
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 18 September 2024
Sec. Pediatric Neurology
Volume 15 - 2024 | https://doi.org/10.3389/fneur.2024.1484752
 Osama Y. Muthaffar1,2†
Osama Y. Muthaffar1,2† Anas S. Alyazidi1,2*†
Anas S. Alyazidi1,2*† Daad Alsowat3Abdulaziz A. Alasiri3Raidah Albaradie4
Daad Alsowat3Abdulaziz A. Alasiri3Raidah Albaradie4 Lamyaa A. Jad5Husam Kayyali6Mohammed M. S. Jan1,2†
Lamyaa A. Jad5Husam Kayyali6Mohammed M. S. Jan1,2† Ahmed K. Bamaga1,2†Mohammed A. Alsubaie2†Rawan Daghistani7
Ahmed K. Bamaga1,2†Mohammed A. Alsubaie2†Rawan Daghistani7 Saleh S. Baeesa8†
Saleh S. Baeesa8† Meshari A. Alaifan2Abdelhakim Makraz6
Meshari A. Alaifan2Abdelhakim Makraz6 Abrar N. Alsharief2
Abrar N. Alsharief2 Muhammad Imran Naseer9,10*†
Muhammad Imran Naseer9,10*†Background: Drug-resistant epilepsy (DRE) impacts a significant portion, one-third, of individuals diagnosed with epilepsy. In such cases, exploring non-pharmacological interventions are crucial, with the ketogenic diet (KD) standing out as a valuable option. KD, a high-fat and low-carb dietary approach with roots dating back to the 1920s for managing DRE, triggers the formation of ketone bodies and modifies biochemistry to aid in seizure control. Recent studies have increasingly supported the efficacy of KD in addressing DRE, showcasing positive outcomes. Furthermore, while more research is needed, limited data suggests that KD May also be beneficial for specific genetic epilepsy syndromes (GESs).
Objective: This study aimed to assess the short-term efficacy of KD among pediatric patients diagnosed with GESs.
Materials and methods: This is a multi-center retrospective analysis of pediatric patients with GESs diagnosed using next-generation sequencing. The enrolled patients followed the keto-clinic protocol, and the KD efficacy was evaluated at 3, 6, and 12-month intervals based on seizure control and compliance. The collection instrument included demographic, baseline, and prognostic data. The collected data was coded and analyzed promptly.
Results: We enrolled a cohort of 77 patients with a mean current age of 7.94 ± 3.83 years. The mean age of seizure onset was 15.5 months. Notably, patients experienced seizures at a younger age tended to have less positive response to diet. Overall, 55 patients responded favorably to the diet (71.4%) while 22 patients (28.6%) showed no improvement. Patients with genetic etiology showed a significantly more favorable responses to the dietary intervention. Patients with Lennox–Gastaut syndrome showed the most significant improvement (14/15) followed by patients with Dravet syndrome (6/8), and West syndrome (3/4). The number of used anti-seizure medications also played a significant role in determining their response to the diet. While some patients experienced mild adverse events, the most common being constipation, these occurrences were not serious enough to necessitate discontinuation of the diet.
Conclusion: The study revealed a high improvement rate in seizure control, especially among younger patients and those with later seizure onset. The success of dietary treatment hinges greatly on early intervention and the patient’s age. Certain genetic mutations responded favorably to the KD, while efficacy varied among various genetic profiles.
Drug-resistant epilepsy (DRE) can constitute a third of all patients with epilepsy (1, 2), and it has been defined as the failure of adequate trials of two tolerated, appropriately chosen and used anti-seizure medication (ASM) schedules as monotherapy or combined therapy to achieve sustained control of seizures (3). In children with DRE, prompt referral to specialized centers for evaluation is required to consider other treatment modalities, including surgical resection of the seizure focus, vagus nerve stimulators, or implantable brain neurostimulators (4, 5). For individuals who are not suitable candidates for surgery, an alternative treatment modality such as the ketogenic diet (KD) can be considered (4, 6).
The KD is a high-fat, low-carbohydrate, and low-protein diet with restricted calories and fluids that mimic the metabolism of the fasting state to induce ketone bodies formation (7–9). Multiple processes that cause biochemical alterations are associated with KD, such as mediators responsible for neuronal hyperexcitability and cellular substrates (6). From a historical perspective, KD has been in clinical use since the early 1920s to control seizure in patients with DRE; Fasting and other dietary treatments have been in use to treat epilepsy since at least 500 BC (7, 10, 11). Furthermore, the Hippocratic collection recorded fasting as a therapeutic measure against epilepsy (12). There has been a notable resurgence in the global and regional adoption of such dietary treatments (10, 13–15). On this burgeoning list of diets, KD has received extensive interest owing to its beneficial effects, with emerging data supporting its use (16, 17). A recent systematic review of 14 randomized controlled trials (RCTs) that included 1,114 children to measure the efficacy of KD in DRE concluded that KD is an effective treatment modality for DRE in children and adolescents (18). Other data suggest a significantly positive outcome in using KD for DRE in children (19–22). Furthermore, within a more targeted context, a recent meta-analysis examined the effectiveness of different types of KD in the short term. The findings indicated that all evaluated diets played a significant role, showcasing distinct variations among the various treatment modalities (23).
Nonetheless, in terms of genetic epilepsy syndromes (GESs), very little data has been presented to assess the efficacy of KD. In one study, KD was particularly effective in patients with specific epilepsy-causing genetic mutations but lacked effectiveness among other epilepsy-causing mutations (24). Additionally, a prospective series of 57 children with genetic, structural, and metabolic abnormalities showed that KD could be an effective intervention for some patients with specific GESs, including Dravet syndrome, Lennox–Gastaut syndrome (LGS), and Wolf-Hirschhorn syndrome, which was responsive despite its highly refractory nature to all other therapies (25). Nevertheless, comprehensive data from larger cohorts can offer more conclusive insights into the suitability of the KD for patients with GESs. This study aims to emphasize the short-term effectiveness of the KD among pediatric patients diagnosed with GESs.
This study involves a multi-center retrospective analysis of pediatric patients (<14 years) diagnosed with GESs and known genetic mutations. Genetic diagnosis was confirmed using next-generation sequencing (NGS) following adherence to appropriate ethical and logistical protocols. Neuroradiological investigations, including electroencephalography (EEG) and magnetic resonance imaging (MRI), were reviewed to assist with epilepsy syndrome diagnosis. EEG was performed in all but three patients. MRI was performed on the majority of patients for various clinical indications. The participating centers, located in the Gulf region, included patients from establishments in Saudi Arabia and Qatar to ensure sample diversity. Selection of centers was based on the availability of KD facilities and programs. Patients were classified according to syndrome and etiology utilizing the 1989 and more recent 2017 International League Against Epilepsy (ILAE) classification systems (26). The medical records of patients with DRE (defined as failure to respond to two or more ASMs) were retrospectively reviewed, and those meeting our eligibility criteria from the period between January and June 2024 were enrolled. The study adhered to strengthening the reporting of observational studies in epidemiology (STROBE) guidelines for retrospective studies (27).
Patients were recruited following the keto-clinic protocol (Figure 1), with exclusion criteria applied to those with absolute contraindications to the diet. The initiation of the KD was gradual, without fasting, for outpatients, except for cases where the patient is under 1 year of age, in critical condition, or the family requires training to monitor the diet’s response. Each patient’s dietary plan, based on their caloric requirements, was carefully crafted using KD food recipes combined with a ketogenic formula. The administration of KD was overseen by a multidisciplinary care team comprising pediatricians neurologists and licensed and specialized KD dietitians. Alongside KD as the primary treatment approach, participants received standard care provided to all patients in the ward. Two key parameters, baseline and prognostic data, were documented to assess the efficacy of KD among the participants. We categorized the effectiveness of KD in the patients into three categories: responders (≥50% seizure control), non-responders (<50% seizure control), and seizure freedom after a follow-up at 3, 6, and 12 months. Patients were labeled as responders whenever achieving ≥50% seizure control at any stage of the KD intervention. The genetic mutations included ion channels (calcium channels: CACNA1E, CACNA1A, CACNA1H, sodium channels: SCN1A, SCN2A, SCN9A, SCN8A, and potassium channels: KCNQ2, KCNB1), transporters and metabolism (ABCA2, SLC2A1) enzymes and enzyme modulators (ATP6, ATP6V1A, AMT, PAFAH1B1, PPP3CA, PRUNE1, RARS2, ACADM), structural and scaffold proteins (ADAM22, ATL1, COL4A2, UNC80, WDR62), receptors and signal transduction (CNTNAP2, GRIN2A), synaptic function and neurotransmission (CREBBP, PIK3CA), developmental and transcription regulators (KIF1A, DOCK7, EXOC8, FGF12, FGF20, KDM5B, KMT2D, POGZ), mitochondrial and metabolic disorders (DNM1L, PDHA1, PEX3), and miscellaneous genes (STRADA, TSC1, TSC2, NCL Type 6, GLS, FAM111A, NR2FA). Clinical syndromes included LGS, Dravet syndrome, West syndrome, GLUT1 deficiency syndrome, tuberous sclerosis, Bosch-Boonstra-Schaaf syndrome, Congenital Lipomatous Overgrowth, Vascular Malformations, Epidermal Nevis, Spinal/Skeletal Anomalies/Scoliosis (CLOVES)/Megalencephaly-Capillary Malformation (MCM) syndrome, Angelman syndrome, Pitt-Hopkins-like syndrome-1, Landau–Kleffner syndrome, White Sutton syndrome, and unclassifiable encephalopathy with genetic mutations.
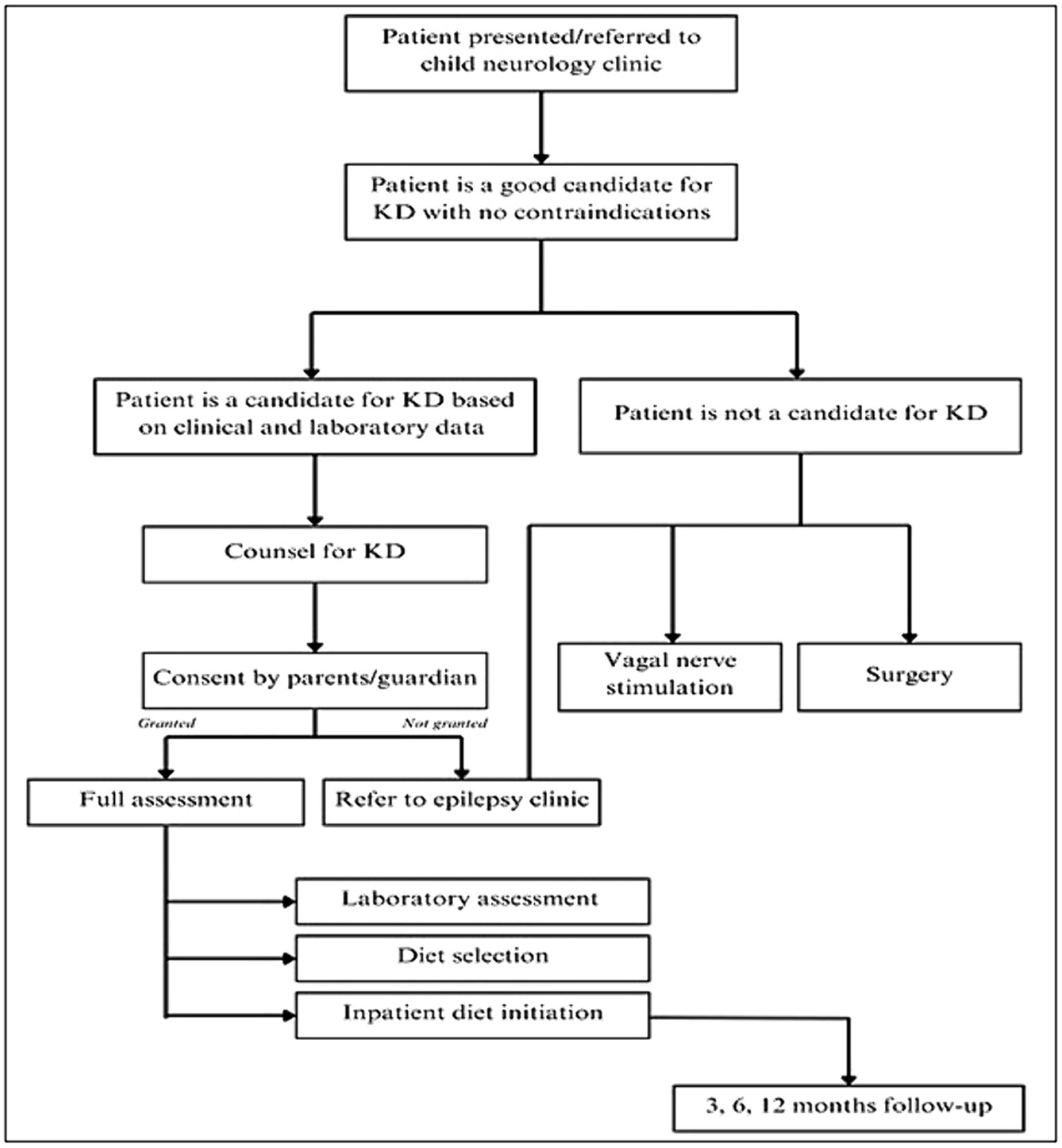
Figure 1. Algorithm of the keto-clinic protocol adopted in the study’s institutions.
Genetic mutations in ion channels, such as CACNA1E, CACNA1A, and CACNA1H (calcium channels), as well as SCN1A, SCN2A, SCN9A, SCN8A (sodium channels), and KCNQ2, KCNB1 (potassium channels), were associated with Dravet syndrome, West syndrome, and LGS. Mutations in transporters and metabolism genes including ABCA2 and SLC2A1 were associated with GLUT1 deficiency syndrome. Enzyme-related genes such as ATP6, ATP6V1A, AMT, PAFAH1B1, PPP3CA, PRUNE1, RARS2, and ACADM were associated with metabolic disorders and unclassifiable encephalopathy. Receptor and signal transduction mutations in CNTNAP2 and GRIN2A were related to Landau–Kleffner syndrome and Pitt-Hopkins-like syndrome-1. Synaptic function and neurotransmission genes like CREBBP and PIK3CA were associated with CLOVES/MCM syndrome and Bosch-Boonstra-Schaaf syndrome. Developmental and transcription regulators such as KIF1A, DOCK7, EXOC8, FGF12, FGF20, KDM5B, KMT2D, and POGZ were implicated in syndromes like Angelman syndrome and White Sutton syndrome. Mitochondrial and metabolic disorder genes, including DNM1L, PDHA1, and PEX3, contributed to various unclassifiable encephalopathies. Lastly, miscellaneous genes like STRADA, TSC1, TSC2, NCL Type 6, GLS, FAM111A, and NR2FA were linked to conditions such as tuberous sclerosis and other genetic encephalopathies.
The assessment of KD compliance was conducted through dietary recall during follow-up visits. The primary outcome focused on changes in seizure frequency at 3, 6, and 12 months compared to the baseline before initiating the KD.
DNA capture probes were used alongside NGS-based copy number variation analysis with Illumina arrays. The coding regions of the gene, 10 bp of flanking intronic sequences, and known pathogenic/likely pathogenic variants within the gene (coding and non-coding) were targeted for analysis. Data analysis, variant calling, and annotation was performed using the Torrent Variant Caller software. The children’s variant inheritance mode was compared to the parents’ exome sequencing results upon availability of the data. Furthermore, after identifying a variant, the interpretation followed the 5-tier classification system recommended by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (28). According to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology classification system, pathogenic or likely pathogenic variants were selected as causative mutations for epileptic activity (28).
The data collection instrument included various sections. The first included patients’ demographic data, which included gender, genetic and clinical diagnosis, current age, age of seizure onset, and the age of the KD initiation. The second section comprised the patients’ baseline clinical data, categorized based on the timing of KD initiation. The variables encompassed seizure etiology as per the ILAE Task Force guidelines for consistency in reporting, seizure frequency, characteristics, prior ineffective ASM treatments (before KD initiation), and the percentage of seizure control achieved. In the third section, the patients’ prognostic data were examined, focusing on the clinical outcomes post-KD initiation. This included assessing parameters such as seizure frequency, semiology, the number of ASMs utilized, and the evaluation of seizure control at 3, 6, and 12 months during follow-up visits. We also assessed the KD feeding route, patient’s adherence and reasons for discontinuation, and adverse events (if any) post-KD initiation. Also, the electrographic (i.e., EEG) and neuroradiological (i.e., MRI) data were collected upon availability for each patient and emphasized abnormal findings.
The collected data was coded before the head, and IBM SPSS Statistics for Windows, version 27 (IBM Corp., Armonk, NY, USA) was used to analyze data. The sample size was not determined precisely because the subjects were n > 12, as previously suggested in the literature (29). In our sampling technique, we adopted Julious formulation (2005), which suggests a minimum sample size of 12 subjects per treatment arm (29). Categorical variables were represented using frequencies and percentages. For continuous variables, measures of central tendency were calculated. A paired samples t-test was employed to compare the mean seizure frequency before the start of the diet and 12 months after the start of the diet within each individual. Univariate binary logistic regression analysis was performed to identify factors significantly associated with the improvement of seizures. Potential confounders, including age at the time of the study, age at keto diet initiation, age at seizure onset, gender, seizure etiology, number of ASMs used, and route of feeding, were included in a multivariate regression model to adjust for their effects on the observed control in seizure frequency. Due to the adjuvant nature of KD in treating children with DRE, medication is unavoidable. As ASMs May be a potential factor influencing the outcome, we considered limiting such confounders in the statistical analysis. The results were presented as odds ratios (OR) with corresponding 95% confidence intervals (CI). A p-value of less than 0.05 was considered statistically significant.
Following our study procedure and protocols, 77 patients were included. Their mean current age is 7.94 ± 3.83 years. The group of responders had a mean current age of 7.58 ± 3.49 years, comprising 55 individuals, which accounted for 71.4% of the total. Notably, the mean age of seizure onset among responders was higher, at 22 months. In contrast, non-responders had a mean current age of 8.82 ± 4.53 years, with 22 individuals making up 28.6% of the group. The mean age of seizure onset for non-responders was 9 months, indicating an earlier onset. There was a slightly higher number of male patients in the study, totaling 42 individuals and representing 54.6% of the total study cohort. Females constituted 35 patients, comprising of 45.5%. In terms of respondents, 30 male patients achieved seizure improvement (≥50 seizure control), comprising 71.4% of the total male patients. There were 25 female patients achieving improvement and 10 with no improvement. The majority of our patients experienced monthly seizures after the start of the diet; of those, 89.3% achieved seizure improvement with the diet and had been experiencing either daily or weekly episodes of seizure prior to the initiation of the diet. Out of the 22 patients with weekly seizures, 59.1% showed improvement in their seizure frequency following the diet. Patients who experienced daily seizures both before and after the dietary intervention had the highest rate of lack of improvement (66.7%), with their seizure frequency remaining unchanged despite the diet. Interestingly, 33.3% of these patients who continued to have daily seizures did report reduction or control in their seizure activity. An inverse relationship was noted between the number of antiseizure medications (ASMs) used and the rate of seizures improvement. Patients who did not use any ASMs or were on a single medication showed a 100% improvement rate on the dietary intervention. Among those on two medications, 80.0% experienced improvement, while patients using three or more medications achieved improvement in 63.0% of cases. We observed a 70.3% improvement in patients with abnormal EEG results, while 73.2% of those with abnormal brain MRI findings showed improvement with the dietary intervention. In Table 1, detailed information on improvement rates based on clinical data are presented (Table 1). The trend of seizure control based on the gender of our patients across the three intervals of our study is demonstrated in Figures 2–4.
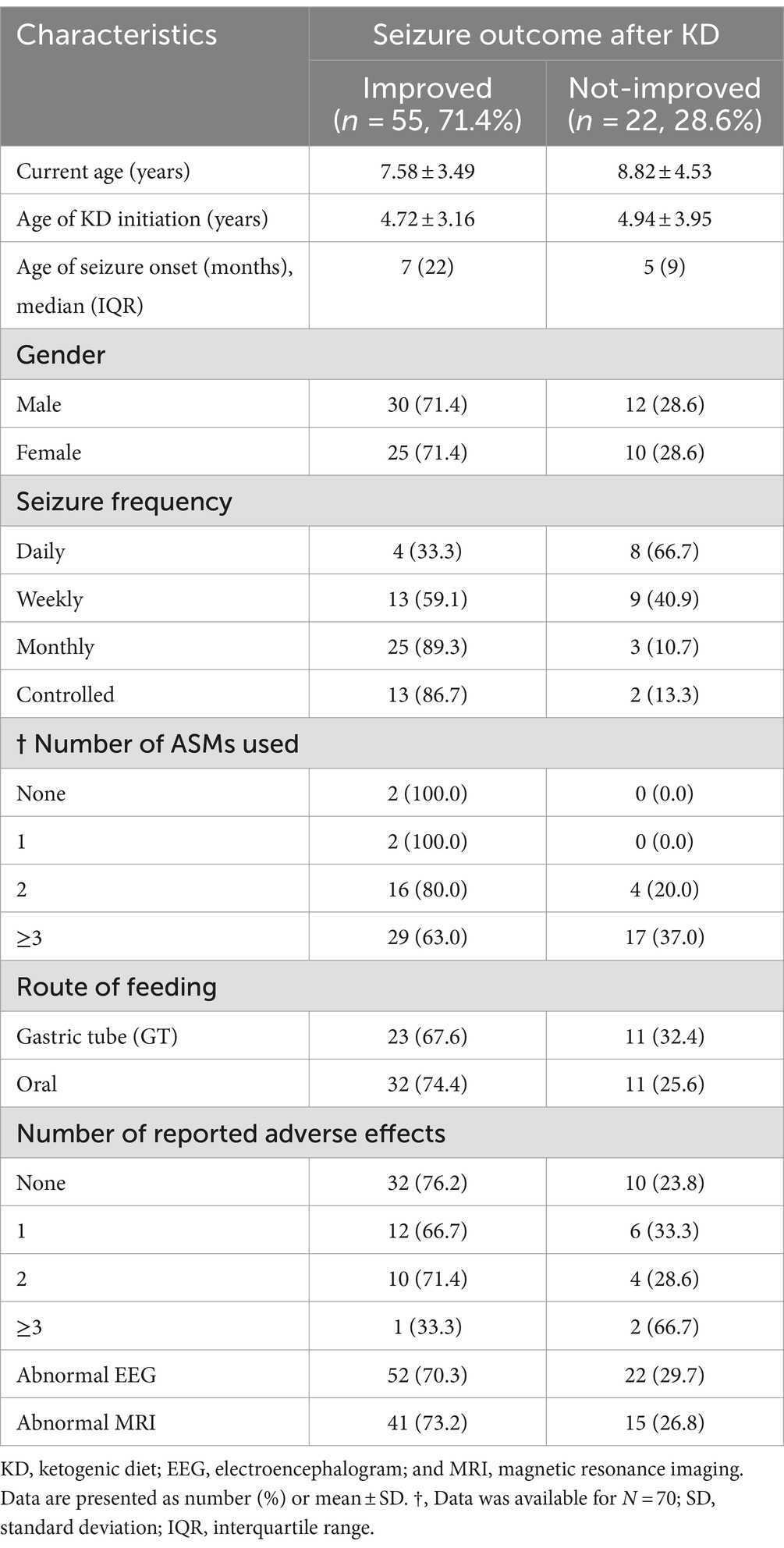
Table 1. Sociodemographic and clinical characteristics of children on KD: improved vs. not-improved seizure outcomes (n = 77).
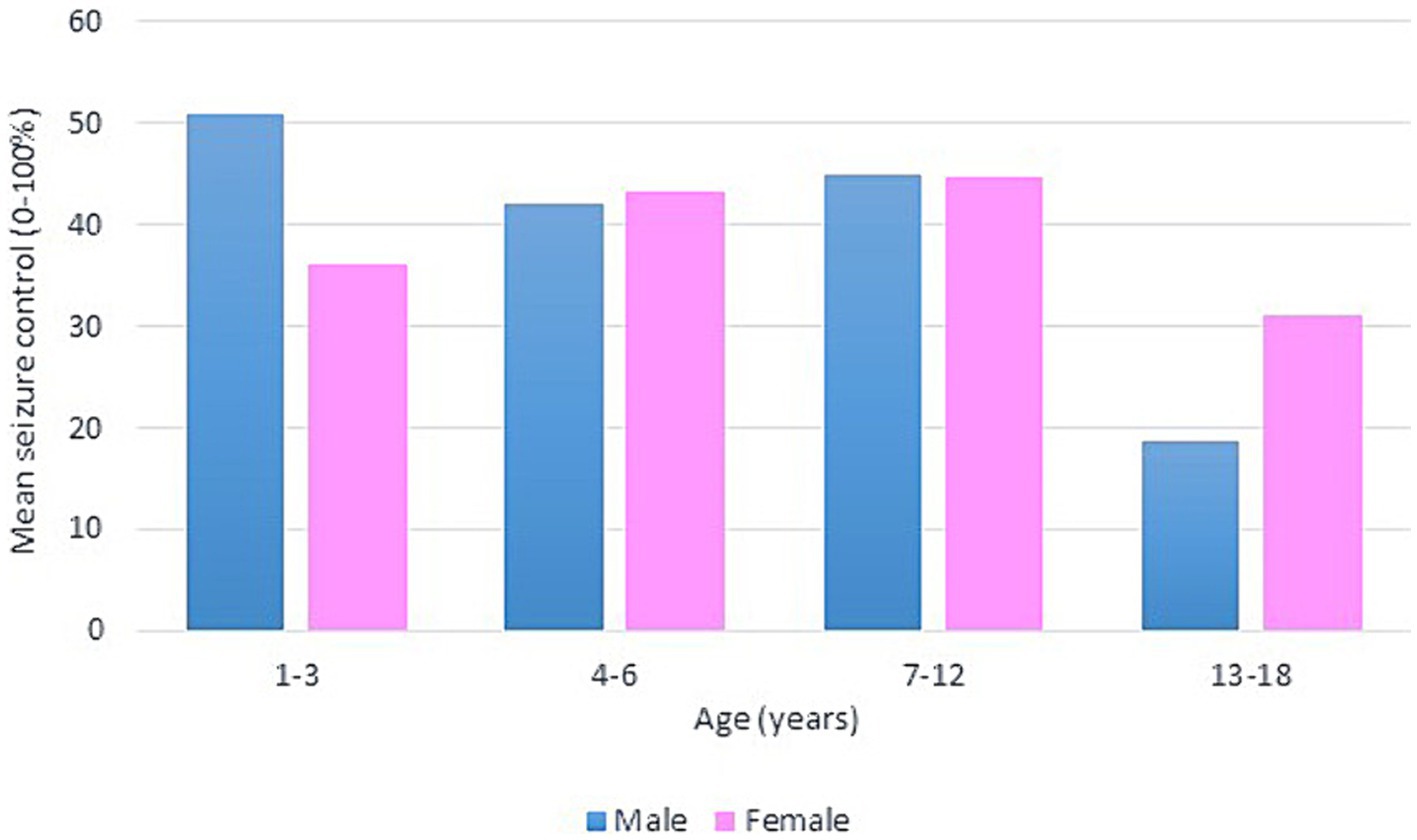
Figure 2. Bar chart of mean seizure control (0–100%) after 3 months of KD, by age group and gender.
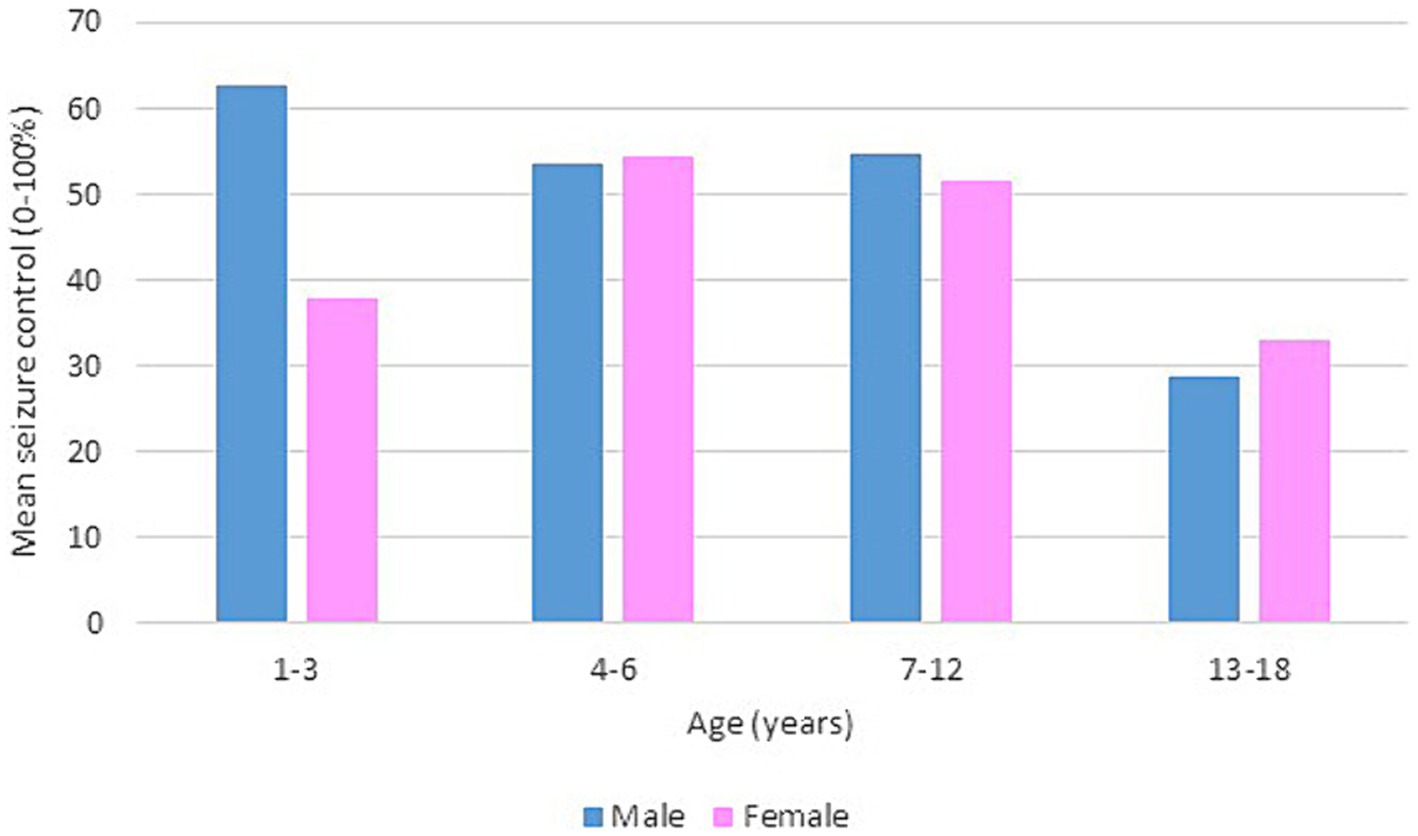
Figure 3. Bar chart of mean seizure control (0–100%) after 6 months of KD, by age group and gender.

Figure 4. Bar chart of mean seizure control (0–100%) after 12 months of KD, by age group and gender.
As to the etiology of the seizure, patients were designated into on the following groups; genetic, structural, metabolic disorder, immune disorder, infectious, or unknown causes. The genetic etiology group had the largest number of patients, with individuals in this group representing 78.8% of the total. Patients with structural etiology showed improvement in 66.7% of the cases, while individuals with metabolic and unknown etiologies experienced a higher rate of no improvement compared to those who showed improvement (Table 2).
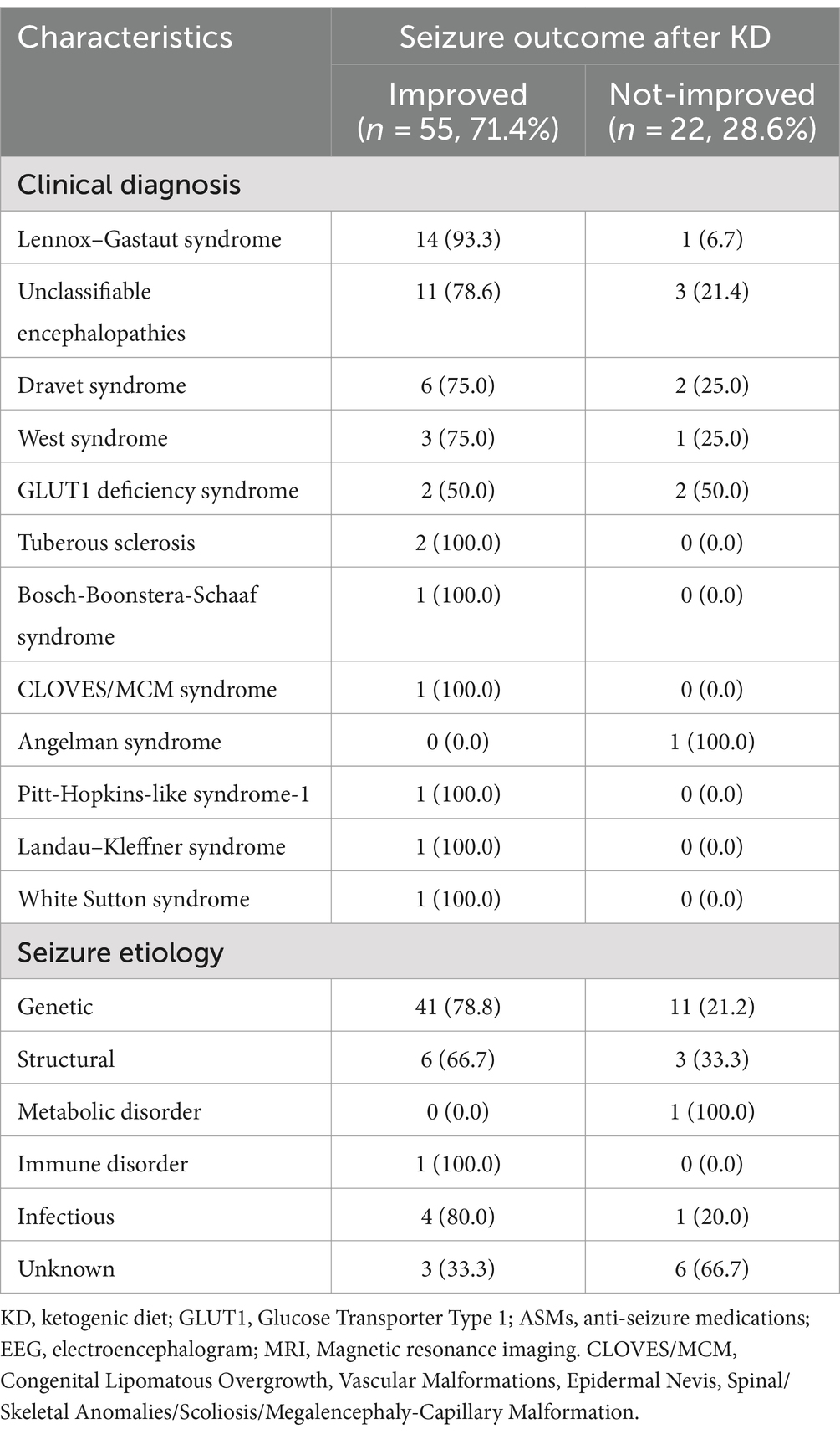
Table 2. Clinical diagnosis and seizure etiology of children on KD: improved vs. not-improved seizure outcomes (n = 77).
Regarding the clinical diagnosis, the majority of patients were diagnosed with LGS (n = 15, 19.5%), followed by unclassifiable encephalopathies (n = 14, 18.1%), Dravet syndrome (n = 8, 10.4%), West syndrome (n = 4, 5.2%), glucose transporter type 1 (GLUT1) deficiency syndrome (n = 4, 5.2%), and others described in Table 2. Regarding improvements to the diet, 93.3% of patients diagnosed with LGS achieved seizure improvement at a specific interval (i.e., 3, 6, or 12 months) of their diet intake. While patients with unclassifiable encephalopathies have a 78.6% improvement, followed closely by patients with Dravet syndrome who had improvement among 75.0% of the patients. Among the conditions where 100.0% of the patients showed improvement were tuberous sclerosis, Bosch-Boonstera-Schaaf syndrome, CLOVES/MCM syndrome, Pitt-Hopkins-like syndrome-1, Landau–Kleffner syndrome, and White Sutton syndrome. However, we only recruited one patient for each condition except for tuberous sclerosis. Other diagnoses are described in Table 2.
When evaluating patient responses based on their genetic background, notable findings emerged, indicating that individuals with LGS exhibited the most significant improvement in seizure control. The assessment was conducted at 3, 6, and 12-month intervals. Initially, 12 out of 15 patients demonstrated improved seizure control during the first interval, as outlined in the methodology. Subsequently, one more patient showed improvement in the second interval, followed by another patient in the third interval. Throughout our observation period, the total number of respondents who experienced improved seizures within the LGS group reached 14 out of 15 patients.
The genetic mutations observed in these patients included SCN8A, CACNA1H, and KIF1A. In the initial interval, 5 out of 8 patients with Dravet syndrome were classified as responders, a proportion that remained consistent in the subsequent two intervals, with 6 out of 8 patients showing positive response to treatment. The primary genetic mutations identified in Dravet syndrome cases were SCN1A, followed by SCN2A and SCN9A. Within the cohort of four patients diagnosed with West syndrome, initially two patients responded to treatment, with a third patient showing a positive response in the subsequent interval. Among the responders with West syndrome, three out of four patients were identified, with mutations detected in EXOC8 and PEX3 genes. In patients with GLUT1 deficiency syndrome, two individuals displayed a positive response without any increase in the number of responders in the following months. More detailed information on other conditions can be found in Table 3.
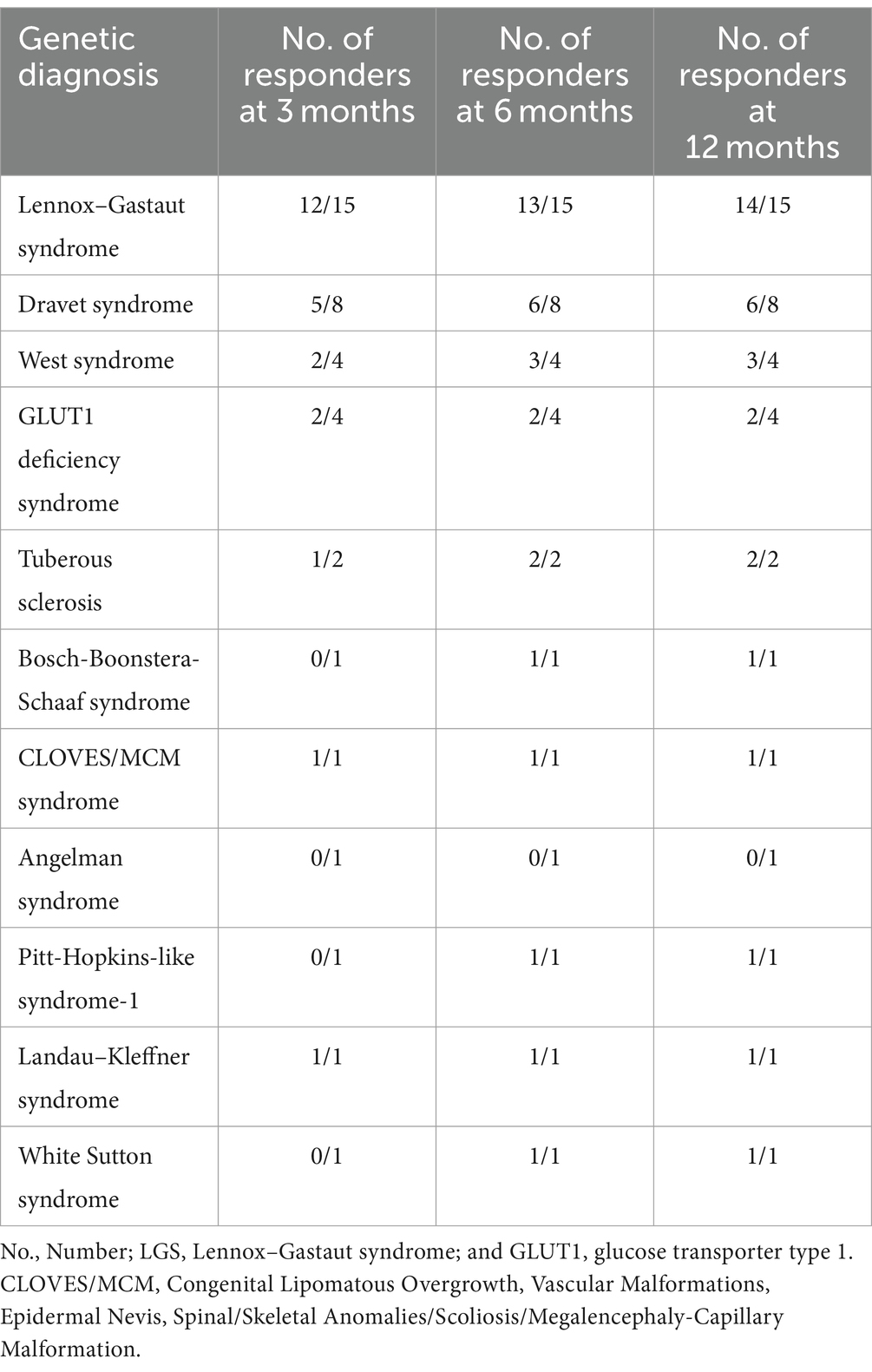
Table 3. Number of responders by genetic diagnosis at 3, 6, and 12 months.
We assessed seizure control across different etiologies in Table 4. A significant p-value was observed in patients with genetic and structural etiologies, p < 0.001 and p < 0.047, respectively. This indicates a positive response to the dietary intervention among patients with these two etiologies. In detail, the mean number of seizures in patients with genetic etiology prior to the diet was 18.752 ± 19.80 percent. Which notably rose to 60.77 ± 26.10 percent after initiating the diet. Similarly, those with structural etiology, had a mean control of 22.00 ± 23.48 percent before the diet, increasing to 55.00 ± 35.43 percent following the diet. Patients with infectious and unknown etiologies did not show significant improvement following the dietary intervention. Specifically, individuals with unknown etiology had mean control rates of 13.33 ± 9.35 percent before the diet initiation, increasing to 30.00 ± 28.72 percent after the diet (p = 0.092). Similarly, patients with infectious etiology displayed mean control rates of 11.00 ± 11.40 percent before the diet, slightly rising to 47.00 ± 30.94 percent after the diet (p = 0.102). The analysis of the KD’s impact on various genetic mutations revealed varying levels of improvement across gene groups. Although most groups exhibited some degree of improvement, none reached statistical significance (Table 5). Specifically, 83.3% of patients with ion channel mutations and all patients in the miscellaneous genes group showed improvement. Similarly, both the receptors and signal transduction, as well as the synaptic function and neurotransmission groups, exhibited 100% improvement, though these groups had limited sample sizes. The enzymes and enzyme modulators group showed a non-significant trend toward fewer improvements (42.9%, p = 0.075).
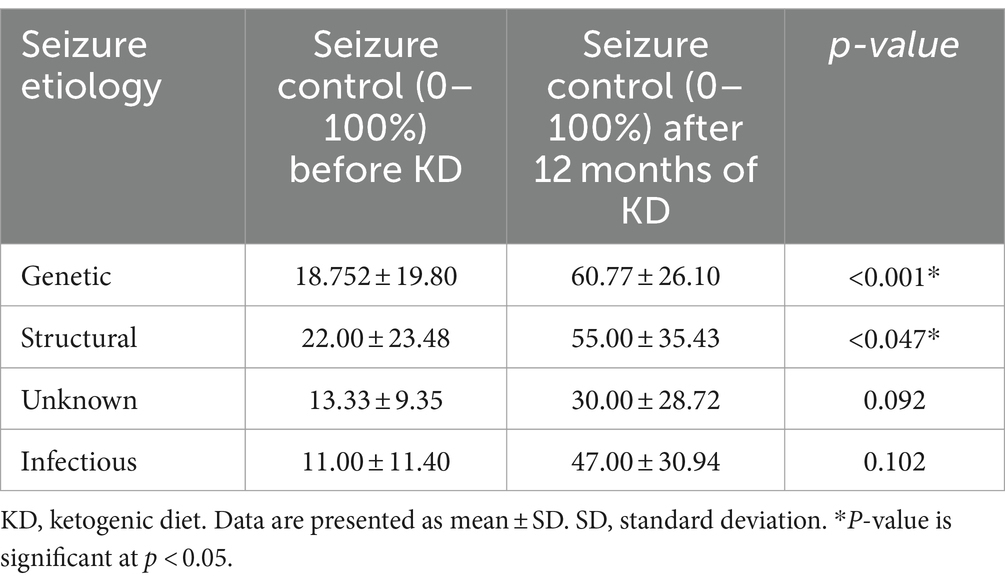
Table 4. Seizure control rate before and after 12 months of the start of KD according to seizure etiology.
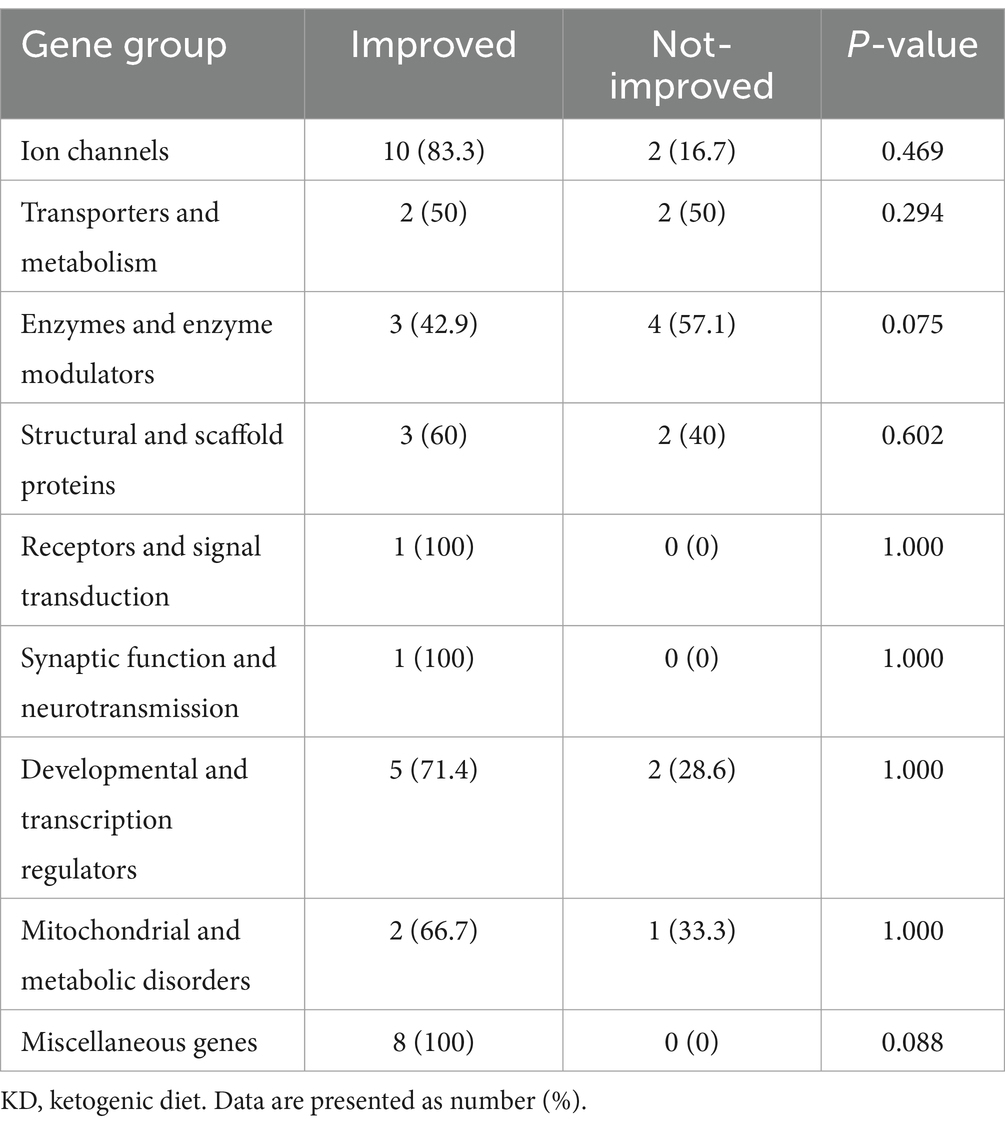
Table 5. Impact of the KD on patient outcomes across various genetic mutations.
We conducted a univariate and multivariate logistic regression analysis of factors contributing to seizure control (Table 6). Among the independent variables were their current age, age of KD initiation, age of seizure onset, male gender, seizures of genetic etiology, the number of used ASMs grouped into patients with none, one, or two medications into one group and patients with three or more medications, and oral feeding as a route of feeding. The groups with a significant seizure control after the diet using the univariate logistic regression included those with seizures of genetic etiology (p = 0.041; OR, 2.929; CI 95%, 1.043–8.226). After adjusting the model for common confounders (age at the time of the study, age of KD initiation, age of seizure onset, gender, seizure etiology, number of ASMs, and route of feeding), we found two independent factors significantly affecting the seizure control. They included seizures of genetic etiology (p = 0.021; OR, 5.633; CI 95%, 1.294–24.531) and the number of used ASMs (p = 0.037; OR, 0.217; CI 95%, 0.052–0.911). There was an inverse relationship between the number of ASMs used and the response to the dietary intervention, with patients taking three or more ASMs showing a higher likelihood of a suboptimal response. Among the 35 patients who reported adverse events, 18 experienced a single adverse event, 14 had two types of adverse events, and three reported three adverse events. The most commonly reported adverse event was constipation (n = 20, 26.0%), followed by irritability (n = 11, 14.3%) and nausea (n = 7, 9.1%). Additional reported events included vomiting, weight loss, fatigue, hypoglycemia, nutritional deficiencies, kidney stones, and pancreatitis.
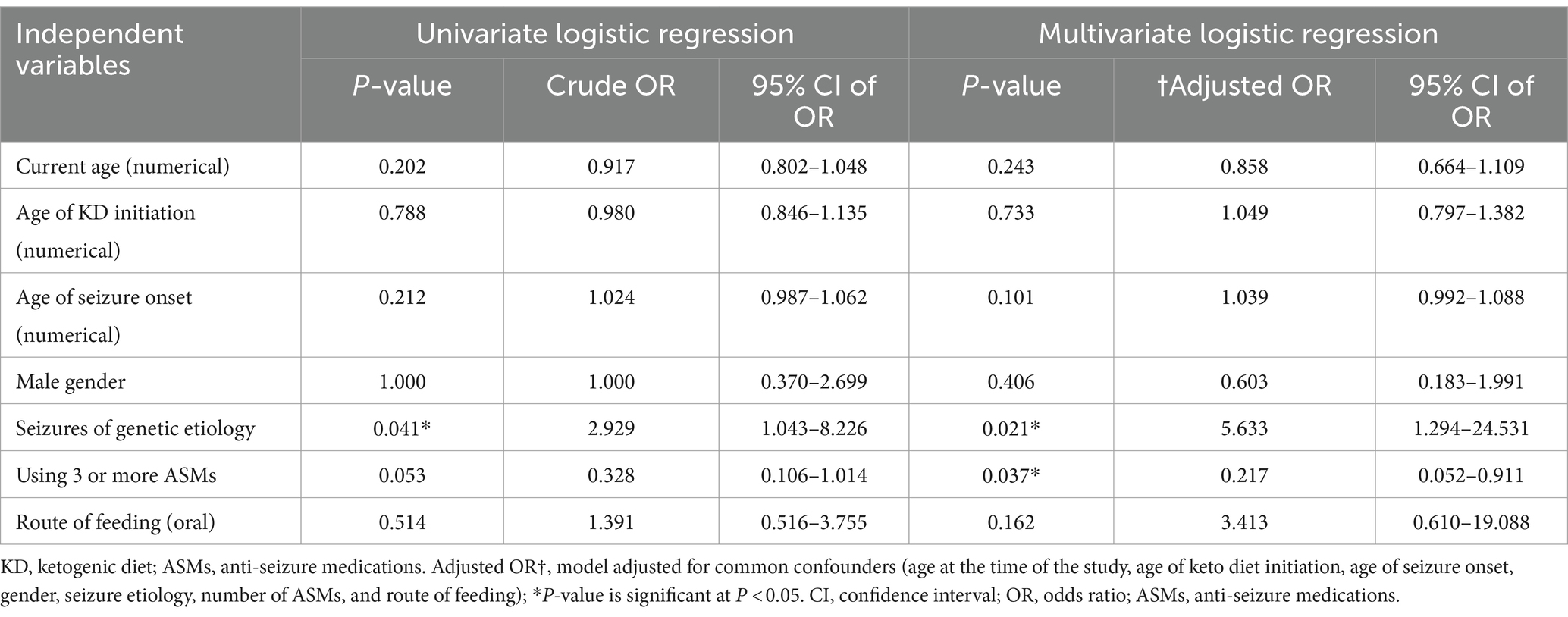
Table 6. Univariate and multivariate logistic regression analysis of factors contributing to seizure control.
To the best of our knowledge, this is the first study conducted in the Arab region to evaluate the effectiveness and tolerability of the KD among patients with GESs. This research focused on analyzing seizure outcomes concerning socio-clinical determinants and diet-related factors. The study encompassed patients from six centers across Saudi Arabia and Qatar, diagnosed with a variety of conditions, such as LGS, Dravet syndrome, West syndrome, GLUT1 deficiency syndrome, tuberous sclerosis, Bosch-Boonstra-Schaaf syndrome, CLOVES/MCM syndrome, Angelman syndrome, Pitt-Hopkins-like syndrome-1, Landau–Kleffner syndrome, and White Sutton syndrome.
Our findings revealed that younger patients exhibited a more positive response to the KD compared to older patients, regardless of their clinical characteristic or the age at which they began the diet. The overall improvement rate, assessed using seizure reduction, among patients after initiating the diet was notably high at 71.43%. This is higher than previously reported studies which suggest 44–66.7% seizure reduction among children with DREs (30, 31).
Additionally, patients who responded favorably to the KD tended to have a later onset of their first seizure episode. This observation is consistent with existing knowledge that an earlier onset of seizures is generally associated with poorer outcomes (32). Our study supports the view that the timing of seizure onset and patient age are crucial factors in predicting the effectiveness of dietary interventions in managing GESs.
Our analysis did not find a significant association between gender and response to the KD. However, gender differences in seizure susceptibility and other epileptogenic processes have been reported in the literature, with males often exhibiting greater seizure susceptibility than females (33, 34). In our study, gender appeared to be a confounding factor that, in combination with other clinical variables, that May contribute to seizure control.
After controlling for potential confounders such as gender, current age, age of seizure onset, age of KD initiation, seizure etiology, number of ASMs used, and route of feeding, two variables—genetic etiology and the number of ASMs used—were significantly associated with higher seizure control (see Table 6). This aligns with findings by Ko et al., who reported the efficacy of KD among GESs, specifically in patients with SCN1A, KCNQ2, STXBP1, and SCN2A mutations, though less effective in those with CDKL5 mutations (24). Similarly, we observed seizure control in specific GESs, including Dravet syndrome (mainly caused by SCN1A mutations) and LGS, which can involve mutations in CDKL5, DNM1, STXBP1, or SCN2A genes. A meta-analysis by Wang et al. further supports KD’s use in GESs, particularly Dravet syndrome (35). However, our sub-analysis of the genetic mutation revealed no statistically significant relationship between specific mutations and seizure improvement after initiating the KD. Nonetheless, this could be attributed to the small number of patients carrying each mutation whereas a larger sample can potentially lead to statistical association. In Table 7, we summarized the literature findings regarding the effectiveness of KD in genetic epilepsies and certain genetic mutations (24, 36–45).
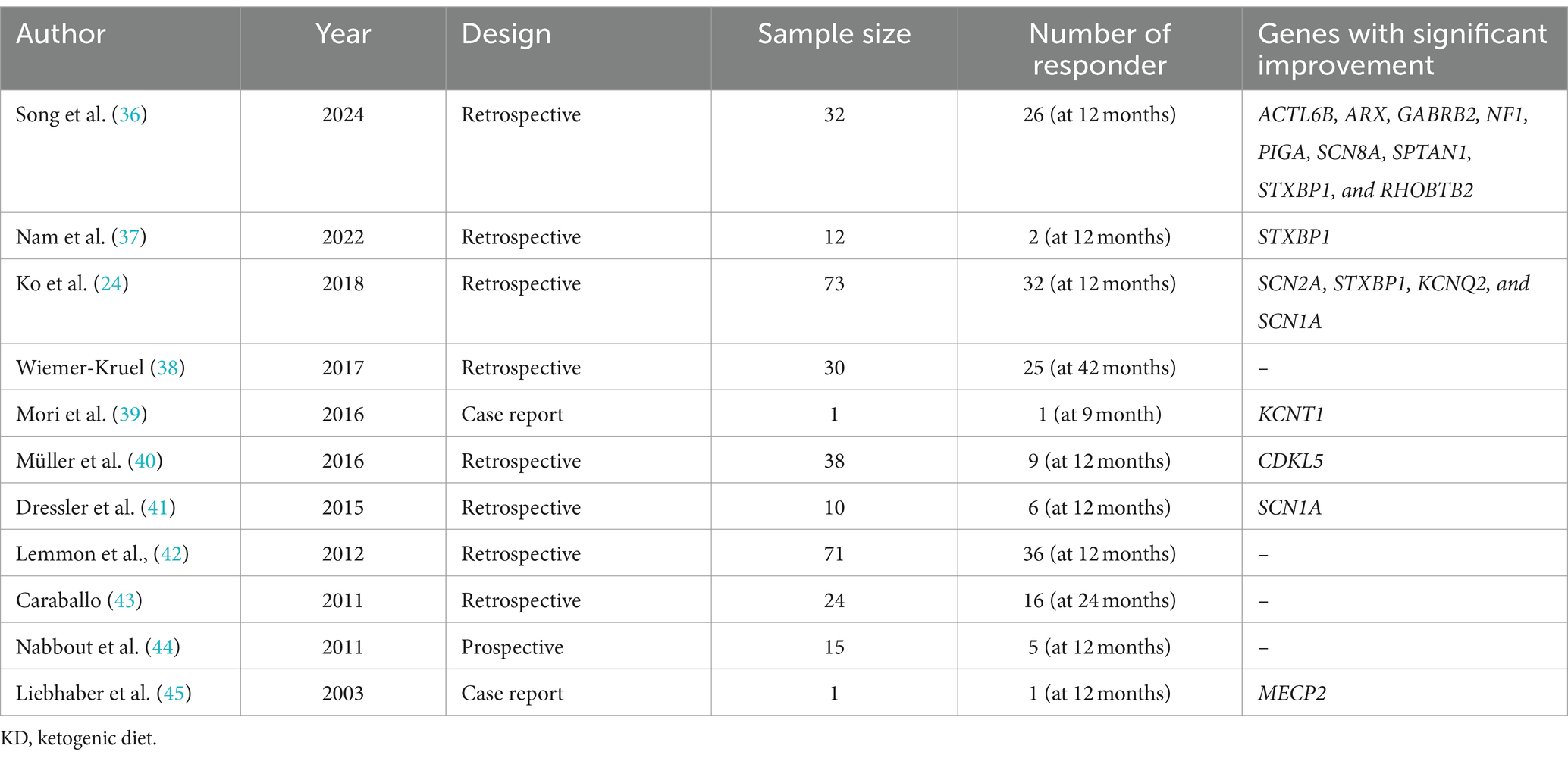
Table 7. Literature studies investigating the efficacy of KD in genetic epilepsies.
Similarly, our study noted a favorable response to KD in 75% of patients with West syndrome at 1 year, consistent with prior observations. For instance, Ko et al. found KD to be more effective in patients with West syndrome lacking CDKL5 mutations when compared to those with the mutation (24).
Comparing seizure rates before and after KD administration, we observed a significant control in seizures among patients with genetic (p < 0.001) and structural etiologies (p < 0.047). This supports other studies’ findings suggesting KD’s efficacy across various etiologies, including developmental and epileptic encephalopathy due to acquired structural etiologies (46). Although surprising, growing literature echoes these results (47).
Another significant factor affecting the response to KD was the number of ASMs used. Children who had tried fewer ASMs before starting KD showed better responses. This is consistent with the literature, indicating that children who have failed multiple ASMs tend to respond less favorably to KD. This could be attributed to the fact that children categorized as having DRE often present with more severe and refractory forms of epilepsy, suggesting a more complex underlying pathophysiology (48, 49).
Like many epilepsy treatments, the KD is associated with adverse events. In our cohort, we observed various adverse events following KD initiation. Specifically, 18 patients experienced one type of adverse event, 14 reported two events, and three reported three adverse events. Despite these occurrences, the overall safety profile of the diet was considered acceptable, as the majority of adverse events being mild in nature. Common adverse effects included constipation, irritability, nausea, vomiting, nutritional deficiencies, hypoglycemia, pancreatitis, and kidney stones. Gastrointestinal issues were the leading complaints, consistent with findings in the literature (50). Vomiting was reported in some of our patients, and previous reports suggest that this complication can affect approximately one-fourth of cases (51). Kidney stones, another significant concern, can be observed in 3–10% of patients (52). This risk can be mitigated through adequate hydration, using oral citrates, and avoiding certain ASMs known to increase the likelihood of stone formation, such as zonisamide, and acetazolamide (53, 54).
The study was limited to a lack of studies nationwide for comparative analysis. Small sample sizes in certain clinical diagnoses prevented definitive conclusions about the diet’s efficacy in these groups. Furthermore, the lack of sufficient number of participants in certain groups including the low number of patients with metabolic and infectious etiologies in Table 2 compared to other etiologies May have impacted the outcome of the related analysis. Recall bias May have skewed reported responses to the diet. The concurrent use of ASMs could impact patient outcomes and responses to the diet, prompting us to regard ASMs as a potential confounding factor in the logistic regression model. To address these issues, we increased our sample size and recruited patients from multiple centers.
This study provides the first comprehensive evaluation of the KD for patients with GESs in the Arab region. Our findings indicate a significant improvement in seizure control, especially among younger patients and those with a later onset of seizures. This highlights the importance of early intervention and age in predicting the success of dietary treatments for GESs. Patients with specific genetic mutations responded well to KD, aligning with previous research, though efficacy varied across genetic profiles. Additionally, the ASMs used before starting KD is crucial in predicting response. Therefore, selecting appropriate candidates is vital, particularly in resource-limited settings.
Our study confirms significant seizure control improvements in patients with genetic and structural etiologies, supporting the broad applicability of KD across various epileptic conditions. KD emerges as an effective treatment option for GESs, advocating its consideration as a viable strategy for managing DRE. This underscores the need for ongoing research to optimize and personalize dietary interventions based on genetic and clinical profiles. Further research is necessary to explore the long-term benefits and potential side effects of KD and to identify additional socio-clinical determinants influencing treatment outcomes. Such efforts are essential for enhancing our understanding of the optimal use of KD in managing genetic epileptic syndromes.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
The studies involving humans were approved by the Unit of Biomedical Ethics Research Committee at the Faculty of Medicine, King Abdulaziz University (reference number 211-23), the primary study location. Other collaborators obtained and ensured adherence to their respective institution's ethical requirements. The study was conducted under the guiding principles of the World Medical Association Declaration of Helsinki. The studies were conducted in accordance with the local legislation and institutional requirements. Patients' data were masked, and written informed consent was waived according to the nature of the study (multi/center retrospective study). All information was kept private and anonymous.
OM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. ASA: Formal analysis, Investigation, Methodology, Project administration, Visualization, Writing – original draft. DA: Formal analysis, Project administration, Resources, Software, Supervision, Writing – original draft. AAA: Formal analysis, Investigation, Supervision, Visualization, Writing – review & editing. RA: Conceptualization, Investigation, Validation, Visualization, Writing – original draft. LJ: Conceptualization, Data curation, Formal analysis, Visualization, Writing – original draft. HK: Investigation, Methodology, Project administration, Visualization, Writing – original draft. MJ: Investigation, Project administration, Resources, Supervision, Writing – original draft. AB: Formal analysis, Methodology, Software, Validation, Visualization, Writing – original draft. MoA: Data curation, Formal analysis, Investigation, Resources, Writing – original draft. RD: Investigation, Methodology, Project administration, Visualization, Writing – original draft. SB: Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – review & editing. MeA: Investigation, Methodology, Project administration, Visualization, Writing – original draft. AM: Data curation, Methodology, Project administration, Software, Visualization, Writing – review & editing. ANA: Conceptualization, Methodology, Project administration, Resources, Supervision, Validation, Writing – review & editing. MN: Conceptualization, Data curation, Formal analysis, Visualization, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors of the study would like to express their deepest gratitude to Sarah Bahowarth for her profound role in organizing the data and assistance in data interpretation. They are also grateful to the Pediatric Neurology Research Group, registered in the Deanship of Scientific Research at King Abdulaziz University (registration number: 67561), for their assistance in issuing the necessary ethical approvals.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Picot, MC, Baldy-Moulinier, M, Daurès, JP, Dujols, P, and Crespel, A. The prevalence of epilepsy and pharmacoresistant epilepsy in adults: a population-based study in a Western European country. Epilepsia. (2008) 49:1230–8. doi: 10.1111/J.1528-1167.2008.01579.X
2. Baeesa, SS, Maghrabi, YE, Baeesa, MS, Jan, FM, and Jan, MM. Publications pattern of clinical epilepsy research in Saudi Arabia. Neurosciences. (2017) 22:255–60. doi: 10.17712/NSJ.2017.4.20170231
3. Kwan, P, Arzimanoglou, A, Berg, AT, Brodie, MJ, Allen Hauser, W, Mathern, G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia. (2010) 51:1069–77. doi: 10.1111/J.1528-1167.2009.02397.X
4. Liu, G, Health, A, Slater, N, and Perkins, A. Epilepsy: treatment options. Am Fam Physician. (2017) 96:87–96.
5. Appleton, RE, Freeman, A, and Cross, JH. Diagnosis and management of the epilepsies in children: a summary of the partial update of the 2012 NICE epilepsy guideline. Arch Dis Child. (2012) 97:1073–6. doi: 10.1136/archdischild-2012-302822
6. D’Andrea Meira, I, Romão, TT, Do Prado, HJP, Krüger, LT, Pires, MEP, and Da Conceição, PO. Ketogenic diet and epilepsy: what we know so far. Front Neurosci. (2019) 13:434220. doi: 10.3389/FNINS.2019.00005/BIBTEX
7. Ułamek-Kozioł, M, Czuczwar, SJ, Pluta, R, and Januszewski, S. Ketogenic Diet and Epilepsy. Nutr. (2019) 11:2510. doi: 10.3390/NU11102510
8. Peterman, MG. The ketogenic diet. JAMA J Am Med Assoc. (1928) 90:1427–9. doi: 10.1001/JAMA.1928.02690450007003
10. Yuen, AWC, and Sander, JW. Rationale for using intermittent calorie restriction as a dietary treatment for drug resistant epilepsy. Epilepsy Behav. (2014) 33:110–4. doi: 10.1016/J.YEBEH.2014.02.026
11. Wheless, JW. History of the ketogenic diet. Epilepsia. (2008) 49:3–5. doi: 10.1111/J.1528-1167.2008.01821.X
12. Cule, J. The falling sickness: a history of epilepsy from the Greeks to the beginnings of modern neurology. Med Hist. (1973) 17:214–5. doi: 10.1017/S0025727300018640
13. Alkhorayef, N, Almutery, FT, Rasheed, Z, Althwab, SA, Aljohani, ASM, Alhawday, YAN, et al. Regulatory effects of ketogenic diet on the inflammatory response in obese Saudi women. J Taibah Univ Med Sci. (2023) 18:1101–7. doi: 10.1016/J.JTUMED.2023.03.006
14. Almaghrabi, MA, Muthaffar, OY, Alahmadi, SA, Abdulsbhan, MA, Bamusa, M, Aljezani, MA, et al. GAMT deficiency among pediatric population: clinical and molecular characteristics and management. Child Neurol Open. (2023) 10:1–15. doi: 10.1177/2329048X231215630
15. Bahassan, NA, and Jan, MM. Ketogenic diet update and application. Neurosciences. (2006) 11:235–40.
16. Stafstrom, CE, and Rho, JM. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front Pharmacol. (2012) 3:59. doi: 10.3389/FPHAR.2012.00059
17. Barañano, KW, and Hartman, AL. The ketogenic diet: uses in epilepsy and other neurologic illnesses. Curr Treat Options Neurol. (2008) 10:410–9. doi: 10.1007/S11940-008-0043-8
18. Desli, E, Spilioti, M, Evangeliou, A, Styllas, F, Magkos, F, and Dalamaga, M. The efficacy and safety of ketogenic diets in drug-resistant epilepsy in children and adolescents: a systematic review of randomized controlled trials. Curr Nutr Rep. (2022) 11:102–16. doi: 10.1007/S13668-022-00405-4
19. Barborka, CJ. Ketogenic diet treatment of epilepsy in adults. JAMA J Am Med Assoc. (1928) 91:73–8. doi: 10.1001/JAMA.1928.02700020007003
20. Neal, EG, Chaffe, H, Schwartz, RH, Lawson, MS, Edwards, N, Fitzsimmons, G, et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. (2008) 7:500–6. doi: 10.1016/S1474-4422(08)70092-9
21. Kverneland, M, Selmer, KK, Nakken, KO, Iversen, PO, and Taubøll, E. A prospective study of the modified Atkins diet for adults with idiopathic generalized epilepsy. Epilepsy Behav. (2015) 53:197–201. doi: 10.1016/j.yebeh.2015.10.021
22. Liu, H, Yang, Y, Wang, Y, Tang, H, Zhang, F, Zhang, Y, et al. Ketogenic diet for treatment of intractable epilepsy in adults: a meta-analysis of observational studies. Epilepsia Open. (2018) 3:9–17. doi: 10.1002/EPI4.12098
23. Devi, N, Madaan, P, Kandoth, N, Bansal, D, and Sahu, JK. Efficacy and safety of dietary therapies for childhood drug-resistant epilepsy: a systematic review and network Meta-analysis. JAMA Pediatr. (2023) 177:258–66. doi: 10.1001/JAMAPEDIATRICS.2022.5648
24. Ko, A, Jung, DE, Kim, SH, Kang, HC, Lee, JS, Lee, ST, et al. The efficacy of ketogenic diet for specific genetic mutation in developmental and epileptic encephalopathy. Front Neurol. (2018) 9:530. doi: 10.3389/fneur.2018.00530
25. Thammongkol, S, Vears, DF, Bicknell-Royle, J, Nation, J, Draffin, K, Stewart, KG, et al. Efficacy of the ketogenic diet: which epilepsies respond? Epilepsia. (2012) 53:e55–e59. doi: 10.1111/J.1528-1167.2011.03394.X
26. Fisher, RS, Cross, JH, French, JA, Higurashi, N, Hirsch, E, Jansen, FE, et al. Operational classification of seizure types by the international league against epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. (2017) 58:522–30. doi: 10.1111/EPI.13670
27. von Elm, E, Altman, DG, Egger, M, Pocock, SJ, Gøtzsche, PC, and Vandenbroucke, JP. The strengthening the reporting of observational studies in epidemiology (STROBE) statement: guidelines for reporting observational studies. J Clin Epidemiol. (2008) 61:344–9. doi: 10.1016/J.JCLINEPI.2007.11.008
28. Richards, S, Aziz, N, Bale, S, Bick, D, das, S, Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/GIM.2015.30
29. Julious, SA. Sample size of 12 per group rule of thumb for a pilot study. Pharm Stat. (2005) 4:287–91. doi: 10.1002/PST.185
30. Baghdadi, LR, Alhomaidi, RA, Alhelal, FS, Alqahtani, AA, Aldosary, SA, Almohammedi, SM, et al. Effect of a ketogenic diet on decrease of seizures in refractory epilepsy among children (infancy to 14 years old) in Saudi Arabia: a cross-sectional study. Transl Pediatr. (2023) 12:1676–89. doi: 10.21037/tp-23-211
31. Gerges, M, Selim, L, Girgis, M, El Ghannam, A, Abdelghaffar, H, and El-Ayadi, A. Implementation of ketogenic diet in children with drug-resistant epilepsy in a medium resources setting: Egyptian experience. Epilepsy Behav Case Reports. (2019) 11:35–8. doi: 10.1016/J.EBCR.2018.11.001
32. Nickels, K. Earlier is not always better: outcomes when epilepsy occurs in early life versus adolescence. Epilepsy Curr. (2020) 20:27–9. doi: 10.1177/1535759719888896
33. Reddy, DS, Thompson, W, and Calderara, G. Molecular mechanisms of sex differences in epilepsy and seizure susceptibility in chemical, genetic and acquired epileptogenesis. Neurosci Lett. (2021) 750:135753. doi: 10.1016/J.NEULET.2021.135753
34. Cepeda, MS, Teneralli, RE, Kern, DM, and Novak, G. Differences between men and women in response to antiseizure medication use and the likelihood of developing treatment resistant epilepsy. Epilepsia Open. (2022) 7:598–607. doi: 10.1002/EPI4.12632
35. Wang, YQ, Fang, ZX, Zhang, YW, Xie, LL, and Jiang, L. Efficacy of the ketogenic diet in patients with Dravet syndrome: a meta-analysis. Seizure. (2020) 81:36–42. doi: 10.1016/J.SEIZURE.2020.07.011
36. Song, T, Deng, J, Chen, C, Wang, X, Han, T, Wang, X, et al. Long-term effectiveness and tolerability of ketogenic diet therapy in patients with genetic developmental and epileptic encephalopathy onset within the first 6 months of life. Epilepsia Open. (2024) 9:643–52. doi: 10.1002/EPI4.12899
37. Nam, JY, Teng, LY, Cho, K, Kang, HC, Lee, JS, Kim, HD, et al. Effects of the ketogenic diet therapy in patients with STXBP1-related encephalopathy. Epilepsy Res. (2022) 186:106993. doi: 10.1016/J.EPLEPSYRES.2022.106993
38. Wiemer-Kruel, A, Haberlandt, E, Hartmann, H, Wohlrab, G, and Bast, T. Modified Atkins diet is an effective treatment for children with Doose syndrome. Epilepsia. (2017) 58:657–62. doi: 10.1111/EPI.13701
39. Mori, T, Imai, K, Oboshi, T, Fujiwara, Y, Takeshita, S, Saitsu, H, et al. Usefulness of ketogenic diet in a girl with migrating partial seizures in infancy. Brain and Development. (2016) 38:601–4. doi: 10.1016/J.BRAINDEV.2015.12.012
40. Müller, A, Helbig, I, Jansen, C, Bast, T, Guerrini, R, Jähn, J, et al. Retrospective evaluation of low long-term efficacy of antiepileptic drugs and ketogenic diet in 39 patients with CDKL5-related epilepsy. Eur J Paediatr Neurol. (2016) 20:147–51. doi: 10.1016/J.EJPN.2015.09.001
41. Dressler, A, Trimmel-Schwahofer, P, Reithofer, E, Mühlebner, A, Gröppel, G, Reiter-Fink, E, et al. Efficacy and tolerability of the ketogenic diet in Dravet syndrome - comparison with various standard antiepileptic drug regimen. Epilepsy Res. (2015) 109:81–9. doi: 10.1016/J.EPLEPSYRES.2014.10.014
42. Lemmon, ME, Terao, NN, Ng, YT, Reisig, W, Rubenstein, JE, and Kossoff, EH. Efficacy of the ketogenic diet in Lennox-Gastaut syndrome: a retrospective review of one institution’s experience and summary of the literature. Dev Med Child Neurol. (2012) 54:464–8. doi: 10.1111/J.1469-8749.2012.04233.X
43. Caraballo, RH. Nonpharmacologic treatments of Dravet syndrome: focus on the ketogenic diet. Epilepsia. (2011) 52:79–82. doi: 10.1111/J.1528-1167.2011.03009.X
44. Nabbout, R, Copioli, C, Chipaux, M, Chemaly, N, Desguerre, I, Dulac, O, et al. Ketogenic diet also benefits Dravet syndrome patients receiving stiripentol: a prospective pilot study. Epilepsia. (2011) 52:e54–7. doi: 10.1111/J.1528-1167.2011.03107.X
45. Liebhaber, GM, Riemann, E, and Matthias Baumeister, FA. Ketogenic diet in Rett syndrome. J Child Neurol. (2003) 18:74–5. doi: 10.1177/08830738030180011801
46. Villaluz, MM, Lomax, LB, Jadhav, T, Cross, JH, and Scheffer, IE. The ketogenic diet is effective for refractory epilepsy associated with acquired structural epileptic encephalopathy. Dev Med Child Neurol. (2018) 60:718–23. doi: 10.1111/DMCN.13687
47. Dou, X, Xu, X, Mo, T, Chen, H, Wang, Z, Li, X, et al. Evaluation of the seizure control and the tolerability of ketogenic diet in Chinese children with structural drug-resistant epilepsy. Seizure. (2022) 94:43–51. doi: 10.1016/J.SEIZURE.2021.11.008
48. Zarnowska, IM. Therapeutic use of the ketogenic diet in refractory epilepsy: what we know and what still needs to be learned. Nutrients. (2020) 12:1–23. doi: 10.3390/NU12092616
49. Herrero, JR, Villarroya, EC, Peñas, JJG, Alcolea, BG, Fernández, BG, Macfarland, LAP, et al. Safety and effectiveness of the prolonged treatment of children with a ketogenic diet. Nutrients. (2020) 12:306. doi: 10.3390/NU12020306
50. el-Rashidy, OF, Nassar, MF, Shokair, WA, and el Gendy, YGA. El Gendy YGA: ketogenic diet for epilepsy control and enhancement in adaptive behavior. Sci Rep. (2023) 13:2102. doi: 10.1038/S41598-023-27373-1
51. Jagadish, S, Payne, ET, Wong-Kisiel, L, Nickels, KC, Eckert, S, and Wirrell, EC. The ketogenic and modified Atkins diet therapy for children with refractory epilepsy of genetic etiology. Pediatr Neurol. (2019) 94:32–7. doi: 10.1016/J.PEDIATRNEUROL.2018.12.012
52. Acharya, P, Acharya, C, Thongprayoon, C, Hansrivijit, P, Kanduri, SR, Kovvuru, K, et al. Incidence and characteristics of kidney stones in patients on ketogenic diet: a systematic review and Meta-analysis. Diseases. (2021) 9:39. doi: 10.3390/diseases9020039
53. Kossoff, EH, Pyzik, PL, Furth, SL, Hladky, HD, Freeman, JM, and Vining, EPG. Kidney stones, carbonic anhydrase inhibitors, and the ketogenic diet. Epilepsia. (2002) 43:1168–71. doi: 10.1046/J.1528-1157.2002.11302.X
54. Kossoff, EH, Zupec-Kania, BA, Auvin, S, Ballaban-Gil, KR, Christina Bergqvist, AG, Blackford, R, et al. Optimal clinical management of children receiving dietary therapies for epilepsy: updated recommendations of the international ketogenic diet study group. Epilepsia Open. (2018) 3:175–92. doi: 10.1002/EPI4.12225
Keywords: drug-resistant epilepsy, ketogenic diet, epilepsy syndromes, dietary therapies, children, seizure
Citation: Muthaffar OY, Alyazidi AS, Alsowat D, Alasiri AA, Albaradie R, Jad LA, Kayyali H, Jan MMS, Bamaga AK, Alsubaie MA, Daghistani R, Baeesa SS, Alaifan MA, Makraz A, Alsharief AN and Naseer MI (2024) Short-term effectiveness and side effects of ketogenic diet for drug-resistant epilepsy in children with genetic epilepsy syndromes. Front. Neurol. 15:1484752. doi: 10.3389/fneur.2024.1484752
Edited by:
Pasquale Striano, Giannina Gaslini Institute (IRCCS), ItalyReviewed by:
Raffaele Falsaperla, Policlinico San Marco, ItalyCopyright © 2024 Muthaffar, Alyazidi, Alsowat, Alasiri, Albaradie, Jad, Kayyali, Jan, Bamaga, Alsubaie, Daghistani, Baeesa, Alaifan, Makraz, Alsharief and Naseer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anas S. Alyazidi, YWx5YXppZGkuYW5hc0BnbWFpbC5jb20=; YWFseWF6aWRpMDAxNUBzdHUua2F1LmVkdS5zYQ==; Muhammad Imran Naseer, bWluYXNlZXJAa2F1LmVkdS5zYQ==; bWltcmFubmFzZWVyQHlhaG9vLmNvbQ==
†ORCID: Osama Y. Muthaffar, http://orcid.org/0000-0002-3458-1697
Anas S. Alyazidi, http://orcid.org/0000-0002-1388-5069
Mohammed M. S. Jan, http://orcid.org/0000-0002-5188-8922
Ahmed K. Bamaga, http://orcid.org/0000-0002-0115-9457
Mohammed A. Alsubaie, http://orcid.org/0000-0002-6098-0704
Saleh S. Baeesa, http://orcid.org/0000-0002-3053-7912
Muhammad Imran Naseer, http://orcid.org/0000-0002-9343-5825
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.