) and the combination of therapies, such as neurotrophic factors and behavioral rehabilitation, promise to be the most effective.
We have emphasized the correlation between neurobiological changes and behavior but we remain relatively ignorant of the mechanisms linking the two. We do know, however, that cerebral injury generally leads to a loss of connections within different neural networks and that treatments are capable both of reversing these losses as well as stimulating the production of new connections in networks not directly affected by the injuries. The latter changes likely reflect the development of compensatory mechanisms, which are essentially a form of learning.
The application of rehabilitation strategies based upon the laboratory animal studies has yet to be realized. Nonetheless, the results of rodent studies suggest that many rehabilitation strategies could be implemented fairly easily. Perhaps the major contributions of the animal studies to date is to demonstrate: (1) that rehabilitation programs can work after early brain injury; and (2) that there are identifiable mechanisms that account for the beneficial effects. The latter contribution is particularly important because most current therapies used with brain-injured people are not based upon an understanding why they might work. Once we understand why treatments work, we are in a better position to identify specific targets to improve the development of new and more effective treatments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adkins, D. L., Campos, P., Quach, D., Borromeo, M., Schallert, K., and Jones, T. A. (2006). Epidural cortical stimulation enhances motor function after sensorimotor cortical infarcts in rats. Exp. Neurol. 200, 356–370.
Adkins, D. L., Hsu, J. E., and Jones, T. A. (2008). Motor cortical stimulation promotes synaptic plasticity and behavioral improvements following sensorimotor cortex lesions. Exp. Neurol. 212, 14–28.
Adkins-Muir, D. L., and Jones, T. A. (2003). Cortical electrical stimulation combined with rehabilitative training: enhanced functional recovery and dendritic plasticity following focal cortical ischemia in rats. Neurol. Res. 25, 780–788.
Alvarez-Buylla, A., and Lim, D. A. (2004). For the long run: maintaining germinal niches in the adult brain. Neuron 41, 683–686.
Arvidsson, A., Collin, T., Kirik, D., Kokaia, Z., and Lindvall, O. (2002). Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 8, 963–970.
Baranauskas, G. (2001). “Pain-induced plasticity in the spinal cord,” in Toward a Theory of Neuroplasticity, eds C. A. Shaw and J. McEachern (Philadelphia, PA: Psychology Press), 373–386.
Bell, H. C., Pellis, S. M., and Kolb, B. (2010). Juvenile peer play experience and the development of the orbitofrontal and medial prefrontal cortex. Behav. Brain Res. 207, 7–13.
Biernaskie, J., and Corbett, D. (2001). Enriched rehabilitative training promotes improved forelimb motor function and enhanced dendritic growth after focal ischemic injury. J. Neurosci. 21, 5272–5280.
Black, J. E., Kodish, I. M., Grossman, A. W., Klintsova, A. Y., Orlovskaya, D., Vostrikov, V., Uranova, N., and Greenough, W. T. (2004). Pathology of layer V pyramidal neurons in the prefrontal cortex of patients with schizophrenia. Am. J. Psychiatry 161, 742–744.
Brown, J. A., Lutsep, H., Cramer, S. C., and Weinand, M. (2003). Motor cortex stimulation for enhancement of recovery after stroke: case report. Neurol. Res. 25, 815–818.
Chen, P., Goldberg, D., Kolb, B., Lanser, and Benowitz, L. (2002). Axonal rewiring and improved function induced by inosine after stroke. Proc. Natl. Acad. Sci. U.S.A. 99, 9031–9036.
Comeau, W., Hastings, E., and Kolb, B. (2007). Differential effect of pre and postnatal FGF-2 following medial prefrontal cortical injury. Behav. Brain Res. 180, 18–27.
Comeau, W. L., McDonald, R., and Kolb, B. (2010). Learning-induced alterations in prefrontal cortical circuitry. Behav. Brain Res. 214, 91–101.
Craig, C. G., D’sa, R., Morshead, C. M., Roach, A., and van der Kooy, D. (1999). Migrational analysis of the constitutively proliferating subependyma population in adult mouse forebrain. Neuroscience 93, 1197–1206.
Dabydeen, L., Thomas, J. E., Aston, T. J., Hartley, H., Sinha, S. K., and Eyre, J. A. (2008). High-energy and -protein diet increases brain and corticospinal tract growth in term and preterm infants after perinatal brain injury. Pediatrics 12, 181–182.
Doetsch, F., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (1999). Subventricular zone astroctyes are neural stem cells in the adult mammalian brain. Cell 97, 703–716.
Doetsch, F., and Hen, R. (2005). Young and excitable: the function of new neurons in the adult mammalian brain. Curr. Opin. Neurobiol. 15, 121–128.
Feeney, D. M., and Sutton, R. L. (1987). Pharmacotherapy for recovery of function after brain injury. Crit. Rev. Neurobiol. 3, 135–197.
Fiorino, D., and Kolb, B. (2003). Sexual experience leads to long-lasting morphological changes in male rat prefrontal cortex, parietal cortex, and nucleus accumbens neurons. Soc. Neurosci. Abs. 29, 402.3.
Gibb, R., Gonzalez, C. L. R., Wegenast, W., and Kolb, B. (2010). Tactile stimulation facilitates recovery following cortical injury in adult rats. Behav. Brain Res. 214, 102–107.
Goldstein, L. B. (2003). Amphetamines and related drugs in motor recovery after stroke. Phys. Med. Rehab. Clinics North Am. 14, S125–S134.
Gonzalez, C. L. R., Gharbawie, O. A., and Kolb, B. (2006). Chronic low-dose administration of nicotine facilitates recovery and synaptic change after focal ischemia in rats. Neuropharmacology 50, 777–787.
Gonzalez, C. R., and Kolb, B. (2003). A comparison of different models of stroke on behavior and brain. Eur. J. Neurosci. 18, 1950–1962.
Greenough, W. T., and Chang, F. F. (1989). “Plasticity of synapse structure and pattern in the cerebral cortex,” in Cerebral Cortex, Vol. 7, eds A. Peters, and E. G. Jones (New York: Plenum Press), 391–440.
Gregg, C., and Weiss, S. (2003). Generation of functional radial glial cells by embryonic and adult forebrain neural stem cells. J. Neurosci. 23, 11587–11601.
Halliwell, C., and Kolb, B. (2003). Vitamin/mineral supplements enhance recovery from perinatal cortical lesions in rats. Soc Neuros. Abst. 29, 459.11.
Harlow, H. F., Gluck, J. P., and Suomi, S. J. (1972). Generalization of behavioral data between nonhuman and human animals. Am. Psychol. 27, 709–716.
Hart, T., Chaimas, N., Moore, R. Y., and Stein, D. G. (1978). Effects of nerve growth factor on behavioral recovery following caudate nucleus lesions in rats. Brain Res. Bull. 3, 245–250.
Jacobs, B., and Scheibel, A. B. (1993). A quantitative dendritic analysis of Wernicke’s area in humans. I. Lifespan changes. J. Comp. Neurol. 327, 383–396.
Jin, K., Sun, Y., Xie, L., Peel, A., Mao, X. O., Batteur, S., and Greenberg, D. A. (2003). Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol. Cell. Neurosci. 24, 171–189.
Johansson, B. B. (1996). Functional outcome in rats transferred to an enriched environment 15 days after focal brain ischemia. Stroke 27, 324–326.
Johansson, B. B. (2000). Brain plasticity and stroke rehabilitation. The Willis lecture. Stroke 31, 223–230.
Kaplan, B. J., Fisher, J. E., Crawford, S. G., Field, C. J., and Kolb, B. (2004). Improved mood and behavior during treatment with a mineral-vitamin supplement: an open-label case series of children. J. Child Adolesc. Psychopharmacol. 4, 115–122.
Kaplan, B. J., Simpson, J. S., Ferre, R. C., Gorman, C. P., McMullen, D. M., and Crawford, S. G. (2001). Effective mood stabilization with a chelated mineral supplement: an open-label trial in bipolar disorder. J. Clin. Psychiatry 62, 936–944.
Keith, J., Wu, Y., Epp, J. R., and Sutherland, R. J. (2007). Fluoxetine and the dentate gyrus: memory, recovery of function, and electrophysiology. Behav.Pharmacol. 18, 521–531.
Kleim, J. A., Bruneau, R., VandenBerg, P., MacDonald, E., Mulrooney, R., and Pocock, D. (2003). Motor cortex stimulation enhances motor recovery and reduces peri-infarct dysfunction following ischemic insult. Neurol. Res. 25, 789–793.
Kolb, B., Cioe, J., and Comeau, W. (2008). Contrasting effects of motor and visual learning tasks on dendritic arborization and spine density in rats. Neurobiol. Learn. Mem. 90, 295–300.
Kolb, B., Cote, S., Ribeiro-da-Silva, A., and Cuello, A. C. (1997). NGF stimulates recovery of function and dendritic growth after unilateral motor cortex lesions in rats. Neuroscience 76, 1139–1151.
Kolb, B., and Elliott, W. (1987). Recovery from early cortical damage in rats. II. Effects of experience on anatomy and behavior following frontal lesions at 1 or 5 days of age. Behav. Brain Res. 26, 47–56.
Kolb, B., Forgie, M., Gibb, R., Gorny, G., and Rowntree, S. (1998). Age, experience, and the changing brain. Neurosci. Biobehav. Rev. 22, 143–159.
Kolb, B., and Gibb, R. (2010). Tactile stimulation facilitates functional recovery and dendritic change after neonatal medial frontal or posterior parietal lesions in rats. Behav. Brain Res. 214, 115–120.
Kolb, B., Gorny, G., Li, Y., Samaha, A. N., and Robinson, T. E. (2003a). Amphetamine or cocaine limits the ability of later experience to promote structural plasticity in the neocortex and nucleus accumbens. Proc. Natl. Acad. Sci. U.S.A. 100, 10523–10528.
Kolb, B., Gorny, G., Sonderpalm, A., and Robinson, T. E. (2003b). Environmental complexity has different effects on the structure of neurons in the prefrontal cortex versus the parietal cortex or nucleus accumbens. Synapse 48, 149–153.
Kolb, B., Gibb, R., and Gorny, G. (2003c). Experience-dependent changes in dendritic arbor and spine density in neocortex vary with age and sex. Neurobiol. Learn. Mem. 79, 1–10.
Kolb, B., Morshead, C, Gonzalez, C., Kim, N., Shingo, T., and Weiss, S. (2007). Growth factor-stimulated generation of new cortical tissue and functional recovery after stroke damage to the motor cortex of rats. J. Cerebral Blood Flow Metab. 27, 983–997.
Kolb, B., and Whishaw, I. Q. (1983). Dissociation of the contributions of the prefrontal, motor and parietal cortex to the control of movement in the rat. Can. J. Psychol. 37, 211–232.
Kramer, A. F., Bherer, L., Colcombe, S. J., Dong, W., and Greenough, W. T. (2004). Environmental influences on cognitive and brain plasticity during aging. J. Gerontol. A Biol. Sci. Med. Sci. 59, M940–M957.
Leker, R. R., Soldner, F., and Velasco, I., Gavin, D. K., Androutsellis-Theotokis, A., and McKay, R. D. (2007). Long-lasting regeneration after ischemia in the cerebral cortex. Stroke 38, 153–161.
Lena, C., and Changeux, J. P. (1999). The role of beta 2-subunit-containing nicotinic acetylcholine receptors in the brain explored with a mutant mouse. Ann. N. Y. Acad. Sci. 868, 611–616.
Lie, D. C., Song, H., Colamarino, S. A., Ming, G. L., and Gage, F. H. (2004). Neurogenesis in the adult brain: new strategies for central nervous system diseases. Annu. Rev. Pharmacol. Toxicol. 44, 399–421.
Lindvall, O., Kokaia, Z., and Martinez-Serrano, A. (2004). Stem cell therapy for human neurodegenerative disorders – how to make it work. Nat. Med. 10, S42–S50.
Mattson, M. P., Duan, W., Chan, S. L., and Guo, Z. (2001). “Modification of brain aging and neurodegenerative disorders by genes, diet, and behavior,” in Toward a Theory of Neuroplasticity, eds C. A. Shaw and J. McEachern (Philadelphia, PA: Psychology Press), 402–426.
McDonald, R. J., Foong, N. S., and Devan, B. D. (2004). The challenges in understanding mammalian cognition and memory-based behaviors: an interactive learning approach. Neurosci. Biobehav. Rev. 28, 719–745.
McEwen, B. S. (2005). Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metab. Clin. Exp. 54(Suppl. 1), 20–23.
Meck, W. H., Smith, R. A., and Williams, C. L. (1988). Pre- and postnatal choline supplementation produces long-term facilitation of spatial memory. Dev. Psychobiol. 21, 339–353.
Meck, W. H., and Williams, C. L. (2003). Metabolic imprinting of choline by its availability during gestation: implications for memory and attentional processing across the lifespan. Neurosci. Biobehav. Rev. 27, 385–399.
Metz, G. A., and Whishaw, I. Q. (2002). Cortical and subcortical lesions impair skilled walking in the variably spaced ladder rung walking task. J. Neurosci. Methods 115, 169–179.
Monfils, M.-H., Driscoll, I., Kamitakahara, H., Wilson, B., Flynn, C., Teskey, G. C., Kleim, J. A., and Kolb, B. (2006). FGF-2-induced cell proliferation stimulates anatomical, neurophysiological, and functional recovery from neonatal motor cortex injury. Eur. J. Neurosci. 24, 739–749.
Monfils, M. H., Driscoll, I., Vandenberg, P. M., Thomas, N. J., Danka, D., Kleim, J. A., and Kolb, B. (2005). Basic fibroblast growth factor stimulates functional recovery after neonatal lesions of motor cortex in rats. Neuroscience 134, 1–8.
Monfils, M.-H., Driscoll, I., Vavrek, R., Kolb, B., and Fouad, K. (2008). FGF-2 induced functional improvement from neonatal motor cortex injury via corticospinal projections. Exp. Brain Res. 185, 453–460.
Monfils, M. H., and Teskey, G. C. (2004). Induction of long-term depression is associated with decreased dendritic length and spine density in layers III and V of sensorimotor neocortex. Synapse 53, 114–121.
Monfils, M. H., VandenBerg, P. M., Kleim, J. A., and Teskey, G. C. (2004). Long-term potentiation induces expanded movement representations and dendritic hypertrophy in layer V of rat sensorimotor neocortex. Cereb. Cortex 14, 586–593.
Moroz, I. A., and Kolb, B. (2005). Amphetamine facilitates recovery of skilled reaching following cortical devascularization in the rat. Soc. Neurosci. Abst. 669.6.
Morshead, C. M., Reynolds, B. A., Craig, C. G., McBurney, M. W., Staines, W. A., Morassutti, D., Weiss, S., and van der Kooy, D. (1994). Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron 13, 1071–1082.
Muhammad, A., Medland, B., Rota, R., Gibb, R., and Kolb, B. (2009) Tactile stimulation during development attenuates drug sensitization in adulthood. Soc. Neurosc. Abst 468.13.
Mumby, D. G. (2005). “Object recognition,” in The Behavior of the Laboratory Rat, eds I. Q. Whishaw and B. Kolb (New York: Oxford), 383–391.
Nudo, R. J., Wise, B. M., SiFuentes, F., and Milliken, G. W. (1996). Neural substrates for the effects of rehabilitative training on motor recovery after ischemic infarct. Science 272, 1791–1794.
Papadopoulos, C., Tsai, S.-Y., Cheatwood, J. L., Bollnow, M. R., Kolb, B., Schwab, M., and Kartje, G. (2006). Dendritic plasticity in the adult rat following middle cerebral artery occlusion and Nogo-A neutralization. Cereb. Cortex 16, 529–536.
Papadopoulos, C. M., Tsai, S. Y., Guillen, V., Ortega, J., Kartje, G. L., and Wolf, W. A. (2009). Motor recovery and axonal plasticity with short-term amphetamine after stroke. Stroke 40, 294–302.
Parent, J. M., Vexler, Z. S., Gong, C., Derugin, N., and Ferriero, D. M. (2002). Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann. Neurol. 52, 802–811.
Plautz, E. J., Barbay, S., Frost, S. B., Friel, K. M., Dancause, N., Zoubina, E. V., Stowe, A. M., Quaney, B. M., and Nudo, R. J. (2003). Post-infarct cortical plasticity and behavioral recovery using concurrent cortical stimulation and rehabilitative training: a feasibility study in primates. Neurol. Res. 25, 801–810.
Racine, R. J. (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294.
Raison, C., Capuron, L., and Miller, A. H. (2006). Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 27, 24–31.
Rampon, C., Jiang, C. H., Dong, H., Tang, Y. P., Lockart, D. J., Schultz, P. G., Tsien, J. Z., and Hu, Y. (2000). Effects of environmental enrichment on gene expression in the brain. Proc. Natl. Acad. Sci. U.S.A. 97, 12880–12884.
Reynolds, B. A., and Weiss, S. (1992). Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255, 1613–1808.
Robinson, T. E., and Kolb, B. (2004). Structural plasticity associated with drugs of abuse. Neuropharmacology 47(Suppl. 1), 33–46.
Sanai, N., Tramontin, A. D., Quiñones-Hinojosa, A., Barbaro, N. M., Gupta, N., Kunwar, S., Lawton, M. T., McDermott, M. W., Parsa, A. T., Manuel-García Verdugo, J., Berger, M. S., and Alvarez-Buylla, A. (2004). Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature 427, 740–744.
Sandstrom, N. J., Loy, R., and Williams, C. L. (2002). Prenatal choline supplementation increase NGF levels in the hippocampus and frontal cortex of young and adult rats. Brain Res. 947, 9–16.
Shaw, C. A., and McEachern, J. C. (eds). (2001). Toward a Theory of Neuroplasticity. Philadelphia, PA: Psychology Press.
Shingo, T., Sorokan, S. T., Shimazaki, T., and Weiss, S. (2001). Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J. Neurosci. 21, 9733–9743.
Silasi, G., and Kolb, B. (2007). Chronic inhibition of cyclooxygenase-2 induces dendritic hypertrophy and limited functional improvement following motor cortex stroke. Neuroscience 144, 1160–1168.
Smith, J. M., Lunga, P., Story, D., Harris, N., Le Belle, J., James, M. F., Pickard, J. D., and Fawcett, J. W. (2007). Inosine promotes recovery of skilled motor function in a model of focal brain injury. Brain 130(Pt 4), 915–925.
Stewart, J., and Kolb, B. (1988). The effects of neonatal gonadectomy and prenatal stress on cortical thickness and asymmetry in rats. Behav. Neural Biol. 49, 344–360.
Sun, D., Bullock, M. R., McGinn, M. J., Zhou, Z., Altemeni, N., Hagood, S., Hamm, R., and Colello, R. J. (2009). Basic fibroblast growth factor-enhanced neurogenesis contributes to cognitive recovery in rats following traumatic brain injury. Exp. Neurol. 216, 56–65.
Sutherland, R. J. (2005). “Cognitive processes,” in The Behavior of the Laboratory Rat, eds I. Q. Whishaw and B. Kolb (New York: Oxford), 422–435.
Teasell, R., Bayona, N., Salter, K., Hellings, C., and Bitensky, J. (2006). Progress in clinical neurosciences: stroke recovery and rehabilitation. Can. J. Neurol. Sci. 33, 357–364.
Tees, R. C., and Mohammadi, E. (1999). The effects of neonatal choline dietary supplementation on adult spatial and configural learning and memory in rats. Dev. Psychobiol. 35, 226–240.
Teskey, G. C. (2001). “Using kindling to model the neuroplastic changes associated with learning and memory, neuropsychiatric disorders, and epilepsy,” in Toward a Theory of Neuroplasticity, eds C. A. Shaw and J. McEachern (Philadelphia, PA: Psychology Press), 347–358.
Teskey, G. C., Flynn, C., Goertzen, C. D., Monfils, M. H., and Young, N. A. (2003). Cortical stimulation improves skilled forelimb use following a focal ischemic infarct in the rat. Neurol. Res. 25,794–800.
Teskey, G. C., Monfils, M. H., Silasi, G., and Kolb, B. (2006). Neocortical kindling is associated with opposing alterations in dendritic morphology in neocortical layer V and striatum from neocortical layer III. Synapse 59, 1–9.
Whishaw, I. Q., and Kolb, B. (eds). (2005). The Behavior of the Laboratory Rat. New York: Oxford University Press.
Will, B., and Kelche, C. (1992). “Environmental approaches to recovery of function from brain damage: a review of animal studies (1981 to 1991),” in Recovery from Brain Damage: Reflections and Directions, eds F. D. Rose and D. A. Johnson (New York: Plenum), 79–104.
Windle, V., and Corbett, D. (2005). Fluoxetine and recovery of motor function after focal ischemia in rats. Brain Res. 1044, 25–32.
Won, S. J., Xie, L., Kim, S. H., Tang, H., Wang, Y., Mao, X., Banwait, S., and Jin, K. (2006). Influence of age on the response to fibroblast growth factor-2 treatment in a rat model of stroke. Brain Res. 1123, 237–244.
Zai, L., Ferrari, C., Subbaiah, S., Havton, L. A., Coppola, G., Strittmater, S., Irwin, N., Geschwind, D., and Benowitz, L. I. (2009). Inosine alters gene expression and axonal projections in neurons contralateral to a cortical infarct and improves skilled use of the impaired limb. J. Neurosci. 29, 8187–8197.
Keywords: brain plasticity, recovery of function, cerebral cortex
Citation: Kolb B, Teskey GC and Gibb R (2010) Factors influencing cerebral plasticity in the normal and injured brain. Front. Hum. Neurosci. 4:204. doi:10.3389/fnhum.2010.00204
Received: 17 May 2010;
Paper pending published: 01 June 2010;
Accepted: 04 October 2010;
Published online: 02 November 2010.
Edited by:
Donald T. Stuss, Baycrest Centre for Geriatric Care, CanadaReviewed by:
Gordon Winocur, Rotman Research Institute, Canada;Copyright: © 2010 Kolb, Teskey and Gibb. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Bryan Kolb, Department of Neuroscience, University of Lethbridge, Lethbridge, AB, Canada T1K 3M4. e-mail:S29sYkB1bGV0aC5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.