 Lassana Cissé1,2
Lassana Cissé1,2 Salia Bamba1,3
Salia Bamba1,3 Seybou H. Diallo1,4
Seybou H. Diallo1,4 Weizhen Ji3
Weizhen Ji3 Mohamed Emile Dembélé1
Mohamed Emile Dembélé1 Abdoulaye Yalcouyé1,5
Abdoulaye Yalcouyé1,5 Toumany Coulibaly6
Toumany Coulibaly6 Ibrahima Traoré4
Ibrahima Traoré4 Lauren Jeffries3
Lauren Jeffries3 Salimata Diarra1,3
Salimata Diarra1,3 Alassane Dit Baneye Maiga1Salimata Diallo4Karamoko Nimaga7
Alassane Dit Baneye Maiga1Salimata Diallo4Karamoko Nimaga7 Amadou Touré8
Amadou Touré8 Oumou Traoré1
Oumou Traoré1 Mahamadou Kotioumbé1
Mahamadou Kotioumbé1 Emily Kathryn Mis3
Emily Kathryn Mis3 Cheick Abdel Kader Cissé1Cheick Oumar Guinto1,6
Cheick Abdel Kader Cissé1Cheick Oumar Guinto1,6 Kenneth H. Fischbeck9
Kenneth H. Fischbeck9 Mustafa K. Khokha3
Mustafa K. Khokha3 Saquib A. Lakhani3
Saquib A. Lakhani3 Guida Landouré1,6*
Guida Landouré1,6*- 1Faculté de Médecine et d’Odontostomatologie, Université des Sciences, des Techniques et des Technologies de Bamako (USTTB), Bamako, Mali
- 2Service de Médecine, Hôpital Nianankoro Fomba de Ségou, Ségou, Mali
- 3Department of Pediatrics, Pediatric Genomics Discovery Program (PGDP), Yale University School of Medicine, New Haven, CT, United States
- 4Service de Neurologie, Centre Hospitalier Universitaire Gabriel Touré, Bamako, Mali
- 5Department of Genetic Medicine, McKusick-Nathans Institute, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 6Service de Neurologie, Centre Hospitalier Universitaire Point G, Bamako, Mali
- 7Clinique médicale Dinandougou, Marka Coungo, Mali
- 8Service de Pédiatrie, Centre Hospitalier Universitaire Gabriel Touré, Bamako, Mali
- 9Neurogenetics Branch, NINDS, NIH, Bethesda, MD, United States
Background and objectives: Progressive myoclonic epilepsy (PME) is a group of neurological disorders characterized by recurrent myoclonic seizures with progressive neurological deterioration. We investigated the genetics of three unrelated patients with PME from Mali, a country in sub-Saharan Africa highly underrepresented in genetic and genomic research.
Methods: Participants were carefully examined and phenotyped. DNA was obtained for genetic analysis including whole exome sequencing (WES). In silico prediction tools and ACMG criteria were used to assess the deleteriousness of putative candidate variants.
Results: Pedigree analysis suggests autosomal recessive inheritance patterns for one family and sporadic forms of PME for the two other cases. WES identified novel homozygous missense variants in all the three patients, one each for NHLRC1, EPM2A, and NEU1. The sequence variants segregated with PME in each family and in silico studies including protein 3D structures, CADD scores and ACMG criteria suggested that they were damaging.
Discussion: PME is a group of clinically heterogeneous neurological disorders. Most reported cases in the literature are from European background with only a few cases described in North Africa. We report here novel pathogenic variants in three different genes causing PME phenotypes in three unrelated Malian patients, suggesting that genetic studies of underrepresented populations may expand the genetic epidemiology of PME. These findings also emphasize the need for inclusive genetic research to ensure a more targeted diagnostic and therapeutic approaches for diverse patient populations.
1 Introduction
Progressive myoclonic epilepsy (PME) is a group of clinically and genetically heterogeneous disorders characterized by recurrent myoclonic seizures, and progressive neurological and cognitive deterioration. PMEs are uncommon disorders and the diagnosis is challenging since symptoms are often non-specific and can mimic other neurological conditions (1). Although the etiology is undetermined in many patients, the gene defects for several PMEs (Unverricht-Lundborg disease, Lafora disease, several forms of neuronal ceroid lipofuscinoses, myoclonic epilepsy with ragged-red fibers [MERRF], and type 1 sialidoses) have been identified, leading to improvement in diagnosis and therapy (2, 3).
Despite this progress, genetically confirmed cases of PME remain scarce in several sub-Saharan Africa (SSA) countries, including Mali, highlighting a significant gap in the exploration of molecular neurological disorders in the region. The limited availability of specialists and genetic diagnostic tools contributes to this knowledge deficit (4). Nevertheless, Mali’s diverse population, characterized by at least 14 distinct ethnic groups, exhibits homogeneous cluster populations with specific phenotypic traits and an increased prevalence of recessive disorders in certain areas (4). This presents a unique opportunity to identify unique PME-associated genotypes. In this study, we present novel findings from three Malian families with PME attributable to rare genetic variants.
2 Methods
2.1 Standard protocol approvals, registrations, and patient consents
This study was done in full compliance with the declaration of Helsinki and approval was obtained from the Ethics Committee of the Faculté de Médecine et d’Odontostomatologie, Université des Sciences, des Techniques et des Technologies de Bamako, Mali. Written informed consent/assent was obtained from all participants with agreement to share data.
2.2 Clinical and laboratory assessment
Patients were examined by neurologists, pediatricians, and medical geneticists. Blood chemistries, computed tomography (CT) scan, magnetic resonance imaging (MRI) and electroencephalography (EEG) were performed in selected available patients to refine the diagnosis or to rule out acquired causes of seizures.
2.3 Genetic analysis
DNA was extracted from peripheral blood using the QIAGEN Puregene Blood DNA kit C (Qiagen, Germantown, MD, USA) following the manufacturer’s instruction. WES was performed in all affected individuals and the available family members (Figure 1A: III.2, III.3, III.4; Figure 1B: V.2, VI.1; Figure 1C: III.3, III.5). Sanger sequencing was performed to confirm the sequence variation and to check for segregation within the families (Family 1: III.2, III.3, III.4; Family 2: V.2, VI.1; Family 3: III.1, III.2, III.3, III.5). Variant calling, annotation, and prioritization as well as prediction for deleteriousness are detailed in Supplementary material 1 (5, 6). Variants were selected based on the suspected inheritance pattern within the family and the number of affected individuals, considering de novo, hemizygous, homozygous, and compound heterozygous variants, according to the following criteria: (i) missense, nonsense, frameshift, non-frameshift, or splicing site variants, (ii) SNPs with a minor allele frequency of <0.0005 in the SNP database were selected and filtered, (iii) In order to assess deleterious effects of variants, bioinformatics tools were applied including combined annotation dependent depletion [CADD pathogenicity prediction scoring >20 predicts the top 1% of deleterious variants1 (7)], sorting intolerant from tolerant,2 MutationTaster,3 protein variation effect analyzer,4 polymorphism phenotyping v2.5 The final candidate variant was evaluated according to ACMG (American College of Medical Genetics and Genomics) variant interpretation guidelines (8).

Figure 1. Phenotypic characteristics and genetic findings in families with PME (Family 1). (A) Pedigree of the family with Lafora disease with no reported consanguinity but two siblings having the disease (the black arrow indicates the proband and asterisks the participants seen in clinic). (B) The amino acid (Phe201) encoded at the variant site was highly conserved among mammals (Using the UCSC Genome Browser). (C) A chromatogram showing the changes, a homozygous C > T change (Phe201Ser). (D) Analyses of the secondary structures (ss) on 3D models revealed several major changes in the mutant (red) side chains when compared to the wild-type (blue) side chains of NHLRC1. Phenotypic characteristics and genetic findings in families with PME (Family 2). (E) Pedigree of the family with Lafora disease with an affected member from parental consanguinity (the black arrow indicates the proband and asterisks the participants seen in clinic). (F) A chromatogram showing the changes, a homozygous splice site variant in EPM2A (c.301 + 1G > C). Phenotypic characteristics and genetic findings in families with PME (Family 3). (G) Pedigree of the family with Sialidosis type 1 with an affected member from a non-consanguineous marriage (the black arrow indicates the proband and asterisks the participants seen in clinic). (H) The amino acid (Arg305) encoded at the variant site was highly conserved among mammals (Using the UCSC Genome Browser). (I) A chromatogram showing the changes, a homozygous G > T change (Arg305Leu). (J) Analyses of the secondary structures (ss) on 3D models revealed several major changes in the mutant (red) side chains when compared to the wild-type (blue) side chains of NEU1.
For genes with predicted nonsynonymous protein coding variants (NHLRC1 and NEU1), secondary protein structures were predicted on PSIPRED Workbench.6 Protein three-dimensional (3D) structures were modeled on Swiss-model server and visualized using Pymol software (9). More details are provided in Supplementary material 1.
3 Cases description
Phenotypic and genetic features are summarized in Table 1.
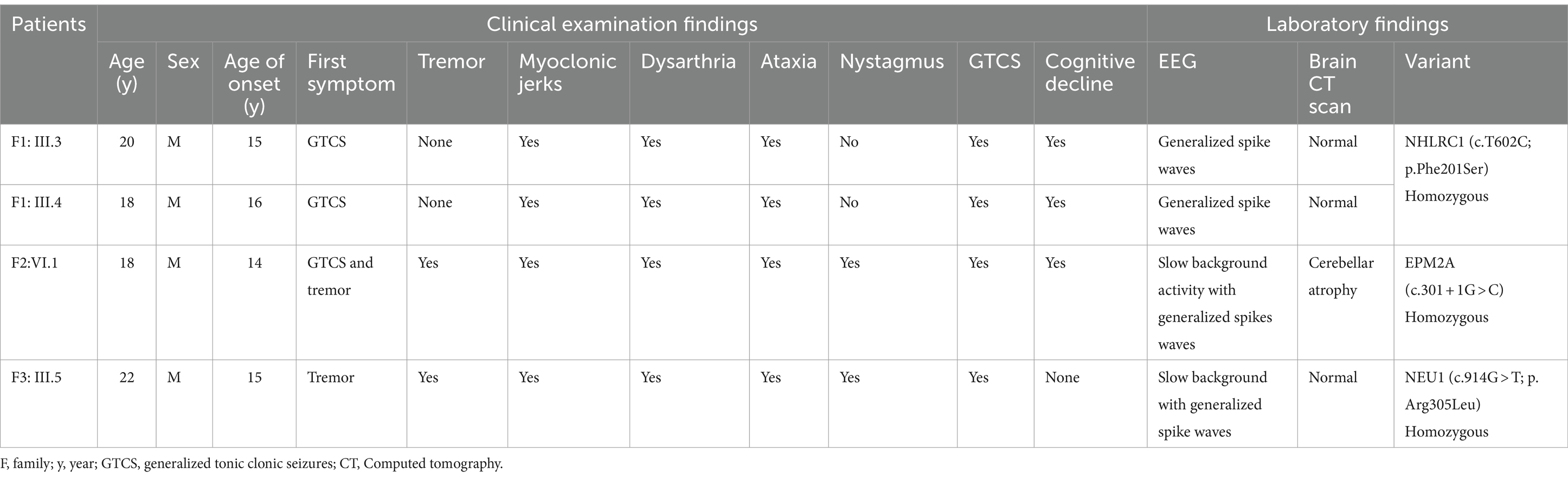
Table 1. Phenotypic and genetic findings in patients with PME.
3.1 Case 1 (Family 1)
This family had two male affected siblings aged 18 and 20 years from Dogon ethnicity who were referred to our clinic for genetic exploration of pharmaco-resistant generalized tonic clonic seizures which started at the ages of 15 and 16 years, respectively (Figure 1A). No parental consanguinity was reported but parents are from the same ethnic background and the same geographical area. Neurological examination found generalized myoclonic jerks with neuropsychiatric symptoms including aggressiveness, slurred speech and cognitive decline. EEG revealed a normal background with generalized spike waves, suggesting generalized epilepsy. Brain CT scan was normal. Despite antiseizure medications including sodium valproate and clonazepam, their neurological condition worsened progressively and died at ages 21 and 23, respectively. WES was performed on both patients to identify the genetic cause of PME. Shared variants between the two patients were selected from sequencing results through rigorous bioinformatics analysis. In silico prediction revealed five deleterious variants. According to ACMG classification, three variants were selected: one likely pathogenic and two variants of uncertain significance (VUS) (Supplementary Table S1). The likely pathogenic variant, classified by ACMG criteria (PM1, PM2, PP1, PP3, PP4), was identified in NHLRC1, c.602 T > C (p.Phe201Ser) in both affected siblings, segregating with the disease in the family. The amino acid (Phe201) which was highly conserved among mammals using the UCSC Genome Browser and the changes, a homozygous C>T change (Phe201Ser) are shown in the Figure 1B,C. Substituting a non-polar Phenylalanine with a polar Serine was predicted to affect protein structure (Figure 1D; Supplementary Figure S1).
3.2 Case 2 (Family 2)
The proband was an 18-year-old male from Soninké ethnicity with myoclonic epilepsy that began at 14 years of age with initial symptoms of mild hand tremors causing objects like pens and bowls to slip (Figure 1E). A chromatogram showing the changes, a homozygous splice site variant in EPM2A (c.301+1G>C) is depicted in Figure 1F. He is from an eventful pregnancy and delivery with consanguineous parents. Clinical examination found intellectual disability, dysarthria, and visual impairment. While muscle strength was preserved, brisk reflexes were noted in the upper and lower limbs, in addition to tremors and clonus in the latter. He had difficulties standing, and was unable to walk. EEG showed a slow background activity with generalized spikes waves (Supplementary Figure S2) and brain imaging showed cerebellar atrophy. Treatment was initiated with sodium valproate, clonazepam and piracetam but myoclonus and generalized tonic clonic seizures recured frequently and the patient consequently died at age 20. WES was performed on the proband and his unaffected mother. Shared monoallelic variants were excluded from the sequencing results through rigorous bioinformatics analysis. In silico prediction revealed 24 deleterious variants. According to ACMG classification, nine variants were selected: one likely pathogenic and eight variants of uncertain significance (VUS) (Supplementary Table S2). The likely pathogenic variant, classified by ACMG criteria (PVS1, PM2, PP4), was identified in EPM2A, c.301 + 1G > C, with a SpliceAI score of 0.96 for donor loss and 0.2 for donor gain.
3.3 Case 3 (Family 3)
The proband was a 22-year-old male with a 7-year history of dysarthria and myoclonic jerks in the peribuccal area (Figure 1G). He was from Bambara ethnicity. There was no parental consanguinity and no familial history of the disease. The patient developed generalized myoclonic jerks that were exacerbated by strong emotions and sensory stimulations, as well as generalized tonic clonic seizures with generalized rigidity and hypertonia leading to severe disability. Reflexes and coordination were difficult to evaluate due to exacerbated myoclonic jerks on attempt. EEG showed slow background with generalized spike waves and brain CT scan was normal. Treatment with sodium valproate and clonazepam led to an initial improvement of myoclonic jerks and seizures. However, his neurological condition worsened gradually and he died 5 years later. WES was performed on the proband and one of his unaffected paternal cousins. Shared monoallelic variants were excluded through rigorous bioinformatics analysis. In silico prediction revealed 28 deleterious variants. According to ACMG classification, 23 variants were selected: one likely pathogenic and 22 variants of uncertain significance (VUS) (Supplementary Table S3). The likely pathogenic variant, classified by ACMG criteria (PM1, PM2, PM5), was identified in NEU1, c.914G > T (p.Arg305Leu). The amino acid (Arg305) conservation among mammals and a chromatogram showing the changes, a homozygous G>T change (Arg305Leu) are depicted in Figure 1H,I. The predicted 3D structure of variant NEU1 showed significant changes, gaining new helical structures from switching positively charged Arg305 to the non-polar Leu305 (Figure 1J; Supplementary Figure S3).
Variant’s pathogenicity criteria is summarized in Supplementary Table S4.
4 Discussion
Progressive Myoclonic epilepsies are a group of clinically and genetically heterogeneous disorders characterized by myoclonus, seizures, and progressive neurological decline with variable onset in childhood or adolescence. Molecularly, several common and rare PMEs including Unverricht-Lundborg disease (ULD), Lafora disease (LD), several forms of neuronal ceroid lipofuscinoses (NCLs), myoclonic epilepsy with ragged-red fibers [MERRF], type 1 sialidose (ST-1), North Sea PME, Acid ceramidase deficiency (Farber disease/SMA-PME) have been characterized with a wide phenotype variability (10). They are mostly transmitted following an autosomal recessive manner with rare cases of mitochondrial and autosomal dominant inheritance (10). Despite significant progress being made to understand the landscape of molecular defects underlying PME, only a few cases have been described in Africa (11, 12). To the best of our knowledge, this is the first report of PME caused by variants in NEU1 and EPM2A on the African continent and the second reported PME caused by variant in NHLRC1 in the Malian population (11–13).
Variants in NHLRC1 and EPM2A are known to cause Lafora disease through synaptic vesicle formation and neurotransmitter release. The NHLRC1 gene encodes malin, an E3-ubiquitin ligase and the EPM2A gene encodes laforin, a dual-specificity protein phosphatase. Mutation of these two genes results in the accumulation of polyglucan bodies called Lafora bodies within neurons and that can cause a range of neurological symptoms (14). Here, we report the second mutation in NHLRC1 and the first mutation in EPM2A in the Malian population. Notably, both the previously reported amino acid change (p.His187Pro) and the (p.Phe201Ser) variant we report here are localized in a highly conserved NHL domain (12). Like the previously reported NHLRC1 variant in Mali, the case we present here showed a severe phenotype with a shorter disease duration while mild disease phenotype have been associated to NHLRC1 variants in other continents (10, 12).
NEU1 is known to encode for a sialidase enzyme that is involved in the breakdown of sialic acid, a sugar found on the surface of cells. Damaging variants in this gene lead to the accumulation of sialic acid in the brain and cause a form of neurodegenerative disease known as sialidosis and characterized by cognitive impairment, seizures, and motor problems. Mutations in this gene were previously associated with neurodegenerative diseases including Gaucher disease and Alzheimer’s disease (15). This is the first mutation in NEU1 reported in the Malian population. The disease duration was 12 years in our patient, however more prolonged durations up to 30 years have been reported in other populations (16). This could be explained by genetic or environmental modifiers. In addition, difficulties in accessing specialized healthcare services could also play a role in our context.
Overall, the discovery of these variants in PME patients from different ethnic backgrounds (Bambara, Dogon, and Soninké) for the first time in Mali or in Africa further supports a role for the identification of genetic variants in African PME. The increasing access to sequencing technologies may unravel genetic variants with significant implications for the understanding and treatment of PME, and will trigger hope for the inclusion of African patients in the future therapeutic developments for this debilitating disease.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/clinvar/ with accession numbers SCV005200387.1, SCV005200391.1 and SCV005200399.1 respectively for variants in NHLRC1 (Case 1), EPM2A (Case 2) and NEU1 (Case 3).
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Faculté de Médecine et d’Odontostomatologie, Université des Sciences, des Techniques et des Technologies de Bamako, Mali. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
LC: Data curation, Investigation, Methodology, Validation, Writing – original draft. SB: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Writing – original draft. SeD: Data curation, Investigation, Methodology, Supervision, Validation, Writing – review & editing. WJ: Formal analysis, Methodology, Validation, Writing – review & editing, Investigation. MD: Data curation, Investigation, Methodology, Validation, Writing – review & editing. AY: Data curation, Investigation, Methodology, Validation, Writing – review & editing. TC: Investigation, Methodology, Validation, Writing – review & editing. IT: Investigation, Methodology, Validation, Writing – review & editing. LJ: Formal analysis, Investigation, Validation, Writing – review & editing. SDiar: Data curation, Investigation, Methodology, Validation, Writing – review & editing. AM: Data curation, Investigation, Methodology, Validation, Writing – review & editing. SDial: Data curation, Investigation, Methodology, Validation, Writing – review & editing. KN: Investigation, Methodology, Supervision, Validation, Writing – review & editing. AT: Investigation, Methodology, Validation, Writing – review & editing. OT: Data curation, Investigation, Methodology, Validation, Writing – review & editing. MKo: Data curation, Investigation, Methodology, Validation, Writing – review & editing. EM: Formal analysis, Investigation, Validation, Writing – review & editing. CC: Formal analysis, Investigation, Validation, Writing – review & editing. CG: Data curation, Investigation, Methodology, Supervision, Validation, Writing – review & editing. KF: Formal analysis, Supervision, Validation, Writing – review & editing. MKh: Formal analysis, Investigation, Supervision, Validation, Writing – review & editing. SL: Conceptualization, Formal analysis, Investigation, Methodology, Supervision, Validation, Writing – review & editing. GL: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Supervision, Validation, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by grant numbers U01HG007044 under the H3Afria initiative and R01NS118522 funded by the National Institute of Neurological Disorders and Stroke (NINDS), NINDS intramural research funds, and the Centre Hospitalier Universitaire du Point “G,” Bamako, Mali. We thank Alice Schindler (Technical assistance for sequencings, Neurogenetics Branch, NINDS, NIH, Bethesda, MD, USA). We are grateful to the patients and their families.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2024.1455467/full#supplementary-material
Supplementary Figure S1 | The predicted 3D structure of variant NHLRC1. The Substituting a non-polar Phenylalanine with a polar Serine was predicted to affect protein structure apparent in the refined 3D structure (A), which is predicted to impact the overall folding of the protein (B, C). In addition, the change of a Non-polar and hydrophobic amino acid (aa) Phenylalanine into a polar aa Serine may impact the binding interaction of the protein.
Supplementary Figure S3 | The 3D structure of the mutant NEU1 revealed additional major changes including a gain of new helical structures (A). Although both wildtype aa R305 and mutant L305 are not involved directly in bonding interaction (B,C), the change of a positively charged aa (Arginine) to a non-Polar and hydrophobic aa (Leucine) is predicted to impact the protein interaction ability.
Abbreviations
ACMG, American College of Medical Genetics and Genomics; EPM2A, Epilepsy, progressive myoclonus type 2A; MERRF, myoclonic epilepsy with ragged-red fibers; NEU1, neuraminidase 1; NHLRC1, NHL repeat containing E3 ubiquitin protein ligase 1; PME, Progressive Myoclonic Epilepsy; SMA, Spinal muscular atrophy
Footnotes
1. ^https://cadd.gs.washington.edu/home.
2. ^SIFT; https://sift.bii.a-star.edu.sg/.
3. ^http://www.mutationtaster.org/.
4. ^PROVEAN; http://provean.jcvi.org/index.php.
5. ^PolyPhen-2; http://genetics.bwh.harvard.edu/pph2/.
References
1. Orsini, A, Valetto, A, Bertini, V, Esposito, M, Carli, N, Minassian, BA, et al. The best evidence for progressive myoclonic epilepsy: a pathway to precision therapy. Seizure. (2019) 71:247–57. doi: 10.1016/j.seizure.2019.08.012
2. Lehesjoki, AE . Molecular background of progressive myoclonus epilepsy. EMBO J. (2003) 22:3473–8. doi: 10.1093/emboj/cdg338
3. Holmes, GL . Drug treatment of progressive myoclonic epilepsy. Pediatr Drugs. (2020) 22:149–64. doi: 10.1007/s40272-019-00378-y
4. Landouré, G, Samassékou, O, Traoré, M, Meilleur, KG, Guinto, CO, Burnett, BG, et al. Genetics and genomic medicine in Mali_ challenges and future perspectives. Mol Genet Genomic Med. (2016) 4:126–34. doi: 10.1002/mgg3.212
5. Koboldt, DC, Larson, DE, and Wilson, RK. Using VarScan 2 for germline variant calling and somatic mutation detection. Curr Protoc Bioinformatics. (2013) 44:15.4.1–17. doi: 10.1002/0471250953.bi1504s44
6. Wang, K, Li, M, and Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
7. Kircher, M, Witten, DM, Jain, P, O'Roak, BJ, Cooper, GM, and Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46:310–5. doi: 10.1038/ng.2892
8. Richards, S, Aziz, N, Bale, S, Bick, D, das, S, Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
9. Venselaar, H, te Beek, TA, Kuipers, RK, Hekkelman, ML, and Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-science approach with life scientist-friendly interfaces. BMC Bioinformat. (2010) 11:548. doi: 10.1186/1471-2105-11-548
10. Zimmern, V, and Minassian, B. Progressive myoclonus epilepsy: a scoping review of diagnostic, phenotypic and therapeutic advances. Genes. (2024) 15:171. doi: 10.3390/genes15020171
11. Esterhuizen, AI, Carvill, GL, Ramesar, RS, Kariuki, SM, Newton, CR, and Poduri, A. Clinical application of epilepsy genetics in Africa: is now the time? Front Neurol. (2018) 9:276. doi: 10.3389/fneur.2018.00276
12. Traoré, M, Landouré, G, Motley, W, Sangaré, M, Meilleur, K, Coulibaly, S, et al. Novel mutation in the NHLRC1 gene in a Malian family with a severe phenotype of Lafora disease. Neurogenetics. (2009) 10:319–23. doi: 10.1007/s10048-009-0190-4
13. Khiari, HM, Lesca, G, Malafosse, A, and Mrabet, A. A novel exon 3 mutation in a Tunisian patient with Lafora’s disease. J Neurol Sci. (2011) 304:136–7. doi: 10.1016/j.jns.2011.02.011
14. Ianzano, L, Zhang, J, Chan, EM, Zhao, XC, Lohi, H, Scherer, SW, et al. Lafora progressive myoclonus epilepsy mutation database-EPM2A and NHLRC1 (EPM2B) genes. Hum Mutat. (2005) 26:397. doi: 10.1002/humu.9376
15. Annunziata, I, Patterson, A, Helton, D, Hu, H, Moshiach, S, Gomero, E, et al. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-β secretion via deregulated lysosomal exocytosis. Nat Commun. (2013) 4:2734. doi: 10.1038/ncomms3734
Keywords: progressive myoclonic epilepsy, genetic, novel variants, Mali, West Africa
Citation: Cissé L, Bamba S, Diallo SH, Ji W, Dembélé ME, Yalcouyé A, Coulibaly T, Traoré I, Jeffries L, Diarra S, Maiga ADB, Diallo S, Nimaga K, Touré A, Traoré O, Kotioumbé M, Mis EK, Cissé CAK, Guinto CO, Fischbeck KH, Khokha MK, Lakhani SA and Landouré G (2024) Genetic profile of progressive myoclonic epilepsy in Mali reveals novel findings. Front. Neurol. 15:1455467. doi: 10.3389/fneur.2024.1455467
Edited by:
Joohyun Park, Institute of Medical Genetics and Applied Genomics Tübingen, GermanyReviewed by:
Francesco Nicita, Bambino Gesù Children’s Hospital (IRCCS), ItalyRomina Combi, University of Milano-Bicocca, Italy
Copyright © 2024 Cissé, Bamba, Diallo, Ji, Dembélé, Yalcouyé, Coulibaly, Traoré, Jeffries, Diarra, Maiga, Diallo, Nimaga, Touré, Traoré, Kotioumbé, Mis, Cissé, Guinto, Fischbeck, Khokha, Lakhani and Landouré. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guida Landouré, Z3VpZGFAaWNlcm1hbGkub3Jn