 Pankaj Bagga
Pankaj Bagga Sudhakar Singh
Sudhakar Singh Gobind Ram
Gobind Ram Subham Kapil
Subham Kapil Avtar Singh
Avtar Singh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 24 May 2024
Sec. Neurorehabilitation
Volume 15 - 2024 | https://doi.org/10.3389/fneur.2024.1368658
This article is part of the Research Topic Integrated Clinical Management and Neurorehabilitation for Lumbosacral Spinal Diseases View all 11 articles
Spinal muscular atrophy (SMA) is an uncommon disorder associated with genes characterized by the gradual weakening and deterioration of muscles, often leading to substantial disability and premature mortality. Over the past decade, remarkable strides have been made in the field of SMA therapeutics, revolutionizing the landscape of patient care. One pivotal advancement is the development of gene-targeted therapies, such as nusinersen, onasemnogene abeparvovec and risdiplam which have demonstrated unprecedented efficacy in slowing disease progression. These therapies aim to address the root cause of SMA by targeting the survival motor neuron (SMN) gene, effectively restoring deficient SMN protein levels. The advent of these innovative approaches has transformed the prognosis for many SMA patients, offering a glimmer of hope where there was once limited therapeutic recourse. Furthermore, the emergence of small molecule compounds and RNA-targeting strategies has expanded the therapeutic arsenal against SMA. These novel interventions exhibit diverse mechanisms of action, including SMN protein stabilization and modulation of RNA splicing, showcasing the multifaceted nature of SMA treatment research. Collective efforts of pharmaceutical industries, research centers, and patient advocacy groups have played an important role in expediting the translation of scientific discoveries into visible clinical benefits. This review not only highlights the remarkable progress achieved in SMA therapeutics but also generates the ray of hope for the ongoing efforts required to enhance accessibility, optimize treatment strategies, rehabilitation (care and therapies) and ultimately pave the way for an improved quality of life for individuals affected by SMA.
Spinal muscular atrophy (SMA) is a rare and debilitating genetic disorder that primarily exerts influence on the motor neurons in the spinal cord, leading to muscle atrophy and weakness. This disorder has garnered significant attention in recent years due to the development of groundbreaking treatments, such as gene therapies, which have the potential to change the prognosis for individuals affected by SMA (1). SMA is a heterogeneous group of inherited neuromuscular disorders characterized by the progressive degeneration of motor neurons in the spinal cord. The condition’s prevalence and incidence can vary significantly based on geographic and ethnic factors, making it a complex subject for epidemiological study (2). The estimated global prevalence of SMA ranges from 1 in 6,000 to one in 10,000 live births, with variations in different populations and regions. This range represents a significant burden for affected families and healthcare systems (3). This condition is an autosomal recessive disorder resulted by mutations in the survival motor neuron 1 (SMN1) gene. The incidence of SMA depends on the carrier frequency of these mutations in a given population (4). SMA is classified into different divisions based on the age of onset and clinical severity, which also have implications for epidemiology. SMA type I, with an early outbreak and severe phenotype, is often the most common, while SMA type IV, with adult onset and milder symptoms, is rarer (5). There may be variations in the prevalence and incidence of SMA across different countries and regions. For example, some studies suggest a higher prevalence in certain European populations. Research is ongoing to understand these variations better (6). Addressing the impact of SMA on families and healthcare systems necessitates a multifaceted approach that includes comprehensive support services, caregiver education and training, psychosocial interventions, financial assistance programs, and healthcare system reforms to improve access to specialized care (7, 8). Collaboration among healthcare providers, lawmakers, advocacy organizations, and community stakeholders is critical for reducing the effect of SMA and improving the well-being of afflicted individuals and their families (9). In this review, we discussed the epidemiology and clinical classification of SMA, shedding light on the latest research, rehabilitation and clinical findings.
In 1891, SMA first came to light through the observations of Guido Werdnig in two infant brothers (10). Over the subsequent nine years, Johann Hoffmann documented an additional seven cases. The traditional framework for classifying SMA was built upon the timing of symptom onset and the highest level of motor function achieved. While the cases they scrutinized had intermediate symptom severity, the term “Werdnig-Hoffmann disease” was coined to denote the more severe manifestations of SMA (11). The year 1899 saw Sylvestre and Beevor describe severe forms of SMA, further delineating the spectrum. In 1964, Dubowitz contributed to the field by detailing intermediate forms of SMA in 12 patients and naming this variant “Dubowitz disease” (11–13). Additionally, in 1955, the discovery of a milder form of SMA was made, with Kugelberg and Welander providing a comprehensive description one year later (14). This division of kinds of SMA is based on the age of symptom when its starts, the highest motor function achieved, and the severity of muscle weakness. Research into the epidemiology of SMA is vital for understanding the wide prevalence of disease, its occurrence, and natural evolution. These studies provide beneficial insights into the biological, ecological, and demographic factors that drive the development and progression of SMA in populations. The International Standard of Care Committee for SMA (ISCCSMA) has classified SMA into five primary types, as shown in the Figure 1, which include:
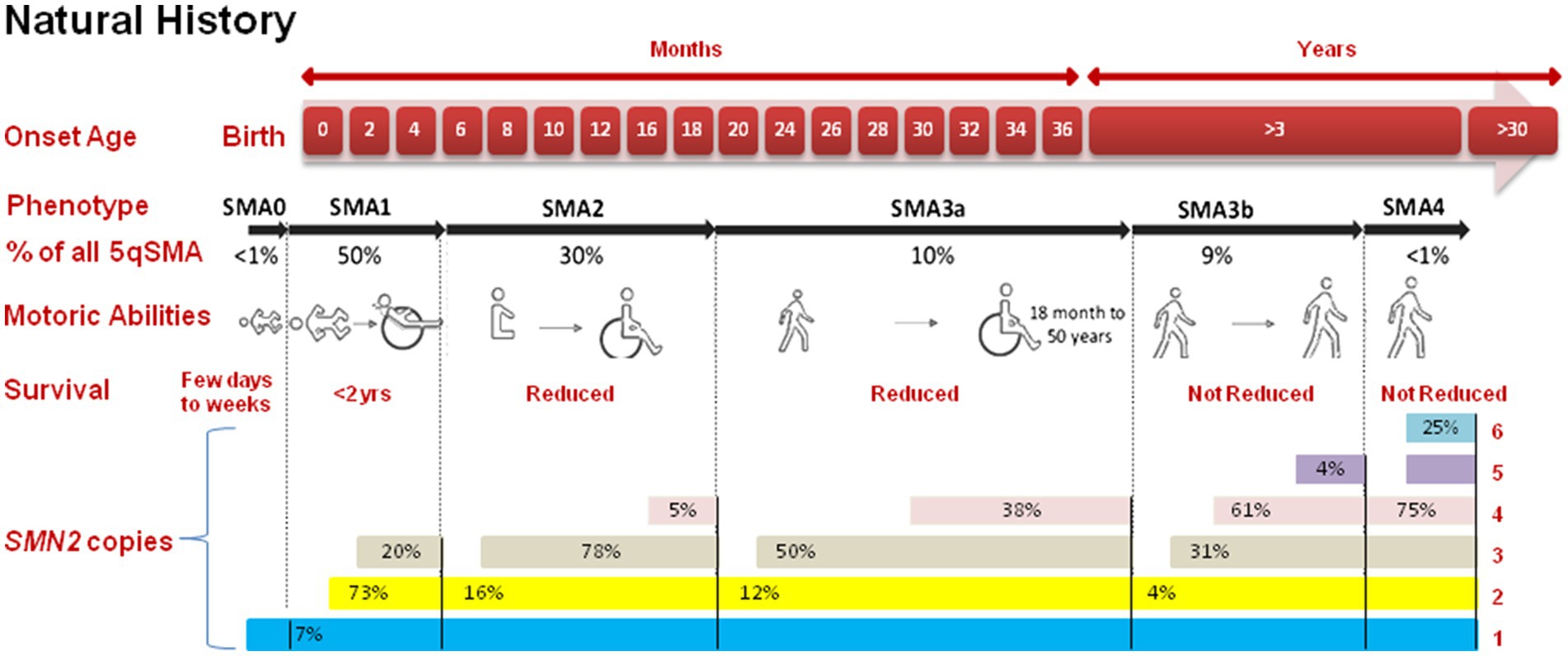
Figure 1. Comprehensive insight into the progression of SMA. The top panel categorizes the five types of SMA (0, 1, 2, 3, 4) which is based on the onset age and achieved motor capabilities. SMA type 3 is further subdivided upto 3a (onset <3 years) and 3b (onset >3 years). Furthermore, the figure presents the total count of SMN2 gene copies for each SMA type.
This is the most drastic and rarest form of SMA, with onset in womb or within the first few days of life. Neonate with SMA type 0 often exhibit severe muscle weakness and may not survive beyond a few months (15).
This is the most common form of SMA. Symptoms typically appear before first six months of life, and affected newborn may never will have the ability to sit independently or stand. SMA type I is characterized by severe muscle weakness, respiratory difficulties, and a shortened lifespan if not treated (16).
This form of SMA has a later onset, typically occurring between six and eighteen months of age. Affected individuals often get the ability to sit but may struggle with standing or walking. The rate of disease progression varies among individuals (12).
This type of disorder has an onset after 18 months of age. Individuals with this form may achieve the ability to walk independently, but muscle weakness and atrophy progress slowly. Some individuals may experience a relatively normal lifespan (17).
This is the mildest form of SMA, with an adult onset of symptoms. Affected individuals may experience muscle weakness, twitching, and exercise intolerance. The progression of the disorder is slow, and life aging is normal (18). Twenty cases with SMA type 4 were found in a Brazilian cohort of 227 SMA patients. This study includes the biggest cohort of SMA type 4 patients and provides practical, genetic, radiological, and neurophysiological aspects that may serve as biomarkers for future SMA-specific genetic therapeutics (19).
The SMN1 gene encodes the survival motor neuron (SMN) protein, which is essential for the normal functioning of motor neurons in the spinal cord (20, 21). SMN, a foundational protein within the SMN-Gemin multiprotein complex, serves as a core component. Additionally, it actively engages in various physiological functions, including responding to cellular stress, facilitating axon transport, regulating cytoskeletal dynamics, modulating mitochondrial and bioenergy pathways, and participating in ubiquitin pathways. Consequently, SMN emerges as a significant molecule, intricately involved in a multitude of essential activities that underpin human existence (22). These SMN genes are found within the 5q13 region, which harbors inverted repeats and multiple gene copies (20, 23, 24). The telomeric version of SMN1, with its nine exons, generates a functional 294-amino acid, 38 kDa SMN protein as shown in Figure 2. Typically, this protein is found in both organalles that is the cytoplasm and nucleus, specifically in the Gemini of coiled bodies compartment, which forms Cajal bodies holding high concentrations of small ribonucleoproteins (snRNPs) along with pre-mRNAs (25). SMN contains crucial and highly conserved domains that are essential for its cellular functions. Any kind of mutations occurring within these domains of SMN1 result in the production of an inefficient protein (26). The SMN2 is a centromeric gene which is a paralog of SMN1, having almost identical sequences with SMN1 except for 5 nucleotide differences. To understand the contributions of the survival motor neuron 2 (SMN2) gene to spinal muscular atrophy (SMA) pathology, it’s important to grasp its differences in alternative splicing compared to SMN1 and how these differences impact disease severity and progression. The one of these changes leads to the exclusion of exon number 7 in approximately 90% of the transcripts through alternative splicing (27). SMN2 is located 875 kb far from SMN1 and originates from a duplication of an ancestral gene which is unique to the human lineage (28). Both SMN1 and SMN2 genes encode the survival motor neuron (SMN) protein, which is crucial for the survival and function of motor neurons. However, a critical difference between SMN1 and SMN2 lies in a single nucleotide difference within exon 7, resulting in a C-to-T transition in SMN2. This single nucleotide change in SMN2 affects the alternative splicing pattern, leading to the exclusion (skipping) of exon 7 in a significant proportion of transcripts. Exon 7 skipping results in the production of an isoform of the SMN protein lacking exon 7 (SMNΔ7), which is less stable and less functional compared to the full-length SMN protein produced by SMN1. The exclusion of exon 7 in a substantial portion of SMN2 transcripts results in reduced levels of functional SMN protein in cells, contributing to the pathogenesis of SMA. While SMN2 can partially compensate for the loss of SMN1 function, the lower levels of full-length SMN protein produced by SMN2 are insufficient to fully support motor neuron survival and function. The severity and progression of SMA are influenced by the number of copies of SMN2 present in the genome. Individuals with fewer copies of SMN2 typically produce lower levels of functional SMN protein and tend to have more severe forms of the disease, whereas those with more copies of SMN2 may produce higher levels of functional SMN protein and exhibit milder symptoms. The unique alternative splicing pattern of SMN2 has made it a primary target for therapeutic interventions aimed at increasing the production of full-length SMN protein. SMA symptoms manifest when there is a deficiency of proper functional SMN protein, usually stemming from minimum one copy of the SMN1 (29). However, around 10% of full-length SMN2 transcripts, often present in multiple copies within the genome, provide some degree of protection against motor neuron degeneration (30). The more SMN2 copies a patient possesses, the more they can compensate for the absence of SMN1 (31). Consequently, in rare cases, individuals with 6 or more copies can exhibit milder symptoms appearing after the age of 30, characterized by mild muscle weakness and retained full mobility. Most type I SMA patient’s carries either one or two SMN2 copies (32). While the number of SMN2 gene copies strongly correlates with disease severity, some studies suggest that it may not always be a definitive indicator of severity, especially in SMA patients who retain one SMN1 allele (33). Additionally, even when SMN is expressed normally, point mutations in SMN can affect protein functionality and stability, leads to the disorder, along with genetic and epigenetic factors, as well as environmental influences, may modulate disease (34). Approaches such as antisense oligonucleotide (ASO) therapy and small molecule drugs target the splicing machinery to promote the inclusion of exon 7 in SMN2 transcripts, thereby increasing the production of functional SMN protein. These therapies aim to augment the levels of functional SMN protein in motor neurons, potentially ameliorating disease symptoms and improving outcomes for individuals with SMA.
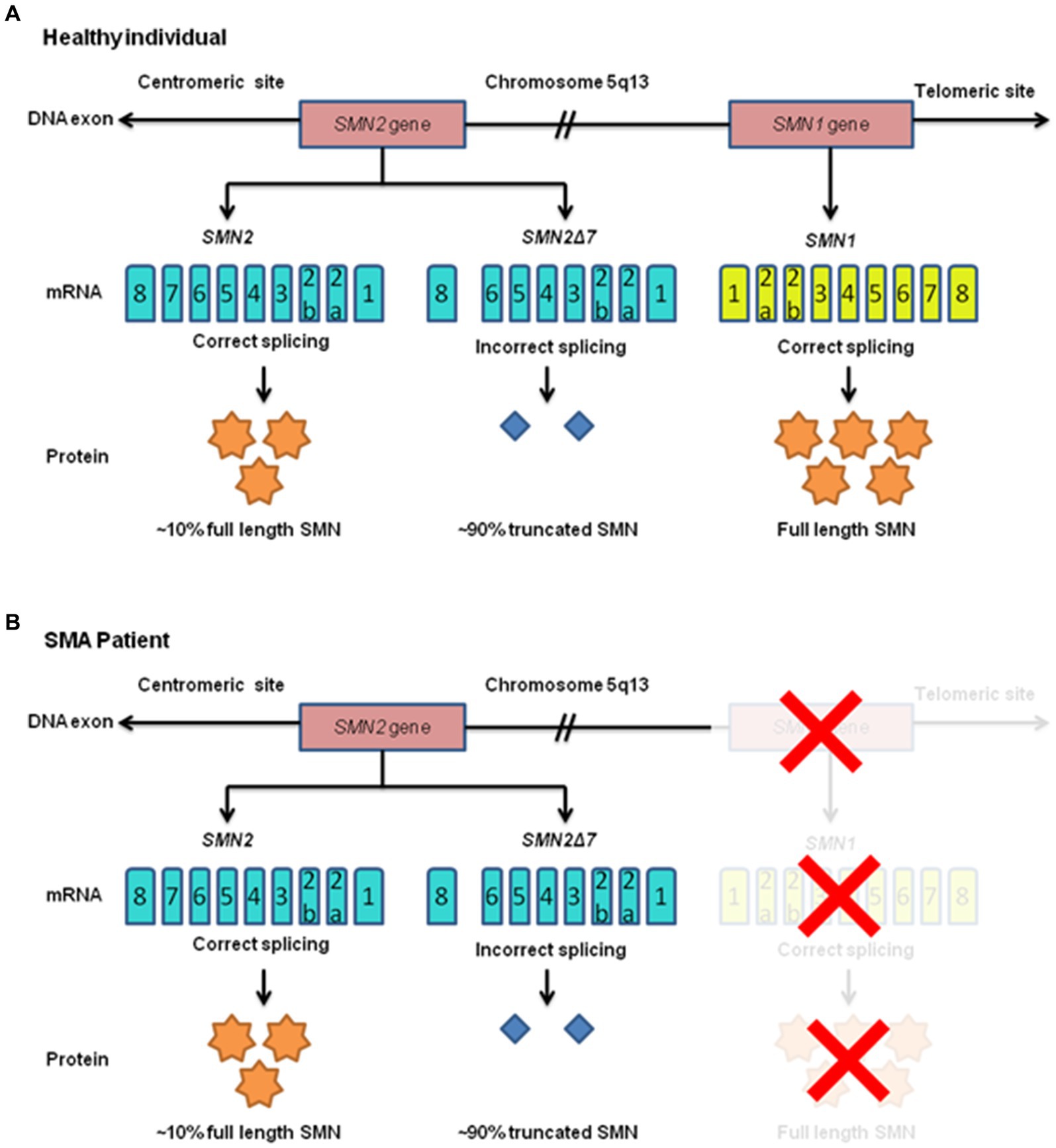
Figure 2. Human survival motor neuron (SMN) gene expression is shown in a schematic diagram for both healthy people and those with SMA. The chromosome 5q13 region (long arm of chromosome 5) has been explicitly identified as the location of the telomeric SMN1 and centromeric SMN2 genes. Full-length, functional SMN (FL-SMN) protein is produced by the SMN1 gene whereas the SMN2 gene, owing to incorrect splicing, produces 90% truncated SMN protein (SMNΔ7) and only 10% of FL-SMN protein. (A) Both SMN genes are present in healthy individuals. (B) The SMN1 gene is absent in SMA patients due to mutations, which prevent SMN1 from producing FL-SMN protein (this condition is denoted by a red “X”). Because production is completely dependent on the SMN2 gene, there is inadequate production.
The underlying molecular mechanisms of SMA revolve around the loss of functional SMN protein and its impact on motor neurons and muscle cells. The reduction in functional SMN protein in motor neurons results in their degeneration (35). Motor neurons are responsible for transmitting signals from the spinal cord to muscles, and without proper functioning SMN protein, these neurons become vulnerable to damage and eventual death. This leads to muscle weakness and atrophy (36). SMN protein plays a crucial role in the assembly of snRNPs, which are essential for mRNA splicing (37). Impaired snRNP assembly due to SMN deficiency leads to widespread splicing defects in various genes, further exacerbating motor neuron dysfunction (38). While SMA is primarily a disorder of motor neurons, the resulting muscle atrophy and weakness are critical clinical features. The lack of neural input from affected motor neurons causes muscle disuse, contributing to muscle wasting (39). The neuromuscular junction, where motor neurons communicate with muscle cells, is also affected in SMA (40). The loss of functional motor neurons disrupts this communication, leading to muscle weakness and contractures (41).
SMA presents with a spectrum of clinical features, ranging from severe, early-onset forms to milder, adult-onset forms. The key clinical manifestations include muscle weakness, atrophy, and hypotonia. Patients with SMA may also exhibit respiratory difficulties, joint contractures, and scoliosis (42). It is essential to recognize these clinical signs early, as timely intervention can significantly impact the prognosis and quality of life for affected individuals.
A thorough clinical evaluation is the initial step in diagnosing SMA. The healthcare provider takes a detailed medical history, conducts a physical examination, and assesses motor function (43). The clinical evaluation includes a review of family history to identify any known cases of SMA or related neuromuscular disorders. It is important to consider that SMA may not be the first suspicion in cases with mild or atypical symptoms (44).
Electromyography (EMG) is a diagnostic technique used to assess the electrical activity of muscles and nerves (45). In SMA, EMG may reveal neurogenic changes, denoting motor neuron dysfunction. EMG can help distinguish SMA from other neuromuscular disorders and provide information about the extent of motor neuron involvement (46).
Nerve conduction studies (NCS) evaluate the function of peripheral nerves. In SMA, NCS can be normal or show mild abnormalities. These tests help rule out other neurological conditions and provide additional information to support the diagnosis of SMA (47).
While muscle biopsy is not the primary diagnostic tool for SMA, it can be used to confirm the absence of muscle pathology, ruling out conditions like muscular dystrophy. Muscle biopsies typically show atrophy and denervation in SMA, supporting the diagnosis (48).
Measuring serum creatine kinase (CK) levels can be useful in differentiating SMA from muscular dystrophies. In SMA, CK levels are usually within the normal range or only mildly elevated, whereas in muscular dystrophies, CK levels are significantly elevated (49).
Newborn screening for spinal muscular atrophy (SMA) is a critical and rapidly evolving aspect of pediatric healthcare aimed at early detection and intervention for this devastating genetic disorder. One of the primary benefits of newborn screening for SMA is the early identification of affected infants. Unlike in the past, when diagnosis often occurred after the onset of symptoms, newborn screening allows for early intervention and treatment (50).
Genetic testing is the gold standard for diagnosing SMA. It provides a definitive diagnosis, identifies the specific genetic mutation, and helps determine the severity of the condition. Genetic testing typically involves the following approaches: (a) the primary genetic test for SMA involves analyzing the SMN1 gene. Most SMA cases result from deletions or mutations in this gene, leading to reduced SMN protein levels. This test is highly specific and sensitive and can diagnose SMA with a high degree of accuracy (51). (b) In addition to SMN1 analysis, counting the number of copies of the SMN2 gene can provide information about the disease severity. SMA patients with more SMN2 copies tend to have milder forms of the disease, while those with fewer copies typically have more severe forms (52).
Next-generation sequencing (NGS) is a powerful tool for identifying rare or typical mutations in the SMN1 gene. It can be especially useful in cases where standard genetic tests do not yield a diagnosis. NGS can also detect other rare genetic conditions that may mimic SMA (53).
Genetic testing can be performed during pregnancy to identify SMA in the fetus. This can be done through chorionic villus sampling or amniocentesis. Early diagnosis allows for informed reproductive decisions and early intervention if the fetus is affected (54). Prenatal testing for spinal muscular atrophy (SMA) raises several ethical considerations, including issues related to informed consent, autonomy, disability rights, and the potential for discrimination. It’s crucial that expectant parents fully understand the purpose, benefits, limitations, and potential consequences of SMA prenatal testing. They should have access to comprehensive information about SMA, including its prognosis, available treatments, and the emotional impact of receiving a positive result. Current recommendations by the American College of Medical Genetics (ACMG) include offering SMA carrier screening to all couples, regardless of race or ethnicity, before conception or early in pregnancy. Current recommendation by the American Congress of Obstetricians and Gynecologists (ACOG) do not advise preconception and prenatal screening for SMA be offered to the general population and advice testing offered to general population (55). Some disability rights advocates argue that prenatal testing for conditions like SMA perpetuates ableism and sends a message that individuals with disabilities have less value or are not worthy of existence. This perspective challenges the notion that certain disabilities should be actively prevented or eliminated through selective abortion.
There are several treatments approaches for the SMA such as (Table 1).
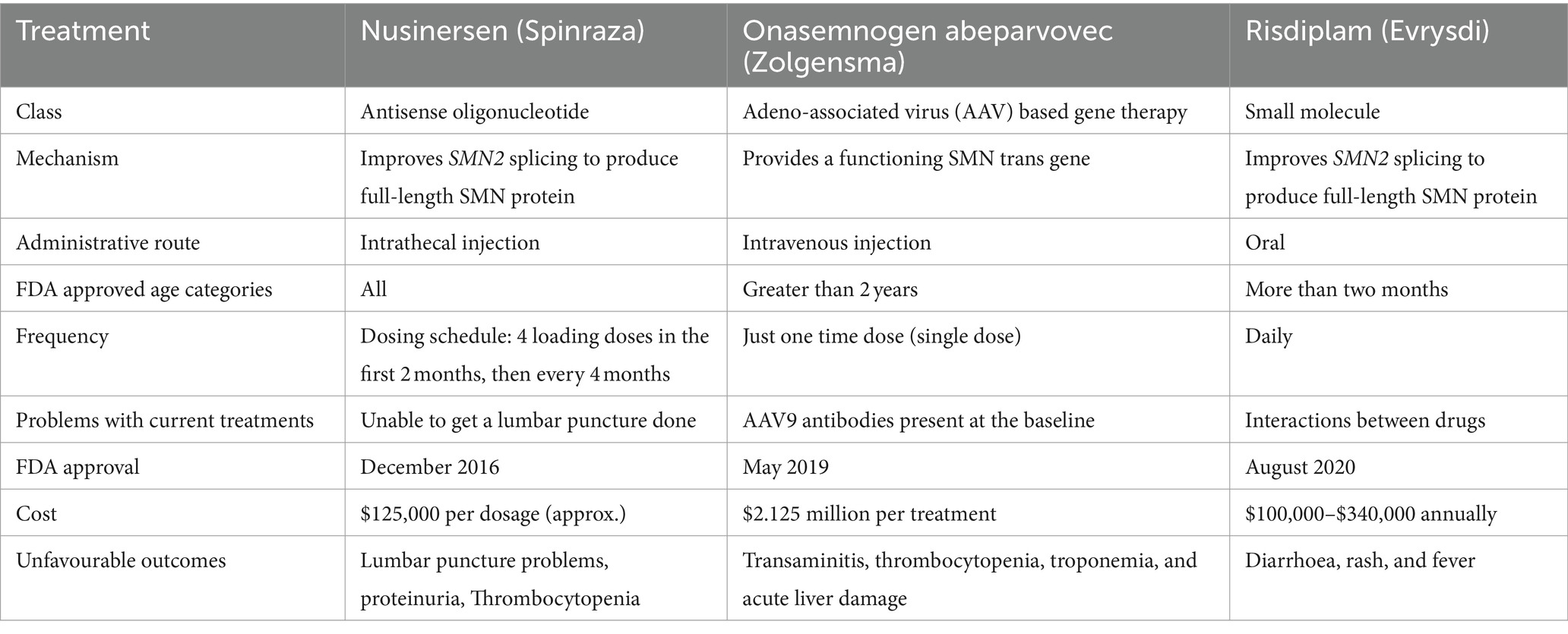
Table 1. Treatment options currently in use for spinal muscular atrophy.
Nusinersen (marketed as Spinraza®) was the first FDA-approved disease-modifying treatment for SMA having obtained approval in December 2016 and by the EMA in 2017 for both infant and adult (56). This innovative therapeutic approach, which involves the intrathecal administration of a 2′-O-methoxyethyl phosphorothioate modified antisense oligonucleotide (ASO), focuses on enhancing the incorporation of exon 7 into mRNA transcripts of SMN2 (57). The intrathecal route of administration is crucial for nusinersen’s effectiveness in treating SMA because it allows for targeted delivery to the site of pathology, bypasses the blood-brain barrier, optimizes concentration at the target site, minimizes systemic side effects, and provides a longer duration of action within the central nervous system (57). By administering nusinersen directly into the central nervous system (CNS) through the intrathecal route, the medication effectively suppresses the activity of certain splice-factors and binds to a specific intronic splice-silencing site within intron 7 of SMN2 (58). This intervention substantially increases the probability of exon 7 being included in the mRNA, ultimately enabling the translation of a more substantial quantity of fully functional SMN protein (27). This enhanced production has demonstrated significant improvements in both survival and the overall condition of various experimental models of SMA. Importantly, nusinersen’s journey to approval and commercialization has been bolstered by a multitude of studies confirming its efficacy, without any notable drug-related adverse events (59). Due to ASOs’ inability to traverse the blood-brain barrier, nusinersen was consistently administered intrathecally in all clinical trials. During the initial loading phase, it was administered four times over two months, and in the maintenance phase, it was given once every four months (60). The standard dosage of nusinersen typically amounts to 12 milligrams (61). Studies on nusinersen have revealed the potential for some patients to regain lost abilities, such as sitting up, standing, and walking, without the need for therapy. Furthermore, early initiation of this treatment has demonstrated positive outcomes in individuals with SMA types I, II, and III (62). It is worth noting that a notable drawback of this therapy is the possibility of side effects, including constipation and upper and lower respiratory tract infections (63).
Onasemnogene abeparvovec (Zolgensma), an advanced gene therapy: In May 2019, the FDA granted approval to AVXS-101, also known as Zolgensma is a gene therapy approved for the treatment of SMA, developed by AveXis, a subsidiary of Novartis (56). This approval followed the release of favorable outcomes from the phase one clinical trial known as START (Identifier: NCT01547871). This trial assessed the drug’s safety and effectiveness when administered as a one-time infusion to infants with SMA symptoms appearing before six months of age. Subsequently, in March 2020, Zolgensma received conditional marketing authorization, and in May 2020, it was granted approval by the European Medicines Agency (EMA) as well (Zolgensma, 2020a; Novartis, 2020) (64). The adeno-associated virus 9 (AAV9) capsid is used to transport the SMN-encoding complementary DNA (cDNA) to the motor neurons that need it (65–67). A single dose of AVV9 administered intravenously (IV) is sufficient to transport a functional copy of the SMN1 gene over the blood-brain barrier and into patient cells, where it may stimulate the production of SMN protein (27). The SMN1 trans gene and synthetic promoter based on AVV9 are also crucial components in maintaining SMN protein synthesis throughout time (68). Although it successfully corrects the underlying molecular defect in SMA, it has a deleterious effect on the liver by elevating serum amino transferase (69). However, prednisone is effective at reducing elevated liver enzymes. Therefore, at least three months after administration, patients should be monitored for liver function (70, 71). The long-term durability of the benefits of Zolgensma (onasemnogene abeparvovec) is still being actively studied and monitored. Zolgensma is a gene therapy approved for the treatment of spinal muscular atrophy (SMA) in pediatric patients, and it has shown remarkable efficacy in improving motor function and survival in clinical trials. While the initial data from clinical trials and real-world experience have shown sustained benefits of Zolgensma treatment over several years, including improvements in motor function and survival, more long-term follow-up is needed to fully understand the duration of these benefits. Clinical trials and observational studies are ongoing to assess the durability of Zolgensma’s effects, including its impact on motor function, respiratory function, quality of life, and survival rates over extended periods. These studies involve monitoring patients treated with Zolgensma for several years to track their progress and detect any potential changes in treatment outcomes over time. It’s important to note that as research continues and more data become available, our understanding of the long-term benefits and potential limitations of Zolgensma treatment will continue to evolve. Patients and caregivers should work closely with healthcare providers to stay informed about the latest research findings and recommendations regarding the use of Zolgensma in the management of SMA (65, 72).
Risdiplam (Evrysdi™) was authorized by the FDA as of 7 August 2020, as the first oral medication for children as young as 2 months old and adults with SMA. It is a collaborative development effort involving Roche, PTC Therapeutics Inc., and the SMA Foundation, aimed at addressing spinal muscular atrophy (56, 73). Risdiplam serves as a modifier of mRNA splicing that leads to an elevation in SMN protein expression (74). It is a tiny molecule that changes the splicing of the SMN2 by binding to two locations in the SMN2 pre-mRNA. These sites are known as the 5′ splice site (5′ ss) of intron number 7 and the exon splicing enhancer 2 (ESE2) of exon 7 (75). Increases in full-length SMN mRNA and protein levels are caused by the unique specificity of binding two sites, which also reduces impact on other pre-mRNA splicing and prevents the likelihood of off-target effects (76). According to preclinical studies, risdiplam can reach the central nervous system and peripheral organs in vivo and can result in a significant increase of SMN protein in the blood, brain, and muscles, as well as an increase in survival in various SMA mouse models (77). While risdiplam’s systemic distribution in preclinical tests with oral administration allowed for the possibility of an impact on other tissues, nusinersen’s intrathecal delivery method mostly limited its effect to motoneurons of the central nervous system (78). Previous studies in human and murine models suggest that SMA may in fact be considered a multi-system disorder involving the neuromuscular junction, cardio-vascular system, lung, gastrointestinal-tract, and liver (79). Risdiplam has shown significant improvements in motor function and has the advantage of being an oral therapy, making it a more convenient option for many patients (80).
SMA treatments have evolved notably in recent years, particularly as previously stated gene therapies like Zolgensma and disease-modifying drugs like Spinraza (nusinersen). Much more significant perspective, which should consider insurance coverage, availability across regions, and efforts to improve access to SMA treatments. While these drugs offer a promising treatment option for children with SMA, they are highly costly, as seen in Table 1. According to studies, certain governments have made efforts to design policies for such disease treatments, such as: The Department of Revenue, Ministry of Finance, govt. of had issued Notification No. 46/2021-Customs dated 30.09.2021, which waives all Basic Customs Duty (BCD) and Integrated Goods and Services Tax (IGST) on drugs imported (personal use only) for the treatment of spinal muscular atrophy (SMA) rare disease, making medicines for SMA rare disease more affordable (81). Government of India also made a provision for financial assistance of up to Rs. 50 lakhs to patients suffering from any category of Rare Diseases such as SMA and for treatment at any of the Centres of Excellence (CoE) identified in the NPRD-2021, outside of the Rashtriya Arogaya Nidhi umbrella scheme (82). The National Health Service (NHS) England, for example, states that Biogen (the pharmaceutical company that manufactures treatment for SMA) will make the treatment for spinal muscular atrophy (SMA) available to the youngest and most severely affected (SMA type 1) patients immediately, with NHS England offering funding contingent on the National Institute for Health and Care Excellence (NICE) publication of final guidance. In Singapore, the Rare Disease pay has been established to pay five drugs to treat three rare diseases. In Malaysia and Australia, qualifying patients are given discounted access to pricey and life-saving medications (83).
In addition to disease-modifying treatments, SMA management often involves a multidisciplinary approach as shown in Figure 3 that focuses on addressing the symptoms and complications associated with the disease. Supportive care strategies aim to improve the quality of life for individuals with SMA and include:
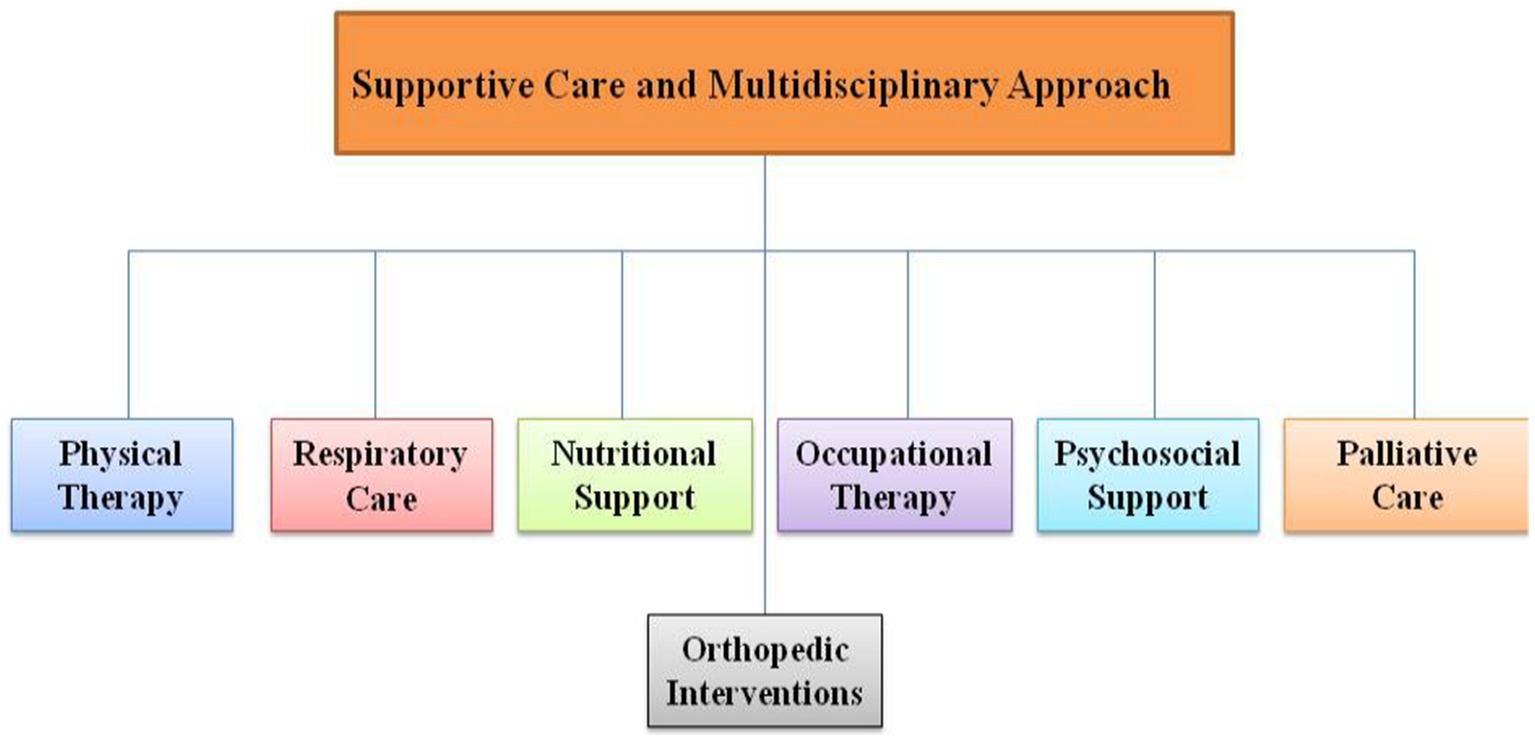
Figure 3. Supportive care and multidisciplinary approach flow chart.
Physical therapy is essential in managing muscle weakness, contractures, and maintaining range of motion (84, 85). People affected by spinal muscular atrophy may experience limitations in their ability to fully articulate their joints due to muscle weakness. This can potentially lead to the development of contractures, characterized by muscle tightness that may become permanent and restrict mobility (86). Physical therapy often incorporates exercises and stretching routines designed to enhance flexibility and overall functionality (87). These interventions aim to minimize the likelihood of joint contractures, mitigate the progression of scoliosis, and promote the maintenance of a healthy weight. Mounting evidence suggests that consistent participation in physical therapy sessions can yield positive outcomes in terms of both function and the progression of spinal muscular atrophy in patients (86, 87).
The management of the respiratory system plays a pivotal role in addressing spinal muscular atrophy (88). Weakness in the chest muscles can hinder one’s capacity to breathe or cough effectively, consequently elevating the likelihood of infections (89, 90). The available respiratory care solutions encompass both non-invasive and invasive methods: (i) non-invasive respiratory care comprise strategies that aim to circumvent or postpone the necessity for invasive procedures (88). Specialized apparatus, such as a ventilator or a bilevel positive airway pressure (BiPAP) machine, can provide a constant airflow to the lungs using a mask that covers the mouth and/or nose. Additionally, a home-based cough assist device may be utilized to facilitate coughing and the clearance of secretions. (ii) Invasive respiratory care establishes a protected passage to the lungs using an endotracheal tube inserted either through the mouth (intubation) or directly into the trachea via a minor neck incision (tracheotomy) (91, 92).
The weakening of muscles can result in some individuals with SMA experiencing a decline in their ability to chew and swallow proficiently. This situation increases the risk of food or liquids being aspirated into the lungs, potentially leading to respiratory infections (93, 94). To address this, a temporary or permanent feeding tube may be inserted to ensure the essential intake of nutrition and hydration (93). Broadly speaking, there are two categories of feeding tubes: those inserted through the nose and those placed in the abdominal area. Nasogastric (NG) tubes are inserted through the nasal passage and deliver nutrition directly into the stomach (95, 96). These are typically employed for patients requiring short-term feeding tube access and are conveniently replaceable (95). Gastrostomy (G) tubes are surgically implanted through the abdominal wall and directly into the stomach (97–99). Due to their ease of maintenance, they are often the favored choice for individuals in need of extended-term feeding assistance.
Orthopedic surgery may be required in cases where there are severe joint contractures or scoliosis (98, 100). Scoliosis, characterized by a spinal curvature, can be a challenge for individuals with spinal muscular atrophy as a consequence of muscle weakness (101). An orthopedic specialist may propose postural support in the form of bracing or recommend surgical intervention to address scoliosis (98, 101).
Occupational therapists work with SMA patients to enhance their ability to perform daily tasks and improve their independence (102, 103). They assess patients’ needs and recommend assistive devices or home modifications to facilitate daily activities. Their role includes: adaptive techniques means teaching individuals adaptive techniques and recommending assistive devices that enable greater independence (104). Home modifications means assessing home environments and suggesting modifications to make daily tasks more manageable (105). Assisting with communication means in cases of severe SMA, occupational therapists can help individuals use communication devices to facilitate interaction with others (106).
SMA affects not only the physical health but also the emotional and psychosocial well-being of patients and their families (107). Ensuring the holistic care of a patient with SMA and their family is insufficient without the monitoring and treatment of their psychosocial welfare. Requirements differ based on the patient’s age and specific sub-types of SMA. Psychosocial health can be influenced by various elements, including social and emotional factors as well as treatment factors such as innovative therapies. Psychosocial care should encompass a wide range of characteristics, including social and cognitive development, quality of life, and the impact on patient and family functioning in various situations such as home, school, or job. The care of SMA should include the involvement of a mental health practitioner, such as a psychologist, psychiatrist, or neuropsychologist, as well as a social worker who has specialised experience in assisting patients with chronic diseases. Assessments should be considered around the time of diagnosis, before entering school, and after a change in functionality. Implementing psychologically informed care and employing a range of interventions has the potential to mitigate psychological morbidity in both children and parents. During every multidisciplinary appointment, it is important to assess the individual’s mental health and quality of life. If deemed essential, the mental health clinician will be involved in evaluating the psychological state of the patient, as well as their parents and siblings (107). Psychosocial support addresses these aspects and may include: (i) counseling: individual and family counseling can help individuals and caregivers cope with the emotional challenges of living with SMA (107, 108). (ii) Support groups: joining support groups, either in person or online, can provide a sense of community and shared experiences. (iii) Mental health services: access to mental health services is essential for addressing anxiety, depression, and stress that may result from the condition (108, 109).
For individuals with advanced SMA or those with severe complications, palliative care can provide symptom management, pain relief, and emotional support to enhance quality of life (110).
The management of SMA requires a multidisciplinary team approach involving various healthcare professionals to address the diverse needs of individuals with the condition. The team may include neurologists, physical and occupational therapists, respiratory therapists, nutritionists, orthopedic surgeons, and social workers. This collaborative approach ensures that the physical, emotional, and psychosocial aspects of SMA are managed comprehensively (111). Supportive care is the fundamental aspect of clinical management in spinal muscular atrophy (SMA). The development of disease-modifying medications such as nusinersen, onasemnogene abeparvovecxioi, and risdiplam has provided improved treatment choices for the most severe forms of the condition. These medications have increased survival rates and brought hope for a longer and better quality of life. Additionally, they have influenced the way healthcare is provided for these patients. Although there have been some improvements in the field, adults living with SMA and those transitioning into adulthood have been somewhat overlooked, despite the emergence of studies and advancements such as enhanced respiratory care, home adaptations, and devices that promote greater independence, like power wheelchairs and voice amplifiers. It is important for everyone to acknowledge and appreciate these achievements. The effects of fragmented care might be intensified for individuals migrating from paediatric to adult care, as they no longer receive the same degree of coordinated assistance provided in paediatric settings. According to reports, individuals who are transferring from paediatric to adult healthcare services face challenges in understanding and interacting with a complex health system and new specialists. They commonly describe this experience as “challenging and intimidating.”
In addition, adults with SMA may have difficulties during this phase of transition, particularly when they need to relocate (e.g., for higher education), as this necessitates them to become part of a new healthcare system. Although not prevalent within the SMA community as a whole, certain jurisdictions have acknowledged this issue and have adopted targeted measures to mitigate its effects. For instance, they have established transitional clinics where medical professionals from both adult and paediatric fields participate in appointments. We advocate for increased implementation of optimal methods and specialised procedures (such as established transitional care clinics, communication paths between paediatric and adult experts, and a nationwide network of specialists) that streamline the transition to adult care and the transfer of knowledge (46).
The use of assistive devices and technology plays a significant role in SMA management. These devices help individuals with SMA lead more independent and fulfilling lives (112). Examples include: wheelchairs, scooters, and other mobility aids provide individuals with the freedom to move and explore their environments (113). Augmentative and alternative communication (AAC) devices assist those with severe SMA in expressing themselves (106). These systems enable individuals to control various aspects of their environment, such as lights, doors, and appliances, through adapted technology. Adapting the home environment with features like ramps, wider doorways, and accessible bathrooms enhances accessibility (105). Adaptive technology and software allow individuals with limited physical mobility to use computers and access the internet (104).
Research into SMA continues, with several promising therapies under investigation. These therapies include small molecule drugs, gene-editing technologies, and exon-skipping therapies, among others. The goal is to further enhance the disease-modifying potential and offer a more comprehensive treatment approach for SMA. Advances in genetic testing have made it easier to diagnose SMA accurately and offer prenatal testing for at-risk pregnancies. These advancements include: NGS has become a powerful tool in identifying rare or atypical mutations in the SMN1 gene and other related genes. It can uncover genetic variations that were previously challenging to detect (114, 115). Techniques like chorionic villus sampling and amniocentesis allow for the diagnosis of SMA in the fetus, enabling informed reproductive decisions and early intervention if the fetus is affected (116, 117).
Emerging gene-editing techniques, such as CRISPR-Cas9, offer the potential to correct genetic mutations directly, providing a curative approach to SMA. These technologies are in the early stages of development and are being explored in preclinical studies. In addition to this, CRISPR technology, which expands the scope of genetic engineering and gene treatments, enables the treatment of a wide range of hereditary illnesses. Some prior research in the literature show that SMA can be treated using the CRISPR method. Homology directed repair (HDR)-based CRISPR technology, which produces a high rate of in-del (insertion-deletion) mutations rather than editing, has been proven unsuitable for therapeutic purposes. CRISPR-prime editing (PE) technology is a novel type of gene editing technique that enables precise genomic alterations without the need for double-strand breaks or donor DNA sequences. The CRISPR-prime editing approach has also been employed in rare disorders like as sickle cell anaemia and Tay–Sachs, and its effectiveness in editing diverse harmful variants has been proven. However, CRISPR Prime Editing-mediated gene editing for spinal muscular atrophy (SMA) has yet to be investigated (114).
Small molecules that target specific pathways involved in SMA are also under investigation. These drugs aim to increase SMN protein production and improve motor function (116).
Researchers are exploring the use of combination therapies, including a mix of SMN2-targeting drugs and other treatments, to enhance the efficacy of SMA management (118). Combining different therapeutic strategies to maximise SMA treatment outcomes is an exciting approach. Limited data supports the efficacy of expensive drug combinations in people, encouraging clinicians and scientists to examine all therapeutic options (56, 119) A combination of SMN-dependent ASO-inducing SMN2 exon inclusion and SMN-independent myostatin inhibition yielded positive results in a SMA animal model (56, 120). A limited sample of patients were treated with a combination of Zolgensma and nusinersen, but the long-term benefits remain unclear. Zolgensma and nusinersen have distinct modes of action, making drug-to-drug interactions less common. Nusinersen targets an intron sequence to increase exon 7 inclusion. The transplanted Zolgensma gene lacks introns and hence should not interfere with nusinersen translation. Zolgensma treatment should be approached with caution due to the reported adverse event of thrombocytopenia associated with nusinersen. Long-term follow-up data, particularly in pre-symptomatic patients, is needed to evaluate the effectiveness and hazards of combination therapy (56). Combination therapies, treatments and advocacy initiatives have played an important role in determining research orientations and legislative changes in the management of spinal muscular atrophy (SMA). Advocacy groups such as cure SMA, SMA foundation, and fight SMA have been instrumental in catalyzing SMA research. By raising awareness, funding research initiatives, and fostering collaborations among scientists, these organizations have accelerated the pace of discovery in understanding the underlying genetic mechanisms of SMA, identifying potential therapeutic targets, and developing innovative treatment strategies.
Research has shown the benefits of early intervention, even before the onset of symptoms, in infants with SMA (121). The nurture study demonstrated that early treatment with nusinersen in pre-symptomatic infants significantly improved motor function and developmental outcomes (122).
SMA patient and caregiver advocacy groups have played a crucial role in raising awareness, driving research, and improving access to care and treatments. Their efforts have been instrumental in advancing the SMA field (123).
Thirty years after the discovery of the SMN gene, global scientific efforts have successfully transformed spinal muscular atrophy (SMA) into a manageable condition. Three distinct drugs utilizing cutting-edge technology—gene therapy, antisense oligonucleotides (ASOs), and small molecules focused on SMN2 splice correction—have gained approval from both the FDA and EMA, demonstrating remarkable enhancements, particularly when administered prior to the onset of symptoms. SMA, known for its substantial economic impact, has become even more financially demanding with the introduction of these novel therapies. Addressing the considerable economic burden associated with these treatments has led to an increasing advocacy for the inclusion of SMA in newborn screening (NBS) programs. While the existing therapies are likely sufficient for immediate administration after birth in cases of intermediate and mild SMA, questions arise regarding whether individuals with only two SMN2 copies will follow a trajectory similar to their age-matched counterparts and if they may require additional SMN-independent therapies. Comprehensive longitudinal studies are imperative to explore potential new phenotypes linked to these innovative therapies. Despite the presence of biomarkers associated with disease progression, further investigations are needed to identify potential non responders, enabling them to transition to alternative therapies. SMA treatments are often expensive, prompting concerns regarding equal access for all patients, especially in low-income areas or nations with limited healthcare resources. Addressing pricing and guaranteeing access to these life-changing medications for all SMA patients worldwide remains a serious concern. Numerous unanswered questions persist, necessitating meticulous future research. Nonetheless, SMA stands as a model illustrating how genetic insights can pave the way for the development of targeted therapies.
PB: Formal analysis, Data curation, Writing – original draft. SS: Writing – review & editing, Supervision, Writing – original draft. GR: Writing – review & editing, Supervision. SK: Writing – original draft, Visualization. AS: Writing – review & editing, Supervision, Formal analysis.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
All authors are thankful to Lovely Professional University for providing labs to perform work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Willis, TA. Therapeutic advances in spinal muscular atrophy. Paediatr Child Health. (2023) 33:23–8. doi: 10.1016/j.paed.2022.12.010
2. Lapp, HS, Freigang, M, Hagenacker, T, Weiler, M, Wurster, CD, and Günther, R. Biomarkers in 5q-associated spinal muscular atrophy—a narrative review. J Neurol. (2023) 270:4157–78. doi: 10.1007/s00415-023-11787-y
3. Toro, W, Yang, M, Georgieva, M, Song, W, Patel, A, Jiang, A, et al. Health care resource utilization and costs for patients with spinal muscular atrophy: findings from a retrospective US claims database analysis. Adv Ther. (2023) 40:4589–605. doi: 10.1007/s12325-023-02621-y
4. Savad, S, Ashrafi, MR, Samadaian, N, Heidari, M, Modarressi, M-H, Zamani, G, et al. A comprehensive overview of SMN and NAIP copy numbers in Iranian SMA patients. Sci Rep. (2023) 13:3202. doi: 10.1038/s41598-023-30449-7
5. Toro, W, Yang, M, Georgieva, M, Anderson, A, LaMarca, N, Patel, A, et al. Patient and caregiver outcomes after onasemnogene abeparvovec treatment: findings from the cure SMA 2021 membership survey. Adv Ther. (2023) 40:5315–37. doi: 10.1007/s12325-023-02685-w
6. Whitney, G, Daniel, EE, Knierbein, N, and Daunter, AK. Prevalence of morbidities across the lifespan for adults with spinal muscular atrophy: a retrospective cohort study. Orphanet J Rare Dis. (2023) 18:258. doi: 10.1186/s13023-023-02872-6
7. McMillan, HJ, Gerber, B, Cowling, T, Khuu, W, Mayer, M, Wu, JW, et al. Burden of spinal muscular atrophy (SMA) on patients and caregivers in Canada. J Neuromuscul Dis. (2021) 8:553–68. doi: 10.3233/JND-200610
8. Murrell, DV, Lotze, TE, Farber, HJ, Crawford, CA, and Wiemann, CM. The experience of families with children with spinal muscular atrophy type I across health care systems. J Child Neurol. (2017) 32:917–23. doi: 10.1177/0883073817716853
9. Chan, SHS, Wong, CKH, Wu, T, Wong, W, Yu, MKL, Au, ICH, et al. Significant healthcare burden and life cost of spinal muscular atrophy: real-world data. Eur J Health Econ. (2023) 24:1373–82. doi: 10.1007/s10198-022-01548-5
10. Werdnig, G. Zwei frühinfantile hereditäre Fälle von progressiver Muskelatrophie unter dem Bilde der Dystrophie, aber anf neurotischer Grundlage. Arch Psychiatr Nervenkr. (1891) 22:437–80. doi: 10.1007/BF01776636
11. Nakevska, Z, and Yokota, T. Challenges and future perspective of antisense therapy for spinal muscular atrophy: a review. Eur J Cell Biol. (2023) 102:151326. doi: 10.1016/j.ejcb.2023.151326
12. Awano, T, Kim, J-K, and Monani, UR. Spinal muscular atrophy: journeying from bench to bedside. Neurotherapeutics. (2014) 11:786–95. doi: 10.1007/s13311-014-0293-y
13. Dubowitz, V. Infantile muscular atrophy a prospective study with particular reference to a slowly progressive variety. Brain. (1964) 87:707–18. doi: 10.1093/brain/87.4.707
14. Wohlfart, G, Fex, J, and Eliasson, S. Hereditary proximal spinal muscular atrophy, a clinical entity simulating progressive muscular dystrophy. Acta Psychiatr Neurol Scand. (1955) 30:395–406.
15. Shimizu-Motohashi, Y, Chiba, E, Mizuno, K, Yajima, H, Ishiyama, A, Takeshita, E, et al. Muscle impairment in MRI affect variability in treatment response to nusinersen in patients with spinal muscular atrophy type 2 and 3: a retrospective cohort study. Brain Dev. (2023) 45:161–70. doi: 10.1016/j.braindev.2022.11.002
16. Cintas, P. Current treatments of spinal muscular atrophy in adults. Rev Neurol. (2023) 179:106–13. doi: 10.1016/j.neurol.2022.12.003
17. Oskoui, M, Day, JW, Deconinck, N, Mazzone, ES, Nascimento, A, Saito, K, et al. Two-year efficacy and safety of risdiplam in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA). J Neurol. (2023) 270:2531–46. doi: 10.1007/s00415-023-11560-1
18. Belter, L, Peterson, I, and Jarecki, J. Evaluating perceived fatigue within an adult spinal muscular atrophy population. Neurol Ther. (2023) 12:2161–75. doi: 10.1007/s40120-023-00552-y
19. Souza, PVS, Pinto, WBVR, Ricarte, A, Badia, BML, Seneor, DD, Teixeira, DT, et al. Clinical and radiological profile of patients with spinal muscular atrophy type 4. Eur J Neurol. (2021) 28:609–19. doi: 10.1111/ene.14587
20. Nishio, H, Niba, ETE, Saito, T, Okamoto, K, Takeshima, Y, and Awano, H. Spinal muscular atrophy: the past, present, and future of diagnosis and treatment. Int J Mol Sci. (2023) 24:11939. doi: 10.3390/ijms241511939
21. Younger, DS, and Mendell, JR. Childhood spinal muscular atrophy In: Handbook of clinical neurology : Elsevier B.V: Elsevier (2023). 43–58. doi: 10.1016/B978-0-323-98817-9.00030-2
22. Du, LL, Sun, JJ, Chen, ZH, Shao, YX, and Wu, LC. NOVA1 promotes SMN2 exon 7 splicing by binding the UCAC motif and increases SMN protein expression. Neural Regen Res. (2022) 17:2530–6. doi: 10.4103/1673-5374.339005
23. Kataoka, M, Sahashi, K, Tsujikawa, K, Takeda, J-i, Hirunagi, T, Iida, M, et al. Dysregulation of Aldh1a2 underlies motor neuron degeneration in spinal muscular atrophy. Neurosci Res. (2023) 194:58–65. doi: 10.1016/j.neures.2023.04.007
24. Lefebvre, S, Bürglen, L, Reboullet, S, Clermont, O, Burlet, P, Viollet, L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. (1995) 80:155–65. doi: 10.1016/0092-8674(95)90460-3
25. Ponomarev, AS, Chulpanova, DS, Yanygina, LM, Solovyeva, VV, and Rizvanov, AA. Emerging gene therapy approaches in the management of spinal muscular atrophy (SMA): an overview of clinical trials and patent landscape. Int J Mol Sci. (2023) 24:13743. doi: 10.3390/ijms241813743
26. Liu, Q, and Dreyfuss, G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J. (1996) 15:3555–65. doi: 10.1002/j.1460-2075.1996.tb00725.x
27. Aslesh, T, and Yokota, T. Restoring SMN expression: an overview of the therapeutic developments for the treatment of spinal muscular atrophy. Cells. (2022) 11:417. doi: 10.3390/cells11030417
28. Chen, X, Sanchis-Juan, A, French, CE, Connell, AJ, Delon, I, Kingsbury, Z, et al. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet Med. (2020) 22:945–53. doi: 10.1038/s41436-020-0754-0
29. Prior, TW, Leach, ME, and Finanger, E. Spinal muscular atrophy In: GeneReviews. Seattle, WA: University of Washington (2020)
30. Chaudhary, R, Agarwal, V, Rehman, M, Kaushik, AS, and Mishra, V. Genetic architecture of motor neuron diseases. J Neurol Sci. (2022) 434:120099. doi: 10.1016/j.jns.2021.120099
31. Arikan, Y, Berker Karauzum, S, Uysal, H, Mihci, E, Nur, B, Duman, O, et al. Evaluation of exonic copy numbers of SMN1 and SMN2 genes in SMA. Gene. (2022) 823:146322. doi: 10.1016/j.gene.2022.146322
32. Blasco-Pérez, L, Paramonov, I, Leno, J, Bernal, S, Alias, L, Fuentes-Prior, P, et al. Beyond copy number: a new, rapid, and versatile method for sequencing the entire SMN2 gene in SMA patients. Hum Mutat. (2021) 42:787–95. doi: 10.1002/humu.24200
33. Yamamoto, T, Sato, H, Lai, PS, Nurputra, DK, Harahap, NIF, Morikawa, S, et al. Intragenic mutations in SMN1 may contribute more significantly to clinical severity than SMN2 copy numbers in some spinal muscular atrophy (SMA) patients. Brain Dev. (2014) 36:914–20. doi: 10.1016/j.braindev.2013.11.009
34. Takarada, T, Ar Rochmah, M, Harahap, NIF, Shinohara, M, Saito, T, Saito, K, et al. SMA mutations in SMN Tudor and C-terminal domains destabilize the protein. Brain Dev. (2017) 39:606–12. doi: 10.1016/j.braindev.2017.03.002
35. Valsecchi, V, Anzilotti, S, Serani, A, Laudati, G, Brancaccio, P, Guida, N, et al. miR-206 reduces the severity of motor neuron degeneration in the facial nuclei of the brainstem in a mouse model of SMA. Mol Ther. (2020) 28:1154–66. doi: 10.1016/j.ymthe.2020.01.013
36. Butchbach, MER. Genomic variability in the survival motor neuron genes (SMN1 and SMN2): implications for spinal muscular atrophy phenotype and therapeutics development. Int J Mol Sci. (2021) 22:7896. doi: 10.3390/ijms22157896
37. Malik, I, Kelley, CP, Wang, ET, and Todd, PK. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat Rev Mol Cell Biol. (2021) 22:589–607. doi: 10.1038/s41580-021-00382-6
38. Singh, NN, O’Leary, CA, Eich, T, Moss, WN, and Singh, RN. Structural context of a critical exon of spinal muscular atrophy gene. Front Mol Biosci. (2022) 9:928581. doi: 10.3389/fmolb.2022.928581
39. Navarrete-Opazo, A, Garrison, S, and Waite, M. Molecular biomarkers for spinal muscular atrophy: a systematic review. Neurol Clin Pract. (2021) 11:e524–36. doi: 10.1212/CPJ.0000000000000872
40. Iyer, SR, Shah, SB, and Lovering, RM. The neuromuscular junction: roles in aging and neuromuscular disease. Int J Mol Sci. (2021) 22:8058. doi: 10.3390/ijms22158058
41. Courtney, NL, Mole, AJ, Thomson, AK, and Murray, LM. Reduced P53 levels ameliorate neuromuscular junction loss without affecting motor neuron pathology in a mouse model of spinal muscular atrophy. Cell Death Dis. (2019) 10:515. doi: 10.1038/s41419-019-1727-6
42. Strauss, KA, Farrar, MA, Muntoni, F, Saito, K, Mendell, JR, Servais, L, et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: the phase III SPR1NT trial. Nat Med. (2022) 28:1381–9. doi: 10.1038/s41591-022-01866-4
43. Mercuri, E, Sumner, CJ, Muntoni, F, Darras, BT, and Finkel, RS. Spinal muscular atrophy. Nat Rev Dis Prim. (2022) 8:52. doi: 10.1038/s41572-022-00380-8
44. Faravelli, I, Meneri, M, Saccomanno, D, Velardo, D, Abati, E, Gagliardi, D, et al. Nusinersen treatment and cerebrospinal fluid neurofilaments: an explorative study on spinal muscular atrophy type 3 patients. J Cell Mol Med. (2020) 24:3034–9. doi: 10.1111/jcmm.14939
45. MAR, DS, Brighente, SF, Massignan, A, Tenório, RB, Makariewicz, LL, Moreira, AL, et al. Accuracy of muscle fasciculations for the diagnosis of later-onset spinal muscle atrophy. Neuromuscul Disord. (2022) 32:763–8. doi: 10.1016/j.nmd.2022.07.395
46. Waldrop, MA, and Elsheikh, BH. Spinal muscular atrophy in the treatment era. Neurol Clin. (2020) 38:505–18. doi: 10.1016/j.ncl.2020.03.002
47. Aziz, I, Davis, M, and Liang, C. Late adult-onset spinal muscular atrophy with lower extremity predominance (SMALED). BMJ Case Rep. (2022) 15:e248297. doi: 10.1136/bcr-2021-248297
48. Pera, MC, Coratti, G, Berti, B, D’Amico, A, Sframeli, M, Albamonte, E, et al. Diagnostic journey in spinal muscular atrophy: is it still an odyssey? PLoS One. (2020) 15:e0230677. doi: 10.1371/journal.pone.0230677
49. Freigang, M, Wurster, CD, Hagenacker, T, Stolte, B, Weiler, M, Kamm, C, et al. Serum creatine kinase and creatinine in adult spinal muscular atrophy under nusinersen treatment. Ann Clin Transl Neurol. (2021) 8:1049–63. doi: 10.1002/acn3.51340
50. Shih, STF, Farrar, MA, Wiley, V, and Chambers, G. Newborn screening for spinal muscular atrophy with disease-modifying therapies: a cost-effectiveness analysis. J Neurol Neurosurg Psychiatry. (2021) 92:1296–304. doi: 10.1136/jnnp-2021-326344
51. Niba, ETE, Nishio, H, Wijaya, YOS, Lai, PS, Tozawa, T, Chiyonobu, T, et al. Clinical phenotypes of spinal muscular atrophy patients with hybrid SMN gene. Brain Dev. (2021) 43:294–302. doi: 10.1016/j.braindev.2020.09.005
52. Milligan, JN, Larson, JL, Filipovic-Sadic, S, Laosinchai-Wolf, W, Huang, Y-W, Ko, T-M, et al. Multisite evaluation and validation of a sensitive diagnostic and screening system for spinal muscular atrophy that reports SMN1 and SMN2 copy number, along with disease modifier and gene duplication variants. J Mol Diagn. (2021) 23:753–64. doi: 10.1016/j.jmoldx.2021.03.004
53. Yang, Y, Xia, C, Song, X, Tang, X, Nie, X, Xu, W, et al. Application of a multiplex ligation-dependent probe amplification-based next-generation sequencing approach for the detection of pathogenesis of Duchenne muscular dystrophy and spinal muscular atrophy caused by copy number aberrations. Mol Neurobiol. (2023) 61:200–11. doi: 10.1007/s12035-023-03572-9
54. Zhang, J, Wang, Y, Ma, D, Sun, Y, Li, Y, Yang, P, et al. Carrier screening and prenatal diagnosis for spinal muscular atrophy in 13,069 Chinese pregnant women. J Mol Diagn. (2020) 22:817–22. doi: 10.1016/j.jmoldx.2020.03.001
55. Carré, A, and Empey, C. Review of spinal muscular atrophy (SMA) for prenatal and pediatric genetic counselors. J Genet Couns. (2016) 25:32–43. doi: 10.1007/s10897-015-9859-z
56. Chen, T-H. New and developing therapies in spinal muscular atrophy: from genotype to phenotype to treatment and where do we stand? Int J Mol Sci. (2020) 21:3297. doi: 10.3390/ijms21093297
57. Jablonka, S, Hennlein, L, and Sendtner, M. Therapy development for spinal muscular atrophy: perspectives for muscular dystrophies and neurodegenerative disorders. Neurol Res Pract. (2022) 4:2–32. doi: 10.1186/s42466-021-00162-9
58. Studzińska, S, Szymarek, J, and Mazurkiewicz-Bełdzińska, M. Improvement of serum sample preparation and chromatographic analysis of nusinersen used for the treatment of spinal muscular atrophy. Talanta. (2024) 267:125173. doi: 10.1016/j.talanta.2023.125173
59. Day, JW, Howell, K, Place, A, Long, K, Rossello, J, Kertesz, N, et al. Advances and limitations for the treatment of spinal muscular atrophy. BMC Pediatr. (2022) 22:632. doi: 10.1186/s12887-022-03671-x
60. Przymuszała, M, Gwit, M, Waśko, J, Morańska, K, and Kajdasz, A. Spinal muscular atrophy: where are we now? Current challenges and high hopes. Postępy Hig Med Dośw. (2022) 76:407–19. doi: 10.2478/ahem-2022-0030
61. Łusakowska, A, Wójcik, A, Frączek, A, Aragon-Gawińska, K, Potulska-Chromik, A, Baranowski, P, et al. Long-term nusinersen treatment across a wide spectrum of spinal muscular atrophy severity: a real-world experience. Orphanet J Rare Dis. (2023) 18:230. doi: 10.1186/s13023-023-02769-4
62. Gavriilaki, M, Moschou, M, Papaliagkas, V, Notas, K, Chatzikyriakou, E, Papagiannopoulos, S, et al. Nusinersen in adults with 5q spinal muscular atrophy: a systematic review and meta-analysis. Neurotherapeutics. (2022) 19:464–75. doi: 10.1007/s13311-022-01200-3
63. Hoy, SM. Nusinersen: first global approval. Drugs. (2017) 77:473–9. doi: 10.1007/s40265-017-0711-7
64. Menduti, G, Rasà, DM, Stanga, S, and Boido, M. Drug screening and drug repositioning as promising therapeutic approaches for spinal muscular atrophy treatment. Front Pharmacol. (2020) 11:1696. doi: 10.3389/fphar.2020.592234
65. Chong, LC, Gandhi, G, Lee, JM, Yeo, WWY, and Choi, S-B. Drug discovery of spinal muscular atrophy (SMA) from the computational perspective: a comprehensive review. Int J Mol Sci. (2021) 22:8962. doi: 10.3390/ijms22168962
66. Hoy, SM. Onasemnogene abeparvovec: first global approval. Drugs. (2019) 79:1255–62. doi: 10.1007/s40265-019-01162-5
67. Hensel, N, Kubinski, S, and Claus, P. The need for SMN-independent treatments of spinal muscular atrophy (SMA) to complement SMN-enhancing drugs. Front Neurol. (2020) 11:45. doi: 10.3389/fneur.2020.00045
68. van Alstyne, M, Tattoli, I, Delestrée, N, Recinos, Y, Workman, E, Shihabuddin, LS, et al. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat Neurosci. (2021) 24:930–40. doi: 10.1038/s41593-021-00827-3
69. Abreu, NJ, and Waldrop, MA. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr Pulmonol. (2021) 56:710–20. doi: 10.1002/ppul.25055
70. Ramdas, S, and Servais, L. New treatments in spinal muscular atrophy: an overview of currently available data. Expert Opin Pharmacother. (2020) 21:307–15. doi: 10.1080/14656566.2019.1704732
71. Gandhi, G, Abdullah, S, Foead, AI, and Yeo, WWY. The potential role of miRNA therapies in spinal muscle atrophy. J Neurol Sci. (2021) 427:117485. doi: 10.1016/j.jns.2021.117485
72. René, CA, and Parks, RJ. Expanding the availability of onasemnogene abeparvovec to older patients: the evolving treatment landscape for spinal muscular atrophy. Pharmaceutics. (2023) 15:1764. doi: 10.3390/pharmaceutics15061764
74. Li, JJ. "Risdiplam (Evrysdi), a small molecule, SMN2-directed RNA splicing modifier for treating spinal muscular atrophy." Current drug synthesis. John Wiley & Sons, Inc: Wiley. (2022): 287–303
75. Paik, J. Risdiplam: a review in spinal muscular atrophy. CNS Drugs. (2022) 36:401–10. doi: 10.1007/s40263-022-00910-8
76. Kwon, JM, Arya, K, Kuntz, N, Phan, HC, Sieburg, C, Swoboda, KJ, et al. An expanded access program of risdiplam for patients with type 1 or 2 spinal muscular atrophy. Ann Clin Transl Neurol. (2022) Wiley: John Wiley & Sons, Inc9:810–8. doi: 10.1002/acn3.51560
77. Markati, T, Fisher, G, Ramdas, S, and Servais, L. Risdiplam: an investigational survival motor neuron 2 (SMN2) splicing modifier for spinal muscular atrophy (SMA). Expert Opin Investig Drugs. (2022) 31:451–61. doi: 10.1080/13543784.2022.2056836
78. Ojala, KS, Reedich, EJ, DiDonato, CJ, and Meriney, SD. In search of a cure: the development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. (2021) 11:194. doi: 10.3390/brainsci11020194
79. Yeo, CJ, and Darras, BT. Overturning the paradigm of spinal muscular atrophy as just a motor neuron disease. Pediatr Neurol. (2020) 109:12–9. doi: 10.1016/j.pediatrneurol.2020.01.003
80. Brandsema, J, and Cappa, R. Genetically targeted therapies for inherited neuromuscular disorders. Pract Neurol. (2021):69–73.
81. Available at: https://pib.gov.in/Pressreleaseshare.aspx?PRID=1846230
82. Available at: https://pib.gov.in/PressReleasePage.aspx?PRID=1846230
84. Dunaway, S, Montes, J, McDermott, MP, Martens, W, Neisen, A, Glanzman, AM, et al. Physical therapy services received by individuals with spinal muscular atrophy (SMA). J Pediatr Rehabil Med. (2016) 9:35–44. doi: 10.3233/PRM-160360
85. Trenkle, J, Brugman, J, Peterson, A, Roback, K, and Krosschell, KJ. Filling the gaps in knowledge translation: physical therapy recommendations for individuals with spinal muscular atrophy compared to standard of care guidelines. Neuromuscul Disord. (2021) 31:397–408. doi: 10.1016/j.nmd.2021.02.011
86. Mirea, A, Leanca, MC, Onose, G, Sporea, C, Padure, L, Shelby, E-S, et al. Physical therapy and nusinersen impact on spinal muscular atrophy rehabilitative outcome. Front Biosci. (2022) 27:179. doi: 10.31083/j.fbl2706179
87. Yi, YG, Shin, H-I, and Jang, D-H. Rehabilitation of spinal muscular atrophy: current consensus and future direction. J Genet Med. (2020) 17:55–61. doi: 10.5734/JGM.2020.17.2.55
88. Bach, JR, Niranjan, V, and Weaver, B. Spinal muscular atrophy type 1: a noninvasive respiratory management approach. Chest. (2000) 117:1100–5. doi: 10.1378/chest.117.4.1100
89. Chen, T-H, Hsu, J-H, and Jong, Y-J. Noninvasive airway approaches for acute neuromuscular respiratory failure in emergency departments. Pediatr Pulmonol. (2017) 52:E55–7. doi: 10.1002/ppul.23693
90. Chen, T-H, Liang, W-C, Chen, CI, Liu, Y-C, Hsu, J-H, and Jong, Y-J. Combined noninvasive ventilation and mechanical insufflator-exsufflator for acute respiratory failure in patients with neuromuscular disease: effectiveness and outcome predictors. Ther Adv Respir Dis. (2019) 13:1753466619875928. doi: 10.1177/1753466619875928
91. Blackwood, B, Tume, LN, Morris, KP, Clarke, M, McDowell, C, Hemming, K, et al. Effect of a sedation and ventilator liberation protocol vs usual care on duration of invasive mechanical ventilation in pediatric intensive care units: a randomized clinical trial. JAMA. (2021) 326:401–10. doi: 10.1001/jama.2021.10296
92. Lewelt, A, Newcomb, TM, and Swoboda, KJ. New therapeutic approaches to spinal muscular atrophy. Curr Neurol Neurosci Rep. (2012) 12:42–53. doi: 10.1007/s11910-011-0240-9
93. Li, Y-J, Chen, T-H, Wu, Y-Z, and Tseng, Y-H. Metabolic and nutritional issues associated with spinal muscular atrophy. Nutrients. (2020) 12:3842. doi: 10.3390/nu12123842
94. Moore, GE, Lindenmayer, AW, McConchie, GA, Ryan, MM, and Davidson, ZE. Describing nutrition in spinal muscular atrophy: a systematic review. Neuromuscul Disord. (2016) 26:395–404. doi: 10.1016/j.nmd.2016.05.005
95. Choi, Y-A, Suh, DI, Chae, J-H, and Shin, H-I. Trajectory of change in the swallowing status in spinal muscular atrophy type I. Int J Pediatr Otorhinolaryngol. (2020) 130:109818. doi: 10.1016/j.ijporl.2019.109818
96. Corsello, A, Scatigno, L, Pascuzzi, MC, Calcaterra, V, Dilillo, D, Vizzuso, S, et al. Nutritional, gastrointestinal and endo-metabolic challenges in the management of children with spinal muscular atrophy type 1. Nutrients. (2021) 13:2400. doi: 10.3390/nu13072400
97. Birnkrant, DJ, Pope, JF, Martin, JE, Repucci, AH, and Eiben, RM. Treatment of type I spinal muscular atrophy with noninvasive ventilation and gastrostomy feeding. Pediatr Neurol. (1998) 18:407–10. doi: 10.1016/S0887-8994(97)00227-0
98. Mercuri, E, Finkel, RS, Muntoni, F, Wirth, B, Montes, J, Main, M, et al. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. (2018) 28:103–15. doi: 10.1016/j.nmd.2017.11.005
99. Wadman, RI, De Amicis, R, Brusa, C, Battezzati, A, Bertoli, S, Davis, T, et al. Feeding difficulties in children and adolescents with spinal muscular atrophy type 2. Neuromuscul Disord. (2021) 31:101–12. doi: 10.1016/j.nmd.2020.12.007
100. Katz, NP, Paillard, FC, and Ekman, E. Determining the clinical importance of treatment benefits for interventions for painful orthopedic conditions. J Orthop Surg Res. (2015) 10:24–11. doi: 10.1186/s13018-014-0144-x
101. Haaker, G, and Fujak, A. Proximal spinal muscular atrophy: current orthopedic perspective. Appl Clin Genet. (2013) 6:113–20. doi: 10.2147/TACG.S53615
102. Shank, TM, and Rahman, T. Home use of an upper extremity exoskeleton in children with SMA: a pilot study. Open J Occup Ther. (2021) 9:1–14. doi: 10.15453/2168-6408.1762
103. Salem, Y, and Gropack, SJ. Aquatic therapy for a child with type III spinal muscular atrophy: a case report. Phys Occup Ther Pediatr. (2010) 30:313–24. doi: 10.3109/01942638.2010.493097
104. Kraskowsky, LH, and Finlayson, M. Factors affecting older adults’ use of adaptive equipment: review of the literature. Am J Occup Ther. (2001) 55:303–10. doi: 10.5014/ajot.55.3.303
105. Renda, M, and Lape, JE. Feasibility and effectiveness of telehealth occupational therapy home modification interventions. Int J Telerehabil. (2018) 10:3–14. doi: 10.5195/ijt.2018.6244
106. van Stormbroek, K, and Buchanan, H. Novice occupational therapists: navigating complex practice contexts in South Africa. Aust Occup Ther J. (2019) 66:469–81. doi: 10.1111/1440-1630.12564
107. Ropars, J, Peudenier, S, Genot, A, Barnerias, C, and Espil, C. Multidisciplinary approach and psychosocial management of spinal muscular atrophy (SMA). Arch Pediatr. (2020) 27:7S45–9. doi: 10.1016/S0929-693X(20)30277-3
108. Inhestern, L, Brandt, M, Driemeyer, J, Denecke, J, Johannsen, J, and Bergelt, C. Experiences of health care and psychosocial needs in parents of children with spinal muscular atrophy. Int J Environ Res Public Health. (2023) 20:5360. doi: 10.3390/ijerph20075360
109. Betancourt, TS, Meyers-Ohki, SE, Charrow, AP, and Tol, WA. Interventions for children affected by war: an ecological perspective on psychosocial support and mental health care. Harv Rev Psychiatry. (2013) 21:70–91. doi: 10.1097/HRP.0b013e318283bf8f
110. Hully, M, Barnerias, C, Chabalier, D, le Guen, S, Germa, V, Deladriere, E, et al. Palliative care in SMA type 1: a prospective multicenter French study based on parents’ reports. Front Pediatr. (2020) 8:4. doi: 10.3389/fped.2020.00004
111. Zingariello, CD, Brandsema, J, Drum, E, Henderson, AA, Dubow, S, Glanzman, AM, et al. A multidisciplinary approach to dosing nusinersen for spinal muscular atrophy. Neurol Clin Pract. (2019) 9:424–32. doi: 10.1212/CPJ.0000000000000718
112. Livingstone, R, and Paleg, G. Enhancing function, fun and participation with assistive devices, adaptive positioning, and augmented mobility for young children with infantile-onset spinal muscular atrophy: a scoping review and illustrative case report. Disabilities. (2021) 1:1–22. doi: 10.3390/disabilities1010001
113. Fujak, A, Kopschina, C, Forst, R, Mueller, LA, and Forst, J. Use of orthoses and orthopaedic technical devices in proximal spinal muscular atrophy. Results of survey in 194 SMA patients. Disabil Rehabil Assist Technol. (2011) 6:305–11. doi: 10.3109/17483107.2010.525292
114. Odabas, SP, Bal, E, Yelgen, G, Metin, AS, Karakaya, E, Gulden, G, et al. (2022). Exon7 targeted CRISPR-prime editing approaches for SMN2 gene editing in spinal muscular atrophy (SMA) disease can increase in vitro SMN expression. bioRxiv. Available at: https://doi.org/10.1101/2022.03.21.484406. [Epub ahead of preprint]
115. Lopez-Lopez, D, Loucera, C, Carmona, R, Aquino, V, Salgado, J, Pasalodos, S, et al. SMN1 copy-number and sequence variant analysis from next-generation sequencing data. Hum Mutat. (2020) 41:2073–7. doi: 10.1002/humu.24120
116. Calder, AN, Androphy, EJ, and Hodgetts, KJ. Small molecules in development for the treatment of spinal muscular atrophy: miniperspective. J Med Chem. (2016) 59:10067–83. doi: 10.1021/acs.jmedchem.6b00670
117. Dangouloff, T, and Servais, L. Clinical evidence supporting early treatment of patients with spinal muscular atrophy: current perspectives. Ther Clin Risk Manag. (2019) 15:1153–61. doi: 10.2147/TCRM.S172291
118. Harada, Y, Rao, VK, Arya, K, Kuntz, NL, DiDonato, CJ, Napchan-Pomerantz, G, et al. Combination molecular therapies for type 1 spinal muscular atrophy. Muscle Nerve. (2020) 62:550–4. doi: 10.1002/mus.27034
119. Poletti, A, and Fischbeck, KH. Combinatorial treatment for spinal muscular atrophy: an editorial for ‘combined treatment with the histone deacetylase inhibitor LBH589 and a splice-switch antisense oligonucleotide enhances SMN2 splicing and SMN expression in spinal muscular atrophy cells’ on page 264. J Neurochem. (2020) 153:146–9. doi: 10.1111/jnc.14974
120. Zhou, H, Meng, J, Malerba, A, Catapano, F, Sintusek, P, Jarmin, S, et al. Myostatin inhibition in combination with antisense oligonucleotide therapy improves outcomes in spinal muscular atrophy. J Cachexia Sarcopenia Muscle. (2020) 11:768–82. doi: 10.1002/jcsm.12542
121. Sampaio, H, Wilcken, B, and Farrar, M. Screening for spinal muscular atrophy. Med J Aust. (2018) 209:147–8. doi: 10.5694/mja17.00772
122. Farrar, MA, Teoh, HL, Carey, KA, Cairns, A, Forbes, R, Herbert, K, et al. Nusinersen for SMA: expanded access programme. J Neurol Neurosurg Psychiatry. (2018) 89:937–42. doi: 10.1136/jnnp-2017-317412
Keywords: spinal muscular atrophy, survival motor neuron, rehabilitation, nusinersen, onasemnogene abeparvovec, risdiplam
Citation: Bagga P, Singh S, Ram G, Kapil S and Singh A (2024) Diving into progress: a review on current therapeutic advancements in spinal muscular atrophy. Front. Neurol. 15:1368658. doi: 10.3389/fneur.2024.1368658
Edited by:
Nicholas Van Halm-Lutterodt, Inspired Spine Health, United StatesReviewed by:
Hisahide Nishio, Kobe Gakuin University, JapanCopyright © 2024 Bagga, Singh, Ram, Kapil and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sudhakar Singh, c3VkaGFrYXJzaW5naDg2QGdtYWlsLmNvbQ==; Avtar Singh, QXZ0YXJzaW5naEBhc3R1LmVkdS5ldA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.