 Brittany L. Adler
Brittany L. Adler Tae Chung
Tae Chung Peter C. Rowe
Peter C. Rowe John Aucott
John Aucott- 1Division of Rheumatology, Johns Hopkins University, Baltimore, MD, United States
- 2Department of Physical Medicine and Rehabilitation, Johns Hopkins University, Baltimore, MD, United States
- 3Department of Pediatrics, Johns Hopkins University, Baltimore, MD, United States
Dysautonomia, or dysfunction of the autonomic nervous system (ANS), may occur following an infectious insult and can result in a variety of debilitating, widespread, and often poorly recognized symptoms. Dysautonomia is now widely accepted as a complication of COVID-19 and is an important component of Post-Acute Sequelae of COVID-19 (PASC or long COVID). PASC shares many overlapping clinical features with other infection-associated chronic illnesses including Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) and Post-Treatment Lyme Disease Syndrome (PTLDS), suggesting that they may share common underlying mechanisms including autonomic dysfunction. Despite the recognition of this complication of Lyme disease in the care of patients with PTLD, there has been a scarcity of research in this field and dysautonomia has not yet been established as a complication of Lyme disease in the medical literature. In this review, we discuss the evidence implicating Borrelia burgdorferi as a cause of dysautonomia and the related symptoms, propose potential pathogenic mechanisms given our knowledge of Lyme disease and mechanisms of PASC and ME/CFS, and discuss the diagnostic evaluation and treatments of dysautonomia. We also outline gaps in the literature and priorities for future research.
Introduction
Dysautonomia, or dysfunction of the autonomic nervous system (ANS), often occurs following an infectious insult and can result in a variety of debilitating, widespread, and often poorly recognized symptoms. Postural orthostatic tachycardia syndrome (POTS) is one of the most common manifestations of dysautonomia, and is characterized by orthostatic and exertional intolerance due to vasomotor dysfunction, or the impaired ability to regulate blood flow (1). Patients frequently have other manifestations of dysautonomia, including but not limited to gastrointestinal dysmotility, sweating dysfunction, temperature dysregulation, sicca syndrome, and urinary and visual symptoms (2).
Over half of patients with POTS report a preceding infection (3). COVID-19 is increasingly recognized as a cause of POTS (4, 5), as well as other infections including Epstein Barr Virus (EBV) (6, 7). Lyme disease, which is caused by the tick-borne spirochete Borrelia burgdorferi (B. burgdorferi), results in chronic symptoms in ~10%−20% of patients after the infection is treated, a syndrome called Post-Treatment Lyme Disease Syndrome (PTLDS) (8). PTLDS shares many clinical similarities with other infection-associated chronic illnesses including Post-Acute Sequelae of COVID-19 (PASC), suggesting that they may share common mechanisms including dysautonomia. Although dysautonomia is reported as a complication of Lyme disease in the patient community and among some physicians, there remains a scarcity of research in this field and dysautonomia has not yet been established as a complication of Lyme disease in the medical literature. In this review, we seek to discuss the evidence implicating B. burgdorferi as a cause of dysautonomia and the related symptoms, propose potential pathogenic mechanisms given our knowledge of Lyme disease and mechanisms of PASC and ME/CFS, discuss the diagnostic evaluation and treatments of dysautonomia, and identify remaining gaps in the literature.
Symptoms and clinicopathologic features of dysautonomia
Dysautonomia is a general term used to describe conditions that involve dysfunction of the ANS. The ANS, which is composed of the sympathetic, parasympathetic, and enteric nervous systems, control involuntary functions such as heart rate, blood pressure, digestion, temperature, sweating, and pupillary function. Symptoms of ANS dysregulation are diverse and include orthostatic intolerance, GI symptoms, urinary dysfunction, temperature intolerance, sicca syndrome, abnormal sweating and visual disturbances (9). Orthostatic intolerance refers to a group of symptoms that are provoked by assuming and maintaining upright posture (both sitting and standing), and many of these symptoms improve with recumbency. Once provoked, some orthostatic symptoms can persist for hours, including fatigue. Orthostatic intolerance is often associated with hemodynamic dysregulation that can lead to disabling symptoms of light-headedness, fatigue, weakness, and cognitive impairment that significantly impair quality of life.
POTS is the most common manifestation of dysautonomia. POTS affects ~1–3 million people in the United States (10–12), although this figure is likely an underestimate as dysautonomia is often overlooked, diagnostic testing is not widely available, and the COVID-19 pandemic has led to a dramatic increase in the incidence of dysautonomia that we are only now beginning to appreciate. POTS is defined as a sustained increase in heart rate >30 beats per minute in adults (or a 40 bpm increase in 12–19 year olds) within 10 min of upright posture, without a drop in blood pressure and associated with chronic orthostatic symptoms (13). Dysautonomia can also manifest as (1) neurally mediated reflex hypotension or syncope, defined as a 25-mm Hg reduction in systolic blood pressure from the baseline supine values, sustained for at least 1 min, with no associated increase in heart rate, and accompanied by symptoms of presyncope (severe weakness, lightheadedness, nausea, or diaphoresis) (14), (2) classical or delayed orthostatic hypotension (OH), defined as a drop in systolic blood pressure >20 mmHg or a drop in diastolic blood pressure >10 mmHg with standing (15, 16) within the first 3 min upright (classical OH) or after that point (delayed OH), (14) or (3) initial orthostatic hypotension, defined as a transient SBP drop ≥40 mmHg within 15 s of standing, with recovery within 45 s (17). Orthostatic intolerance confirmed by significant reductions in cerebral blood flow as measured by extracranial Doppler ultrasound can also be present even in the absence of abnormalities in heart rate and blood pressure (18).
There are several pathophysiologic subtypes of POTS which are not mutually exclusive and frequently overlap in an individual patient. These include hypovolemic POTS from reduced blood volume, hyperadrenergic POTS from increased catecholamines which can result in increased blood pressure with standing, excessive venous pooling from connective tissue laxity (19), and neuropathic POTS. Neuropathic POTS is associated with reduced intraepidermal and/or sudomotor (i.e., sweat gland) nerve fiber density on skin biopsy indicative of a small-fiber sensory or autonomic neuropathy. It is hypothesized that damage to sympathetic vasomotor nerves impairs the compensatory increase in systemic vascular resistance during orthostatic stress and/or during exercise, resulting in splanchnic and/or lower limb pooling and reduced cerebral blood flow. This results in typical POTS symptoms including dizziness, cognitive dysfunction (colloquially known as “brain fog”), and fatigue. At the same time, compensatory activation of the sympathetic nervous system in response to reduced cardiac preload results in tachycardia.
Dysautonomia and small-fiber neuropathy often occur as a complication of certain infections
Causes of dysautonomia are heterogeneous and include infections, autoimmune disease, trauma, and neurodegenerative diseases such as multiple system atrophy (MSA). Approximately half of patients with POTS report symptoms of a preceding infection (3). Infectious causes of POTS include EBV, herpes viruses, flavivirus, enterovirus 71, retroviruses (HIV), and SARS-CoV-2 (7). Patients with infection-associated dysautonomia often report similar clinical symptoms regardless of the inciting infection. A small-fiber neuropathy has been observed across the spectrum of POTS regardless of the underlying etiology, suggesting that there may be a final common pathway that results in nerve damage (20). However, few studies have directly compared different infection-associated dysautonomia syndromes to determine if there are different clinical presentations of dysautonomia depending on the type of infection (6).
Acute infection with B. burgdorferi can alter the autonomic nervous system
Left untreated, B. burgdorferi can infect the nervous system (neuroborreliosis) which can have a wide spectrum of presentations, the most common of which are aseptic meningitis, cranial neuropathy and radiculopathy (21, 22). Despite reports suggesting that the autonomic nervous system can also be affected, this complication of Lyme disease has received little attention. Case reports of autonomic disorders that have been reported after acute B. burgdorferi infection (23, 24) include intestinal pseudo-obstruction (25–27), constipation (28–30), unexplained urinary retention (28, 29), postural orthostatic tachycardia syndrome (24, 31), and reflex sympathetic dystrophy (32), which is an autonomic disorder characterized by regional sympathetic hyperactivity. Although there are other causes of urinary retention, the co-existence with enteroparesis in five of these reports implicates dysfunction of the autonomic nervous system. Another study found that 18 patients with serologically positive acute Lyme disease (IgM positive) had lower cardiac vagal tone in response to deep breathing compared to 18 healthy controls (33). In addition to autonomic symptoms, painful small-fiber neuropathy, including reduced intra-epidermal nerve fiber density (EINFD) and reduced sweat gland nerve fiber density (SGNFD), have also been documented during acute B. burgdorferi infection (34) and symptoms improved with antibiotics (35).
Although intriguing, these case reports are limited in number and must be interpreted with caution. In most cases, the patients had a compelling history of Lyme disease being causative given the temporal association of symptoms with an erythema migrans rash, positive Lyme serologies, and improvement with antibiotics. Several of these patients also had other symptoms that were more specific for Lyme disease such as cranial neuropathies. However, not all of the reports used strict criteria to diagnose Lyme disease. For example, several studies did not have a confirmed history of an erythema migrans rash or other objective manifestations of Lyme disease and only used serologies to diagnose Lyme disease which is not diagnostic of an active infection (31, 33). Most patients in these reports were often treated concurrently with other medications in addition to antibiotics, so it is difficult to conclude that the antibiotics alone led to symptom improvement. Furthermore, without confirmation that the patient did not experience orthostatic symptoms prior to Lyme disease and had a normal response to orthostatic stress, we cannot be sure whether dysautonomia followed the infection or preceded it. Although these studies are not without their limitations, collectively they demonstrate that untreated B. burgdorferi might be associated with autonomic dysregulation and small-fiber neuropathy that is potentially reversible.
Much of our understanding of the effects of B. burgdorferi on the autonomic nervous system come from autopsy studies in the early reports of Lyme disease as well as primate studies. Autopsy studies of patients with Lyme disease have identified lymphoplasmocellular infiltrates in the autonomic ganglia and the interstitium of the longitudinal nerves (36, 37). This is accompanied by thickening of the perineural blood vessels, which at times are surrounded by inflammatory cells. Spirochetes have never been directly visualized in the autonomic ganglia or peripheral nerves in humans, although it is likely that they are directly involved during the acute infection (38).
Studies in Rhesus monkeys have confirmed the direct effect of B. burgdorferi on ganglia. The sensory ganglia of macaques that were infected with B. burgdorferi demonstrated cellular apoptosis and necrosis of neuronal and satellite glial cells (39, 40), and the ganglia also stained positive for a lipoprotein expressed on B. burgdorferi suggesting that it is directly infected (39). Furthermore, rhesus DRG tissue explants exposed to live B. burgdorferi induced an inflammatory response and neural apoptosis in the DRG (41). These studies demonstrate that B. burgdorferi can directly infect the dorsal root ganglia and induce neuronal and glial cell apoptosis during acute, untreated Lyme disease. However, a limitation of this study is that it focused on the dorsal root ganglia and did not examine the sympathetic chain ganglia.
PTLDS is a syndrome that may in some cases involve dysautonomia
Approximately 10%−20% of patients with Lyme disease develop chronic symptoms after the acute B. burgdorferi infection despite appropriate treatment with antibiotics (42). This syndrome is called Post-Treatment Lyme Disease Syndrome (PTLDS) and is defined as life-altering symptoms of fatigue, musculoskeletal pain, and/or cognitive difficulties that start within 6 months of the acute infection and persist for more than 6 months (8). The most common symptoms of PTLDS are fatigue, joint and muscle pain, and difficulty with concentrating and sleep, all of which are also common symptoms of dysautonomia. There is a small but notable subset of patients with PTLDS with other symptoms that are more specific to peripheral autonomic neuropathy, including difficulty with urination and nausea (43, 44), however few studies have examined this subset of patients to confirm if they have an autonomic neuropathy.
Despite the significant overlap between symptoms of PTLDS and dysautonomia, data on the role of peripheral nerve dysfunction and dysautonomia in PTLDS are lacking. In two separate case series, a total of seven patients who were treated for Lyme disease later developed new symptoms of POTS 6 months−12 years after the acute infection (45, 46). Another study of 10 patients with well-defined PTLDS who met the IDSA proposed case definition for PTLD demonstrated on tilt table test reduced cerebral blood flow velocity and either sympathetic or parasympathetic dysfunction in all participants (47). Although these reports suggest that PTLDS may be linked to dysautonomia, there are no prospective studies demonstrating a higher risk of dysautonomia in patients who previously had Lyme disease, and there are no temporal studies demonstrating a higher incidence of dysautonomia closer to the time of the acute B. burgdorferi infection.
Small-fiber sensory and autonomic neuropathy on skin biopsy are often found in patients with dysautonomia, and have also been linked with PTLDS. The small-fiber neuropathy provides pathologic evidence of the sensory and autonomic involvement in patients with PTLDS. In the same case series that demonstrated autonomic dysfunction in patients with PTLDS, all 10 patients had evidence of abnormal IENFD and/or SGNFD on skin biopsy (47). Reduced unmyelinated sub-basal nerve fibers in the cornea has also been described in a patient with PTLDS, and is suggestive of small-fiber neuropathy. On corneal microscopy, this patient had abundant dendritic cells, a finding which is typically seen in autoimmune diseases or chronic systemic inflammation (48).
Potential mechanisms of infection-associated dysautonomia
The mechanisms of infection-associated dysautonomia remain unknown and research is still evolving, but studies in PTLDS and other infection-associated chronic illnesses such as PASC and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) have led to several proposed hypotheses which may be applicable to dysautonomia after Lyme disease (see Figure 1). The sensory and autonomic neuropathy that is often present on skin biopsy suggests that, at least in a subset of patients, these infection-associated chronic syndromes result from a neuropathic process. The small-fiber neuropathy is often in a non-length-dependent pattern, suggesting autonomic or dorsal root ganglia as the target tissue. Although B. burgdorferi and SARS-CoV-2 are both known to affect the autonomic ganglia during the acute infection, there are no histopathological studies to date that have examined the ganglia or other neural tissue of patients with PASC or PTLDS. Vascular endothelial dysfunction has also been proposed as a mechanism of PASC, and more research is needed investigating the endothelium in PTLDS.
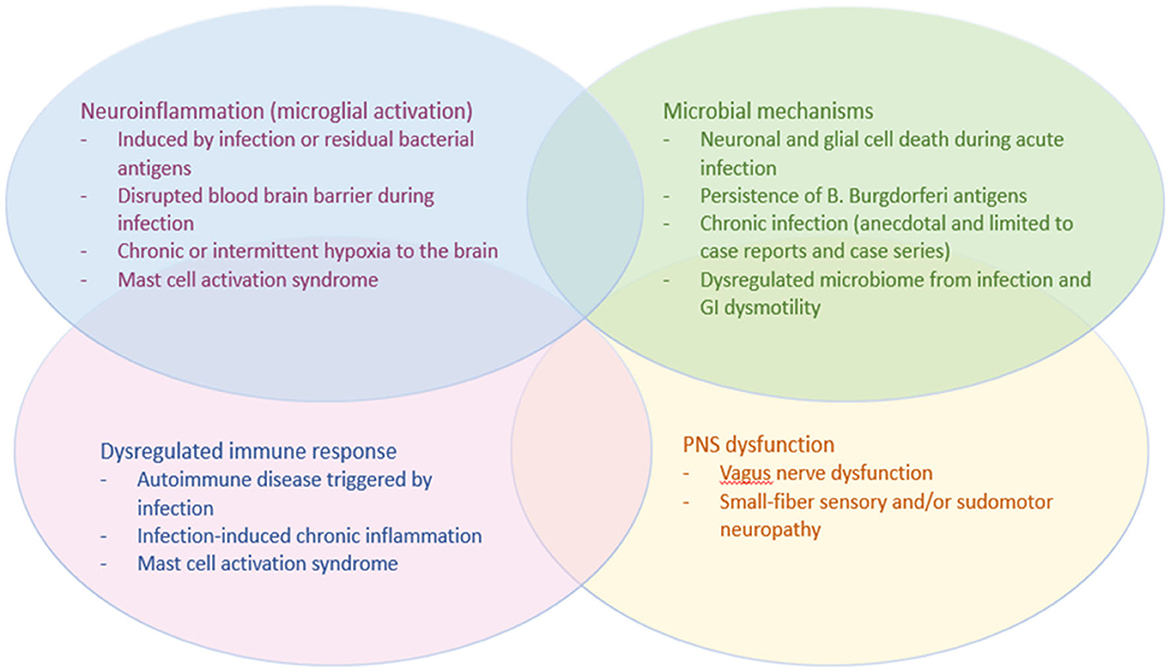
Figure 1. Potential mechanisms of dysautonomia in PTLDS.
An important confounder in Lyme disease research that needs to be acknowledged when discussing mechanism is that patients can be infected with other tick-borne pathogens in addition to B. burgdorferi. Two percent of patients with erythema migrans have Anaplasma phagocytophilum and Babesia microti co-infections, respectively, and other infections such as Bartonella and Powassan virus have also been reported (49, 50). Some of these co-infections have been previously linked to POTS (51) and may confound human studies of PTLDS. Animal and in vitro studies of B. burgdorferi are needed to isolate the effects of B. burgdorferi, and human studies of Lyme disease should evaluate for tick-borne co-infections and other concurrent infections like EBV so the complete infectious profile is understood.
Neuronal and glial cell apoptosis during the acute infection
Neuroborreliosis is well-documented, and accumulating evidence demonstrates that B. burgdorferi can cause neuronal and glial cell death (52). Injury to the ANS during acute infection may cause an autonomic neuropathy that is not reversible or that may take months for the nerves to regenerate. Although no studies have been performed using tissue from autonomic ganglia, in vitro and in vivo studies have demonstrated that B. burgdorferi can cause apoptosis of neurons and satellite glial cells in the CNS and dorsal root ganglia (41, 53–55). Neuronal cell death is thought to be caused in part by inflammatory mediators released by glial cells in response to the spirochete (56, 57). These studies suggest that B. burgdorferi is neurotoxic. However, pathologic studies are needed to confirm that neuronal cell death is the primary cause of chronic symptoms in PTLDS.
Chronic B. burgdorferi infection in the nervous system
Persistent infection has also been invoked as a cause of PTLD-associated dysautonomia. Although previous studies in non-human primates and other animal models have demonstrated persistent B. burgdorferi infection after adequate antibiotic treatment (58, 59), reports of microbial persistence after antibiotic therapy in humans are anecdotal and limited (60–63). A recent study of microbial antigen persistence in late Lyme arthritis has identified B. burgdorderi peptidoglycan, a key component of the spirochete cell wall, in the synovial fluid of patients with antibiotic refractory late Lyme arthritis. These studies need to be confirmed by other groups and with larger cohorts of patients. Even if there is persistent infection, re-treatment with intravenous ceftriaxone followed by doxycycline does not provide dramatic or sustained improvement of symptoms in PTLDS (64). It is possible that a different antibiotic regimen is needed to treat a persistent infection and/or possible co-infections (65), as animal models have demonstrated differences in the efficacy of different antibiotics at treating B. burgdorferi (66). Studies of different antibiotic regimens for PTLDS are needed and should account for the presence of co-infections.
Chronic neuroinflammation
Neuroinflammation, or inflammation of the nervous system, can arise from an infection and persist even after the infection has resolved. Imaging studies in PTLDS have identified clear evidence of microglial activation in the CNS (67), similar to what has been observed in ME/CFS myalgic encephalomyelitis (68). Research is evolving in this field to understand the mechanisms of infection-associated neuroinflammation and how it contributes to chronic symptoms (69). One proposed hypothesis is that immune system activation in response to an infection leads to the release of various cytokines and chemokines. These inflammatory mediators can cross the blood-brain barrier and activate CNS immune cells such as microglia and astrocytes, which in turn release their own inflammatory molecules and reactive oxygen species (ROS) and further propagate the immune response. It is also possible that the inflammatory response can disrupt the blood-brain barrier and allow various cytokines and chemokines to access the CNS, causing further microglial activation and neuroinflammation. Other studies suggest that non-viable spirochetal residues left-over after treatment with antibiotics may persist and retain their ability to induce inflammation and neuronal cell death (70).
Few studies have examined the relationship between autonomic dysfunction and neuroinflammation. Chronic or intermittent hypoxia in sleep apnea is associated with microglial activation in the CNS (71, 72), and is a plausible mechanism underlying neuroinflammation in patients with infection-associated dysautonomia syndromes. Studies using either extracranial Doppler ultrasound of the internal carotid and vertebral arteries or transcranial Doppler ultrasound in ME/CFS and PASC have revealed reduced cerebral perfusion in the upright position, leading to chronic positional hypoxia (18, 73–75). However, no studies have correlated cerebral perfusion with microglial activation in the infection-associated chronic syndromes. Studies in other syndromes such as fibromyalgia, which shares overlapping features with the infection-associated chronic syndromes, suggest that neuroinflammation can also involve the peripheral nervous system including the dorsal root ganglia (76). However, no studies to date have examined neuroinflammation in the autonomic ganglia of patients with infection-associated dysautonomia.
Mast cell activation is another mechanism that may contribute to neuroinflammation in patients with dysautonomia in the setting of PTLDS. POTS has been associated with mast cell activation syndrome (MCAS) (77), a poorly understood multisystem disorder of inflammation, with or without allergic phenomena or tissue growth/development anomalies (78). There is a strong bi-directional relationship between mast cells and the brain, and they serve as intermediaries between the immune system and the nervous system. Mast cells are located next to nociceptors/neurons and reside in the brain (79, 80), and when activated release neuroactive inflammatory mediators that activate microglia and can contribute to neuroinflammation (81, 82). Although MCAS is widely recognized to afflict some patients with dysautonomia, no studies to date have examined the presence of MCAS in patients with PTLDS.
Autoimmunity triggered by the pathogen
Another hypothesis for the chronic symptoms experienced by many patients after Lyme disease is that the spirochete triggers an autoimmune response directed against a neural antigen through molecular mimicry. This hypothesis is supported by the reported lag between the acute infection and the onset of chronic symptoms, which can be 6 months or longer after the initial infection. One study found that patients with PTLDS have significantly higher anti-neural antibodies (49%) compared to healthy controls (15%) or individuals who had Lyme disease and returned to health (18.5%) (83). The specificity of these autoantibodies remains unknown, and it is not clear if these antigens are present in the central nervous system (CNS), the autonomic ganglia, the cranial nerves, or in other nervous system tissue. Antibodies which recognize lysoganglioside as well as enolase γ, an antigen present throughout the nervous system including in the ganglia, have been reported in case reports of PTLDS (84, 85). More research is needed to identify the specificities of anti-neural antibodies in PTLDS and determine if these antibodies are pathogenic.
Autoantibodies targeting specific autonomic receptors or ganglionic nicotinic acetylcholine receptors have been identified in patients with other dysautonomia syndromes and are thought to define an autoimmune subset (86). These autoantibodies include ganglionic, adrenergic, and muscarinic acetylcholine receptor antibodies (87), and correlate with autonomic symptom burden, including GI dysfunction, fatigue, muscle pain, and exercise tolerance (88). These autoantibodies are present in a subset of patients with PASC (89), although their prevalence in PTLDS remains unknown. More research is needed to validate these autoantibodies and determine their specificity for dysautonomia. It is also unclear if these autoantibodies are directly pathogenic, or whether they are a marker of autoimmunity without a specific pathophysiological effect, as the presence of various autoantibodies may simply reflect a dysregulated immune response.
Accumulating evidence suggests that a subset of patients with dysautonomia respond to the immunomodulator intravenous immunoglobulin (IVIG). There are numerous reports of patients with POTS and PASC responding to IVIG (90, 91), although data are scarce in PTLDS. The only published case report of a patient with PTLDS responding to IVIG was a patient with PTLDS and polyneuropathy who had a full recovery with subcutaneous immunoglobulin (92). Randomized clinical trials of IVIG in POTS are ongoing and are also needed in patients with PTLDS and dysautonomia.
Vascular and endothelial damage
While it may be tempting to conclude that PTLDS is a neurologic disorder given the neurotropism of B. burgdorferi, research in other infection-associated chronic illnesses such as PASC have identified endothelial dysfunction as a potential driver of disease. Similar mechanisms may be applicable to other infection-associated chronic illnesses such as PTLDS. SARS-CoV-2 can directly infect endothelial cells, causing cellular damage and dysfunction that triggers an inflammatory cascade and promotes hypercoagulability, microvascular thrombosis and further endothelial dysfunction (93, 94). Fibrin amyloid microclots and platelet hyperactivation have been identified in patients with PASC and are proposed to obstruct capillaries and prevent oxygen delivery to tissues (95). More studies are needed to confirm these findings, and it remains unclear if any endothelial dysfunction results from direct viral infection or is secondary to a dysregulated immune response or an autoimmune phenomena. Similarly, B. burgdorferi spirochetes can adhere to and penetrate endothelial cells in vitro, and activate the vascular endothelium to promote transendothelial migration of neutrophils to induce an inflammatory response (96, 97). However, no studies to date have examined endothelial dysfunction in humans with PTLDS.
Vagal nerve dysregulation
Borrelia burgdorferi is known to infect the cranial nerves, including the vagus nerve (CN X) (21). The vagus nerve plays a vital role in regulating numerous bodily functions and maintaining homeostasis. It is involved in the parasympathetic division of the autonomic nervous system, and is responsible for promoting rest, relaxation, and digestion, while also regulating heart rate, breathing, digestion and other involuntary processes. The vagus nerve also carries sensory information such as taste, touch and pain to the brain, and it carries motor signals from the brain to the muscles of the esophagus and digestive system.
The vagus nerve also has an important role in immune regulation. The vagus nerve transmits signals regarding peripheral inflammation to the CNS, thereby increasing brain cytokines, inducing neuro-inflammation and activating microglia (98–100), all of which have been observed in PTLDS (67, 101, 102). Moreover, the efferent pathways of the vagus nerve have a profound anti-inflammatory effect, mediated by vagus nerve-mediated cholinergic signaling (103, 104). Involvement of the vagus nerve during an acute B. burgdorferi infection and subsequent nerve damage may be a potential mechanism that contributes to dysautonomia and neuro-inflammation in PTLDS. Vagal nerve stimulation can ameliorate microglial activation and cytokine production (105–107), and is currently being studied as a promising therapy to improve symptoms in other infection-associated chronic syndromes including PASC and ME/CFS (108–110).
Dysregulation of gut microbiota homeostasis
Infections may also cause dysautonomia by disrupting the gut microbiome and leading to dysregulation of the gut-brain axis. Patients with PTLDS have evidence of altered gut microbiome compared to controls with and without a history of antibiotic exposure (111). Metabolites produced by the gut microbiome, such as short-chain fatty acids, are important modulators of immune regulation, and disruption of these bacteria may contribute to neuroinflammation and autonomic dysfunction (112, 113). Conversely, GI dysmotility is common in dysautonomia and can lead to small intestinal bacterial overgrowth (SIBO) and further disrupt the gut microbiome (114, 115).
Genetic contribution
Dysautonomia frequently runs in families, suggesting that there may be a genetic component to this syndrome which could lead to subclinical disease which is unmasked or worsened by an infection such as COVID-19 or Lyme disease. The association with joint hypermobility/Ehlers Danlos Syndrome provides further support for a genetic predisposition. Although a few pathogenic mutations have been identified in dysautonomia, such as mutations in genes encoding the norepinephrine transporter (116) or in gain of function mutations in the sodium channel (117, 118), confirmed genetic mutations are not common. Genetic polymorphisms may also play a role in the development of dysautonomia or how the disease presents clinically. Polymorphisms in endothelial nitric oxide synthase are enriched in patients with POTS (119) and polymorphisms in the beta-2 adrenoreceptor may modulate the hemodynamic profile of patients with POTS (120).
Deconditioning
Cardiovascular deconditioning has been proposed as a mechanism underlying infection-associated dysautonomia. Over 90% of patients with POTS or orthostatic intolerance have reduced maximum oxygen uptake during exercise, which is used as a surrogate for deconditioning (121). However, there is no relationship between severity of deconditioning and cerebral blood flow reduction with tilt among ME/CSF patients, which suggests that deconditioning does not explain orthostatic intolerance (122). Furthermore, the development of orthostatic intolerance in elite athletes suggest that deconditioning is a secondary phenomenon and is not a primary driver of disease (123).
Clinical evaluation of dysautonomia in PTLDS and potential treatments
The diagnosis of dysautonomia can be challenging, as symptoms are often nonspecific and overlap with other conditions. A comprehensive evaluation should include a detailed medical history, physical examination, and laboratory tests to rule out other medical conditions that can present similarly. Although dysautonomia has not yet been definitively established as a complication of Lyme disease, given the clinical similarities between PTLDS and other infection-associated chronic illnesses which often present with dysautonomia, we propose that patients with PTLDS should be asked about autonomic symptoms and any symptoms of dysautonomia should be rigorously evaluated with formal testing. Identifying dysautonomia in PTLDS is important because there are specific therapies that can improve autonomic symptoms, including medications that optimize hemodynamics, increase gut motility, or enhance tear and saliva production.
An autonomic evaluation includes screening for autonomic symptoms and checking orthostatic vital signs during a 10 min active or passive stand test. Symptoms of dysautonomia include orthostatic or exertional intolerance (lightheadedness or syncope, palpitations, fatigue, cognitive impairment, blurred vision or muscle weakness/pain worse with standing or activity), GI symptoms, urinary symptoms, sicca, sweating abnormalities and temperature dysregulation. Validated questionnaires such as the Composite Autonomic Symptom Score (COMPASS-31) (124), the Survey of Autonomic Symptoms (SAS) (125), or the Malmo POTS Symptom Score (126) can be used to assess autonomic symptom burden. Symptoms of dysautonomia may be further investigated with more specialized testing, including the tilt table test with or without extracranial or transcranial doppler ultrasound, heart rate variability on deep breathing, Valsalva maneuver, Quantitative Sudomotor Axon Reflex Testing (QSART) and/or thermoregulatory sweat test (TST). A skin biopsy can also be obtained to evaluate for the presence of a small-fiber sensory or sudomotor neuropathy. Table 1 highlights the most common clinically available autonomic tests.
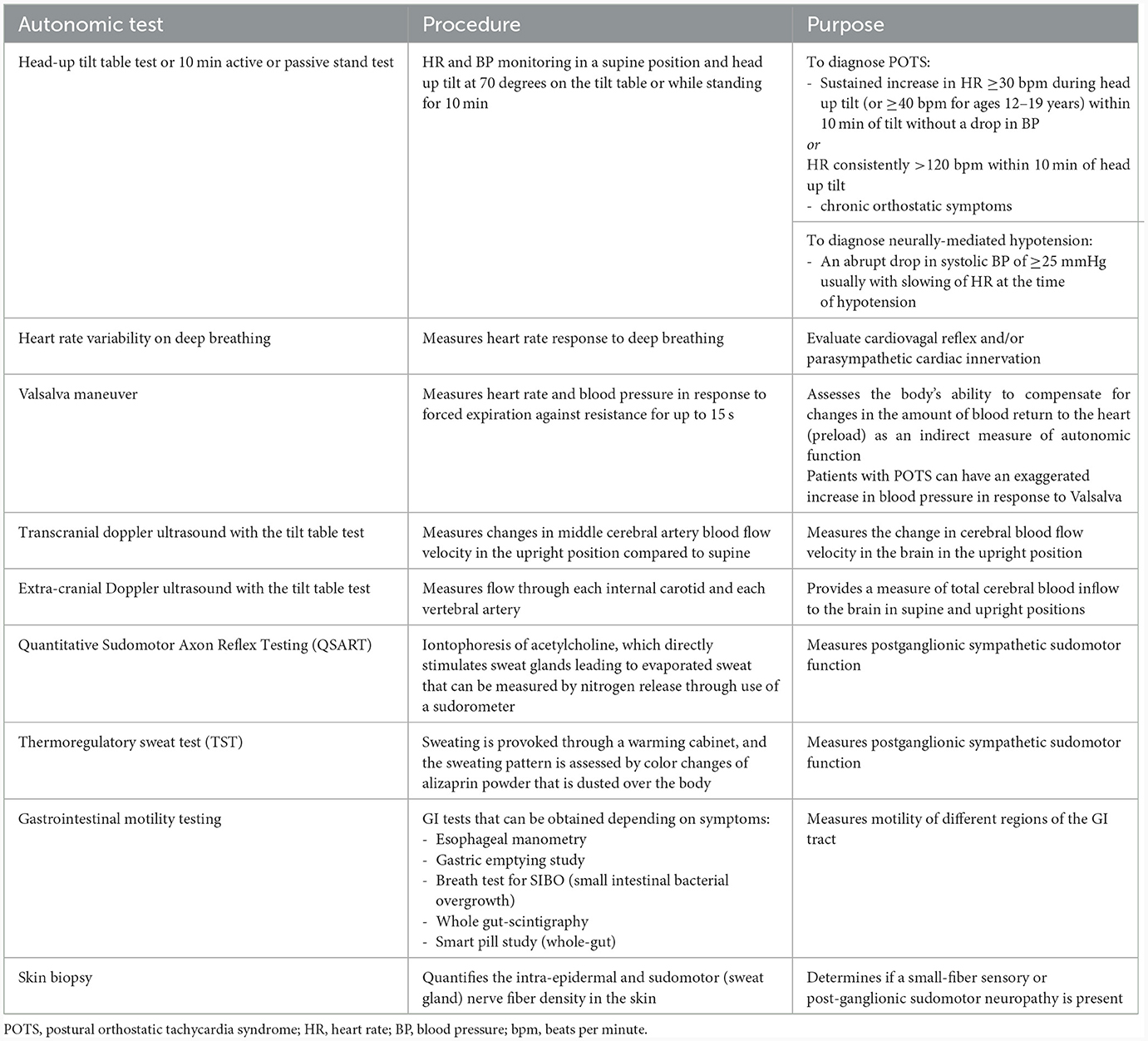
Table 1. Diagnostic tests clinically available for the evaluation of dysautonomia.
There are numerous effective non-pharmacologic and pharmacologic treatments for dysautonomia, and a comprehensive review of these treatments is beyond the scope of this review. Non-pharmacologic treatments for POTS or neurogenic orthostatic hypotension include increased fluid and salt intake, use of compression stockings and an abdominal binder, and gradual increases in activity as tolerated (127). A number of pharmacologic treatments are also available for dysautonomia. For patients with hemodynamic dysregulation from POTS, heart rate can be reduced with beta-blockers or ivabradine, an inhibitor of the hyperpolarization-activated cyclic-nucleotide gate funny (If) current (128). Fludrocortisone is a mineralocorticoid that leads to improved sodium reabsorption in the distal tubule and an early improvement in blood volume, although late effects may be secondary to improved endothelial responses to circulating vasoconstrictors. Midodrine, an α1 agonist, is used to improve vasoconstriction and thereby increase venous return to the heart; it can raise blood pressure in patients with hypotension. Pyridostigmine, a peripherally acting acetylcholinesterase inhibitor increases neurotransmission of acetylcholine to improve cardiovascular dysautonomia (16). For refractory cases, intravenous fluids can be considered for symptom management. Prokinetic agents can be used to stimulate GI motility, including serotonin receptor agonists (i.e., prucalopride), dopamine-receptor antagonists (metoclopramide, domperidone), and the acetylcholinesterase inhibitor pyridostigmine (129). Table 2 summarizes the general categories of treatments that can be considered for a patient with dysautonomia after Lyme disease.
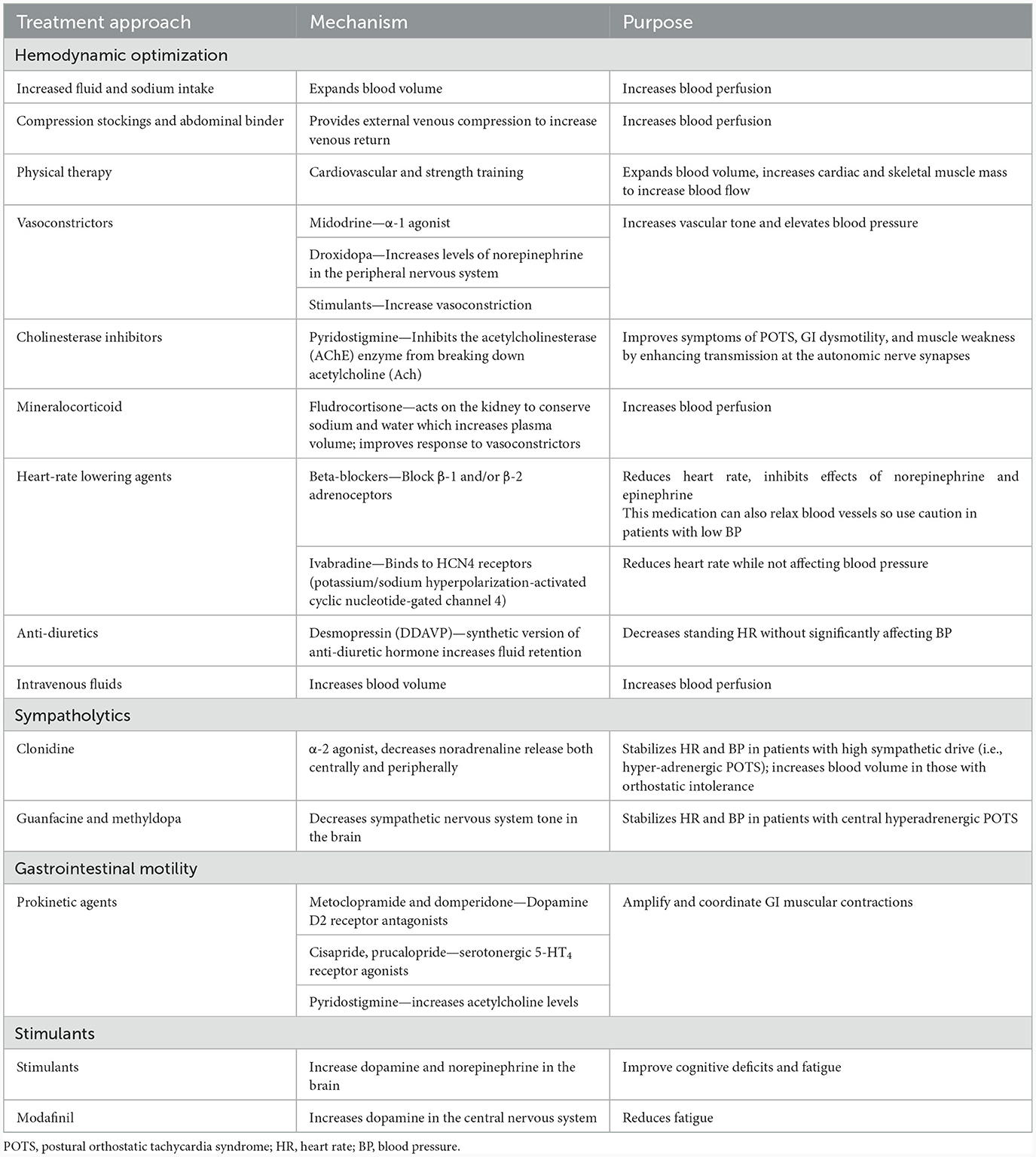
Table 2. Available treatments for dysautonomia.
There are many clinical trials for PASC, some of which are specifically targeted at treating POTS after COVID-19. If successful, these therapies may be translated to treat dysautonomia after other infections. Many case series suggest that intravenous immunoglobulin (IVIG) is effective for PASC and POTS (90, 130), and trials of IVIG are currently underway. Efgartigimod, a neonatal Fc receptor inhibitor which leads to degradation and reduced amounts of circulating IgG, is also being studied for POTS after COVID-19 (NCT05633407). If these drugs are effective, it will strongly implicate autoimmunity as an underlying mechanism for infection-associated chronic illness. Antivirals for PASC are also being studied, which will test the hypothesis that chronic infection is an important mechanism in infection-associated chronic illnesses (131). Other potential treatments for PASC that have been proposed are beyond the scope of this review, and include the CCR5 antagonist Miraviroc along with Pravastatin (132) and neuromodulation using transcranial stimulation to improve fatigue (133). These studies in PASC, among many others, will provide insights into the mechanisms of infection-associated chronic illness that may be applied to PTLDS in the future.
Unmet research needs
Autonomic research in PASC and ME/CFS have clearly demonstrated the important role of dysautonomia in contributing to infection-associated chronic symptoms. Because of the significant clinical similarities with PTLDS, a reasonable hypothesis is that Lyme disease, even after treatment, may be associated with dysautonomia. Although there is a sound biologic basis for this which we have reviewed and there are hints of this association in the literature, there currently are no studies clearly demonstrating this association. There are large gaps in the literature that need to be addressed to better understand the role of dysautonomia in PTLDS.
• Define the true prevalence of small-fiber neuropathy and autonomic dysfunction in PTLDS using objective autonomic testing and skin biopsies in cohorts of patients with well-defined Lyme disease.
• Understand the temporal relationship between the acute B. burgdorferi infection and development of small-fiber neuropathy and autonomic dysfunction.
• Expand on prior histopathological studies of involvement of B. burgdorferi in the autonomic nervous system using human autopsies or animal models.
• Understand the contribution of co-infections in contributing to PTLDS and dysautonomia.
• Identify distinct clinical and pathologic differences in PTLDS compared to other infection-associated chronic illnesses.
Conclusion
Although dysautonomia is reported in case reports and case series to be a complication of treated Lyme disease, there is limited literature clearly establishing this association. Given the significant clinical similarities between post-treatment Lyme disease syndrome (PTLDS) and other dysautonomia syndromes, and the sound biological basis for the involvement of B. burgdorferi in the autonomic nervous system, this field of study needs more research studying this potential complication. If dysautonomia is established as a complication of Lyme disease that contributes to PTLDS, it will lead to new treatments that may improve quality of life for patients affected by this debilitating syndrome.
Author contributions
BA: Conceptualization, Writing – original draft, Writing – review & editing. TC: Writing – review & editing. PR: Writing – review & editing. JA: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Jerome L. Greene Foundation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Arnold AC, Ng J, Raj SR. Postural tachycardia syndrome – diagnosis, physiology, and prognosis. Auton Neurosci. (2018) 215:3–11. doi: 10.1016/j.autneu.2018.02.005
2. Mehr SE, Barbul A, Shibao CA. Gastrointestinal symptoms in postural tachycardia syndrome: a systematic review. Clin Auton Res. (2018) 28:411–21. doi: 10.1007/s10286-018-0519-x
3. Shaw BH, Stiles LE, Bourne K, Green EA, Shibao CA, Okamoto LE, et al. The face of postural tachycardia syndrome – insights from a large cross-sectional online community-based survey. J Intern Med. (2019) 286:438–48. doi: 10.1111/joim.12895
4. Blitshteyn S, Whitelaw S. Postural orthostatic tachycardia syndrome (POTS) and other autonomic disorders after COVID-19 infection: a case series of 20 patients. Immunol Res. (2021) 69:205–11. doi: 10.1007/s12026-021-09185-5
5. Miglis MG, Prieto T, Shaik R, Muppidi S, Sinn DI, Jaradeh S. A case report of postural tachycardia syndrome after COVID-19. Clin Auton Res. (2020) 30:449–51. doi: 10.1007/s10286-020-00727-9
6. van Campen CLMC, Visser FC. Orthostatic intolerance in long-haul COVID after SARS-CoV-2: a case-control comparison with post-EBV and insidious-onset myalgic encephalomyelitis/chronic fatigue syndrome patients. Healthcare. (2022) 10:2058. doi: 10.3390/healthcare10102058
7. Carod-Artal FJ. Infectious diseases causing autonomic dysfunction. Clin Auton Res. (2018) 28:67–81. doi: 10.1007/s10286-017-0452-4
8. Aucott JN. Posttreatment Lyme disease syndrome. Infect Dis Clin. (2015) 29:309–23. doi: 10.1016/j.idc.2015.02.012
9. Goldstein DS, Robertson D, Esler M, Straus SE, Eisenhofer G. Dysautonomias: clinical disorders of the autonomic nervous system. Ann Intern Med. (2002) 137:753–63. doi: 10.7326/0003-4819-137-9-200211050-00011
10. Low PA, Sandroni P, Joyner M, Shen WK. Postural tachycardia syndrome (POTS). J Cardiovasc Electrophysiol. (2009) 20:352–8. doi: 10.1111/j.1540-8167.2008.01407.x
11. Evenson S. “But You Don't Look Sick”: Medical Gaslighting and Disability Identity Among Individuals Living with POTS and ME/CFS. Thesis. (2021). Available online at: https://scholarship.tricolib.brynmawr.edu/handle/10066/23717 (accessed September 9, 2022).
12. Grubb BP. Postural tachycardia syndrome. Circulation. (2008) 117:2814–7. doi: 10.1161/CIRCULATIONAHA.107.761650
13. Freeman R, Wieling W, Axelrod FB, Benditt DG, Benarroch E, Biaggioni I, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Auton Neurosci. (2011) 161:46–8. doi: 10.1016/j.autneu.2011.02.004
14. Rowe PC, Calkins H, DeBusk K, McKenzie R, Anand R, Sharma G, et al. Fludrocortisone acetate to treat neurally mediated hypotension in chronic fatigue syndrome: a randomized controlled trial. JAMA. (2001) 285:52–9. doi: 10.1001/jama.285.1.52
15. Low PA, Opfer-Gehrking TL, Textor SC, Schondorf R, Suarez GA, Fealey RD, et al. Comparison of the postural tachycardia syndrome (POTS) with orthostatic hypotension due to autonomic failure. J Auton Nerv Syst. (1994) 50:181–8. doi: 10.1016/0165-1838(94)90008-6
16. Fedorowski A. Postural orthostatic tachycardia syndrome: clinical presentation, aetiology and management. J Intern Med. (2019) 285:352–66. doi: 10.1111/joim.12852
17. Hira R, Baker JR, Siddiqui T, Ranada SI, Soroush A, Karalasingham K, et al. Objective hemodynamic cardiovascular autonomic abnormalities in post-acute sequelae of COVID-19. Can J Cardiol. (2023) 39:767–75. doi: 10.1016/j.cjca.2023.09.005
18. van Campen CLMC, Rowe PC, Visser FC. Cerebral blood flow remains reduced after tilt testing in myalgic encephalomyelitis/chronic fatigue syndrome patients. Clin Neurophysiol. Pract. (2021) 6:245–55. doi: 10.1016/j.cnp.2021.09.001
19. Roma M, Marden CL, De Wandele I, Francomano CA, Rowe PC. Postural tachycardia syndrome and other forms of orthostatic intolerance in Ehlers-Danlos syndrome. Auton Neurosci. (2018) 215:89–96. doi: 10.1016/j.autneu.2018.02.006
20. Gibbons CH, Bonyhay I, Benson A, Wang N, Freeman R. Structural and functional small fiber abnormalities in the neuropathic postural tachycardia syndrome. PLoS ONE. (2013) 8:e0084716. doi: 10.1371/journal.pone.0084716
21. Chaturvedi A, Baker K, Jeanmonod D, Jeanmonod R. Lyme disease presenting with multiple cranial nerve deficits: report of a case. Case Rep Emerg Med. (2016) 2016:7218906. doi: 10.1155/2016/7218906
22. Halperin J. Nervous System Lyme Disease. UpToDate (2021). Available online at: https://www.uptodate.com/contents/nervous-system-lyme-disease (Retrieved July 18, 2023).
23. Petrun AM, Sinkovič A. Borreliosis presenting as autonomic nervous dysfunction, phrenic nerve palsy with respiratory failure and myocardial dysfunction — a case report. Cent Eur J Med. (2013) 8:463–7. doi: 10.2478/s11536-013-0172-7
24. Younger DS, Orsher S. Lyme neuroborreliosis: preliminary results from an urban referral center employing strict CDC criteria for case selection. Neurol Res Int. (2010) 2010:525206. doi: 10.1155/2010/525206
25. Chatila R, Kapadia CR. Intestinal pseudoobstruction in acute Lyme disease: a case report. Am J Gastroenterol. (1998) 93:1179–80. doi: 10.1111/j.1572-0241.1998.00361.x
26. Schefte DF, Nordentoft T. Intestinal pseudoobstruction caused by chronic lyme neuroborreliosis. A case report. J Neurogastroenterol Motil. (2015) 21:440. doi: 10.5056/jnm14118
27. Autschbach R, Zaremba A, Ulrich B. Pseudo-obstruction of the colon following tick bite injury. Chirurg. (1989) 60:365–7.
28. Leone M, Iqbal A, Hugo Bonatti JR, Anwar S, Feaga C. A patient with SIADH, urinary retention, constipation, and Bell's palsy following a tick bite. Case Rep Nephrol. (2022) 2022:5937131. doi: 10.1155/2022/5937131
29. Shamim EA, Shamim SA, Liss G, Nylen E, Pincus JH, Yepes M. Constipation heralding neuroborreliosis: an atypical tale of 2 patients. Arch Neurol. (2005) 62:671–3. doi: 10.1001/archneur.62.4.671
30. Hansen BA, Finjord T, Bruserud Ø. Autonomous dysfunction in Lyme neuroborreliosis. A case report. Clin Case Rep. (2018) 6:901. doi: 10.1002/ccr3.1494
31. Burman M, Nguyen HL, Murthy V, Gupta PS, Davies C, Wragg A, et al. Severe orthostatic hypotension in a diabetic patient may not be due to diabetic autonomic neuropathy. Clin Med. (2011) 11:290. doi: 10.7861/clinmedicine.11-3-290
32. Gila L, Guerrero A, Astarloa R, Martí P, Gutiérrez JM. Reflex sympathetic dystrophy. A new manifestation of Lyme disease? Enferm Infecc Microbiol Clin. (1990) 8:32–5.
33. Puri BK, Shah M, Monro JA, Kingston MC, Julu PO. Respiratory modulation of cardiac vagal tone in Lyme disease. World J Cardiol. (2014) 6:502. doi: 10.4330/wjc.v6.i6.502
34. Hughes PJ, Lane RJ, Cutler SJ, Wright DJ, Abraham RM, Wade JP, et al. Small-fiber dysfunction in a Borrelia burgdorferi infection. Muscle Nerve. (1993) 16:221–2.
35. Feuer N, Alaedini A. Resolution of pain in the absence of nerve regeneration in small fiber neuropathy following treatment of Lyme disease (P06.228). Neurology. (2013) 80(7 Supplement):P06.228. doi: 10.1212/WNL.80.7_supplement.P06.228
36. Duray PH. Histopathology of clinical phases of human Lyme disease. Rheum Dis Clin North Am. (1989) 15:691–710. doi: 10.1016/S0889-857X(21)01023-1
37. Duray PH. The surgical pathology of human Lyme disease: an enlarging picture. Am J Surg Pathol. (1987) 11:47–60. doi: 10.1097/00000478-198700111-00005
38. Duray PH, Steere AC. Clinical pathologic correlations of Lyme disease by stage. Ann NY Acad Sci. (1988) 539(1 Lyme Disease):65–79. doi: 10.1111/j.1749-6632.1988.tb31839.x
39. Roberts ED, Bohm RP Jr, Cogswell FB, Lanners HN, Lowrie RC Jr, Povinelli L, et al. Chronic Lyme disease in the rhesus monkey. Lab Invest. (1995) 72:146–60.
40. Ramesh G, Borda JT, Gill A, Ribka EP, Morici LA, Mottram P, et al. Possible role of glial cells in the onset and progression of Lyme neuroborreliosis. J Neuroinflammation. (2009) 6:1–16. doi: 10.1186/1742-2094-6-23
41. Ramesh G, Santana-Gould L, Inglis FM, England JD, Philipp MT. The Lyme disease spirochete Borrelia burgdorferi induces inflammation and apoptosis in cells from dorsal root ganglia. J Neuroinflammation. (2013) 10:1–14. doi: 10.1186/1742-2094-10-88
42. Aucott JN, Yang T, Yoon I, Powell D, Geller SA, Rebman AW. Risk of post-treatment Lyme disease in patients with ideally-treated early Lyme disease: a prospective cohort study. Int J Infect Dis. (2022) 116:230–7. doi: 10.1016/j.ijid.2022.01.033
43. Rebman AW, Bechtold KT, Yang T, Mihm EA, Soloski MJ, Novak CB, et al. The clinical, symptom, and quality-of-life characterization of a well-defined group of patients with posttreatment Lyme disease syndrome. Front Med. (2017) 4:224. doi: 10.3389/fmed.2017.00224
44. Puri BK, Shah M, Julu PO, Kingston MC, Monro JA. Urinary bladder detrusor dysfunction symptoms in Lyme disease. Int Neurourol J. (2013) 17:127. doi: 10.5213/inj.2013.17.3.127
45. Kanjwal K, Karabin B, Kanjwal Y, Grubb BP. Postural orthostatic tachycardia syndrome following Lyme disease. Cardiol J. (2011) 18:63–6. doi: 10.1097/MJT.0b013e3181da0763
46. Noyes AM, Kluger J. A tale of two syndromes: Lyme disease preceding postural orthostatic tachycardia syndrome. Ann Noninvasive Electrocardiol. (2015) 20:82–6. doi: 10.1111/anec.12158
47. Novak P, Felsenstein D, Mao C, Octavien NR, Zubcevik N. Association of small fiber neuropathy and post treatment Lyme disease syndrome. PLoS ONE. (2019) 14:e0212222. doi: 10.1371/journal.pone.0212222
48. Kheirkhah A, Darabad RR, Cruzat A, Hajrasouliha AR, Witkin D, Wong N, et al. Corneal epithelial immune dendritic cell alterations in subtypes of dry eye disease: a pilot in vivo confocal microscopic study. Invest Ophthalmol Vis Sci. (2015) 56:7179–85. doi: 10.1167/iovs.15-17433
49. Horowitz RI, Freeman PR. Precision medicine: retrospective chart review and data analysis of 200 patients on dapsone combination therapy for chronic Lyme disease/post-treatment Lyme disease syndrome: part 1. Int J Gen Med. (2019) 12:101–19. doi: 10.2147/IJGM.S193608
50. Mietze A, Strube C, Beyerbach M, Schnieder T, Goethe R. Occurrence of Bartonella henselae and Borrelia burgdorferi sensu lato co-infections in ticks collected from humans in Germany. Clin Microbiol Infect. (2011) 17:918–20. doi: 10.1111/j.1469-0691.2010.03363.x
51. Mayne PJ. Clinical determinants of Lyme borreliosis, babesiosis, bartonellosis, anaplasmosis, and ehrlichiosis in an Australian cohort. Int J Gen Med. (2014) 8:15–26. doi: 10.2147/IJGM.S75825
52. Logigian EL, Steere AC. Clinical and electrophysiologic findings in chronic neuropathy of Lyme disease. Neurology. (1992) 42:303–303. doi: 10.1212/WNL.42.2.303
53. Myers TA, Kaushal D, Philipp MT. Microglia are mediators of Borrelia burgdorferi–induced apoptosis in SH-SY5Y neuronal cells. PLoS Pathog. (2009) 5:e1000659. doi: 10.1371/journal.ppat.1000659
54. Ramesh G, Alvarez AL, Roberts ED, Dennis VA, Lasater BL, Alvarez X, et al. Pathogenesis of Lyme neuroborreliosis: Borrelia burgdorferi lipoproteins induce both proliferation and apoptosis in rhesus monkey astrocytes. Eur J Immunol. (2003) 33:2539–50. doi: 10.1002/eji.200323872
55. Ramesh G, Didier PJ, England JD, Santana-Gould L, Doyle-Meyers LA, Martin DS, et al. Inflammation in the pathogenesis of Lyme neuroborreliosis. Am J Pathol. (2015) 185:1344–60. doi: 10.1016/j.ajpath.2015.01.024
56. Ramesh G, Borda JT, Dufour J, Kaushal D, Ramamoorthy R, Lackner AA, et al. Interaction of the Lyme disease spirochete Borrelia burgdorferi with brain parenchyma elicits inflammatory mediators from glial cells as well as glial and neuronal apoptosis. Am J Pathol. (2008) 173:1415–27. doi: 10.2353/ajpath.2008.080483
57. Ramesh G, Meisner OC, Philipp MT. Anti-inflammatory effects of dexamethasone and meloxicam on Borrelia burgdorferi-induced inflammation in neuronal cultures of dorsal root ganglia and myelinating cells of the peripheral nervous system. J Neuroinflammation. (2015) 12:1–12. doi: 10.1186/s12974-015-0461-y
58. Embers ME, Hasenkampf NR, Jacobs MB, Tardo AC, Doyle-Meyers LA, Philipp MT, et al. Variable manifestations, diverse seroreactivity and post-treatment persistence in non-human primates exposed to Borrelia burgdorferi by tick feeding. PLoS ONE. (2017) 12:e0189071. doi: 10.1371/journal.pone.0189071
59. Embers ME, Barthold SW, Borda JT, Bowers L, Doyle L, Hodzic E, et al. Persistence of Borrelia burgdorferi in rhesus macaques following antibiotic treatment of disseminated infection. PLoS ONE. (2012) 7:e29914. doi: 10.1371/journal.pone.0029914
60. Rudenko N, Golovchenko M, Vancova M, Clark K, Grubhoffer L, Oliver JH. Isolation of live Borrelia burgdorferi sensu lato spirochaetes from patients with undefined disorders and symptoms not typical for Lyme borreliosis. Clin Microbiol Infect. (2016) 22:267.e9–15. doi: 10.1016/j.cmi.2015.11.009
61. Hudson BJ, Stewart M, Lennox VA, Fukunaga M, Yabuki M, Macorison H, et al. Culture-positive Lyme borreliosis. Med J Aust. (1998) 168:500–2. doi: 10.5694/j.1326-5377.1998.tb141415.x
62. Oksi J, Marjamaki M, Nikoskelainen J, Viljanen MK. Bowelia burgorferi detected by culture and PCR in clinical relapse of disseminated Lyme borreliosis. Ann Med. (1999) 31:225–32. doi: 10.3109/07853899909115982
63. Hadžavdić SL, Bartolić L, Bradamante M. Granulomatous hepatitis associated with chronic Borrelia burgdorferi infection: a case report. Research. (2014) 1:875. doi: 10.13070/rs.en.1.875
64. Klempner MS, Hu LT, Evans J, Schmid CH, Johnson GM, Trevino RP, et al. Two controlled trials of antibiotic treatment in patients with persistent symptoms and a history of Lyme disease. N Engl J Med. (2001) 345:85–92. doi: 10.1056/NEJM200107123450202
65. Trouillas P, Franck M. Complete remission in paralytic late tick-borne neurological disease comprising mixed involvement of Borrelia, Babesia, Anaplasma, and Bartonella: use of long-term treatments with antibiotics and antiparasitics in a series of 10 cases. Antibiotics. (2023) 12:1021. doi: 10.3390/antibiotics12061021
66. Alruwaili Y, Jacobs MB, Hasenkampf NR, Tardo AC, McDaniel CE, Embers ME. Superior efficacy of combination antibiotic therapy versus monotherapy in a mouse model of Lyme disease. Front Microbiol. (2023) 14:1293300. doi: 10.3389/fmicb.2023.1293300
67. Coughlin JM, Yang T, Rebman AW, Bechtold KT, Du Y, Mathews WB, et al. Imaging glial activation in patients with post-treatment Lyme disease symptoms: a pilot study using [11C]DPA-713 PET. J Neuroinflammation. (2018) 15:1–7. doi: 10.1186/s12974-018-1381-4
68. Nakatomi Y, Mizuno K, Ishii A, Wada Y, Tanaka M, Tazawa S, et al. Neuroinflammation in patients with chronic fatigue syndrome/myalgic encephalomyelitis: an 11C-(R)-PK11195 PET study. J Nucl Med. (2014) 55:945–50. doi: 10.2967/jnumed.113.131045
69. Casselli T, Divan A, Vomhof-DeKrey EE, Tourand Y, Pecoraro HL, Brissette CA. A murine model of Lyme disease demonstrates that Borrelia burgdorferi colonizes the dura mater and induces inflammation in the central nervous system. PLoS Pathog. (2021) 17:e1009256. doi: 10.1371/journal.ppat.1009256
70. Parthasarathy G, Fevrier HB, Philipp MT. Non-viable Borrelia burgdorferi induce inflammatory mediators and apoptosis in human oligodendrocytes. Neurosci Lett. (2013) 556:200–3. doi: 10.1016/j.neulet.2013.10.032
71. Gnoni V, Ilic K, Drakatos P, Petrinovic MM, Cash D, Steier J, et al. Obstructive sleep apnea and multiple facets of a neuroinflammatory response: a narrative review. J Thorac Dis. (2022) 14:564. doi: 10.21037/jtd-21-1231
72. Yang Q, Wang Y, Feng J, Cao J, Chen B. Intermittent hypoxia from obstructive sleep apnea may cause neuronal impairment and dysfunction in central nervous system: the potential roles played by microglia. Neuropsychiatr Dis Treat. (2013) 9:1077–86. doi: 10.2147/NDT.S49868
73. Novak P. Post COVID-19 syndrome associated with orthostatic cerebral hypoperfusion syndrome, small fiber neuropathy and benefit of immunotherapy: a case report. eNeurologicalSci. (2020) 21:100276. doi: 10.1016/j.ensci.2020.100276
74. Novak P, Mukerji SS, Alabsi HS, Systrom D, Marciano SP, Felsenstein D, et al. Multisystem involvement in post-acute sequelae of coronavirus disease 19. Ann Neurol. (2022) 91:367–79. doi: 10.1002/ana.26286
75. Campen CLMC, Rowe PC, Visser FC. Orthostatic symptoms and reductions in cerebral blood flow in long-haul COVID-19 patients: similarities with myalgic encephalomyelitis/chronic fatigue syndrome. Medicina. (2022) 58:28. doi: 10.3390/medicina58010028
76. Matsuda M, Huh Y, Ji RR. Roles of inflammation, neurogenic inflammation, and neuroinflammation in pain. J Anesth. (2019) 33:131–9. doi: 10.1007/s00540-018-2579-4
77. Kohno R, Cannom DS, Olshansky B, Xi SC, Krishnappa D, Adkisson WO, et al. Mast cell activation disorder and postural orthostatic tachycardia syndrome: a clinical association. J Am Heart Assoc. (2021) 10:e021002. doi: 10.1161/JAHA.121.021002
78. Afrin LB, Ackerley MB, Bluestein LS, Brewer JH, Brook JB, Buchanan AD, et al. Diagnosis of mast cell activation syndrome: a global “consensus-2”. Diagnosis. (2021) 8:137–52. doi: 10.1515/dx-2020-0005
79. Blennerhassett MG, Tomioka M, Bienenstock J. Formation of contacts between mast cells and sympathetic neurons in vitro. Cell Tissue Res. (1991) 265:121–8. doi: 10.1007/BF00318146
80. Theoharides TC. Mast cells: the immune gate to the brain. Life Sci. (1990) 46:607–17. doi: 10.1016/0024-3205(90)90129-F
81. Dong H, Zhang X, Qian Y. Mast cells and neuroinflammation. Med Sci Monit Basic Res. (2014) 20:200. doi: 10.12659/MSMBR.893093
82. Skaper SD, Giusti P, Facci L. Microglia and mast cells: two tracks on the road to neuroinflammation. FASEB J. (2012) 26:3103–17. doi: 10.1096/fj.11-197194
83. Chandra A, Wormser GP, Klempner MS, Trevino RP, Crow MK, Latov N, et al. Anti-neural antibody reactivity in patients with a history of Lyme borreliosis and persistent symptoms. Brain Behav Immun. (2010) 24:1018–24. doi: 10.1016/j.bbi.2010.03.002
84. Maccallini P, Bonin S, Trevisan G. Autoimmunity against a glycolytic enzyme as a possible cause for persistent symptoms in Lyme disease. Med Hypotheses. (2018) 110:1–8. doi: 10.1016/j.mehy.2017.10.024
85. Fallon BA, Strobino B, Reim S, Stoner J, Cunningham MW. Anti-lysoganglioside and other anti-neuronal autoantibodies in post-treatment Lyme disease and erythema migrans after repeat infection. Brain Behav Immunity Health. (2020) 2:100015. doi: 10.1016/j.bbih.2019.100015
86. Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. (2000) 343:847–55. doi: 10.1056/NEJM200009213431204
87. Loebel M, Grabowski P, Heidecke H, Bauer S, Hanitsch LG, Wittke K, et al. Antibodies to β adrenergic and muscarinic cholinergic receptors in patients with chronic fatigue syndrome. Brain Behav Immun. (2016) 52:32–9. doi: 10.1016/j.bbi.2015.09.013
88. Freitag H, Szklarski M, Lorenz S, Sotzny F, Bauer S, Philippe A, et al. Autoantibodies to vasoregulative g-protein-coupled receptors correlate with symptom severity, autonomic dysfunction and disability in myalgic encephalomyelitis/chronic fatigue syndrome. J Clin Med. (2021) 10:3675. doi: 10.3390/jcm10163675
89. Wallukat G, Hohberger B, Wenzel K, Fürst J, Schulze-Rothe S, Wallukat A, et al. Functional autoantibodies against G-protein coupled receptors in patients with persistent Long-COVID-19 symptoms. J Transl Autoimmun. (2021) 4:100100. doi: 10.1016/j.jtauto.2021.100100
90. Rodriguez B, Hoepner R, Salmen A, Kamber N. Z'Graggen WJ. Immunomodulatory treatment in postural tachycardia syndrome: a case series. Eur J Neurol. (2021) 28:1692–7. doi: 10.1111/ene.14711
91. Kesterson K, Schofield J, Blitshteyn S. Immunotherapy with subcutaneous immunoglobulin or plasmapheresis in patients with postural orthostatic tachycardia syndrome (POTS). J Neurol. (2023) 270:233–9. doi: 10.1007/s00415-022-11344-z
92. Theoharides TC, Stewart JM. Post-Lyme syndrome–associated polyneuropathy treated with immune immunoglobulin and a luteolin-containing formulation. J Clin Psychopharmacol. (2016) 36:290. doi: 10.1097/JCP.0000000000000504
93. Charfeddine S, Ibn Hadj Amor H, Jdidi J, Torjmen S, Kraiem S, Hammami R, et al. Long COVID 19 syndrome: is it related to microcirculation and endothelial dysfunction? Insights From TUN-EndCOV Study. Front Cardiovasc Med. (2021) 8:745758. doi: 10.3389/fcvm.2021.745758
94. Bonaventura A, Vecchié A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. (2021) 21:319–29. doi: 10.1038/s41577-021-00536-9
95. Pretorius E, Venter C, Laubscher GJ, Kotze MJ, Oladejo SO, Watson LR, et al. Prevalence of symptoms, comorbidities, fibrin amyloid microclots and platelet pathology in individuals with long COVID/post-acute sequelae of COVID-19 (PASC). Cardiovasc Diabetol. (2022) 21:1–17. doi: 10.1186/s12933-022-01579-5
96. Sellati TJ, Burns MJ, Ficazzola MA, Furie MB. Borrelia burgdorferi upregulates expression of adhesion molecules on endothelial cells and promotes transendothelial migration of neutrophils in vitro. Infect Immun. (1995) 63:4439–47. doi: 10.1128/iai.63.11.4439-4447.1995
97. Sellati TJ, Abrescia LD, Radolf JD, Furie MB. Outer surface lipoproteins of Borrelia burgdorferi activate vascular endothelium in vitro. Infect Immun. (1996) 64:3180–7. doi: 10.1128/iai.64.8.3180-3187.1996
98. Goehler LE, Relton JK, Dripps D, Kiechle R, Tartaglia N, Maier SF, et al. Vagal paraganglia bind biotinylated interleukin-1 receptor antagonist: a possible mechanism for immune-to-brain communication. Brain Res Bull. (1997) 43:357–64. doi: 10.1016/S0361-9230(97)00020-8
99. Goehler LE, Gaykema RPA, Opitz N, Reddaway R, Badr N, Lyte M. Activation in vagal afferents and central autonomic pathways: early responses to intestinal infection with Campylobacter jejuni. Brain Behav Immun. (2005) 19:334–44. doi: 10.1016/j.bbi.2004.09.002
100. Gallaher ZR Ryu V, Herzog T, Ritter RC, Czaja K. Changes in microglial activation within the hindbrain, nodose ganglia, and the spinal cord following subdiaphragmatic vagotomy. Neurosci Lett. (2012) 513:31–6. doi: 10.1016/j.neulet.2012.01.079
101. Aucott JN, Soloski MJ, Rebman AW, Crowder LA, Lahey LJ, Wagner CA, et al. CCL19 as a chemokine risk factor for posttreatment Lyme disease syndrome: a prospective clinical cohort study. Clin Vaccine Immunol. (2016) 23:757–66. doi: 10.1128/CVI.00071-16
102. Jacek E, Fallon BA, Chandra A, Crow MK, Wormser GP, Alaedini A. Increased IFNα activity and differential antibody response in patients with a history of Lyme disease and persistent cognitive deficits. J Neuroimmunol. (2013) 255:85–91. doi: 10.1016/j.jneuroim.2012.10.011
103. Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. (2007) 117:289–96. doi: 10.1172/JCI30555
104. Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex—linking immunity and metabolism. Nat Rev Endocrinol. (2012) 8:743–54. doi: 10.1038/nrendo.2012.189
105. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. (2000) 405:458–62. doi: 10.1038/35013070
106. Koopman FA, Chavan SS, Miljko S, Grazio S, Sokolovic S, Schuurman PR, et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc Nat Acad Sci. (2016) 113:8284–9. doi: 10.1073/pnas.1605635113
107. Huffman WJ, Subramaniyan S, Rodriguiz RM, Wetsel WC, Grill WM, Terrando N. Modulation of neuroinflammation and memory dysfunction using percutaneous vagus nerve stimulation in mice. Brain Stimul. (2019) 12:19–29. doi: 10.1016/j.brs.2018.10.005
108. Badran BW, Huffman SM, Dancy M, Austelle CW, Bikson M, Kautz SA, et al. A pilot randomized controlled trial of supervised, at-home, self-administered transcutaneous auricular vagus nerve stimulation (taVNS) to manage long COVID symptoms. Bioelectron Med. (2022) 8:1–10. doi: 10.1186/s42234-022-00094-y
109. Natelson BH, Blate M, Soto T. Transcutaneous vagus nerve stimulation in the treatment of long COVID-chronic fatigue syndrome. medRxiv [Preprint]. (2022) 2022–11. doi: 10.1101/2022.11.08.22281807
110. Rodriguez L, Pou C, Lakshmikanth T, Zhang J, Mugabo CH, Wang J, et al. Achieving symptom relief in patients with Myalgic encephalomyelitis by targeting the neuro-immune interface and inducing disease tolerance. bioRxiv [Preprint]. (2020) 2020–02. doi: 10.1101/2020.02.20.958249
111. Morrissette M, Pitt N, González A, Strandwitz P, Caboni M, Rebman AW, et al. A distinct microbiome signature in posttreatment Lyme disease patients. mBio. (2020) 11:e02310–20. doi: 10.1128/mBio.02310-20
112. O'Riordan KJ, Collins MK, Moloney GM, Knox EG, Aburto MR, Fülling C, et al. Short chain fatty acids: microbial metabolites for gut-brain axis signalling. Mol Cell Endocrinol. (2022) 546:111572. doi: 10.1016/j.mce.2022.111572
113. Haase S, Haghikia A, Wilck N, Müller DN, Linker RA. Impacts of microbiome metabolites on immune regulation and autoimmunity. Immunology. (2018) 154:230–8. doi: 10.1111/imm.12933
114. Do T, Diamond S, Green C, Warren M. Nutritional implications of patients with dysautonomia and hypermobility syndromes. Curr Nutr Rep. (2021) 10:324–33. doi: 10.1007/s13668-021-00373-1
115. Cheney AM, Costello SM, Pinkham NV, Waldum A, Broadaway SC, Cotrina-Vidal M, et al. Gut microbiome dysbiosis drives metabolic dysfunction in Familial dysautonomia. Nat Commun. (2023) 14:1–12. doi: 10.1038/s41467-023-35787-8
116. Robertson D, Flattem N, Tellioglu T, Carson R, Garland E, Shannon JR, et al. Familial orthostatic tachycardia due to norepinephrine transporter deficiency. Ann N Y Acad Sci. (2001) 940:527–44. doi: 10.1111/j.1749-6632.2001.tb03703.x
117. Hoeijmakers JG, Han C, Merkies IS, Macala LJ, Lauria G, Gerrits MM, et al. Small nerve fibres, small hands and small feet: a new syndrome of pain, dysautonomia and acromesomelia in a kindred with a novel NaV17 mutation. Brain. (2012) 135:345–58. doi: 10.1093/brain/awr349
118. Bartholomew F, Lazar J, Marqueling A, Lee-Messer C, Jaradeh S, Teng JMC. Channelopathy: a novel mutation in the SCN9A gene causes insensitivity to pain and autonomic dysregulation. Br J Dermatol. (2014) 171:1268–70. doi: 10.1111/bjd.13096
119. Garland EM, Winker R, Williams SM, Jiang L, Stanton K, Byrne DW, et al. Endothelial NO synthase polymorphisms and postural tachycardia syndrome. Hypertension. (2005) 46:1103–10. doi: 10.1161/01.HYP.0000185462.08685.da
120. Jacob G, Garland EM, Costa F, Stein CM, Xie HG, Robertson RM, et al. β2-Adrenoceptor genotype and function affect hemodynamic profile heterogeneity in postural tachycardia syndrome. Hypertension. (2006) 47:421–7. doi: 10.1161/01.HYP.0000205120.46149.34
121. Parsaik A, Allison TG, Singer W, Sletten DM, Joyner MJ, Benarroch EE, et al. Deconditioning in patients with orthostatic intolerance. Neurology. (2012) 79:1435–9. doi: 10.1212/WNL.0b013e31826d5f95
122. van Campen CLMC, Rowe PC, Visser FC. Deconditioning does not explain orthostatic intolerance in ME/CFS (myalgic encephalomyelitis/chronic fatigue syndrome). J Transl Med. (2021) 19:193. doi: 10.1186/s12967-021-02819-0
123. Petracek LS, Eastin EF, Rowe IR, Rowe PC. Orthostatic intolerance as a potential contributor to prolonged fatigue and inconsistent performance in elite swimmers. BMC Sports Sci Med Rehabil. (2022) 14:1–11. doi: 10.1186/s13102-022-00529-8
124. Sletten DM, Suarez GA, Low PA, Mandrekar J, Singer W. COMPASS 31: a refined and abbreviated composite autonomic symptom score. Mayo Clin Proc. (2012) 87:1196. doi: 10.1016/j.mayocp.2012.10.013
125. Zilliox L, Peltier AC, Wren PA, Anderson A, Smith AG, Singleton JR, et al. Assessing autonomic dysfunction in early diabetic neuropathy: the survey of autonomic symptoms. Neurology. (2011) 76:1099–105. doi: 10.1212/WNL.0b013e3182120147
126. Spahic JM, Hamrefors V, Johansson M, Ricci F, Melander O, Sutton R, et al. Malmö POTS symptom score: assessing symptom burden in postural orthostatic tachycardia syndrome. J Intern Med. (2023) 293:91–9. doi: 10.1111/joim.13566
127. De Wandele I, Low D, Rowe P, Simmonds JV. Exercise guidelines for postural tachycardia syndrome. In: Postural Tachycardia Syndrome: a Concise and Practical Guide to Management and Associated Conditions (2021). p. 207–15.
128. Taub PR, Zadourian A, Lo HC, Ormiston CK, Golshan S, Hsu JC, et al. Randomized trial of ivabradine in patients with hyperadrenergic postural orthostatic tachycardia syndrome. J Am Coll Cardiol. (2021) 77:861–71. doi: 10.1016/j.jacc.2020.12.029
129. Longo WE, Vernava AM. Prokinetic agents for lower gastrointestinal motility disorders. Dis Colon Rectum. (1993) 36:696–708. doi: 10.1007/BF02238599
130. Thompson JS, Thornton AC, Ainger T, Garvy BA. Long-term high-dose immunoglobulin successfully treats long COVID patients with pulmonary, neurologic, and cardiologic symptoms. Front Immunol. (2023) 13:1033651. doi: 10.3389/fimmu.2022.1033651
131. McCarthy MW. Paxlovid as a potential treatment for long COVID. Expert Opin Pharmacother. (2023) 24:1839–43. doi: 10.1080/14656566.2023.2262387
132. Patterson BK, Yogendra R, Guevara-Coto J, Mora-Rodriguez RA, Osgood E, Bream J, et al. Case series: maraviroc and pravastatin as a therapeutic option to treat long COVID/Post-acute sequelae of COVID (PASC). Front Med. (2023) 10:1122529 doi: 10.3389/fmed.2023.1122529
Keywords: dysautonomia, post-treatment Lyme disease (PTLD), postural orthostatic tachycardia syndrome (POTS), Borrelia (Borreliella) burgdorferi, Lyme disease
Citation: Adler BL, Chung T, Rowe PC and Aucott J (2024) Dysautonomia following Lyme disease: a key component of post-treatment Lyme disease syndrome? Front. Neurol. 15:1344862. doi: 10.3389/fneur.2024.1344862
Received: 26 November 2023; Accepted: 22 January 2024;
Published: 08 February 2024.
Edited by:
Ilene Sue Ruhoy, Mount Sinai South Nassau, United StatesReviewed by:
Etheresia Pretorius, Stellenbosch University, South AfricaBruce Patterson, IncellDx Inc, United States
Timothy Flanigan, Brown University, United States
Nevena Zubcevik, Independent Researcher, Palo Alto, CA, United States
Tania Dempsey, Aim Center for Personalized Medicine, United States
Copyright © 2024 Adler, Chung, Rowe and Aucott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brittany L. Adler, YnJpdC5hZGxlckBqaG1pLmVkdQ==