 Huaqi Liu1,2†
Huaqi Liu1,2† Li Yu
Li Yu- 1Department of Trauma and Microsurgery Orthopedics, Zhongnan Hospital of Wuhan University, Wuhan, China
- 2Clinical Research Center for Microsurgical Orthopedics of Hubei Province, Zhongnan Hospital of Wuhan University, Wuhan, China
Introduction: We present a case of autosomal dominant Charcot–Marie–Tooth disease type 4B3 (CMT4B3) in a family caused by a novel SBF1 missense mutation.
Methods: Two patients, a mother and daughter, were recruited from our hospital. Both exhibited early-onset symptoms, including distal muscle atrophy of the limbs, without cranial nerve involvement. Electromyography was performed to assess nerve amplitudes and conduction velocities. Whole-exome sequencing (WES) and Sanger sequencing were performed to identify genetic mutations.
Results: Electromyography revealed a significant decline in nerve amplitudes, while the nerve conduction velocities (NCVs) remained normal in the extremities. Sequencing identified a novel missense mutation (c.1398C > A, p.H466Q) in exon 13 of the SET binding factor 1 (SBF1) gene in both patients, indicating an autosomal dominant inheritance pattern.
Discussion: Pathogenicity and protein predictions suggest that the myotubularin-related protein 5 (MTMR5), encoded by the mutated SBF1, may possess an altered structure, resulting in disease. These findings will help expand the phenotypic and genetic spectrum of CMT4B3.
1 Introduction
Charcot–Marie–Tooth disease (CMT), also known as hereditary motor and sensory neuropathy (HMSN), is a rare, genetically heterogeneous group of disorders characterized by peripheral neuropathies. These disorders involve progressive atrophy of the muscles in the extremities and a decline in motor and sensory functions (1, 2). Based on the differences in nerve conduction velocities (NCVs) detected by electrophysiological studies, CMT is classified into three types: demyelinating (CMT1), axonal (CMT2), and intermediate (DI-CMT) (3, 4). Autosomal recessive forms of CMT1, referred to as CMT4 (AR-CMT1), include a group of subtypes known as CMT4B, which are associated with genes of the myotubularin-related protein (MTMR) family, namely, CMT4B1 (MTMR2, OMIM*603557) (5), CMT4B2 (MTMR13/SBF2, OMIM*607697) (6), and CMT4B3 (MTMR5/SBF1, OMIM*603560) (7). These genes are primarily involved in regulating endo-lysosomal trafficking (8). Mutations in these genes can lead to focal myelin out-folding and secondary axonal loss in peripheral nerves (4). To date, only seven families with SBF1 mutations have been described (7, 9–16), all of which have autosomal recessive inheritance. In this study, a novel missense mutation in the SBF1 gene was identified in a heterozygote mother and daughter with complex peripheral neuropathy, demonstrating that this mutation could alter the inheritance pattern and induce autosomal dominant inheritance of CMT4B3.
2 Methods
This study was performed in accordance with the principles of the Declaration of Helsinki. Approval was granted by the Medical Ethics Committee of Zhongnan Hospital of Wuhan University (No. 2023296 K). Two patients (Patient II2 and III2) with a mother-daughter relationship (Figure 1A) were first seen at Zhongnan Hospital of Wuhan University in July 2019. Written informed consent was obtained from all the family members for all procedures. Senior doctors specializing in CMT diseases performed clinical assessments, including medical histories and neurological examinations. Electromyography was used to assess motor and sensory conduction velocities of various nerves in the upper and lower limbs in both patients. Brain and spinal MRIs were performed to investigate potential central nervous system impairment in patient III2. Patient III2 was admitted to the hospital for the treatment of bilateral Achilles tendon contractures. During the Achilles tendon release procedure, a small piece of the gastrocnemius muscle (approximately 8 mm3) was excised for further biopsy. The procedure was carried out by two senior orthopedic surgeons with over 10 years of surgical experience. Muscle biopsies were analyzed using hematoxylin and eosin (H&E), ATPase, NADH-TR, and modified Gomori trichrome staining techniques. Genetic analysis, including pathogenicity and protein prediction, was conducted by collecting blood samples from all family members to identify underlying genetic causes. Details of the Whole-exome sequencing (WES) (conducted by We-Health Biomed. Tech. Co., LTD), Sanger sequencing (conducted by Berry Genomics Co., LTD), and protein analysis are demonstrated in Supplementary material 1.
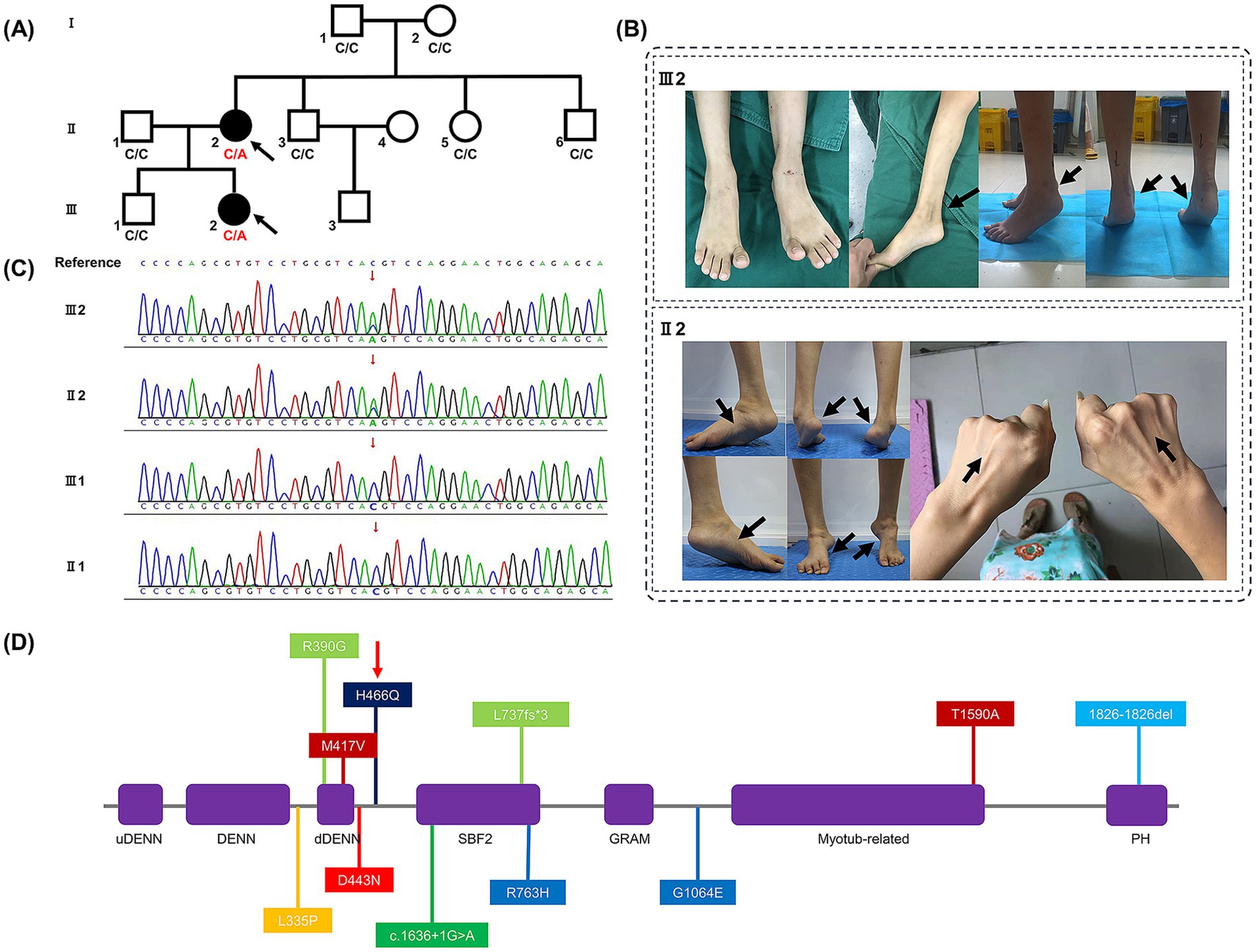
Figure 1. (A) Family pedigree demonstrating the mutated SBF1 gene was inherited dominantly from patient II2 to III2 (affected members are shown in black, and the probands are indicated by black arrows). (B) Clinical phenotypes of patients II2 and III2. After first surgery of patient III2, her Achilles tendon contracture progressively still rendered both heels of her feet difficult to land, as indicated by black arrows. Symmetric atrophy of the lower limbs with high arches deformity and clubfeet, and symmetric atrophy of the upper limbs of patient II2, are also indicated by black arrows. (C) Results of Sanger sequencing in family members II1, II2, III1, and III2. (D) Scheme of the MTMR5 protein secondary structure. Abbreviations: uDENN, upstream DENN; DENN, differentially expressed in normal and neoplastic cells; dDENN, downstream DENN; GRAM, glucosyltransferases, Rab-like GTPase activators and Myotubularins; PH, Pleckstrin homology. Colors: Purple, domains; deep red, Korean family; red, Saudi family; orange, Syrian family; light green, Spanish family; green, Bedouin family; light blue, British family; blue, Italian family; deep blue, our variant (indicated by a red narrow).
3 Results
3.1 Clinical features
3.1.1 Patient II2
Here, we present the case of a 33-year-old female patient with an uneventful birth and normal early development. Her parents’ marriage was non-consanguineous. An abnormal gait was first detected at the age of 5 years, which progressively led to muscle weakness in the distal lower limbs and clubfoot. She could walk without any assistance for a maximum of 20 min. Physical examination revealed muscle atrophy above the elbow and knee (Figure 1B), proximal muscle weakness above the elbow and knee flexion and extension, decreased tendon reflex, mild sensory deficit, and additional symptoms including high arches, stiff gait, Gower’s maneuver, clubfeet, and gastroparesis.
3.1.2 Patient III2
The patient was first examined in our consulting room around the age of 6 years. She had experienced an unsteady gait and heel lift since the age of 2. At the age of 5, she underwent surgery to externally release her Achilles tendon. Seven months after surgery, difficulty with heel contact to the ground gradually emerged (Figure 1B). We conducted several physical examinations and found that while her activities of daily living and sensory perception were normal, she exhibited other symptoms, including weakness in distal muscles (< 4 levels), muscle atrophy below the elbow and knee, high arches, Gower’s maneuver, clubfoot, and gastroparesis.
3.2 Electrophysiological findings
Electromyography demonstrated that the median and ulnar NCVs were > 38 m/s in both patients (Supplementary Table 1). The motor and sensory amplitudes of the upper and lower nerves showed a prominent decrease in both patients. No abnormalities were detected in the F-waves of the median and tibial nerves in either patient.
3.3 Imaging investigations
No obvious lesions were observed in the brain and spinal MRI of patient III2 (Figure 2), particularly at the level of the mesencephalon and pons, where the “fork and bracket” signs were previously reported in Syrian (16) and Bedouin (9) families.
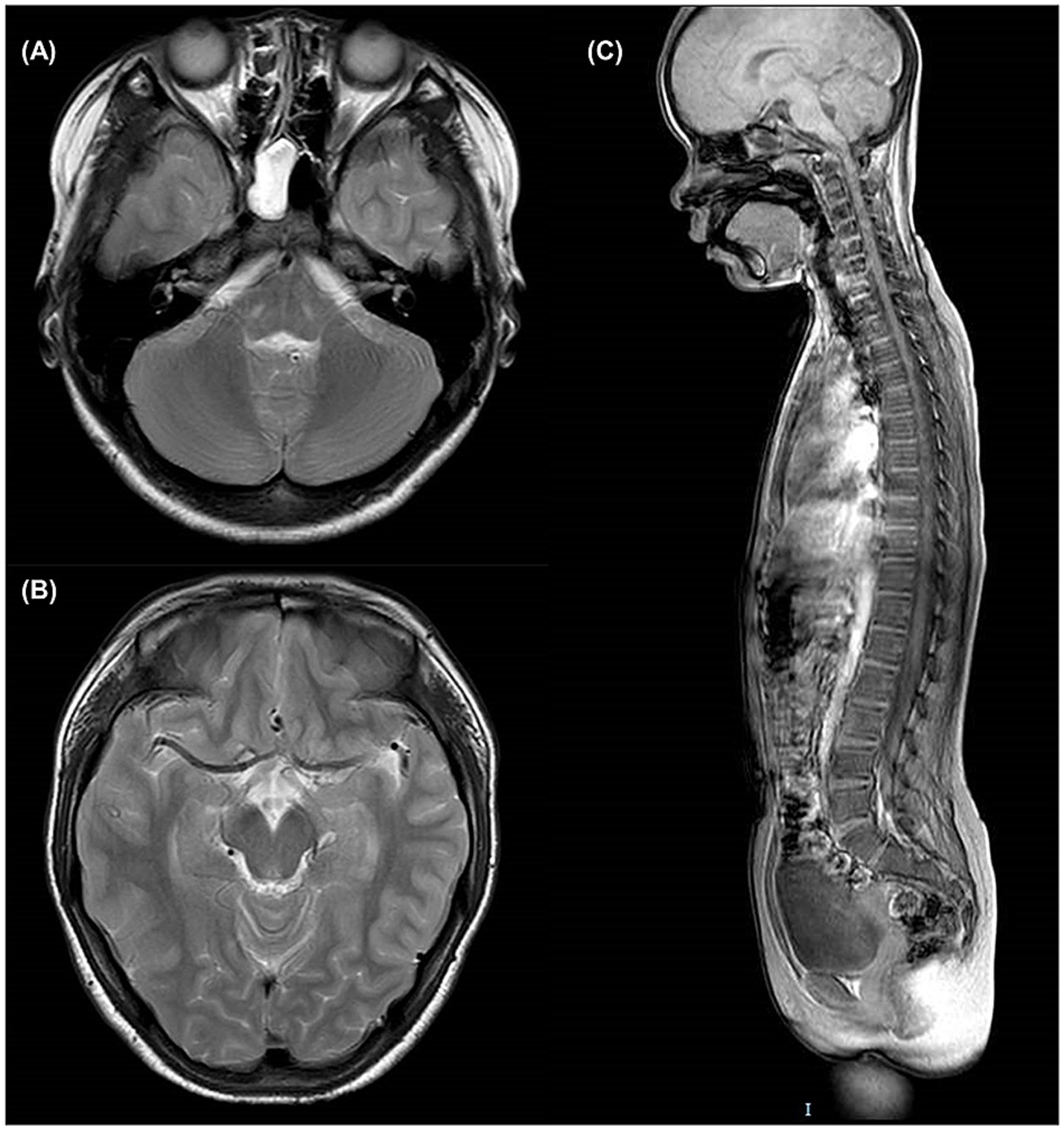
Figure 2. Brain and spinal MRI of patient III2. (A) T2-weighted axial slice at the level of pons. (B) T2-weighted axial slice at the level of mesencephalon. (C) T1-weighted sagittal slice of the entire spine.
3.4 Histological study
Muscle biopsies of the gastrocnemius from patient III2 (Figure 3) revealed small groups of muscle fiber atrophy (red arrows) and fibrosis (black arrows), with variations in myofiber diameter and fiber splitting with cavity formation observed in the H&E and modified Gomori trichrome staining analyses. An increase in the number and aggregation of myonuclear cells (green arrows) was also noted. The color depth variation of muscle fibers in ATPase staining reflected the differentiation between slow- and fast-twitch fibers. Both slow and fast myofiber atrophies indicated the presence of neurogenic lesions. NADH-TR staining showed dispersed muscle fibers, with one or several large vacuoles (blue arrow) appearing in the middle, indicating muscle fiber damage.
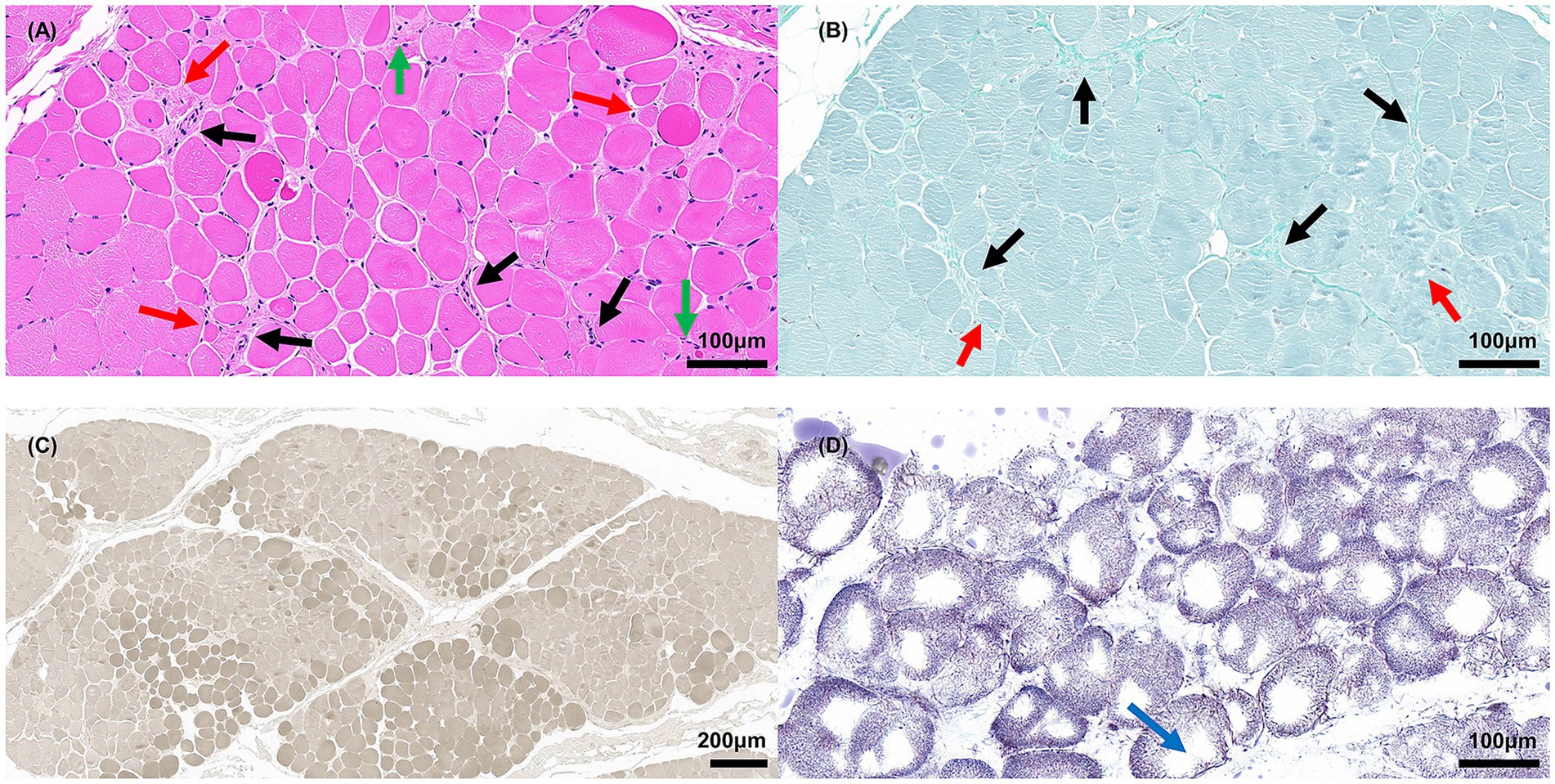
Figure 3. Muscle biopsies of gastrocnemius from patient III2. (A) H&E staining; red arrow indicates atrophic myofibers, black arrow indicates fibrosis, green arrow indicates aggregative myonuclei, scale bar = 100 μm. (B) Modified Gomori trichrome staining; the red arrow indicates atrophic myofibers; the black arrow indicates fibrosis, scale bar = 100 μm. (C) ATPase staining (pH 9.8), scale bar = 200 μm. (D) NADH-TR staining; blue arrow indicates vacuoles, scale bar = 100 μm.
3.5 Genetic analysis and pathogenicity prediction
After conducting tests and analysis, the mean total sequencing yield of WES samples was approximately 14.63 gigabases per sample, with a 99.62% coverage rate for the targeted exon regions (≥10×). Compared to other family members without relevant symptoms, only 3 mutated genes have been identified in patient II2 and III2 in WES test, which were SCN10A, PRDM12, SBF1, respectively. Given to the clinical features and electromyographic study, mutated SBF1 has been considered the main pathogenesis incurring to CMT4B3, and the remaining two mutations might be synonymous or non-pathogenic. Patient III2 and II2 were found to have a novel SBF1 non-synonymous missense mutation (c.1398 Cytosine to Adenine [c.1398C > A] in exon 13 of mRNA) on one allele (chr22:50903281). This mutation resulted in a change at the 466th amino acid of the protein, transitioning from histidine (His) to glutamine (Gln) (p.H466Q). Sanger sequencing also confirmed the mutation at this site (Figure 1C), which has not been previously reported in the literature or listed in dbSNP yet. This mutation was not indexed in the Genome Aggregation Database in East Asian race (gnomAD-EAS), Exome Aggregation Consortium (ExAC), or 1,000 Genomes Project (1000G) databases. Base on the guideline of American College of Medical Genetics and Genomics (ACMG) (17), this mutation has met the pathogenic criteria: 1 moderate (PM2) and 2 supporting (PP2, PP3), which should be classified as a Variant of Uncertain Significance (VUS). According to the scores of in silico predictive analysis tools: FATHMM [−2.26], PolyPhen-2 [0.994], MutationTaster [1], PROVEAN [−7.14], and SIFT [0.002], this mutation was predicted to be detrimental. Given the clinical features and electromyographic findings, the mutated SBF1 gene was considered to be the primary pathogenic factor of CMT4B3. And according to Mendel’s laws of inheritance, this type of mutation should be a dominant mutation.
3.6 Protein analysis
We found that the SBF1 gene was highly conserved at the mutation site in all tested species (Figure 4A). The PhastCons score (1.000) and the phyloP score (4.449) also indicated high conservation. According to the results of PSIPRED 4.0, the 466th mutated amino acid in MTMR5 formed an alpha helix and was located between the dDENN and SBF2 domains (Figure 4B), similar to the wild-type MTMR5 (data source: www.uniprot.org/uniprotkb/O95248/feature-viewer). The three-dimensional structures of the proteins are shown in Figures 4C,D. Amino acid residue 466th may be structurally interconnected with the surrounding amino acid residues (residues nos. 370–377, 454–446, 463–471), whereas the α-helices formed by the latter two sets of residues form a triangular pocket structure with the α-helix formed by the first set residues of dDENN domain, which are located on the outer side of the MTMR5 protein, and may co-exercise dDENN domain function.
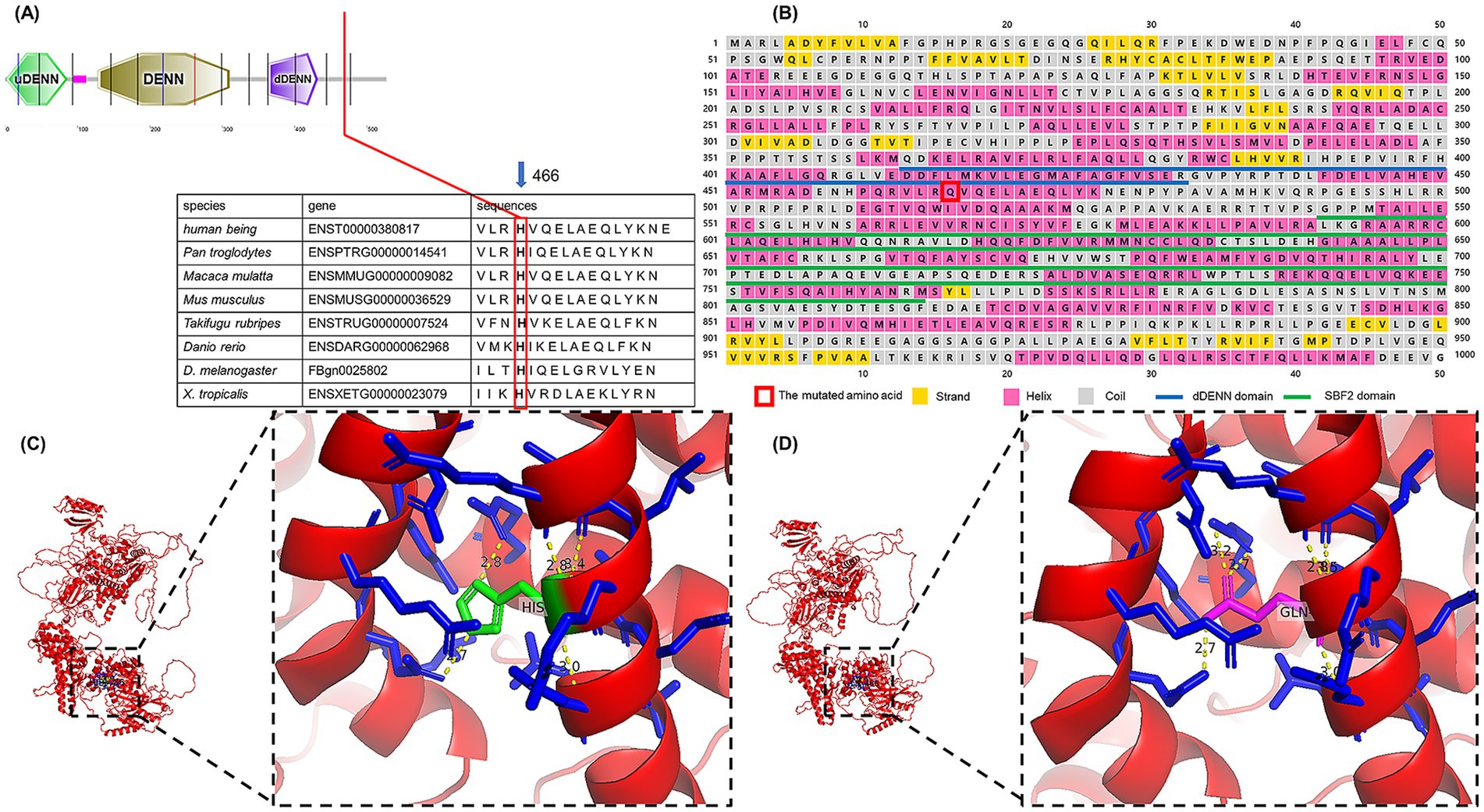
Figure 4. (A) Alignment of amino acid sequences across species. (B) The first 1,000 amino acids were extracted to predict the secondary structure and domain. (C) Wild-type MTMR5 protein; red indicates the protein body, blue indicates residues around the 466th amino acid, green indicates the 466th amino acid, namely His; yellow indicates hydrogen bonds. (D) Mutant MTMR5 protein; the red color indicates the protein body, the blue color indicates the residues around the 466th amino acid, the magenta color indicates the 466th amino acid, namely Gln; the yellow color indicates the hydrogen bonds.
4 Discussion
Here, we describe two patients, a mother and daughter, who suffered from dominant CMT4B3 caused by a novel SBF1 missense mutation. Both patients exhibited muscle atrophy and weakness in the extremities, particularly in the lower limbs, as confirmed by muscle biopsies and electromyography. The mutation site c.1398C > A (p.H466Q) in the SBF1 gene, present on one allele in heterozygotes, was identified using WES and Sanger sequencing. This mutation was predicted to be detrimental, likely affecting the protein’s structure.
Several families with SBF1 variants have been reported to exhibit significant clinical manifestations. Specific information about the mutation is provided in Supplementary Table 2. Both patients in this study were found to have autosomal recessive inheritance and heterogeneous clinical manifestations. Here, we summarize the sites of mutant amino acids in seven previously reported cases along with our variant, as shown in Figure 1D.
In our study, the clinical features of atrophy and weakness in the upper and lower limbs in two patients corresponded to those of CMT4 reported by Nakhro et al. in Korean family (7). However, their electrophysiological findings were distinct. NCVs >38 m/s in the median and ulnar nerves, along with markedly decreased amplitudes in both patients, more closely resembled the phenotype of the axonal form (CMT2) (18, 19). This is consistent with the CMT4B3 cases in other families, except for the Korean family. In addition, cranial nerve neuropathies and syndactyly observed in several familial cases are not consistent with the peripheral neuropathy due to demyelination seen in CMT4B3. Gene mutations in the Korean family may be predicted to be benign or tolerable, suggesting only a mild impact on protein function, whereas mutations in most reported cases were severe enough to cause polyneuropathy of the central nervous system. This large discrepancy in the genotype–phenotype correlation is likely due to specific SBF1 mutations. Moreover, mutations affecting the dDENN motif or nearby amino acid residues have been identified in two families (p.D443N, p.L335P) with syndromic forms of axonal neuropathy (13–16). The novel mutation p.H466Q identified in our study is located near the dDENN motif, which may have contributed to the formation of a similar phenotype (20, 21).
It is unusual for the mutated SBF1 gene to show a pattern of inheritance that deviates from the autosomal recessive manner observed in the seven cases reported before our research. The explanation for dominant inheritance is not clear currently. From a molecular perspective, MTMR5 functions as an adapter for the phosphatase MTMR2, forming a heterodimer through its coiled-coil domain (the C-terminal domain). This interaction regulates MTMR2’s catalytic activity on phosphatidylinositol 3-phosphate [PI (3)P] and influences its subcellular localization (22). It is possible that the mutant MTMR5 forms a heterodimer with MTMR2 that is difficult to depolymerize, producing a dominant negative effect leading to inhibition of the wild-type protein function (23). Additionally, MTMR5 may function as a guanine nucleotide exchange factor (GEF) mediated by the DENN domain, which activates Rab28 by promoting the exchange of GDP to GTP. This process converts inactive GDP-bound Rab proteins into their active GTP-bound forms (24), thereby regulating specific membrane trafficking and vesicular trafficking events (25, 26). The original DENN domain with more divergent domains encircling on its both sides, called uDENN and dDENN, may function together (27). DENNs form a conserved family of Rab GEFs. Based on the predicted protein structure, the mutated amino acid may impact Rab28 protein activation by changing the spatial structure around the DEMM domain, which in turn influences the transportation and conveyance of substances inside and outside the neuron cell (28). Exact pathogenesis requires further investigations in subsequent studies.
5 Conclusion
Here, we report a novel SBF1 missense mutation that causes the autosomal dominant inheritance of CMT4B3 disease within a family. The mother and daughter exhibited neuropathy primarily in the extremities with muscle atrophy, but not in the cranial nerves. Genetic and protein analyses indicate that this SBF1 missense mutation is the main cause of the disease; however, the mechanism underlying its action remains unknown. The identification of autosomal dominant inheritance will expand the genetic spectrum of CMT4B3.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Medical Ethics Committee Zhongnan hospital of Wuhan University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
HL: Conceptualization, Investigation, Software, Writing – original draft. JD: Writing – original draft, Data curation, Validation. ZX: Funding acquisition, Methodology, Resources, Writing – review & editing. LY: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Science and Technology Innovation Special International Science and Technology Cooperation Project of the Hubei Region (2023EHA043), the Medical Technology Innovation Platform Support Project of Zhongnan Hospital (PTXM2021005), and the National Natural Science Foundation of China (81902180).
Acknowledgments
We thank We-Health Biomed. Tech. Co., LTD and Berry Genomics Co., LTD for assistance in sequencing. And we also appreciate Yi Huang, Zhida Shen, Wenjing Chen, Nimra Ghafar Shah for assistance in accomplishment of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2024.1495711/full#supplementary-material
Abbreviations
CMT, Charcot–Marie–Tooth disease; NCV, Nerve conduction velocity; WES, Whole-exome sequencing; SBF1, SET binding factor 1; MTMR, Myotubularin-related protein; OMIM, Online Mendelian Inheritance in Man.
References
1. Tazir, M, Bellatache, M, Nouioua, S, and Vallat, JM. Autosomal recessive Charcot-Marie-tooth disease: from genes to phenotypes. J Peripher Nerv Syst. (2013) 18:113–29. doi: 10.1111/jns5.12026
2. Hoyle, JC, Isfort, MC, Roggenbuck, J, and Arnold, WD. The genetics of Charcot-Marie-tooth disease: current trends and future implications for diagnosis and management. Appl Clin Genet. (2015) 8:235–43. doi: 10.2147/tacg.S69969
3. Bird, TD. Charcot-Marie-tooth hereditary neuropathy overview. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, bean LJH, et al., editors. GeneReviews(®). Seattle, WA: University of Washington, Seattle (1993).
4. Previtali, SC, Quattrini, A, and Bolino, A. Charcot-Marie-tooth type 4B demyelinating neuropathy: deciphering the role of MTMR phosphatases. Expert Rev Mol Med. (2007) 9:1–16. doi: 10.1017/s1462399407000439
5. Bolino, A, Muglia, M, Conforti, FL, LeGuern, E, Salih, MA, Georgiou, DM, et al. Charcot-Marie-tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet. (2000) 25:17–9. doi: 10.1038/75542
6. Senderek, J, Bergmann, C, Weber, S, Ketelsen, UP, Schorle, H, Rudnik-Schöneborn, S, et al. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-tooth neuropathy type 4B2/11p15. Hum Mol Genet. (2003) 12:349–56. doi: 10.1093/hmg/ddg030
7. Nakhro, K, Park, JM, Hong, YB, Park, JH, Nam, SH, Yoon, BR, et al. SET binding factor 1 (SBF1) mutation causes Charcot-Marie-tooth disease type 4B3. Neurology. (2013) 81:165–73. doi: 10.1212/WNL.0b013e31829a3421
8. Mammel, AE, Delgado, KC, Chin, AL, Condon, AF, Hill, JQ, Aicher, SA, et al. Distinct roles for the Charcot-Marie-tooth disease-causing endosomal regulators Mtmr5 and Mtmr13 in axon radial sorting and Schwann cell myelination. Hum Mol Genet. (2022) 31:1216–29. doi: 10.1093/hmg/ddab311
9. Flusser, H, Halperin, D, Kadir, R, Shorer, Z, Shelef, I, and Birk, OS. Novel SBF1 splice-site null mutation broadens the clinical spectrum of Charcot-Marie-tooth type 4B3 disease. Clin Genet. (2018) 94:473–9. doi: 10.1111/cge.13419
10. Gang, Q, Bettencourt, C, Holton, J, Lovejoy, C, Chelban, V, Oconnor, E, et al. A novel frameshift deletion in autosomal recessive SBF1-related syndromic neuropathy with necklace fibres. J Neurol. (2020) 267:2705–12. doi: 10.1007/s00415-020-09827-y
11. Berti, B, Longo, G, Mari, F, Doccini, S, Piccolo, I, Donati, MA, et al. Bi-allelic variants in MTMR5/SBF1 cause Charcot-Marie-tooth type 4B3 featuring mitochondrial dysfunction. BMC Med Genet. (2021) 14:157. doi: 10.1186/s12920-021-01001-1
12. Manole, A, Horga, A, Gamez, J, Raguer, N, Salvado, M, San Millán, B, et al. SBF1 mutations associated with autosomal recessive axonal neuropathy with cranial nerve involvement. Neurogenetics. (2017) 18:63–7. doi: 10.1007/s10048-016-0505-1
13. Romani, M, Mehawej, C, Mazza, T, Mégarbané, A, and Valente, EM. "fork and bracket" syndrome expands the spectrum of SBF1-related sensory motor polyneuropathies. Neurol Genet. (2016) 2:e61. doi: 10.1212/nxg.0000000000000061
14. Bohlega, S, Alazami, AM, Cupler, E, Al-Hindi, H, Ibrahim, E, and Alkuraya, FS. A novel syndromic form of sensory-motor polyneuropathy is linked to chromosome 22q13.31-q13.33. Clin Genet. (2011) 79:193–5. doi: 10.1111/j.1399-0004.2010.01524.x
15. Alazami, AM, Alzahrani, F, Bohlega, S, and Alkuraya, FS. SET binding factor 1 (SBF1) mutation causes Charcot-Marie-tooth disease type 4B3. Neurology. (2014) 82:1665–6. doi: 10.1212/wnl.0000000000000331
16. Mégarbané, A, Dorison, N, Rodriguez, D, and Tamraz, J. Multiple cranial nerve neuropathies, microcephaly, neurological degeneration, and "fork and bracket sign" in the MRI: a distinct syndrome. Am J Med Genet A. (2010) 152a:2297–300. doi: 10.1002/ajmg.a.33417
17. Richards, S, Aziz, N, Bale, S, Bick, D, Das, S, Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
18. Pareyson, D, and Marchesi, C. Diagnosis, natural history, and management of Charcot-Marie-tooth disease. Lancet Neurol. (2009) 8:654–67. doi: 10.1016/s1474-4422(09)70110-3
19. Züchner, S, and Vance, JM. Molecular genetics of autosomal-dominant axonal Charcot-Marie-tooth disease. NeuroMolecular Med. (2006) 8:63–74. doi: 10.1385/nmm:8:1-2:63
20. Verhoeven, K, de Jonghe, P, Coen, K, Verpoorten, N, Auer-Grumbach, M, Kwon, JM, et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-tooth type 2B neuropathy. Am J Hum Genet. (2003) 72:722–7. doi: 10.1086/367847
21. McCray, BA, and Scherer, SS. Axonal Charcot-Marie-tooth disease: from common pathogenic mechanisms to emerging treatment opportunities. Neurotherapeutics. (2021) 18:2269–85. doi: 10.1007/s13311-021-01099-2
22. Kim, SA, Vacratsis, PO, Firestein, R, Cleary, ML, and Dixon, JE. Regulation of myotubularin-related (MTMR)2 phosphatidylinositol phosphatase by MTMR5, a catalytically inactive phosphatase. Proc Natl Acad Sci USA. (2003) 100:4492–7. doi: 10.1073/pnas.0431052100
23. Veitia, RA. Dominant negative factors in health and disease. J Pathol. (2009) 218:409–18. doi: 10.1002/path.2583
24. Yoshimura, S, Gerondopoulos, A, Linford, A, Rigden, DJ, and Barr, FA. Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors. J Cell Biol. (2010) 191:367–81. doi: 10.1083/jcb.201008051
25. Zerial, M, and McBride, H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. (2001) 2:107–17. doi: 10.1038/35052055
26. Behnia, R, and Munro, S. Organelle identity and the signposts for membrane traffic. Nature. (2005) 438:597–604. doi: 10.1038/nature04397
27. Levivier, E, Goud, B, Souchet, M, Calmels, TPG, Mornon, J-P, and Callebaut, I. uDENN, DENN, and dDENN: Indissociable domains in Rab and MAP kinases signaling pathways. Biochem Biophys Res Commun. (2001) 287:688–95. doi: 10.1006/bbrc.2001.5652
Keywords: CMT4B3, SBF1, MTMR5, autosomal dominant, gene mutation
Citation: Liu H, Dong J, Xie Z and Yu L (2024) A novel SBF1 missense mutation causes autosomal dominant Charcot–Marie–Tooth disease type 4B3. Front. Neurol. 15:1495711. doi: 10.3389/fneur.2024.1495711
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Chiara Reale, IRCCS Carlo Besta Neurological Institute Foundation, ItalyFrancesca Luisa Conforti, University of Calabria, Italy
Copyright © 2024 Liu, Dong, Xie and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xie Zhe, eGllemhlQHpuaG9zcGl0YWwuY29t; Yu Li, eXVsaXh4QDE2My5jb20=
†These authors have contributed equally to this work