 Dilbar Mammadova1Cornelia Kraus2Thomas Leis1
Dilbar Mammadova1Cornelia Kraus2Thomas Leis1 Bernt Popp2,3Christiane Zweier2,4Andre Reis2,5
Bernt Popp2,3Christiane Zweier2,4Andre Reis2,5 Regina Trollmann1,5*
Regina Trollmann1,5*- 1Department of Pediatrics, Pediatric Neurology, Friedrich-Alexander Universität Erlangen-Nürnberg, Erlangen, Germany
- 2Institute of Human Genetics, Friedrich-Alexander Universität Erlangen-Nürnberg, Erlangen, Germany
- 3Center of Functional Genomics, Berlin Institute of Health at Charité, Universitätsmedizin Berlin, Berlin, Germany
- 4Department of Human Genetics, Inselspital, University of Bern, Bern, Switzerland
- 5Centre for Rare Disorders Erlangen, University Hospital Erlangen, Erlangen, Germany
Pathogenic heterozygous variants in CACNA1A are associated with familial hemiplegic migraine, episodic ataxia type 2 and spinocerebellar ataxia type 6, and more recently, neurodevelopmental disorders. We describe a severe, early-onset phenotype including severe muscular hypotonia, early-onset epileptic seizures, apnoea, optic atrophy and dysphagia in three siblings carrying compound heterozygous frameshift variants in CACNA1A. Two male patients died at the age of 5 or 14 months of suspected SIDS or severe developmental epileptic encephalopathy (DEE) with refractory seizures and apnoea. A male child (index patient) developed severe early-onset DEE including seizures and ictal apnoea at the age of 4 weeks. Another male child developed generalized epilepsy and mild intellectual impairment in late infancy, and his mother and his maternal uncle were identified as carriers of a known CACNA1A pathogenic variant [c.2602delG heterozygous, p. (Ala868Profs*24)] with a diagnosis of episodic ataxia type 2. This maternal pathogenic variant c.2602delG was detected in the index patient and child 2. Trio-Exome sequencing identified an additional heterozygous pathogenic variant in the CACNA1A gene, c.5476delC, p.(His1826Thrfs*30) in the index patient and child 2, which was inherited from the asymptomatic father. In conclusion, the novel compound heterozygosity for two frameshift pathogenic variants, maternally [c.2602delG, p.(Ala868Profs*24)] and paternally [c.5476delC, p.(His1826Thrfs*3)] is associated with a severe phenotype of early-onset DEE. This observation highlights the necessity of additional analyses to clarify unusual phenotypes even if a pathogenic variant has already been identified, and expands the clinical spectrum of CACNA1A-related disorders.
Introduction
Pathogenic, heterozygous variants in the CACNA1A gene, which encodes the alpha-1 subunit of a voltage-gated P/Q-type calcium channel, are known to be involved in a heterogeneous spectrum of autosomal-dominant, neurological disorders such as episodic ataxia type 2 (MIM# 108500), spinocerebellar ataxia type 6 (MIM#183086), familial hemiplegic migraine with or without progressive cerebellar ataxia (MIM#141500), and a developmental and epileptic encephalopathy (MIM#617106) (1, 2). The neurodevelopmental part of the spectrum is broad and includes mild forms of epilepsy such as absence epilepsy or focal epilepsy as well as severe epileptic phenotypes such as early-onset refractory focal epilepsy and developmental and epileptic encephalopathy (DEE) (3–7). Furthermore, bi-allelic pathogenic variants in the CACNA1A gene have been reported in several infants with epileptic encephalopathy of early-onset and refractory course, severe muscular hypotonia and progressive cerebral, cerebellar, and optic nerve atrophy (4, 5, 7). However, little is still known about genotype–phenotype correlations in CACNA1A-related neurodevelopmental disorders and DEE (3, 8).
Here, we report a rapidly progressive and fatal course of early-onset CACNA1A associated developmental and epileptic encephalopathy in three siblings, of which two were identified to harbor compound heterozygous frameshift variants in CACNA1A and one is considered to have the same genotype. The heterozygous variants segregated in the family with milder, variable neurological phenotypes, respectively.
Materials and methods
Clinical data including findings from general and neurological examination, seizure types and frequency, response to antiseizure medication (ASM), family history, and results of (long-term) video EEG and cerebral MRI scans were obtained from patient records. All children were treated regularly in the interdisciplinary outpatient center of the Division of Pediatric Neurology at the University Hospital of Erlangen. Neuropsychological evaluation was performed by experienced occupational therapists and psychologists.
Exome sequencing of three siblings and their parents was performed at the local Institute of Human Genetics on an Illumina HiSeq 2,500 sequencer using the Twist Human Core Exome Enrichment Technology (Twist Bioscience Inc.) and processed as previously described (9). In total, 1,454 established NDD and 1,224 additional candidate genes from the SysNDD database (version from April 2021) were evaluated (10). Variants were analyzed regarding their occurrence and frequencies in population or variant databases, their segregation and their consequences as well as regarding their plausibility in light of the phenotype. Exome-based copy number variant (CNV) analysis was performed using the ExomeDepth Software (GATK). The parents’ informed consent was obtained.
Results
Clinical characteristics of the four affected male siblings are summarized in Table 1. The leading clinical symptoms in the index case (II-4) and two of his siblings (II-1, II-2) were severe muscular hypotonia (floppy infant), plus refractory early-onset epileptic seizures and deep apneic spells, as well as optic atrophy.
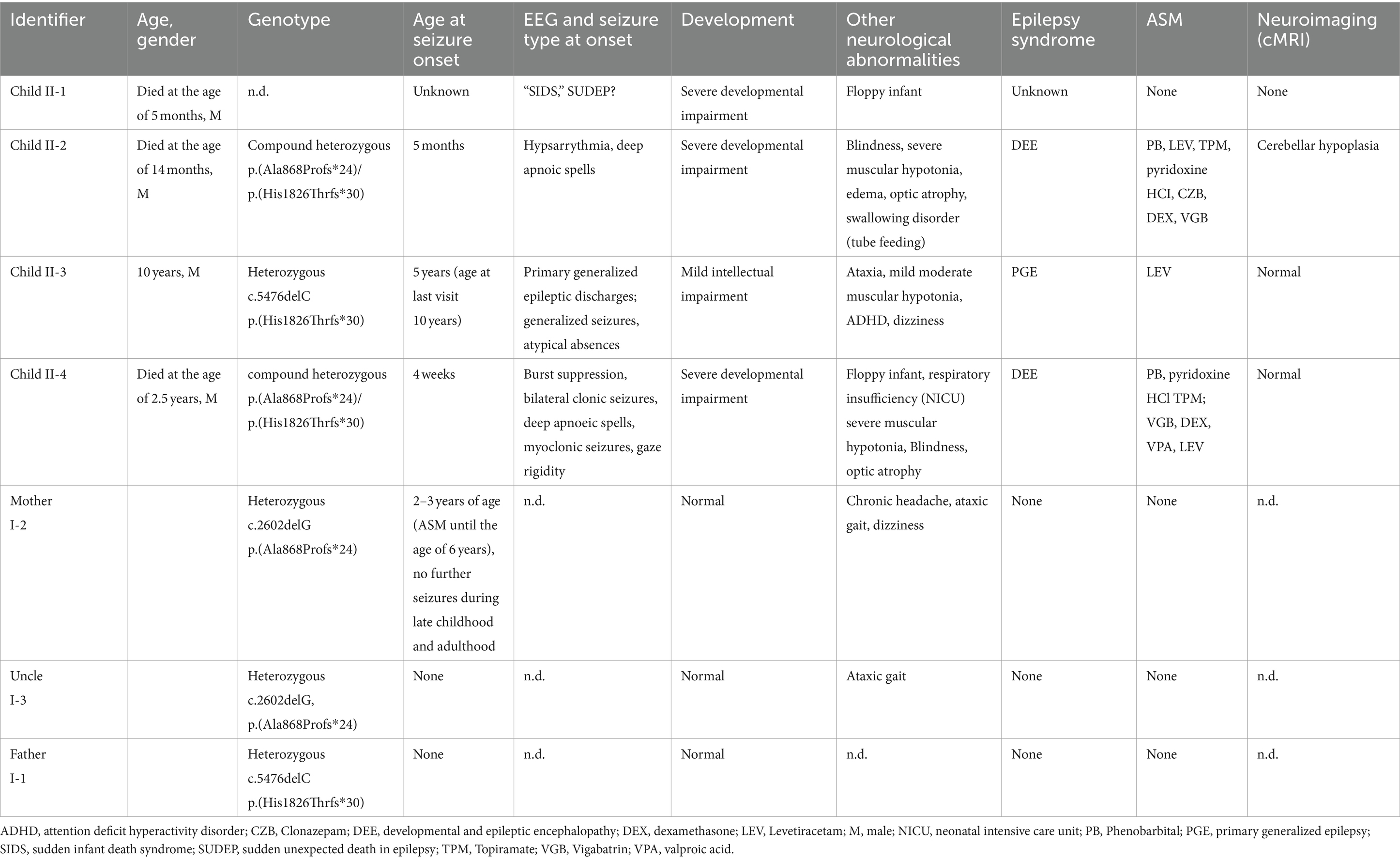
Table 1. Patient characteristics.
Child II-1
The male infant born as the first child of seemingly healthy young, non-consanguineous parents died at the age of 5 months with suspicion of sudden infant death of infancy (SIDS). Neonatal-onset muscular hypotonia was noted, but diagnostic work-up was not performed during early infancy nor post mortem.
Child II-2
The male infant was born after an uneventful pregnancy. Muscular hypotonia was present from first week of life. The boy was admitted at the age of 5 months for further diagnostics and treatment of epileptic seizures, respiratory problems as well as dysphagia and severe muscular hypotonia. EEG showed hypsarrhythmia during awake state as well as during sleep. Brain MRI, metabolic work-up, karyotyping and Chromosomal Microarray Analysis did not indicate structural, metabolic or a specific genetic etiology. Based on clinical symptoms, PEHO (progressive encephalopathy, edema, hypsarrhythmia, optic atrophy) syndrome was suspected. Antiseizure medication (phenobarbital, benzodiazepines, levetiracetam, topiramate, Pyridoxin HCl, dexamethasone, vigabatrin) for prolonged and deep apneic spells and subtle seizures was without lasting effects. Tube feeding and transient mechanical ventilation were necessary. The patient died at the age of 14 months.
Child II-3
The male child, born after an uneventful pregnancy, was referred at the age of 5 years for evaluation of an episodic, unsteady gait, dizziness and decreased vigilance. Clinical evaluation showed mild intellectual impairment and learning disability. Cranial MRI and metabolic screening findings were normal. EEG revealed primary generalized epileptic discharges. Treatment with levetiracetam, which was initiated for primary generalized epilepsy of most probably genetic origin, was successful and led to freedom from further seizures.
Child II-4 (index patient)
The male infant was born showing a normal postnatal adaptation, however, muscular hypotonia was presented. At the age of 4 weeks, first epileptic seizures occurred, and led to the initiation of antiseizure medication with levetiracetam. However, treatment effect was transient, and the boy developed signs of an early-onset epileptic encephalopathy showing severe muscular hypotonia with absent deep tendon reflexes, respiratory and swallowing insufficiency, as well as highly frequent seizures. The semiology of the multiple daily seizures was characterized by gaze rigidity, paleness, and bilateral clonic seizures and apneic spells, as well as myoclonic seizures. Electroencephalography revealed burst suppression (Figure 1A). Cerebral MRI and metabolic work-up did not indicate any specific etiology. Antiseizure medication (including topiramate, vigabatrin, dexamethasone, valproic acid, benzodiazepines, phenobarbital) led to only transient improvement of seizure frequency with persisting severe global developmental delay and muscular hypotonia (tube feeding). EEG remained severely abnormal with generalized monomorphic background slowing and multifocal sharp waves (Figure 1B). Home care under multidisciplinary support was possible. The patient died at the age of 2 years due to respiratory insufficiency.
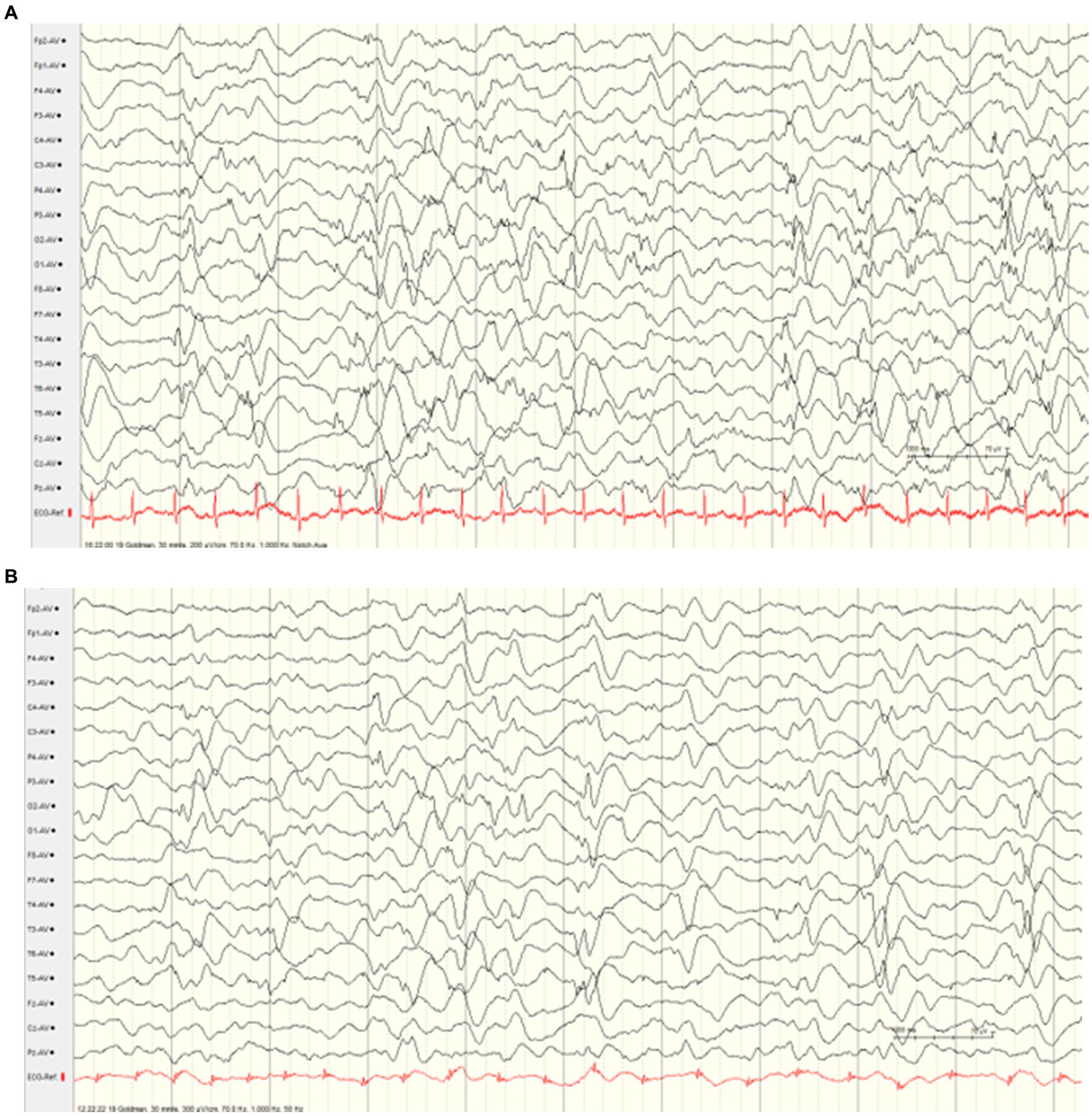
Figure 1. Interictal electroencephalograms (index case). (A) EEG at the age of 4 weeks showing burst suppression. (B) EEG at the age of 5 months showed generalized monomorphic background slowing and multifocal sharp waves.
Genetic results
Some years after birth of child II-3, episodic ataxia type 2 was diagnosed in the mother and one of her brothers due to a heterozygous frameshift pathogenic variant in the CACNA1A gene [c.2602delG, p.(Ala868Profs*24)]. The mother reported mild tremor of the hands and poor gait balance. In addition, mild intellectual deficits were apparent but not formally followed-up. The father had no specific neurological symptoms. He was heterozygous for a pathogenic variant in the CACNA1A gene [c.5476delC heterozygous, p.(His1826Thrfs*30)].
The summary of genetic results is given in Figure 2. Exome sequencing revealed compound heterozygosity for two pathogenic CACNA1A (NM_001127222.1) variants in the index patient (II-4). Exome sequencing also confirmed these two variants in material from deceased child II-2. Child II-3 was found to be heterozygous for the maternal CACNA1A variant c.2602delG, p.(Ala868Profs*24). Material of child II-1 was not available for testing.
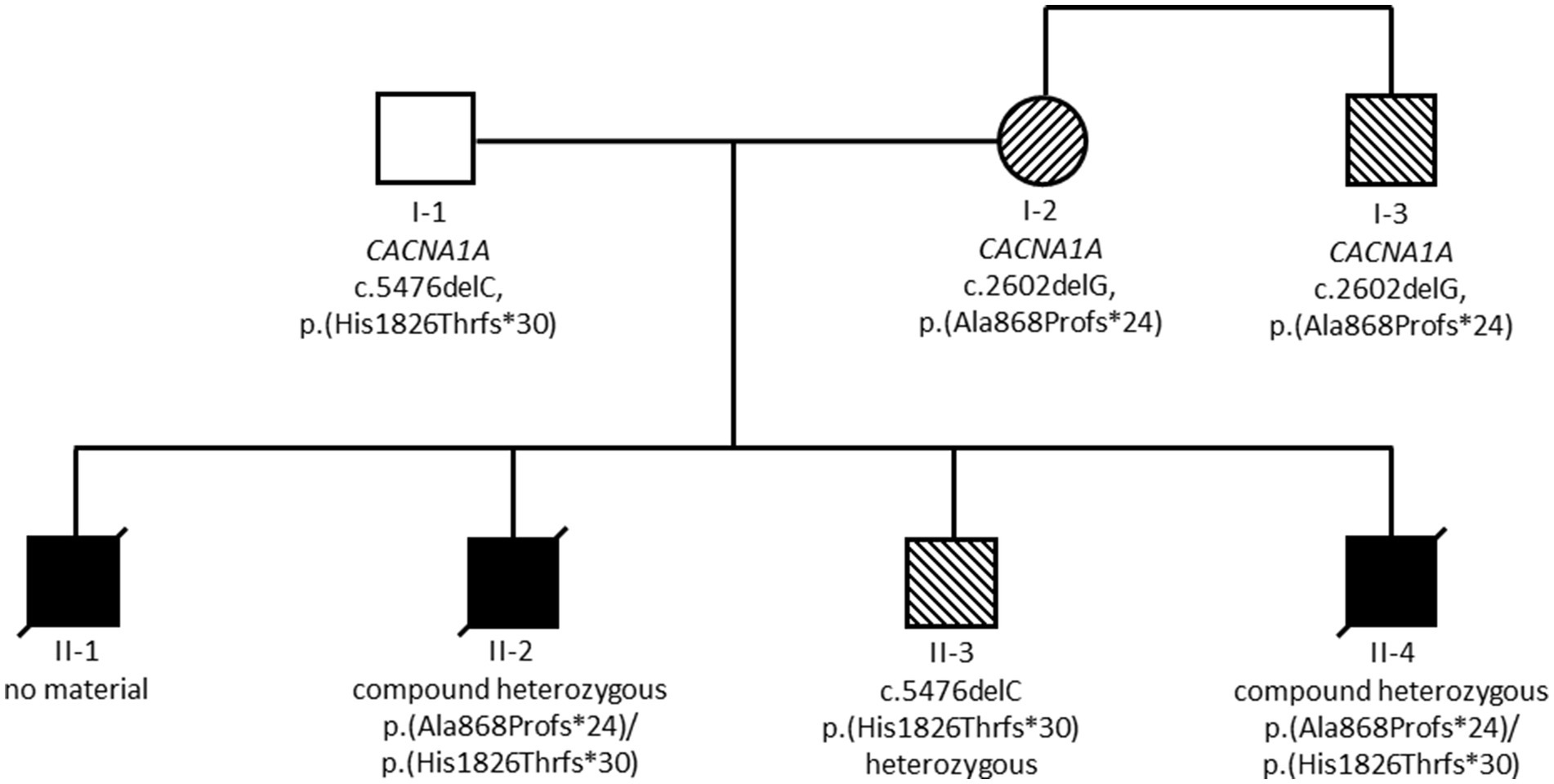
Figure 2. Summary of genetic results. Black symbols indicate individuals with severe and fatal developmental and epileptic encephalopathy, striped symbols indicate individuals with milder epilepsy and/or ataxia phenotypes.
None of the two variants was included in gnomAD (11) or in ClinVar (12) databases, at this point. Both variants are predicted to result in loss of function, either by truncation or nonsense mediated mRNA decay. They were therefore classified as pathogenic according to ACMG criteria PVS1, PM2 and PP4 (13).
Discussion
The present data indicate that the compound heterozygous frameshift pathogenic variants, namely c.2602delG, p.(Ala868Profs*24) and c.5476delC, p.(His1826Thrfs*3), cause refractory early-onset epileptic encephalopathies with a rapidly progressive course and early death. Main clinical symptoms of the severe epileptic encephalopathy in the index case [child II-4, p.(Ala868Profs*24)/p.(His1826Thrfs*30)] and in part in two of his brothers [II-1, no material; II-2, p.(Ala868Profs*24)/p.(His1826Thrfs*30)] were severe muscular hypotonia (floppy infant), refractory early-onset epileptic seizures and deep apneic spells, as well as optic atrophy. So far, only nine individuals from four families with bi-allelic variants in CACNA1 have been reported in literature (5, 7, 30, 32). Interestingly, a homozygous nonsense variant in four siblings resulted in a similarly devastating clinical course with hypotonia, epileptic encephalopathy and lethality in the first 6 months of life (32), while compound heterozygosity for a missense and a truncating variant in three other individuals, respectively, was associated with hypotonia, early-onset epilepsy, profound neurodevelopmental delay but long-time survival in two individuals (age at last assessments 10 and 5 years, respectively) (5, 7). A homozygous in-frame variant in two adult siblings was associated with progressive myoclonic epilepsy with a late onset >50 years (30) (Table 2). Our finding of bi-allelic truncating variants being associated with a severe, early onset epileptic encephalopathy with fatal outcome supports a genotype–phenotype correlation regarding deleteriousness of bi-allelic LOF variants and severity of the phenotype.
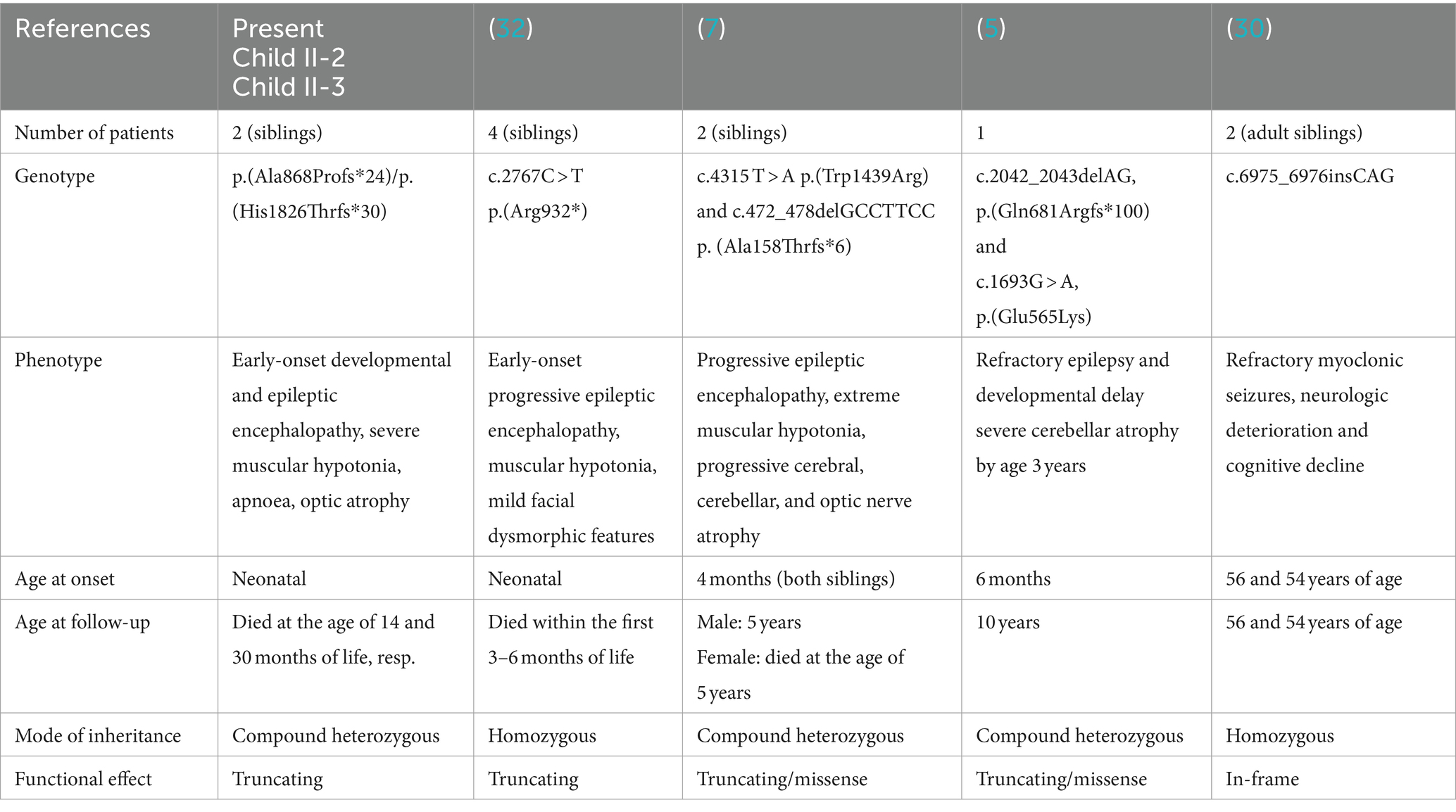
Table 2. Genotype–phenotype correlations in our patients and in previously reported biallelic CACNA1A variants in epilepsy patients.
Similarly to previous reports with early onset epileptic encephalopathy (5, 7, 32), heterozygous parents and one heterozygous sibling in the herewith reported family were either asymptomatic or presented with milder neurological and/or cognitive symptoms. CACNA1 therefore belongs to the growing list of genes with co-dominantly or both autosomal dominantly and autosomal recessively inherited diseases (32).
In our patients, ictal apneic spells associated with subtle orofacial (child II-2) or epileptic spasms and generalized clonic seizures (child II-4) were the most prominent seizure type and most probably the cause of death in both brothers with the proven pathogenic variants. We also suggest that child II-1, who was diagnosed with sudden infant death syndrome (SIDS), probably died of ictal apnoea. The mechanisms of seizure-related apnoea, which is associated with a high risk of death and SUDEP (sudden unexpected death in epilepsy), including irreversible brain stem dysfunction, are not fully understood. However, animal studies suggest a lethal effect of seizures on brain stem function and respiration in adult Cacna1aS218L mice (14). These authors showed, that fatal apnea in the transgenic mice functionally characterized by gain-of-function of voltage-gated CaV2.1 Ca2+ channels is the consequence of cardiorespiratory dysfunction, which is induced by brainstem seizure-related spreading depolarization (SD) reaching respiratory nuclei in the ventrolateral medulla (14). Interestingly, the application of the NMDA receptor antagonists MK-801 and memantine was effective in preventing the medullary spreading depolarization and the fatal outcome (14). Data on developing transgenic mice is missing.
EEG findings were similar in the herewith reported siblings, showing a refractory suppression burst pattern from early infancy without any improvement in response to antiseizure medication. After several months, there was generalized background slowing accompanied by diffuse epileptic discharges. Similar EEG patterns were previously reported in two sisters with compound heterozygous missense and frameshift variants (c.4315 T > A), (p.Trp1439Arg); del c.472_478delGCCTTCC (p.Ala158Thrfs*6) (7), consistent with severe developmental and epileptic encephalopathy.
Pathogenicity of the identified CACNA1 variants is strongly supported by the fact that dysfunction of the alpha-1A subunit of the Cav2.1 P/Q-type voltage-gated calcium channel causes profound functional dysregulations in various developmental neuronal pathways (15–17). The expression level of CaV2.1 α1 subunit modulates synaptic strength, efficacy of synaptic transmission and synaptic plasticity (18). While little is known about global CaV2.1 knock-out (KO) mice lacking the α1 subunit Cacna1a gene product due to early postnatal lethality, studies in a conditional Cacna1a knock-in mouse model revealed a deficiency of P/Q-type calcium channel protein and currents within the first month after birth, which is associated with altered spontaneous firing of Purkinje cells in the cerebellum and forebrain and impaired neurotransmission (17). As shown in a forebrain-specific Ca(V)2.1 KO mouse model, Ca(V)2.1 function crucially modifies complex learning and memory functions (15), and deficits are based on impaired synaptic transmission at hippocampal glutamatergic synapses.
According to gnomAD constraint scores (11), CACNA1 is intolerant toward both missense and truncating variants (pLI = 1, z = 3.95). Accordingly, in the ClinVar database (12) both truncating and missense variants are reported across all CACNA1A-related phenotypes. For several missense variants, either loss-of-function or gain-of-function effects have been demonstrated, both resulting in a similar phenotype of developmental and epileptic encephalopathy (19).
Heterozygous CACNA1A loss-of-function pathogenic variants are rarely associated with epilepsy or epileptic encephalopathy (6, 8, 20, 21). As published by the Epi4K consortium in 2016, among 531 individuals with a variety of unresolved epileptic encephalopathy, targeted sequencing identified six infants with CACNA1A pathogenic variants (including an affected sibling), which were de novo in 3/5 families (21). Five of these infants showed first clinical seizure activity during the neonatal period including myoclonic and tonic seizure types. In addition, atypical Rett syndrome with early-onset epilepsy was associated with a de novo variant in the S6 transmembrane segment of domain III of the P/Q type calcium channel, CACNA1A (c.2128G > A, p.Ala710Thr) (6). Indicating intrafamilial heterogeneity, the mild cognitive impairment and childhood-onset generalized epilepsy in child II-3 was associated with a CACNA1A haploinsufficiency inherited from the EA2 affected mother. This is in line with previous observations of generalized absence epilepsy (8, 22–26), refractory focal epilepsy or developmental and epileptic encephalopathy (8, 21, 27) in patients with heterozygous CACNA1A loss-of-function pathogenic variants. Jung et al. (22) found childhood-onset refractory absence epilepsy and focal epilepsy with secondary generalized tonic–clonic seizures in 19% of individuals with proven CACNA1A loss-of-function pathogenic variants from four unrelated families (N = 3/16). A further 6 patients (n = 6/15, 40%) had febrile seizures during childhood. In addition to epilepsy, developmental delay, intellectual disability or executive dysfunction were found in all patients, often with ADHD (11/16) and autism spectrum disorder (4/16), as well as downbeat nystagmus and episodic ataxia in several cases. Case reports on antiseizure medication response in patients with heterozygous CACNA1A loss-of-function pathogenic variants show variable results ranging from seizure-free in response to pyridoxin (28), VPA and LTG (23), to partial response to VPA and LEV (22) and no response to LTG (29). The heterogeneous phenotype associated with CACNA1A variants is supported by experimental data on loss of presynaptic and somatodendritic CaV2.1 channel function, suggesting dysregulation of a variety of cortical and cerebellar neuronal cell types (20, 30), as well as hippocampal, thalamic and cerebellar networks (16). The present case of childhood-onset CACNA1A-related epilepsy with the leading symptoms of mild generalized epilepsy, mild intellectual impairment and gait ataxia highlights the variable intrafamilial phenotypic spectrum of CACNA1A-related diseases.
In conclusion, our observation of compound heterozygous frameshift variants in three siblings with an early-onset fatal developmental and epileptic encephalopathy further delineates the phenotype associated with bi-allelic CACNA1 variants resulting in complete loss of function. Heterozygous presence of either variant in several family members was associated with either no or with milder, but very variable neurological phenotypes. This case highlights the need for additional analyses to clarify unusual phenotypes, even when a pathogenic variant has already been identified, and expands the clinical spectrum of CACNA1A-related disorders.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
DM: Investigation, Writing – review & editing. CK: Investigation, Methodology, Writing – review & editing. TL: Investigation, Writing – review & editing. BP: Investigation, Methodology, Writing – review & editing. CZ: Investigation, Methodology, Writing – review & editing. AR: Methodology, Writing – review & editing. RT: Conceptualization, Investigation, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank the family for the agreement to comprehensive genetic analysis and for their cooperation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kinder, S, Ossig, C, Wienecke, M, Beyer, A, von der Hagen, M, Storch, A, et al. Novel frameshift mutation in the CACNA1A gene causing a mixed phenotype of episodic ataxia and familiar hemiplegic migraine. Eur J Paediatr Neurol. (2015) 19:72–4. doi: 10.1016/j.ejpn.2014.10.005
2. Mantuano, E, Romano, S, Veneziano, L, Gellera, C, Castellotti, B, Caimi, S, et al. Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J Neurol Sci. (2010) 291:30–6. doi: 10.1016/j.jns.2010.01.010
3. Kessi, M, Chen, B, Pang, N, Yang, L, Peng, J, He, F, et al. The genotype-phenotype correlations of the CACNA1A-related neurodevelopmental disorders: a small case series and literature reviews. Front Mol Neurosci. (2023) 16:1222321. doi: 10.3389/fnmol.2023.1222321
4. Niu, X, Yang, Y, Chen, Y, Cheng, M, Liu, M, Ding, C, et al. Genotype-phenotype correlation of CACNA1A variants in children with epilepsy. Dev Med Child Neurol. (2022) 64:105–11. doi: 10.1111/dmcn.14985
5. Wong-Spracklen, VMY, Kolesnik, A, Eck, J, Sabanathan, S, Spasic-Boskovic, O, Maw, A, et al. Biallelic CACNA1A variants: review of literature and report of a child with drug-resistant epilepsy and developmental delay. Am J Med Genet A. (2022) 188:3306–11. doi: 10.1002/ajmg.a.62960
6. Epperson, MV, Haws, ME, Standridge, SM, and Gilbert, DL. An atypical Rett syndrome phenotype due to a novel missense mutation in CACNA1A. J Child Neurol. (2018) 33:286–9. doi: 10.1177/0883073818754987
7. Reinson, K, Õiglane-Shlik, E, Talvik, I, Vaher, U, Õunapuu, A, Ennok, M, et al. Biallelic CACNA1A mutations cause early onset epileptic encephalopathy with progressive cerebral, cerebellar, and optic nerve atrophy. Am J Med Genet A. (2016) 170:2173–6. doi: 10.1002/ajmg.a.37678
8. Li, XL, Li, ZJ, Liang, XY, Liu, DT, Jiang, M, Gao, LD, et al. CACNA1A mutations associated with epilepsies and their molecular sub-regional implications. Front Mol Neurosci. (2022) 15:860662. doi: 10.3389/fnmol.2022.860662
9. Bosch, E, Hebebrand, M, Popp, B, Penger, T, Behring, B, Cox, H, et al. BDV syndrome: an emerging syndrome with profound obesity and neurodevelopmental delay resembling Prader-Willi syndrome. J Clin Endocrinol Metab. (2021) 106:3413–27. doi: 10.1210/clinem/dgab592
10. Kochinke, K, Zweier, C, Nijhof, B, Fenckova, M, Cizek, P, Honti, F, et al. Systematic Phenomics analysis Deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am J Hum Genet. (2016) 98:149–64. doi: 10.1016/j.ajhg.2015.11.024
11. Chen, S, Francioli, LC, Goodrich, JK, Collins, RL, Kanai, M, Wang, Q, et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature. (2024) 625:92–100. doi: 10.1038/s41586-023-06045-0
12. Landrum, MJ, Lee, JM, Riley, GR, Jang, W, Rubinstein, WS, Church, DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. (2014) 42:D980–5. doi: 10.1093/nar/gkt1113
13. Richards, S, Aziz, N, Bale, S, Bick, D, Das, S, Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
14. Mallmann, RT, Elgueta, C, Sleman, F, Castonguay, J, Wilmes, T, van den Maagdenberg, A, et al. Ablation of ca(V)2.1 voltage-gated Ca2+ channels in mouse forebrain generates multiple cognitive impairments. PLoS One. (2013) 8:e78598. doi: 10.1371/journal.pone.0078598
15. Maejima, T, Wollenweber, P, Teusner, LU, Noebels, JL, Herlitze, S, and Mark, MD. Postnatal loss of P/Q-type channels confined to rhombic-lip-derived neurons alters synaptic transmission at the parallel fiber to purkinje cell synapse and replicates genomic Cacna1a mutation phenotype of ataxia and seizures in mice. J Neurosci. (2013) 33:5162–74. doi: 10.1523/JNEUROSCI.5442-12.2013
16. Lübbert, M, Goral, RO, Keine, C, Thomas, C, Guerrero-Given, D, Putzke, T, et al. CaV2.1 α1 subunit expression regulates presynaptic CaV2.1 abundance and synaptic strength at a central synapse. Neuron. (2019) 101:260–273.e6. doi: 10.1016/j.neuron.2018.11.028
17. Rajakulendran, S, Graves, TD, Labrum, RW, Kotzadimitriou, D, Eunson, L, Davis, MB, et al. Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J Physiol. (2010) 588:1905–13. doi: 10.1113/jphysiol.2009.186437
18. Damaj, L, Lupien-Meilleur, A, Lortie, A, Riou, É, Ospina, LH, Gagnon, L, et al. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur J Hum Genet. (2015) 23:1505–12. doi: 10.1038/ejhg.2015.21
19. Mark, MD, Maejima, T, Kuckelsberg, D, Yoo, JW, Hyde, RA, Shah, V, et al. Delayed postnatal loss of P/Q-type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1a mutations. J Neurosci. (2011) 31:4311–26. doi: 10.1523/JNEUROSCI.5342-10.2011
20. Du, X, Chen, Y, Zhao, Y, Luo, W, Cen, Z, and Hao, W. Dramatic response to pyridoxine in a girl with absence epilepsy with ataxia caused by a de novo CACNA1A mutation. Seizure. (2017) 45:189–91. doi: 10.1016/j.seizure.2016.12.020
21. Zaitsev, AV, Povysheva, NV, Lewis, DA, and Krimer, LS. P/Q-type, but not N-type, calcium channels mediate GABA release from fast-spiking interneurons to pyramidal cells in rat prefrontal cortex. J Neurophysiol. (2007) 97:3567–73. doi: 10.1152/jn.01293.2006
22. Jung, J, Testard, H, Tournier-Lasserve, E, Riant, F, Vallet, AE, Berroir, S, et al. Phenotypic variability of episodic ataxia type 2 mutations: a family study. Eur Neurol. (2010) 64:114–6. doi: 10.1159/000315145
23. Imbrici, P, Jaffe, SL, Eunson, LH, Davies, NP, Herd, C, Robertson, R, et al. Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain. (2004) 127:2682–92. doi: 10.1093/brain/awh301
24. Chen, Y, Lu, J, Pan, H, Zhang, Y, Wu, H, Xu, K, et al. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol. (2003) 54:239–43. doi: 10.1002/ana.10607
25. Hayashida, T, Saito, Y, Ishii, A, Yamada, H, Itakura, A, Minato, T, et al. CACNA1A-related early-onset encephalopathy with myoclonic epilepsy: a case report. Brain Dev. (2018) 40:130–3. doi: 10.1016/j.braindev.2017.08.006
26. Epi4K Consortium; Epilepsy Phenome/Genome ProjectAllen, AS, Berkovic, SF, Cossette, P, Delanty, N, Dlugos, D, et al. De novo mutations in epileptic encephalopathies. Nature. (2013) 501:217–21. doi: 10.1038/nature12439,
27. Jouvenceau, A, Eunson, LH, Spauschus, A, Ramesh, V, Zuberi, SM, Kullmann, DM, et al. Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet. (2001) 358:801–7. doi: 10.1016/S0140-6736(01)05971-2
28. Chioza, B, Wilkie, H, Nashef, L, Blower, J, McCormick, D, Sham, P, et al. Association between the alpha(1a) calcium channel gene CACNA1A and idiopathic generalized epilepsy. Neurology. (2001) 56:1245–6. doi: 10.1212/wnl.56.9.1245
29. Jiang, X, Lupien-Meilleur, A, Tazerart, S, Lachance, M, Samarova, E, Araya, R, et al. Remodeled cortical inhibition prevents motor seizures in generalized epilepsy. Ann Neurol. (2018) 84:436–51. doi: 10.1002/ana.25301
30. Lv, Y, Wang, Z, Liu, C, and Cui, L. Identification of a novel CACNA1A mutation in a Chinese family with autosomal recessive progressive myoclonic epilepsy. Neuropsychiatr Dis Treat. (2017) 13:2631–6. doi: 10.2147/NDT.S145774
31. Jiang, X, Raju, PK, D'Avanzo, N, Lachance, M, Pepin, J, Dubeau, F, et al. Both gain-of-function and loss-of-function de novo CACNA1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox-Gastaut syndrome. Epilepsia. (2019) 60:1881–94. doi: 10.1111/epi.16316
Keywords: early-onset epileptic encephalopathy, apneic spells, optic nerve atrophy, burst suppression, developmental epileptic encephalopathy, generalized epilepsy
Citation: Mammadova D, Kraus C, Leis T, Popp B, Zweier C, Reis A and Trollmann R (2024) Intrafamilial neurological phenotypic variability due to either biallelic or monoallelic pathogenic variants in CACNA1A. Front. Neurol. 15:1458109. doi: 10.3389/fneur.2024.1458109
Edited by:
Carlotta Spagnoli, Santa Maria Nuova Hospital, ItalyReviewed by:
Nikolaos Marinakis, National and Kapodistrian University of Athens, GreeceTakahito Wada, Kyoto University, Japan
Copyright © 2024 Mammadova, Kraus, Leis, Popp, Zweier, Reis and Trollmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Regina Trollmann, cmVnaW5hLnRyb2xsbWFubkB1ay1lcmxhbmdlbi5kZQ==