 Reem M. Alhammad
Reem M. Alhammad Yafa Alshamlan
Yafa Alshamlan Ruwa Alneseyan
Ruwa Alneseyan Talal M. Al-Harbi
Talal M. Al-Harbi Ali Alhijab
Ali Alhijab Mohammed H. Alanazy
Mohammed H. Alanazy
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 13 December 2024
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 15 - 2024 | https://doi.org/10.3389/fneur.2024.1525155
Background: Lambert–Eaton myasthenic syndrome (LEMS) is an autoimmune disorder of the presynaptic neuromuscular junction associated with antibody mediated dysfunction of voltage-gated calcium channels (VGCCs). LEMS can exist as a paraneoplastic syndrome, paraneoplastic-LEMS (P-LEMS), when associated with tumors, most commonly, small cell lung carcinoma (SCLC) or as a non-paraneoplastic condition (NP-LEMS) when no malignancies are detected.
Methods: A retrospective chart review was conducted in 3 tertiary hospitals in Saudi Arabia for patients diagnosed with LEMS between January 2010 and January 2020. Patients meeting all the following criteria were included: (1) weakness or fatigability of one or more extremity or oculo-bulbar muscles, (2) 60% or higher increment of compound muscle action potential (CMAP) amplitudes immediately following isometric exercise, and (3) positive serum P/Q type VGCC antibodies. Clinical, laboratory, and electrophysiologic features, as well as radiologic imaging modalities performed for tumor screening were reviewed.
Results: The study included six patients diagnosed with LEMS, split evenly between P-LEMS and NP-LEMS. Fatigability, particularly in the lower extremities, and dyspnea on exertion were commonly reported symptoms. Low CMAP amplitudes were more frequently seen in NP-LEMS as compared to P-LEMS when recorded from both abductor pollicis brevis and abductor digiti minimi muscles. An incremental response above 60% in post activation CMAPs was detected at similar rates following variable durations of isometric exercise (10, 15, and 20 s). Tumor types detected in 3 patients with P-LEMS are SCLC, breast carcinoma, colon adenocarcinoma, and prostate acinar adenocarcinoma. Triple malignancy was detected in one patient.
Conclusion: This is the first study to describe clinical and electrophysiologic features of LEMS in an Arab ethnic cohort. Early recognition of LEMS has a significant impact on prognosis, especially given the aggressive nature of associated cancers such as SCLC.
Lambert–Eaton myasthenic syndrome (LEMS) is an autoimmune disorder of the presynaptic neuromuscular junction associated with antibody mediated dysfunction of the alpha 1A subunit of the voltage-gated calcium channel (VGCC). Frequent clinical features include proximal muscle weakness, fatigability, autonomic dysfunction, and reduced or absent deep tendon reflexes (1–5). Neurophysiologic findings include the classic triad of low baseline compound muscle action potential (CMAP) amplitudes, a decremental response on low frequency repetitive nerve stimulation (RNS), and an incremental response following isometric exercise or high frequency RNS (6). LEMS can exist as a paraneoplastic syndrome, i.e., paraneoplastic LEMS (P-LEMS) when an associated malignancy (50–60% of patients) is detected, most commonly, small cell lung carcinoma (SCLC) (7–9), or it may occur without detected malignancies, i.e., non-paraneoplastic LEMS (NP-LEMS) (10). Improving knowledge on the clinical and electrophysiologic features of LEMS can guide early diagnosis and detection of associated malignancies (11). Herein, we aim to assess clinical and electrophysiologic features, as well as tumor types detected in six patients with LEMS. To our knowledge, there are no studies describing LEMS in Arab populations, elucidating these features is highly relevant for improved recognition of this rare disease with profound impacts on patient prognosis and quality of life.
A retrospective chart review was conducted in three tertiary hospitals in Saudi Arabia to search for cases diagnosed with LEMS between January 2010 and January 2020. Search terms used include Lambert–Eaton myasthenic syndrome, myasthenic syndrome, or presynaptic neuromuscular disorder. Patients were included in analysis if they met all the following criteria: (1) presented with weakness or fatigability of one or more muscle groups in the extremities, ocular, or bulbar muscles, (2) nerve conduction studies reveal 60% or higher increment of CMAP amplitudes immediately following isometric exercise, and (3) positive serum VGCC-P/Q antibodies.
Demographic characteristics, clinical and electrophysiologic features, as well radiologic images performed for the purpose of malignancy screening were reviewed. Laboratory results collected include HbA1c, ESR, CRP, VGCC antibodies, and Sry like high-mobility group box protein 1 antibodies (SOX1) antibodies.
A positive response to immune-modulating or symptomatic therapy was defined as muscle strength improvement by one or more Medical Research Council (MRC) grades in one or muscle groups. Patients with pathologically confirmed malignancy are designated as P-LEMS, while patients with no detected malignancy are designated NP-LEMS.
Supramaximal stimulation of the peroneal, tibial, median, and ulnar nerves at distal stimulation sites was performed to record baseline CMAPs from the extensor digitorum brevis (EDB), abductor hallucis longus (AHL), abductor pollicis brevis (APB), and abductor digiti minimi (ADM) muscles, respectively. A single post-activation CMAP was recorded from the ADM and APB muscles following variable durations of isometric exercise (either 10, 15, or 20 s). Low frequency (3 Hz) RNS (LF RNS) of the ulnar and median nerves was performed at distal stimulation sites to record a single train of 5 CMAPs from the ADM and APB muscles, respectively. The baseline-to-negative peak amplitudes of CMAPs before and following isometric exercise were measured. Percent increment in the post-activation CMAP was measured as: 100 X (amplitude highest CMAP-baseline CMAP amplitude/baseline CMAP amplitude). A decremental response to LF RNS was calculated as: 100 X (amplitude baseline CMAP – lowest CMAP amplitude/baseline CMAP amplitude). A positive decremental response was defined as more than 10%. Skin temperature of the extremities was controlled at or above 32°C.
Descriptive statistics are used to summarize baseline characteristics, median time to diagnosis, median time to tumor detection in all LEMS cases, and electrophysiologic findings.
An initial chart review captured eight patients from 3 tertiary hospitals with positive P/Q type VGCC antibodies. Two patients were excluded as they showed less than 60% increment in post activation CMAPs. The remaining six patients from 2 tertiary hospitals were included for analysis. Half the patients are male with a median age of 50 years. The most frequently reported symptom is fatigability in the lower extremity, followed by fatigability in upper extremity and dyspnea on exertion. Dry mouth was reported by one patient only. All patients reported dyspnea during exertion or at rest. Forced vital capacity testing was performed in 3 patients, 2 of whom (one NP-LEMS and one P-LEMS) had values less than 80% of predicted for age, gender, height, and weight. Maximal pressures were tested in 2 patients (both NP-LEMS), these show low inspiratory pressures in both (23 and 57% of predicted) with normal maximal expiratory pressures. Other clinical signs and laboratory findings are summarized in Table 1 and Supplementary Table S1.
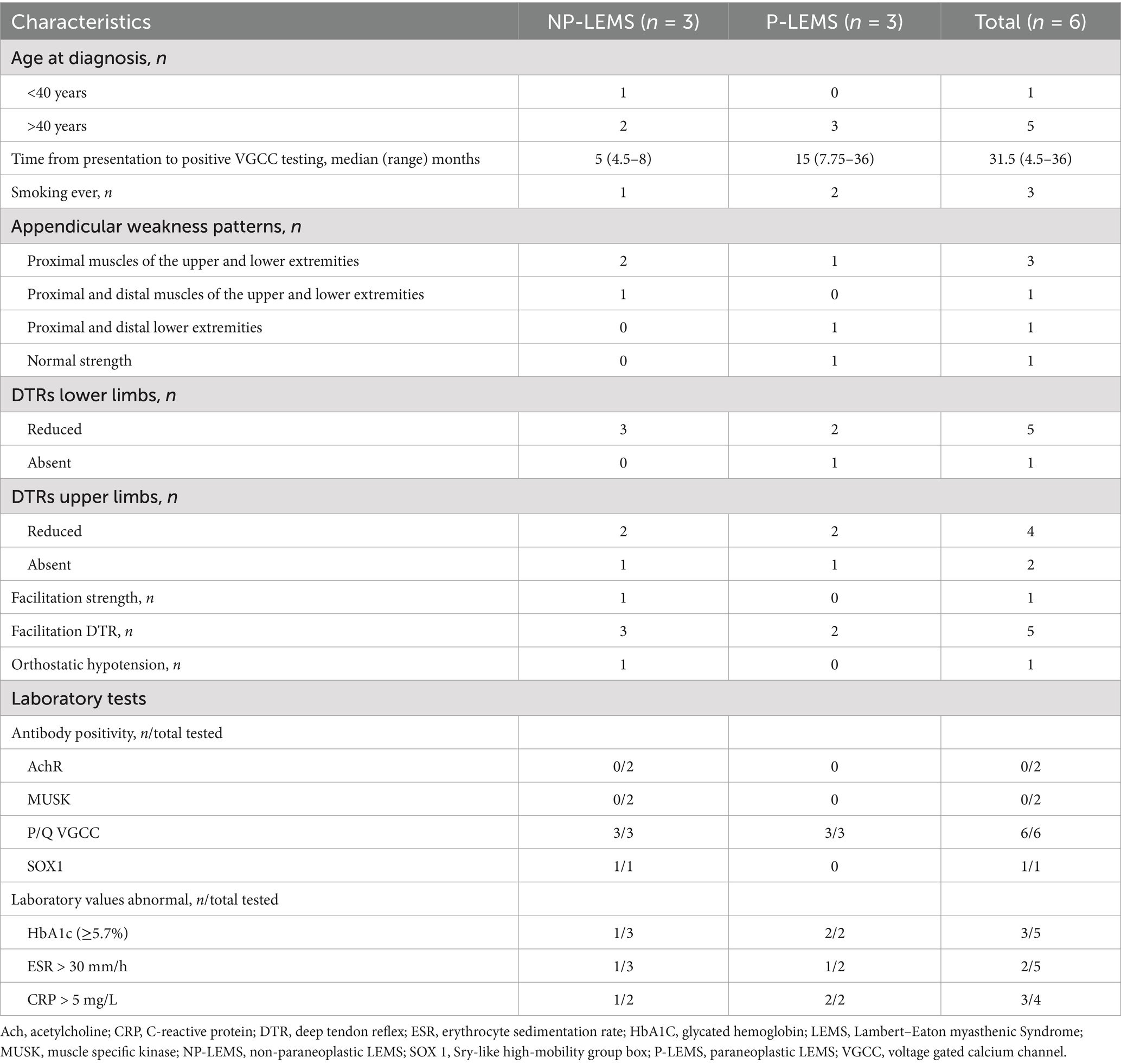
Table 1. Clinical and laboratory features of LEMS patients.
These are summarized in Table 2 and Supplementary Table S2. Low baseline CMAP amplitudes were seen in the majority of upper or lower extremity muscles tested. A decremental response above 10% in CMAP amplitudes following LF RNS was detected in 4 out of 4 tests in the ADM muscle and 2 out of 4 tests in the APB muscle. The ADM muscle was more frequently tested for post activation incremental response in CMAPs as compared to the APB muscle (5 tests vs. 3 tests respectively). An incremental response above 60% in post-activation CMAP amplitudes following isometric exercise was present in all cases and was detected following 10, 15, and 20 s of isometric exercise in two patients each. A positive incremental response (above 60%) in post-activation CMAP amplitudes was detected in 5 out 5 tests in the ADM muscle and 3 out of 3 tests in the APB muscle. An incremental response above 100% in post-activation CMAPs following isometric exercise was present in all but 2 patients. High frequency RNS (HF RNS) (50 Hz) was not performed for any of our patients.
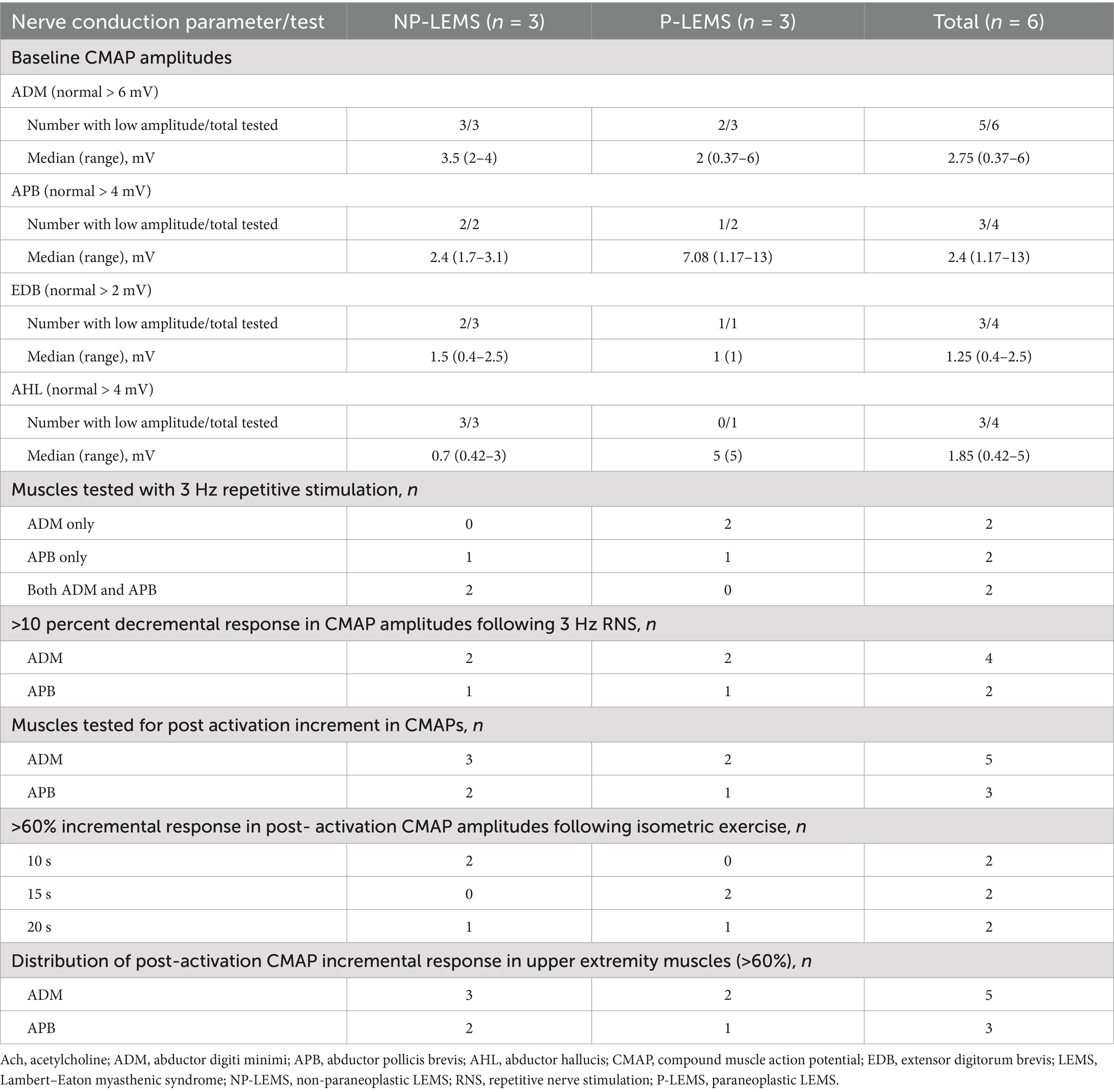
Table 2. Nerve conduction study findings in LEMS patients.
Out of 3 patients with P-LEMS, 2 had been diagnosed with malignancy prior to diagnosis with LEMS (breast cancer, SCLC), time intervals between tumor diagnosis and diagnosis of LEMS were 3 years and 4 weeks, respectively. The third patient with P-LEMS was diagnosed with three separate tumors: prostate acinar adenocarcinoma (gleason score 7 [4 + 3], grade group 3) was diagnosed historically, 3 years prior to the diagnosis with LEMS, then SCLC was detected prospectively by CT scan and fluorodeoxyglucose positron-emission tomography (FDG-PET) scan 12 weeks following diagnosis of LEMS, and the third malignancy was colon adenocarcinoma detected prospectively by FDG-PET 1 year following diagnosis of LEMS. Figure 1 depicts imaging studies performed for tumor screening and malignancies detected in our LEMS cohort.
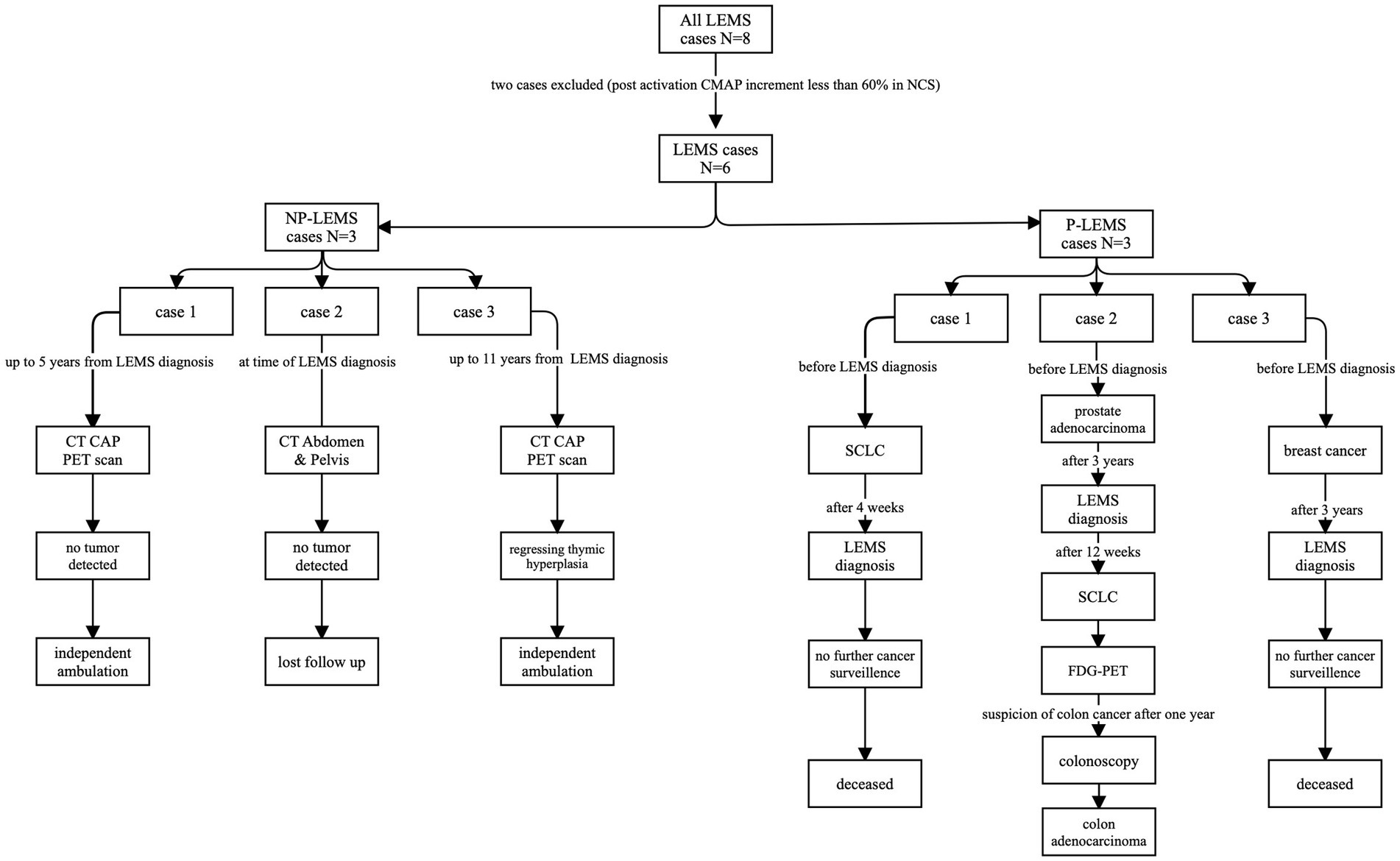
Figure 1. Flowchart of radiologic studies performed, and malignancies detected in our LEMS cohort. LEMS, CT, computerized tomography; CAP, chest-abdomen-pelvis; Lambert–Eaton myasthenic syndrome; NP-LEMS, non-paraneoplastic LEMS; P-LEMS, paraneoplastic LEMS; PET, positron-emission tomography; SCLC, small cell lung carcinoma.
Manual muscle strength testing improvement of >1 MRC grade was documented for one patient following oral prednisolone treatment and for 2 patients following Intravenous Immunoglobulin treatment (Table 3).
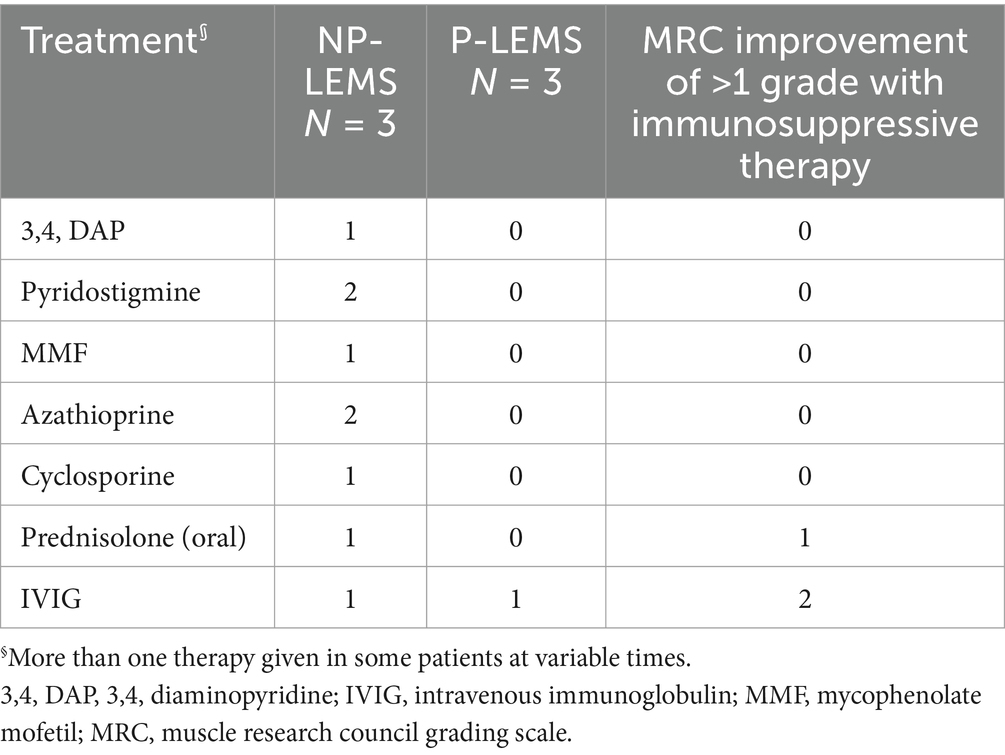
Table 3. Symptomatic and immune-modulating therapies administered to LEMS patients.
Long-term follow up was documented for two patients with NP-LEMS: one patient with associated Hashimoto’s thyroiditis was followed for 5 years, while the other patient with associated SOX1 antibodies was followed for 11 years following LEMS symptom onset. Frequency of various imaging modalities performed for tumor screening, radiologic findings at last follow up visit, and functional outcomes in these two patients are summarized in Supplementary Table S3.
Chemotherapy agents used for the treatment of SCLC were documented for 2 of our P-LEMS patients. These include carboplatin/etoposide for one patient, and Atezolizumab, an anti-programed cell death-ligand 1 (PD-L1) antibody (immune check point inhibitor), in another patient. The latter patient developed symptoms of LEMS prior to initiation of Atezolizumab.
This is the first study to describe the clinical spectrum, neurophysiologic findings, as well as oncologic outcomes in LEMS from an Arab population. To our knowledge, this is also the first study to describe the association of triple malignancy with LEMS and compare the frequency of low baseline CMAP amplitudes in P-LEMS and NP-LEMS.
Similar to prior reports, median age at onset of symptoms in our series was 50 years (11, 12). The most frequent presenting symptoms in our patients is fatigability in proximal upper and lower extremity muscles in addition to dyspnea on exertion. Bulbar symptoms are more prevalent in P-LEMS in our series, consistent with previous studies (12).
Symptoms of autonomic dysfunction are reported in all NP-LEMS patients and were not documented for P-LEMS patients in our cohort. Approximately 80–96% of LEMS cases have autonomic dysfunction which may be the earliest manifestations of this disorder. Dry mouth is the most common symptom followed by erectile dysfunction in men and constipation, while orthostatic hypotension and altered patterns of perspiration are less frequent (12, 13). Dry mouth was reported by only one NP-LEMS patient in our series.
The most frequently affected muscle groups during manual muscle strength assessment in our patients are proximal lower extremity muscles, predominantly involving hip flexion and knee extension, in keeping with previous reports of proximal lower extremity muscle affection in 87% and 90% of NP-LEMS and P-LEMS, respectively (14–16). Deep tendon reflexes were absent or reduced in all our patients, similar to previous studies (13, 17, 18). Postexercise improvement in DTRs was seen in all but one of our patients, more frequently than observed in previous reports (19–21). A single patient in our series (P-LEMS) had normal strength testing with no facilitation detected in muscle strength or DTRs. Such clinical presentations can be diagnostically challenging and require clinical vigilance and a low threshold for electrophysiologic and serological testing for LEMS.
Time from symptom onset to diagnosis was longer in P-LEMS (median 15 months) as compared to NP-LEMS (5 months). This is in contrast to previous reports showing earlier diagnosis in P-LEMS as compared to NP-LEMS with overall intervals from symptom onset to diagnosis ranging between 6 months and 36 years (15, 22).
All of our patients revealed a classic triad of abnormalities in NCS; low baseline CMAP amplitudes, decremental response on low frequency RNS (LF RNS), and incremental response above 60% following isometric exercise. This pattern was the most common of six patterns of NCS abnormalities seen in 71 percent of LEMS cases reported by Oh (6). We used a cutoff of 60% increment or higher in post activation CMAP amplitudes as an inclusion criterion for LEMS based on the findings of Oh et al. (21), who found this cutoff value to be a reasonable alternative to the 100% increment for the diagnosis of LEMS in view of its high diagnostic sensitivity (97%) and specificity (99%). In the current series, an incremental response above 60% in post activation CMAP amplitudes was present in all studies performed on the ADM and APB muscles (5 and 3 tests respectively) and was detected at equal rates following variable durations of isometric exercise, seen in one third of patients following 10, 15, and 20 s of isometric exercise each. Hatanaka and Oh et al. (19) reported post- activation CMAP amplitude increments three times higher following exercise durations of 10-s as compared to 30-s. Increment above 100% was observed in post-activation CMAP amplitudes in all but 2 of our patients (Supplementary Table S2). Maddison et al. (23) studied the distribution of RNS abnormalities in 10 patients with LEMS in the ADM, APB, anconeus, biceps, and trapezius muscles and concluded that the most sensitive muscles for detecting the characteristic abnormalities (low CMAP amplitudes and increment above 100% after 10 s exercise) were the ADM and anconeus muscles. This contrasts to the higher sensitivity of APB muscle in detecting decremental responses in Myasthenia Gravis (24, 25). The presence of normal baseline CMAP amplitudes and lack of incremental response following isometric exercise or HF RNS in Myasthenia Gravis differentiates this disorder from LEMS (6).
Similar to findings reported by Maddison et al. (23), we detected a decremental response following LF RNS more frequently in the ADM muscle as compared to the APB muscle (4 out of 4 compared to 2 out of 4 tests respectively). To our knowledge, no studies have compared the frequency of low baseline CMAP amplitudes in P-LEMS and NP-LEMS. We observed low baseline CMAP amplitudes more frequently in our NP-LEMS patients as compared to P-LEMS when recorded from both APB (2/2 tested vs. 1/2 tested respectively) and ADM (3/3 tested vs. 2/3 tested respectively) muscles. These observations are limited by the small number of cases in our cohort.
All of our patients are VGCC-P/Q seropositive as this was an inclusion criterion, these antibodies are highly sensitive and specific, detected in 90% of LEMS, more frequently in P-LEMS (26–28). None of our patients were tested for VGCC-N. In a study of neurologic accompaniments of 236 VGCC seropositive patients, LEMS was present in 2.5% of patients, all of whom were seropositive for VGCC-P/Q and none had detectable VGCC-N antibodies (28). The significance of VGCC-P/Q or VGCC-N antibodies must be interpreted in the context of clinical and paraclinical findings as these antibodies were detected in diverse autoimmune neurologic phenotypes other than LEMS and in 4% of neurologically asymptomatic patients with lung cancer (28). A single patient with NP-LEMS in our cohort was tested for SOX1 antibody and was seropositive. SOX1 antibodies are more frequent in P-LEMS but have been detected in up to 6% of NP-LEMS (5).
Half of our patients with LEMS had associated tumors (P-LEMS), with a predominance in males, similar to previous reports (12, 14, 29, 30). Tumors detected in our cohort are similar to those previously reported in association with LEMS (26, 31–33): SCLC (detected in two patients), breast carcinoma (one patient), colon adenocarcinoma (one patient), and prostate acinar adenocarcinoma (one patient). The presence of triple malignancy in association with LEMS in one of our patients is not previously reported to our knowledge and underscores individualized cancer surveillance in P-LEMS. Tumor screening can also be guided by the Dutch-English LEMS Tumor Association Prediction (DELTA-P) score, developed and validated by Titulaer et al. (34). No malignancies were detected following comprehensive cancer surveillance in the single patient in our series with NP-LEMS who was seropositive for SOX1 up to 11 years from the diagnosis of LEMS. This patient had evidence of thymic hyperplasia from the initial PET-CT image which later regressed and was not detectable in the last FDG-PET scan performed (Supplementary Table S3).
Treatment of LEMS involves treating the underlying tumor in P-LEMS as well as symptomatic and immune-modulating therapies for both P-LEMS and NP-LEMS. Symptomatic treatment with 3,4-diaminopyridine (3,4-DAP) in LEMS patients showed clinically significant improvements in QMG scores (physician-rated quantitative assessment score) as well as improved resting CMAP amplitudes compared to placebo in a recent meta-analysis (35). 3,4-DAP is a potassium channel blocker that prolongs depolarization of the presynaptic terminal by increasing the influx of calcium through the VGCC, thus increasing the release of acetylcholine manifested as improved muscle function and autonomic symptoms (1, 36). Acetylcholinesterase inhibitors such as pyridostigmine have been used in combination with 3,4 DAP with some studies showing improved benefit above 3,4 DAP monotherapy (37).
Immune-modulating treatments prescribed to our patients include prednisolone, cyclosporine, azathioprine, and IVIG. IVIG is mostly used to treat exacerbations and is equally effective for seronegative and seropositive LEMS (38). The combination of prednisolone and azathioprine improved muscle strength and resting CMAP amplitudes recorded from the ADM in a combined retrospective and prospective study of 47 patients with NP-LEMS (39).
A single patient in our cohort was treated with Atezolizumab, an immune check point inhibitor (ICI), for the management of SCLC. Symptoms of LEMS in this patient developed prior to initiating Atezolizumab. LEMS has been reported to develop as an immune related adverse event (irAE) secondary to multiple ICIs (nivolumab, ipilimumab, 668 atezolizumab, and pembrolizumab) with some cases showing clinical improvement following treatment with oral prednisolone or IVIg (40–43). The safety of ICI administration to patients with LEMS is unknown with reports of both worsening and stability of neurologic symptoms in patients with pre-existing LEMS treated with ICIs (44, 45).
The LEMS has a considerable impact on health status with up to 75% of patients reporting partial or total restrictions in their activities of daily living (ADLs) (46). The two patients with NP-LEMS in our series had suboptimal MG-ADL scores at last follow-up visits (scores of 3 and 11 at 5 and 11 years following LEMS diagnosis respectively) but remained independent for self-care and ambulation, consistent with overall stable disease course seen in long-term observational studies (47).
Limitations of this study include the retrospective design, the small sample size, and tertiary care setting of participating centers, all limiting generalizability of our findings. The small number of cases could reflect the condition’s true rarity or under-recognition of LEMS symptoms within the limited population screened in this study.
In conclusion, this is the first report of LEMS in patients of Arab ethnicity elucidating detailed clinical and NCS findings. Early clinical recognition of LEMS can guide tumor screening and early tumor detection, profoundly impacting therapy and prognosis. This is especially crucial in SCLC, an aggressive tumor with poor prognosis. Studies with larger numbers of patients are needed to assess efficacy of various symptomatic and immunosuppressive therapies.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the Institutional Review Board Subcommittee at King Saud University-College of Medicine (Reference number: E-20-5251). The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by-product of routine care or industry. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional.
RAlh: Conceptualization, Methodology, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing. YA: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft. RAln: Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. TA-H: Data curation, Investigation, Methodology, Resources, Writing – review & editing. AA: Investigation, Methodology, Resources, Writing – review & editing. MA: Conceptualization, Methodology, Project administration, Resources, Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2024.1525155/full#supplementary-material
ADM, Abductor digiti minimi; AHL, abductor hallucis longus; APB, Abductor Pollicis Brevis; CMAP, compound muscle action potential; CT, Computed Tomography; CAP, Chest, Abdomen and Pelvis; 3,4-DAP, 3,4-diaminopyridine; DTR, deep tendon reflex; EDB, extensor digitorum brevis; LEMS, Lambert–Eaton myasthenic syndrome; NP-LEMS, non-paraneoplastic LEMS; P-LEMS, Paraneoplastic LEMS; PET, positron emission tomography; RNS, repetitive nerve stimulation; SCLC, small cell lung carcinoma; SOX1, Sry like high-mobility group box protein 1; P/Q VGCC, P/Q type Voltage-gated calcium channels
1. Sanders, DB, Massey, JM, Sanders, LL, and Edwards, LJ. A randomized trial of 3,4-diaminopyridine in Lambert-Eaton myasthenic syndrome. Neurology. (2000) 54:603–3. doi: 10.1212/wnl.54.3.603
2. Tsao, CY, Mendell, JR, Friemer, ML, and Kissel, JT. Lambert-Eaton myasthenic syndrome in children. J Child Neurol. (2002) 17:74–6. doi: 10.1177/088307380201700123
3. Portaro, S, Parisi, D, Polizzi, A, Ruggieri, M, Andreetta, F, Bernasconi, P, et al. Long-term follow-up in infantile-onset lambert-Eaton myasthenic syndrome. J Child Neurol. (2014) 29:NP58–61. doi: 10.1177/0883073813499970
4. Raja, SM. Lambert-Eaton Myasthenic syndrome and botulism. Continuum. (2022) 28:1596–614. doi: 10.1212/CON.0000000000001205
5. Titulaer, MJ, Lang, B, and Verschuuren, JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. (2011) 10:1098–107. doi: 10.1016/S1474-4422(11)70245-9
6. Oh, SJ. Distinguishing features of the repetitive nerve stimulation test between Lambert-Eaton Myasthenic syndrome and myasthenia gravis, 50-year reappraisal. J Clin Neuromuscul Dis. (2017) 19:66–75. doi: 10.1097/CND.0000000000000190
7. Morrell, D, Drapkin, B, Shechter, G, and Grebla, R. P2.31-03 Lambert-Eaton Myasthenic syndrome is underrecognized in small cell lung cancer: an analysis of real-world data. J Thorac Oncol. (2023) 18:S407–8. doi: 10.1016/j.jtho.2023.09.729
8. Payne, M, Bradbury, P, Lang, B, Vincent, A, Han, C, Newsom-Davis, J, et al. Prospective study into the incidence of Lambert Eaton myasthenic syndrome in small cell lung cancer. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer. (2010) 5:34–8. doi: 10.1097/JTO.0b013e3181c3f4f1
9. Titulaer, MJ, Wirtz, PW, Willems, LNA, van Kralingen, KW, Smitt, PAES, and Verschuuren, JJGM. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol Off J Am Soc Clin Oncol. (2008) 26:4276–81. doi: 10.1200/JCO.2008.17.5133
10. Kesner, VG, Oh, SJ, Dimachkie, MM, and Barohn, RJ. Lambert-Eaton Myasthenic syndrome. Neurol Clin. (2018) 36:379–94. doi: 10.1016/j.ncl.2018.01.008
11. Ivanovski, T, and Miralles, F. Lambert-Eaton Myasthenic syndrome: early diagnosis is key. Degener Neurol Neuromuscul Dis. (2019) 9:27–37. doi: 10.2147/DNND.S192588
12. Tim, RW, Massey, JM, and Sanders, DB. Lambert-Eaton myasthenic syndrome (LEMS). Clinical and electrodiagnostic features and response to therapy in 59 patients. Ann N Y Acad Sci. (1998) 841:823–6. doi: 10.1111/j.1749-6632.1998.tb11024.x
13. Mantegazza, R, Meisel, A, Sieb, JP, Le Masson, G, Desnuelle, C, and Essing, M. The European LEMS registry: baseline demographics and treatment approaches. Neurol Ther. (2015) 4:105–24. doi: 10.1007/s40120-015-0034-0
14. Gutmann, L, Phillips, LH, and Gutmann, L. Trends in the association of Lambert-Eaton myasthenic syndrome with carcinoma. Neurol Int. (1992) 42:848–8. doi: 10.1212/WNL.42.4.848
15. Wirtz, PW, Smallegange, TM, Wintzen, AR, and Verschuuren, JJ. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. (2002) 104:359–63. doi: 10.1016/S0303-8467(02)00054-9
16. Titulaer, MJ, Wirtz, PW, Kuks, JBM, Schelhaas, HJ, van der Kooi, AJ, Faber, CG, et al. The Lambert-Eaton myasthenic syndrome 1988-2008: a clinical picture in 97 patients. J Neuroimmunol. (2008) 201-202:153–8. doi: 10.1016/j.jneuroim.2008.05.025
17. Eaton, LM, and Lambert, EH. Electromyography and electric stimulation of nerves in diseases of motor unit: observations on myasthenic syndrome associated with malignant tumors. JAMA J Am Med Assoc. (1957) 163:1117–24. doi: 10.1001/jama.1957.02970480021005
18. Yoshikawa, H, Adachi, Y, Nakamura, Y, Kuriyama, N, Murai, H, Nomura, Y, et al. Nationwide survey of Lambert-Eaton myasthenic syndrome in Japan. BMJ Neurol Open. (2022) 4:e000291. doi: 10.1136/bmjno-2022-000291
19. Hatanaka, Y, and Oh, SJ. Ten-second exercise is superior to 30-second exercise for post-exercise facilitation in diagnosing Lambert-Eaton myasthenic syndrome. Muscle Nerve. (2008) 37:572–5. doi: 10.1002/mus.20979
20. Odabasi, Z, Demirci, M, Kim, DS, Lee, DK, Ryan, HF, Claussen, GC, et al. Postexercise facilitation of reflexes is not common in Lambert-Eaton myasthenic syndrome. Neurology. (2002) 59:1085–7. doi: 10.1212/WNL.59.7.1085
21. Oh, SJ, Kurokawa, K, Claussen, GC, and Ryan, HF. Electrophysiological diagnostic criteria of Lambert-Eaton myasthenic syndrome. Muscle Nerve. (2005) 32:515–20. doi: 10.1002/mus.20389
22. Pellkofer, HL, Armbruster, L, Linke, R, Schumm, F, and Voltz, R. Managing non-paraneoplastic Lambert-Eaton myasthenic syndrome: clinical characteristics in 25 German patients. J Neuroimmunol. (2009) 217:90–4. doi: 10.1016/j.jneuroim.2009.09.017
23. Maddison, P, Newsom-Davis, J, and Mills, K. Distribution of electrophysiological abnormality in Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. (1998) 65:213–7. doi: 10.1136/jnnp.65.2.213
24. Pike-Lee, T, Higginbotham, D, and Li, Y. Direct comparison of median and ulnar repetitive nerve stimulation in generalized myasthenia gravis. Muscle Nerve. (2021) 64:490–3. doi: 10.1002/mus.27366
25. Vahabi, Z, Nazari, F, Fatehi, F, Bayegi, V, Saffarian, Z, Saffarian, F, et al. Electrophysiological studies in patients with seropositive/seronegative myasthenia gravis. Curr J Neurol. (2021) 20:120. doi: 10.18502/cjn.v20i3.7686
26. Lennon, VA, Kryzer, TJ, Griesmann, GE, O’Suilleabhain, PE, Windebank, AJ, Woppmann, A, et al. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. (1995) 332:1467–75. doi: 10.1056/NEJM199506013322203
27. Motomura, M, Lang, B, Johnston, I, Palace, J, Vincent, A, and Newsom-Davis, J. Incidence of serum anti-P/O-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J Neurol Sci. (1997) 147:35–42. doi: 10.1016/S0022-510X(96)05303-8
28. Zalewski, N, Lennon, VA, Lachance, DH, Klein, CJ, Pittock, SJ, and McKeon, A. P/q- and n-type calcium channel antibodies: oncological, neurological and serological accompaniments. Muscle Nerve. (2016) 54:220–7. doi: 10.1002/mus.25027
29. Gilhus, NE. Lambert-Eaton Myasthenic syndrome; pathogenesis, diagnosis, and therapy. Autoimmune Dis. (2011) 2011:973808. doi: 10.4061/2011/973808
30. Hiroaki, Y, Yumi, A, Yosikazu, N, Nagato, K, Hiroyuki, M, Yoshiko, N, et al. Makoto Matsui - Nationwide survey of Lambert-Eaton myasthenic syndrome in Japan. BMJ Neurology Open (2022) 4:e000291.
31. Têtu, B, Ro, JY, Ayala, AG, Ordóñez, NG, Logothetis, CJ, and von Eschenbach, AC. Small cell carcinoma of prostate associated with myasthenic (Eaton-lambert) syndrome. Urology. (1989) 33:148–52. doi: 10.1016/0090-4295(89)90017-4
32. Galton, C, Thomson, D, and Boyle, R. Lambert-Eaton myasthenic syndrome and non-pulmonary small cell carcinoma. J Neurol Neurosurg Psychiatry. (1998) 64:819–20. doi: 10.1136/jnnp.64.6.819
33. Monteiro, C, Moreira, I, Lima, JL, and Santos, E. Lambert–Eaton myasthenic syndrome and prostatic adenocarcinoma. Neurol Sci. (2015) 36:2145–6. doi: 10.1007/s10072-015-2315-x
34. Titulaer, MJ, Maddison, P, Sont, JK, Wirtz, PW, Hilton-Jones, D, Klooster, R, et al. Clinical Dutch-English Lambert-Eaton Myasthenic syndrome (LEMS) tumor association prediction score accurately predicts small-cell lung Cancer in the LEMS. J Clin Oncol. (2011) 29:902–8. doi: 10.1200/JCO.2010.32.0440
35. Zhang, N, Hong, D, Ouyang, T, Meng, W, Huang, J, Li, M, et al. 3,4-diaminopyridine treatment for Lambert-Eaton myasthenic syndrome in adults: a meta-analysis of randomized controlled trials. BMC Neurol. (2021) 21:371. doi: 10.1186/s12883-021-02405-3
36. Oh, SJ, Claussen, GG, Hatanaka, Y, and Morgan, MB. 3,4-Diaminopyridine is more effective than placebo in a randomized, double-blind, cross-over drug study in LEMS. Muscle Nerve. (2009) 40:795–800. doi: 10.1002/mus.21422
37. Tim, RW, Massey, JM, and Sanders, DB. Lambert-Eaton myasthenic syndrome: electrodiagnostic findings and response to treatment. Neurology. (2000) 54:2176–8. doi: 10.1212/WNL.54.11.2176
38. Okada, A, Koike, H, Nakamura, T, Motomura, M, and Sobue, G. Efficacy of intravenous immunoglobulin for treatment of Lambert-Eaton myasthenic syndrome without anti-presynaptic P/Q-type voltage-gated calcium channel antibodies: a case report. Neuromuscul Disord NMD. (2015) 25:70–2. doi: 10.1016/j.nmd.2014.08.006
39. Maddison, P, Lang, B, Mills, K, and Newsom-Davis, J. Long term outcome in Lambert-Eaton myasthenic syndrome without lung cancer. J Neurol Neurosurg Psychiatry. (2001) 70:212–7. doi: 10.1136/jnnp.70.2.212
40. Nakatani, Y, Tanaka, N, Enami, T, Minami, S, Okazaki, T, and Komuta, K. Lambert-Eaton Myasthenic syndrome caused by Nivolumab in a patient with squamous cell lung Cancer. Case Rep Neurol. (2018) 10:346–52. doi: 10.1159/000494078
41. Agrawal, K, and Agrawal, N. Lambert-Eaton Myasthenic syndrome secondary to Nivolumab and Ipilimumab in a patient with small-cell lung Cancer. Case Rep Neurol Med. (2019) 2019:1–4. doi: 10.1155/2019/5353202
42. Anderson, CJ, Guidon, AC, Khan, FB, Thomas, AA, Riehle, C, Hehir, M, et al. Case report of Lambert Eaton Myasthenic syndrome in a patient with small cell lung Cancer on immune checkpoint inhibitor therapy. OBM Neurobiol. (2021) 5:1–9. doi: 10.21926/obm.neurobiol.2101086
43. Kunii, E, Owaki, S, Yamada, K, Yoshihara, M, Yamaba, Y, Takakuwa, O, et al. Lambert-Eaton Myasthenic syndrome caused by Atezolizumab in a patient with small-cell lung Cancer. Intern Med Tokyo Jpn. (2022) 61:1739–42. doi: 10.2169/internalmedicine.8387-21
44. Sakaguchi, T, Kokubo, Y, Furuhashi, K, Nakamura, Y, Suzuki, Y, Ito, K, et al. An extensive-stage small-cell lung Cancer case with preexisting Lambert-Eaton Myasthenic syndrome successfully treated with an immune checkpoint inhibitor. Clin Lung Cancer. (2022) 23:e273–5. doi: 10.1016/j.cllc.2021.09.001
45. Dohrn, MF, Schöne, U, Küppers, C, Christen, D, Schulz, JB, Gess, B, et al. Immunoglobulins to mitigate paraneoplastic Lambert Eaton Myasthenic syndrome under checkpoint inhibition in Merkel cell carcinoma. Neurol Res Pract. (2020) 2:52. doi: 10.1186/s42466-020-00099-5
46. Harms, L, Sieb, JP, Williams, AE, Graham, R, Shlaen, R, Claus, V, et al. Long-term disease history, clinical symptoms, health status, and healthcare utilization in patients suffering from Lambert Eaton myasthenic syndrome: results of a patient interview survey in Germany. J Med Econ. (2012) 15:521–30. doi: 10.3111/13696998.2012.660897
Keywords: Lambert–Eaton myasthenic syndrome, neuromuscular junction, voltage-gated calcium channels, small cell lung carcinoma, compound muscle action potential (CMAP)
Citation: Alhammad RM, Alshamlan Y, Alneseyan R, Al-Harbi TM, Alhijab A and Alanazy MH (2024) Clinical presentations, electrophysiologic features, and long-term follow-up in Lambert–Eaton myasthenic syndrome: a series of six patients. Front. Neurol. 15:1525155. doi: 10.3389/fneur.2024.1525155
Edited by:
Corrado Italo Angelini, University of Padua, ItalyReviewed by:
Satish Vasant Khadilkar, Bombay Hospital, IndiaCopyright © 2024 Alhammad, Alshamlan, Alneseyan, Al-Harbi, Alhijab and Alanazy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reem M. Alhammad, cmFsaGFtYWRAa3N1LmVkdS5zYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.