 Dapeng Cai
Dapeng Cai Haohao Wu
Haohao Wu Kang Du
Kang Du
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 18 April 2024
Sec. Neurogenetics
Volume 15 - 2024 | https://doi.org/10.3389/fneur.2024.1349861
This article is part of the Research Topic Case Reports in Neurogenetics, volume III - 2023 View all 20 articles
This study reported a case of early-onset parkinsonism associated with a novel variant of the PLA2G6 gene. The boy first started showing symptoms at the age of 11, with gait instability and frequent falls. As the disease progressed, his gait instability worsened, and he developed difficulties with swallowing and speaking, although there was no apparent decline in cognitive function. An MRI of the head revealed significant atrophy of the cerebellum. The initial diagnosis for the boy was early-onset parkinsonism, classified as Hoehn-Yahr grade 5.Genomic sequencing of the patient indicated that he had compound heterozygous variations in the PLA2G6 gene: c.1454G>A (p.Gly485Glu) and c.991G>T (p.Asp331Tyr). Pedigree analysis revealed that his younger brother also carried the same variant, albeit with milder symptoms. The patient's unaffected mother was found to be a carrier of the c.991G>T variant. Additionally, this study reviewed 62 unrelated families with PLA2G6 gene-related early-onset parkinsonism. The analysis showed a higher proportion of female probands, with a mean age of onset of ~23.0 years. Primary symptoms were predominantly bradykinesia and psychosis, with tremors being relatively rare. Cerebellar atrophy was observed in 41 patients (66.1%). Among the reported mutations, the most common mutation was c.991G>T, presenting in 21 families (33.9%), followed by c.2222G>A in eight families (12.9%). Other mutations were less common. Notably, the c.991G>T mutation was exclusive to Chinese families and was a prevalent mutation among this population. The initial symptoms varied significantly among patients with different mutations.
Early-onset parkinsonism (EOP) is a neurodegenerative disease related to genetic factors. PLA2G6 gene mutation is considered to be one of the pathogenic genes involved in the development of EOP (1). Autosomal recessive EOP caused by mutations in the PLA2G6 gene is called PLA2G6-associated Neurodegeneration (PLAN) (2, 3). These include Infantile neuroaxonal dystrophy (INAD), Atypical neuroaxonal dystrophy (ANAD), and EOP (4). In this study, a case of EOP caused by a novel PLA2G6 gene mutation was reported, and previous reports of EOP related to this gene were reviewed.
A 22-year-old male patient was admitted to the hospital due to gait abnormality and frequent falls. The patient developed the above symptoms at the age of 11, and his motor development was slightly worse than that of his peers. After that, the patient's gait instability was aggravated, dysphagia and dysarthria gradually appeared, without obvious cognitive decline, and no special diagnosis and treatment were given. The patient's anomalies of gait and weakness of extremities were further aggravated, manifested as frequent falls, requiring bed rest or wheelchair. The patient's articulation disorder and deglutition disorders were aggravated compared with the previous ones, and the cognitive decline was presented, but he could still communicate normally with his family members. The proband's father died of trauma. Prior to his death, he denied the anomalies of gait, muscle weakness and other symptoms. The proband's younger brother began to have gait instability at the age of 11, and his motor development since childhood was slightly worse than that of his peers. At the age of 20, he can still walk normally, live independently, but his muscle tension is symmetrically increased. The proband's mother had a head trembling a few months ago, without other special discomforts. The patient's cranial magnetic resonance imaging (MRI) examination in September 2022 revealed cerebellar atrophy. The diagnosis of Parkinson's syndrome was made, and the Hoehn-Yahr grade was 5.
The nervous system physical examination revealed that the patient had normal vital signs, clear mind, but had severe dysarthria. The orientation of time, character and place was normal, the calculation and comprehension were decreased. The cranial nerves examination did not reveal any abnormalities. The distal and proximal muscle strength of both upper limbs was grade 3, the distal and proximal muscle strength of both lower limbs was grade 2, the symmetry of muscle tension of both upper limbs was increased, the muscle tension of both lower limbs was decreased, the tendon reflex of both upper limbs was brisk, the tendon reflex of both lower limbs was absent, the Rossolimo's sign of both upper limbs was positive, the pathological sign of both lower limbs was negative, the meningeal irritation sign was negative, and the patient had no sensory abnormalities and ataxia signs in the physical examination. Wide base gait, slow movement, reduced swing arms of both hands, and unilateral assistance during walking. The patient could not cooperate to complete the bilateral finger-nose test, heel-knee-tibia test, and pull-back test.
The results of auxiliary examination suggest that: there were no abnormalities in hematuria, stool routine, biochemical indicators, homocysteine, ceruloplasmin, hepatitis, syphilis, HIV, coagulation function, autoimmune antibody spectrum, and cardiac ultrasound. MRI plain scan and contrast-enhancement of the head indicated brain atrophy, especially in the bilateral cerebellar hemispheres (Figure 1C).

Figure 1. (A) Family diagram of the proband; (B) Another complex heterozygous variation of proband c.1454G>A with Sanger sequencing; (C) Cerebellar atrophy can be seen in the sagittal T1WI phase of the proband's head MRI; (D) The conservative analysis of this variant suggests that it is highly conservative.
The patient's whole genome sequencing suggested that the PLA2G6 gene compound heterozygous variants c.1454G > A (p.Gly485Glu), c.991G > T (p.Asp331Tyr) (Figures 1A, B). The results of pedigree verification suggested that the proband's brother was consistent with the proband's results. The proband's mother was a asymptomatic carrier of the variant of PLA2G6 gene c.991G > T. The proband's father failed to perform pedigree verification due to unexpected death, which was consistent with the role of family co-segregation. The variant of c.991G > T has been previously reported as a pathogenic variation (5), however, the variant of c.1454G > A has not been reported. The Mutation Taster software predicted it as a pathogenic variation, and the probability was 0.9999; this variant was not found in ExAC and Thousand Human Genome Database, and the conservation analysis suggested that it was highly conserved (Figure 1D).
The patient was finally diagnosed with PLA2G6 gene-related early-onset Parkinson's syndrome. The patient was treated with madopa 62.5 mg tid gradually increased to 125 mg tid orally. After 2 months of follow-up, the patient's gait abnormality was slightly improved compared with the previous one, and the disease did not progress significantly.
In this study, the keywords “PLA2G6,” “parkinsonism,” and “Parkinson” were searched through “Pubmed,” “Wanfang Medicine,” and “China National Knowledge Infrastructure” databases. The literature of PLA2G6 gene-related EOP patients reported in Chinese and English was reviewed. A total of 62 families, including 30 Chinese families, were reviewed, and the clinical and genetic characteristics of the probands in all families were summarized (Table 1).
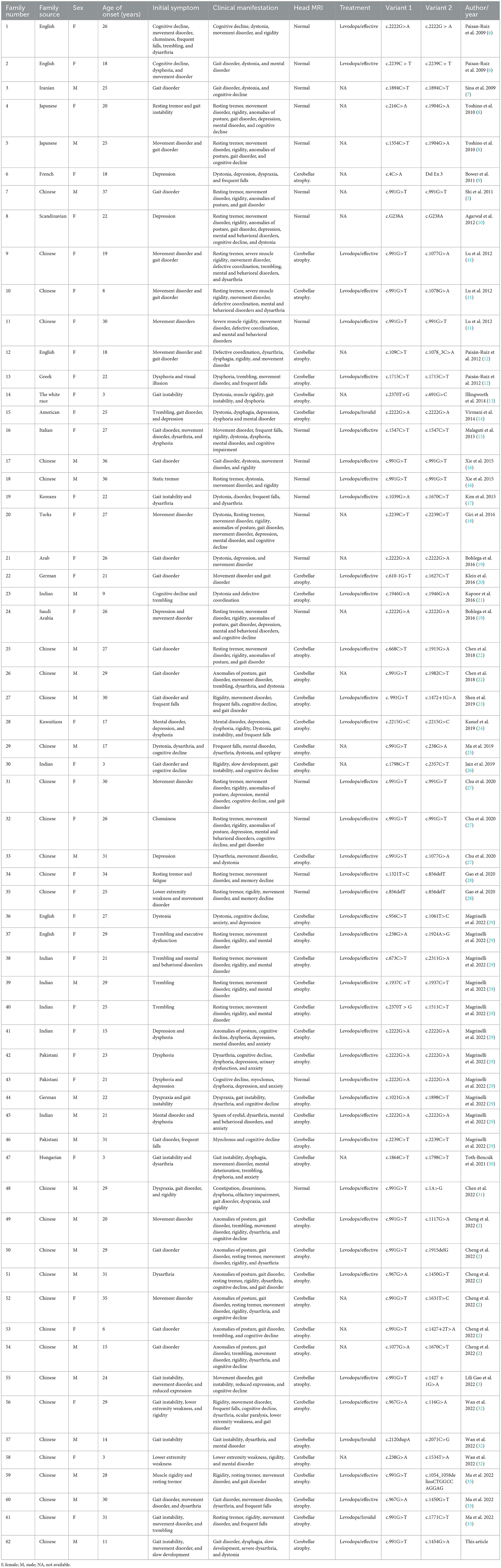
Table 1. A retrospective analysis of PLA2G6 gene-related EOP literature has been reported.
By reviewing the literature, it was found that among the 62 probands, the male to female ratio was 2: 3, and the average onset age of male patients was 22.9 ± 8.7 years old. The average age of onset in women was 23.0 ± 8.5 years. There were 51 cases (82.2%) with movement disorder as the initial symptom, including 27 cases (43.5%) with gait disorder, 10 cases (16.1%) with gait instability, and 11 cases (17.7%) with limb trembling. There were 14 cases (22.6%) with depression, dysphoria, and other emotional instability as the initial symptoms. All probands were examined by cranial MRI, and 41 (66.1%) patients had cerebellar atrophy.
Among the 30 cases of national probands, the male to female ratio was 3:2, and the average onset age of male patients was 22.9 ± 8.9 years. The average age of onset in women was 22.9 ± 8.8 years. The most common mutation was c.991 G > T mutation in 21 families (70%), followed by c.967 G > A mutation in three families (10 %), c.1077 G > A mutation in three families (10%), and other mutations were rare. The most common clinical manifestations were movement disorder in 30 cases (100%), including gait disorder in 17 cases (56.7%), limb trembling in 19 cases (63.3%), and mental and behavioral disorders in 24 cases (80%). Among them, movement disorder was the most common symptom, with 28 cases (93.3%).
Among the 15 European and American probands, the ratio of male to female was 1:14. The most common clinical manifestations were movement disorder in 15 cases (100 %) and mental and behavioral disorders in 14 cases (93.3 %). Among them, mental and behavioral disorders were the most common symptoms, a total of 14 cases (93.3 %).
Of the 14 probands from the Middle East and western Asia, the ratio of male to female was 5: 9. The most common clinical symptoms were movement disorder (10 cases, 71.4%) and mental and behavioral disorders (13 cases, 92.9%). The most common symptoms were movement disorder (nine cases, 64.3%) and mental and behavioral disorders (seven cases, 50%).
Among the reported mutations, the most common mutation was c.991G > T in 21 families (33.9%), followed by c.2222G > A in eight families (12.9%), and other mutation types were rare. Among them, the c.991 G > T mutation only was found in Chinese, and the c.2222 G > A mutation was mainly distributed in the Middle East, western Asia and other countries. Among them, the ratio of male to female in the proband with c.991G > T mutation was 13:8. There were 15 cases (71%) of compound heterozygous mutations and six cases (29%) of homozygous mutations. The average age of onset of patients with compound heterozygous mutations was 23.1 ± 9.2 years old, and the most common initial symptom was movement disorder in seven cases (46.7%). There were 14 cases (93.3%) with dystonia and 10 cases (66.7%) with mental and behavioral disorders. The average age of patients with homozygous mutations was 32.5 ± 4.5 years old, and the initial symptoms were atypical, including one case of gait disorder (16.7%) and one case of movement disorder (16.7%). There were six cases (100%) of dystonia and three cases (50%) of mental and behavioral disorders. Among the probands with compound heterozygous mutations, there were 14 cases (93%) of cerebellar atrophy, and no cerebellar atrophy was found in the probands with homozygous mutations. The male to female ratio of the proband with c.2222G > A mutation was 1:7, all of which were homozygous mutations. The average age of onset was 23.3 ± 7.6 years. The most common initial symptoms were mental and behavioral disorders in seven cases (87.5%), including dysphoria, depression and other symptoms in 6 cases (75.0%). There were five cases (62.5%) of dystonia and eight cases (100%) of mental and behavioral disorders. A total of four probands had cerebellar atrophy (50%).
In addition, among all the probands, 9 (33%) patients with homozygous mutations had cerebellar atrophy, and 36 (85%) patients with compound heterozygous mutations had cerebellar atrophy.
PLA2G6 gene is located in 22q13.1 region, about 6.0 Mb, containing 17 exons, encoding 85 ku cytosolic Ca2+ independent phospholipids A2 (iPLA2). There are two forms of iPLA2-A and iPLA2-β. The iPLA2-β enzyme is closely related to neurodegenerative diseases, and different mutation sites can lead to different degrees of changes in iPLA2-β enzyme. This leads to different clinical phenotypes of PLAN (31). The mutation types of this gene include missense mutation, truncation mutation and copy number variation, but the specific mechanism of this mutation is not clear (31). Previous studies have shown that the pathogenesis of EOP caused by PLA2G6 gene may be the loss of iPLA2 enzyme protein function caused by PLA2G6 gene mutation, which in turn causes phospholipid metabolism disorder of nerve cell membrane, intracellular iron deposition, lipid peroxidation, mitochondrial inner membrane damage, and Golgi morphological changes, eventually leading to a large number of apoptosis of dopaminergic neurons, decreased dopamine secretion, and the presence of Lewy bodies formed by misfolding and aggregation of α-synuclein in surviving neurons, leading to the occurrence of EOP (34, 35).
After reviewing the literature, this study showed that there were slightly more female patients with PLA2G6 gene-related EOP than male patients, and all of them had similar age of onset. The average age of onset was about 22 years old. The patients of EOP usually had gait disorder and movement disorder as the initial symptoms, but the resting tremor was relatively rare. As the disease progressed, it might be accompanied by symptoms such as rigidity, cognitive decline, mental and behavioral disorders (36–38). The majority of patients responded well to levodopa preparations, but the incidence of dyskinesia and symptom fluctuations reported in the literature was high and occurred earlier (34).
Based on retrospective analysis, it was found that the most common mutation in Chinese people was c.991G > T. One of the mutations reported in this study was also this variant. The mutation accounted for more than half of the Chinese pedigrees reported. It was further confirmed that the c.991G > T was the hot spot mutation of the PLA2G6 gene in China (2, 27), suggesting that this mutation had a founder effect in Chinese patients. The most common mutation reported abroad was c.2222G > A, which was mainly found in the Middle East and western Asia, including Arab, Saudi Arabia, India, Pakistan and other countries. The most common symptoms of c.991G > T mutation-related patients were movement disorder and gait disorder. Patients with c.2222G > A mutation usually had cognitive impairment, anxiety, depression, dysphoria, and other mental disorders, accompanied by a small amount of movement disorders. It was found that among the EOP probands caused by PLA2G6 gene, the probands with mental and behavioral disorders in Europe and America, western Asia and the Middle East were significantly higher than those in Chinese probands, which further confirmed the correlation between the clinical phenotype of EOP and different genotypes (29). However, Cheng et al. suggested that it might also be due to the complex phenotypic characteristics of Chinese patients, which could easily cover up symptoms such as myoclonus, cognitive decline and mental and behavioral disorders (2), suggesting that the evaluation of cognitive and mental disorders in EOP patients should be strengthened in clinical work.
In this study, 21 probands with c.991G > T mutation reported previously were further analyzed. It was found that patients with c.991G > T mutation usually had movement disorder, gait disorder and other symptoms as the first symptoms, followed by aggravation of symptoms and dystonia, resting tremor and other motor symptoms and non-motor symptoms. Among them, patients with c.991 G > T homozygous mutation occurred about 10 years later than those with compound heterozygous mutation. Furthermore, the initial symptoms were atypical and the clinical manifestations were milder. All of them were sensitive to levodopa treatment, which was consistent with previous studies (3, 11, 16). In addition, this study found that almost all of the probands with compound heterozygous mutations at this variant had cerebellar atrophy, while no cerebellar atrophy occurred in the six homozygous mutant probands, further suggesting that the clinical manifestations of patients with homozygous mutations at this variant were relatively mild. Previous in vitro cell experiments showed that c.991G > T mutant cells still retained 30% iPLA2β enzyme activity compared with wild-type cells, but the iPLA2β enzyme activity in H597fx69 cells expressing frameshift mutations only retained 6% (39). Because different mutation sites have different effects on iPLA2β enzyme activity, the reason for the difference between the two may be that another heterozygous mutation site outside the c.991 G > T mutation site has a greater effect on iPLA2β enzyme activity. The PLA2G6 protease activity of patients is higher than that of patients with heterozygous mutations, but more in vitro experiments of non-frameshift mutations are needed for further verification in the future. Therefore, we hypothesize that heterozygous and homozygous mutations in the PLA2G6 gene have different effects on the activity of iPLA2β enzyme, and the proportion of iPLA2β enzyme activity loss can partially explain that homozygous mutation probands have relatively benign clinical and neuroimaging phenotypes compared with heterozygous mutation probands (16).
This study also found that 64.8% of the probands showed brain atrophy on head MRI, but most studies showed that only a small number of EOP probands showed iron deposition on head MRI (31, 37, 40). From a pathological point of view, PLAN is characterized by the depletion of neurons in the cerebellar cortex, accompanied by astrocyte proliferation, axonal spheroids in the central and peripheral nervous system, and progressive brain iron deposition (2), cerebellar atrophy is the earliest sign on head MRI, while the signs of brain iron deposition in the basal ganglia often appear later. This may be the reason why MRI cerebellar atrophy signs are common and iron deposition signs are rare in EOP probands (2). Some researchers found that pro-inflammatory cytokines were significantly up-regulated, microglial activation, and reactive astrocyte proliferation were found in the pathological tissues of patients. Therefore, it is believed that inflammatory response is involved in the pathological process of cerebellar atrophy, and it is speculated that early anti-inflammatory treatment may help to delay the progression of cerebellar atrophy in patients with Parkinson's syndrome (41).
Another compound heterozygous mutation c.1454G > A in the PLA2G6 gene of the proband in this study has not been reported. Like most other EOP patients with PLA2G6 compound heterozygous mutations, the symptoms of PLA2G6 gene-related EOP in this patient were basically similar. The onset of the disease was 11 years old, with gait disorder, and the clinical manifestation was dystonia-Parkinson syndrome. The genetic test results of the proband's younger brother were consistent with those of the proband, and the age of onset was 11 years old. However, the clinical symptoms of the proband's younger brother were significantly lighter than those of the proband. The initial symptoms were anomalies of gait, and he could walk normally and take care of himself with the progression of the disease, and were not accompanied by symptoms such as dysphonia and dysphagia. It was speculated whether the proband had more susceptible genes than his younger brother, such as GBA, MAPT, SNCA, etc., leading to more severe clinical symptoms (42). In addition, different hormone levels could also affect the progression of Parkinson's disease (29). Considering that the living environment and habits of the proband and the proband's brother were roughly the same, this might also be one of the reasons for the clinical differences between the two.
Although the incidence of EOP is not high, the morbidity and mortality are very high (38), and most patients have a good response to treatment such as madopar 5–10 years after onset (3). This study also found that the vast majority of PLA2G6 gene-related EOP responds to levodopa treatment, but the delayed use of levodopa will increase the incidence of dyskinesia, and the switching period fluctuation is more obvious (33). Therefore, early diagnosis is of great significance for early initiation of anti-Parkinson therapy.
Because the sample size of most studies on PLA2G6 gene-related EOP in Chinese population is relatively small (3, 16, 27), this leads to limitations in the clinical and phenotypic comparison of different PLA2G6 gene mutation reviews in this study. The clinical and genetic characteristics of PLA2G6 gene-related EOP patients in China will be more clear in future multicenter large sample studies.
In conclusion, this study reported a case of early-onset parkinsonism caused by a novel variant of PLA2G6 gene and reviewed previous reports. This expands the genetic pedigree of the disease and increased clinicians' understanding of the clinical and genetic characteristics of early-onset parkinsonism.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
The studies involving humans were approved by the Ethics Committee of Qujing First People's Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
DC: Conceptualization, Data curation, Formal analysis, Writing – original draft. HW: Conceptualization, Methodology, Supervision, Writing – review & editing. BH: Conceptualization, Methodology, Supervision, Writing – review & editing. WX: Data curation, Methodology, Writing – review & editing. KD: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Software, Supervision, Validation, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Yunnan Provincial Department of Education Science Research Funding (No. 2023Y0702).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Riboldi GM, Frattini E, Monfrini E, Frucht SJ, Di Fonzo A. A practical approach to early-onset Parkinsonism. J Parkinsons Dis. (2022) 12:1–26. doi: 10.3233/JPD-212815
2. Cheng HL, Chen YJ, Xue YY, Wu ZY, Li HF, Wang N. Clinical characterization and founder effect analysis in Chinese patients with phospholipase A2-associated neurodegeneration. Brain Sci. (2022) 12:50517. doi: 10.3390/brainsci12050517
3. Gao L, Shi C, Lin Q, Wu Y, Hu L, Wang M, et al. Case report: a case of PLA2G6 gene-related early-onset Parkinson's disease and review of literature. Front Neurosci. (2022) 16:1064566. doi: 10.3389/fnins.2022.1064566
4. Deng X, Yuan L, Jankovic J, Deng H. The role of the PLA2G6 gene in neurodegenerative diseases. Ageing Res Rev. (2023) 89:101957. doi: 10.1016/j.arr.2023.101957
5. Shi CH, Tang BS, Wang L, Lv ZY, Wang J, Luo LZ, et al. PLA2G6 gene mutation in autosomal recessive early-onset parkinsonism in a Chinese cohort. Neurology. (2011) 77:75–81. doi: 10.1212/WNL.0b013e318221acd3
6. Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. (2009) 65:19–23. doi: 10.1002/ana.21415
7. Sina F, Shojaee S, Elahi E, Paisan-Ruiz C. R632W mutation in PLA2G6 segregates with dystonia-parkinsonism in a consanguineous Iranian family. Eur J Neurol. (2009) 16:101–4. doi: 10.1111/j.1468-1331.2008.02356.x
8. Yoshino H, Tomiyama H, Tachibana N, Ogaki K, Li Y, Funayama M, et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology. (2010) 75:1356–61. doi: 10.1212/WNL.0b013e3181f73649
9. Bower MA, Bushara K, Dempsey MA, Das S, Tuite PJ. Novel mutations in siblings with later-onset PLA2G6-associated neurodegeneration (PLAN). Mov Disord. (2011) 26:1768–9. doi: 10.1002/mds.23617
10. Agarwal P, Hogarth P, Hayflick S, MacLeod P, Kuriakose R, McKenzie J, et al. Imaging striatal dopaminergic function in phospholipase A2 group VI-related parkinsonism. Mov Disord. (2012) 27:1698–9. doi: 10.1002/mds.25160
11. Lu CS, Lai SC, Wu RM, Weng YH, Huang CL, Chen RS, et al. PLA2G6 mutations in PARK14-linked young-onset parkinsonism and sporadic Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet. (2012) 159B:183–91. doi: 10.1002/ajmg.b.32012
12. Paisán-Ruiz C, Li A, Schneider SA, Holton JL, Johnson R, Kidd D, et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging. (2012) 33:814–23. doi: 10.1016/j.neurobiolaging.2010.05.009
13. Illingworth MA, Meyer E, Chong WK, Manzur AY, Carr LJ, Younis R, et al. PLA2G6-associated neurodegeneration (PLAN): further expansion of the clinical, radiological and mutation spectrum associated with infantile and atypical childhood-onset disease. Mol Genet Metab. (2014) 112:183–9. doi: 10.1016/j.ymgme.2014.03.008
14. Virmani T, Thenganatt MA, Goldman JS, Kubisch C, Greene PE, Alcalay RN. Oculogyric crises induced by levodopa in PLA2G6 parkinsonism-dystonia. Parkinsonism Relat Disord. (2014) 20:245–7. doi: 10.1016/j.parkreldis.2013.10.016
15. Malaguti MC, Melzi V, Di Giacopo R, Monfrini E, Di Biase E, Franco G, et al. A novel homozygous PLA2G6 mutation causes dystonia-parkinsonism. Parkinsonism Relat Disord. (2015) 21:337–9. doi: 10.1016/j.parkreldis.2015.01.001
16. Xie F, Cen Z, Ouyang Z, Wu S, Xiao J, Luo W. Homozygous pD331Y mutation in PLA2G6 in two patients with pure autosomal-recessive early-onset parkinsonism: further evidence of a fourth phenotype of PLA2G6-associated neurodegeneration. Parkinsonism Relat Disord. (2015) 21:420–2. doi: 10.1016/j.parkreldis.2015.01.012
17. Kim YJ, Lyoo CH, Hong S, Kim NY, Lee MS. Neuroimaging studies and whole exome sequencing of PLA2G6-associated neurodegeneration in a family with intrafamilial phenotypic heterogeneity. Parkinsonism Relat Disord. (2015) 21:402–6. doi: 10.1016/j.parkreldis.2015.01.010
18. Giri A, Guven G, Hanagasi H, Hauser AK, Erginul-Unaltuna N, Bilgic B, et al. PLA2G6 mutations related to distinct phenotypes: a new case with early-onset Parkinsonism. Tremor Other Hyperkinet Mov. (2016) 6:363. doi: 10.5334/tohm.289
19. Bohlega SA, Al-Mubarak BR, Alyemni EA, Abouelhoda M, Monies D, Mustafa AE, et al. Clinical heterogeneity of PLA2G6-related Parkinsonism: analysis of two Saudi families. BMC Res Notes. (2016) 9:295. doi: 10.1186/s13104-016-2102-7
20. Klein C, Lochte T, Delamonte SM, Braenne I, Hicks AA, Zschiedrich-Jansen K, et al. PLA2G6 mutations and Parkinsonism: long-term follow-up of clinical features and neuropathology. Mov Disord. (2016) 31:1927–9. doi: 10.1002/mds.26814
21. Kapoor S, Shah MH, Singh N, Rather MI, Bhat V, Gopinath S, et al. Genetic analysis of PLA2G6 in 22 Indian families with infantile neuroaxonal dystrophy, atypical late-onset neuroaxonal dystrophy and dystonia Parkinsonism complex. PLoS ONE. (2016) 11:e155605. doi: 10.1371/journal.pone.0155605
22. Chen YJ, Chen YC, Dong HL, Li LX, Ni W, Li HF, et al. Novel PLA2G6 mutations and clinical heterogeneity in Chinese cases with phospholipase A2-associated neurodegeneration. Parkinsonism Relat Disord. (2018) 49:88–94. doi: 10.1016/j.parkreldis.2018.02.010
23. Shen T, Hu J, Jiang Y, Zhao S, Lin C, Yin X, et al. Early-onset Parkinson's disease caused by PLA2G6 compound heterozygous mutation, a case report and literature review. Front Neurol. (2019) 10:915. doi: 10.3389/fneur.2019.00915
24. Kamel WA, Al-Hashel JY, Abdulsalam AJ, Damier P, Al-Mejalhem AY. PLA2G6-related parkinsonism presenting as adolescent behavior. Acta Neurol Belg. (2019) 119:621–2. doi: 10.1007/s13760-018-1003-z
25. Ma LM, Zhao J, Shi YY, Chen ZZ, Ren ZX, Zhang JW. PLA2G6 compound complicated mutation in an atypical neuroaxonal dystrophy pedigree. Zhonghua Yi Xue Za Zhi. (2019) 99:354–8. doi: 10.3760/cma.j.issn.0376-2491.2019.05.007
26. Jain S, Bhasin H, Romani M, Valente EM, Sharma S. Atypical childhood-onset neuroaxonal dystrophy in an Indian girl. J Pediatr Neurosci. (2019) 14:90–3. doi: 10.4103/jpn.JPN_91_18
27. Chu YT, Lin HY, Chen PL, Lin CH. Genotype-phenotype correlations of adult-onset PLA2G6-associated neurodegeneration: case series and literature review. BMC Neurol. (2020) 20:101. doi: 10.1186/s12883-020-01684-6
28. Gao C, Huang T, Chen R, Yuan Z, Tian Y, Zhang Y, et al. Han Chinese family with early-onset Parkinson's disease carrying novel frameshift mutation and compound heterozygous mutation of PRKN appearing incompatible with MDS clinical diagnostic criteria. Front Neurol. (2020) 11:582323. doi: 10.3389/fneur.2020.582323
29. Magrinelli F, Mehta S, Di Lazzaro G, Latorre A, Edwards MJ, Balint B, et al. Dissecting the phenotype and genotype of PLA2G6-related Parkinsonism. Mov Disord. (2022) 37:148–61. doi: 10.1002/mds.28807
30. Toth-Bencsik R, Balicza P, Varga ET, Lengyel A, Rudas G, Gal A, et al. New insights of phospholipase A2 associated neurodegeneration phenotype based on the long-term follow-up of a large Hungarian family. Front Genet. (2021) 12:628904. doi: 10.3389/fgene.2021.628904
31. Chen T, Chang Y, Cui Z, Yin X, Wang M, Gao Z, et al. Two cases of PLA2G6-associated young onset Parkinson disease. Chin J Nerv Mental Dis. (2022) 48:746–9. doi: 10.3969/j.issn.1002-0152.2022.12.008
32. Wan Y, Jiang Y, Xie Z, Ling C, Du K, Li R, et al. Novel PLA2G6 pathogenic variants in Chinese patients with PLA2G6-associated neurodegeneration. Front Neurol. (2022) 13:922528. doi: 10.3389/fneur.2022.922528
33. Ma J, Wang X, Wang C. PLA2G6 gene related early onset Parkinson syndrome with cerebellar atrophy: 3 cases report. Chin J Neurol. (2022) 55:1292–7. doi: 10.3760/cma.j.cn113694-20220720-00563
34. Jankovic J, Tan EK. Parkinson's disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry. (2020) 91:795–808. doi: 10.1136/jnnp-2019-322338
35. Ramanadham S, Ali T, Ashley JW, Bone RN, Hancock WD, Lei X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J Lipid Res. (2015) 56:1643–68. doi: 10.1194/jlr.R058701
36. Zhao Y, Qin L, Pan H, Liu Z, Jiang L, He Y, et al. The role of genetics in Parkinson's disease: a large cohort study in Chinese mainland population. Brain. (2020) 143:2220–34. doi: 10.1093/brain/awaa167
37. Liu H, Wang Y, Pan H, Xu K, Jiang L, Zhao Y, et al. Association of rare heterozygous PLA2G6 variants with the risk of Parkinson's disease. Neurobiol Aging. (2021) 101:295–7. doi: 10.1016/j.neurobiolaging.2020.11.003
38. de Oliveira P, Montanaro V, Carvalho D, Martins B, Ferreira A, Cardoso F. Severe early-onset Parkinsonian syndrome caused by PLA2G6 heterozygous variants. Mov Disord Clin Pract. (2021) 8:794–6. doi: 10.1002/mdc3.13230
39. Gui YX, Xu ZP, Wen L, Liu HM, Zhao JJ, Hu XY. Four novel rare mutations of PLA2G6 in Chinese population with Parkinson's disease. Parkinsonism Relat Disord. (2013) 19:21–6. doi: 10.1016/j.parkreldis.2012.07.016
40. Chan DK, Mok V, Ng PW, Yeung J, Kwok JB, Fang ZM, et al. PARK2 mutations and clinical features in a Chinese population with early-onset Parkinson's disease. J Neural Transm. (2008) 115:715–9. doi: 10.1007/s00702-007-0011-6
41. Blanchard H, Taha AY, Cheon Y, Kim HW, Turk J, Rapoport SI. iPLA2beta knockout mouse, a genetic model for progressive human motor disorders, develops age-related neuropathology. Neurochem Res. (2014) 39:1522–32. doi: 10.1007/s11064-014-1342-y
Keywords: PLA2G6, early-onset parkinsonism, cerebellar atrophy, hot spot mutation, heterogeneity
Citation: Cai D, Wu H, Huang B, Xiao W and Du K (2024) A novel variant of PLA2G6 gene related early-onset parkinsonism: a case report and literature review. Front. Neurol. 15:1349861. doi: 10.3389/fneur.2024.1349861
Received: 07 December 2023; Accepted: 18 March 2024;
Published: 18 April 2024.
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Barbara Garavaglia, IRCCS Carlo Besta Neurological Institute Foundation, ItalyCopyright © 2024 Cai, Wu, Huang, Xiao and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kang Du, ZHVrYW5neW5AMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.