
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 31 January 2024
Sec. Pediatric Neurology
Volume 15 - 2024 | https://doi.org/10.3389/fneur.2024.1340458
This article is part of the Research TopicConsanguinity and Rare Genetic Neurological DiseasesView all 9 articles
 Qi Yang1,2†
Qi Yang1,2† Qiang Zhang1,2†
Qiang Zhang1,2† Sheng Yi1,2
Sheng Yi1,2 Shujie Zhang1,2
Shujie Zhang1,2 Shang Yi1,2Xunzhao Zhou1,2Zailong Qin1,2Biyan Chen1,2Jingsi Luo1,2,3*
Shang Yi1,2Xunzhao Zhou1,2Zailong Qin1,2Biyan Chen1,2Jingsi Luo1,2,3*Kleefstra syndrome (KLEFS) refers to a rare inherited neurodevelopmental disorder characterized by intellectual disability (ID), language and motor delays, behavioral abnormalities, abnormal facial appearance, and other variable clinical features. KLEFS is subdivided into two subtypes: Kleefstra syndrome-1 (KLEFS1, OMIM: 610253), caused by a heterozygous microdeletion encompassing the Euchromatic Histone Lysine Methyltransferase 1 (EHMT1) gene on chromosome 9q34.3 or pathogenic variants in the EHMT1 gene, and Kleefstra syndrome-2 (KLEFS2, OMIM: 617768), caused by pathogenic variants in the KMT2C gene. More than 100 cases of KLEFS1 have been reported with pathogenic variants in the EHMT1 gene. However, only 13 patients with KLEFS2 have been reported to date. In the present study, five unrelated Chinese patients were diagnosed with KLEFS2 caused by KMT2C variants through whole-exome sequencing (WES). We identified five different variants of the KMT2C gene in these patients: c.9166C>T (p.Gln3056*), c.9232_9247delCAGCGATCAGAACCGT (p.Gln3078fs*13), c.5068dupA (p.Arg1690fs*10), c.10815_10819delAAGAA (p.Lys3605fs*7), and c.6911_6912insA (p.Met2304fs*8). All five patients had a clinical profile similar to that of patients with KLEFS2. To analyze the correlation between the genotype and phenotype of KLEFS2, we examined 18 variants and their associated phenotypes in 18 patients with KLEFS2. Patients carrying KMT2C variants presented with a wide range of phenotypic defects and an extremely variable phenotype. We concluded that the core phenotypes associated with KMT2C variants were intellectual disability, facial dysmorphisms, language and motor delays, behavioral abnormalities, hypotonia, short stature, and weight loss. Additionally, sex may be one factor influencing the outcome. Our findings expand the phenotypic and genetic spectrum of KLEFS2 and help to clarify the genotype–phenotype correlation.
Kleefstra syndrome (KLEFS) is a rare inherited neurodevelopmental disorder (NDD) characterized by neuropsychiatric anomalies, developmental delay (DD), mild-to-severe intellectual disability (ID), childhood hypotonia, speech delay, and craniofacial abnormalities (1, 2). Globally, the prevalence stands between 1:25,000 and 1:35,000 individuals. KLEFS is primarily caused by a heterozygous microdeletion encompassing the Euchromatin Histone Methyltransferase 1 (EHMT1) gene on chromosome 9q34.3 or a pathogenic variant in the EHMT1 gene (3, 4). Additionally, pathogenic variants in KMT2C have been found in patients with a clinical diagnosis of Kleefstra syndrome-2. KMT2C (KMT2C; MIM 606833, NM_170606) is located on chromosome 7q36 and consists of 59 exons, encoding lysine-N-methyltransferase 2C, which catalyzes histone 3 lysine 4 (H3K4) methylation and epigenetically regulates gene transcription through chromatin modifications (5). Dysregulated H3K4 methylation can cause ID, epilepsy, autism spectrum disorder (ASD), schizophrenia, and other neurodevelopmental and neuropsychiatric disorders (6, 7). Recently, novel or de novo pathogenic variants in the KMT2C gene have been reported to cause a variety of developmental abnormalities, including ID, ASD, schizophrenia, and non-syndromic primary tooth eruption failure (8–13). To date, only 13 cases of KLEFS2 have been reported in the literature (8, 14–18). Currently, there is no clear association between the clinical phenotype and variant sites. Thus, additional reports would help us better understand the phenotype spectrum of this disease and explore the relationship between genotypes and phenotypes.
In this study, we present five additional patients diagnosed with Kleefstra syndrome-2 by whole-exome sequencing with a highly heterogeneous phenotype, including mild-to-moderate ID, facial dysmorphism, language and motor delays, short stature, weight loss, microcephaly, and behavioral abnormalities. Molecular analyses identified five novel KMT2C variants. Additionally, we have reviewed the literature to summarize the genotypes, phenotypes, and clinical features of KLEFS2.
For this study, five unrelated patients with confirmed KMT2C variants, who were diagnosed with neuropsychiatric anomalies, intellectual disability, global developmental delay, and craniofacial abnormalities, were recruited from the Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region (Table 1).
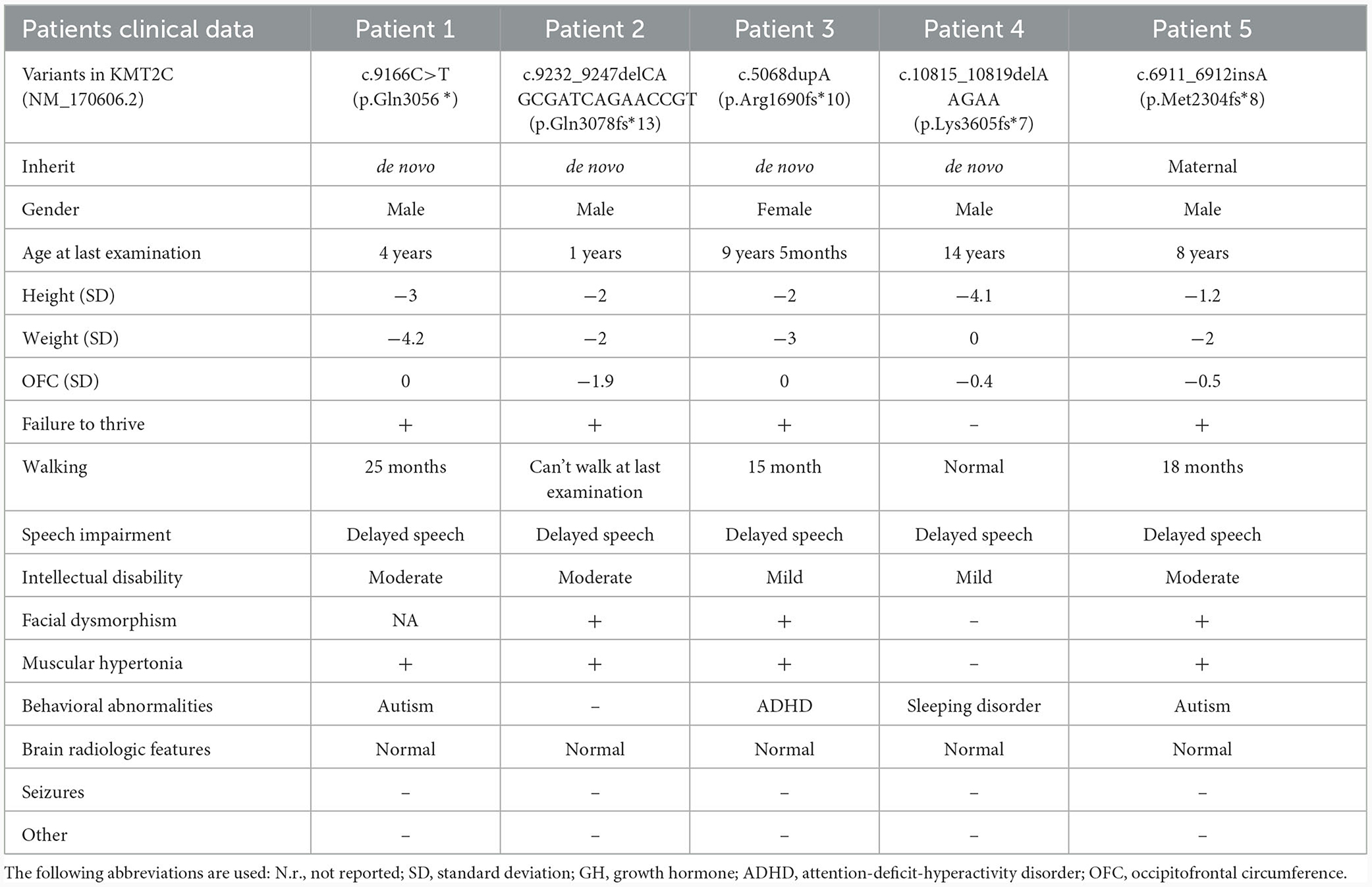
Table 1. Clinical features of the patients with KMT2C mutations.
A total of five individuals (four male patients and one female patient) with NDD from unrelated families underwent WES (Figure 1). Genomic DNA was extracted from 5 ml of peripheral blood of the patients and their family members using a Lab-Aid DNA kit (Zeesan Biotech Co., Ltd., Xiamen, China). Whole-exome capture was performed using an Agilent SureSelect V5 enrichment capture kit (Agilent Technologies, Santa Clara, CA, USA), followed by sequencing on the Hiseq2000 platform to generate 100-bp paired-end reads. The sequence reads were aligned to the human genome assembly GRCh37 using the Burrows–Wheeler aligner (BWA-MEM, version 0.7.10). TGEX was utilized to annotate and classify all the variants. The variants were filtered based on specific criteria, including (a) the exclusion of non-rare (MAF ≥ 0.01) variants in the public database (e.g., 1,000 Genomes Project, dbSNP, and Genome Aggregation Database), (b) mutations in the exonic region and splice regions, and (c) predicted deleterious effects by MutationTaster, CADD, SIFT, or PolyPhen2. The pathogenicity of identified variants was interpreted and classified according to the American College of Medical Genetics (ACMG)/Association of Molecular Pathology (AMP) guidelines (19).
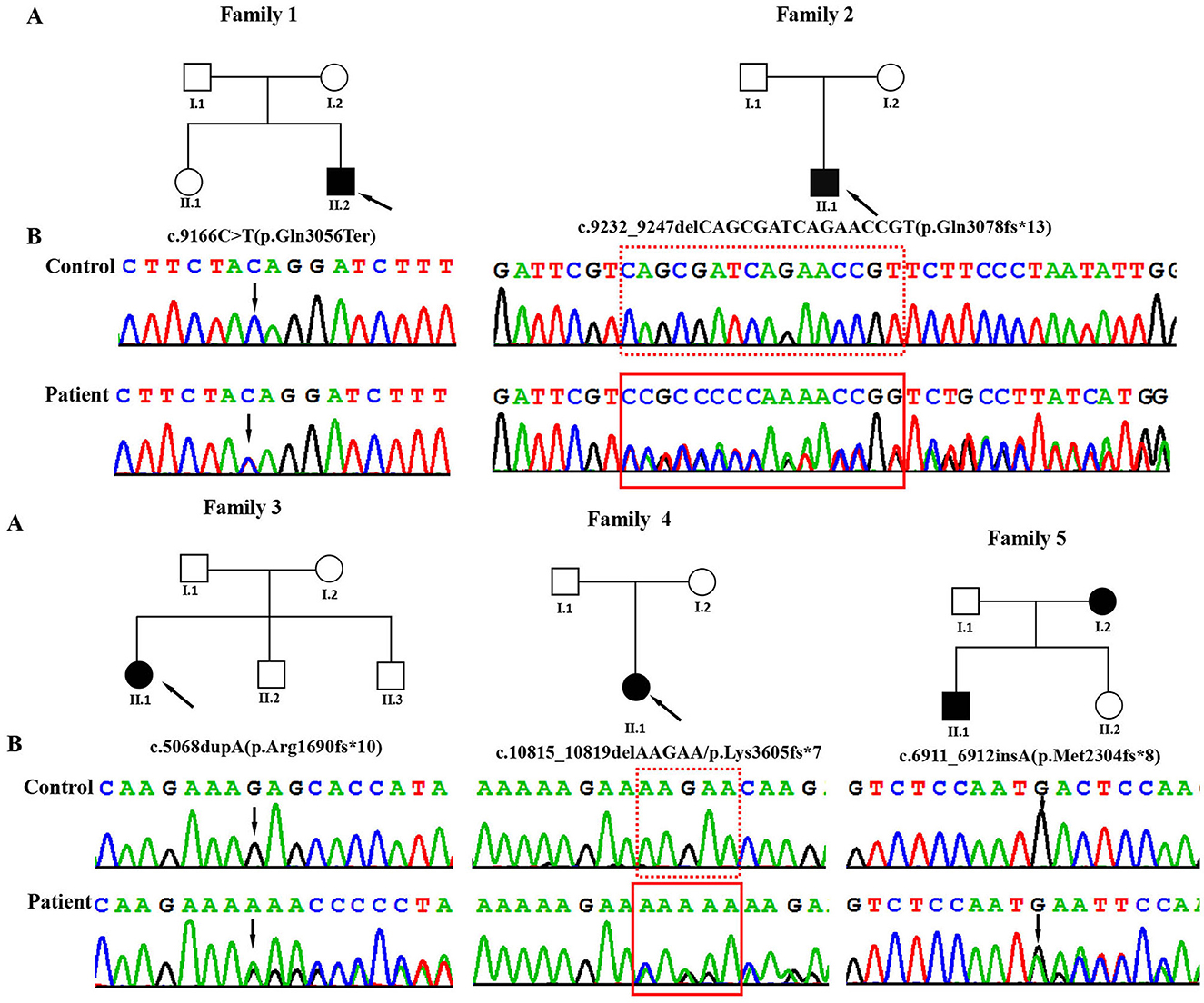
Figure 1. (A) Pedigrees of the affected families. Probands are denoted by arrows. (B) The Sanger chromatograms of the detected variants in probands of families 1–5. Among them, P1 [NM_170606.3:c.9166C>T (p.Gln3056Ter)] had a non-sense variant, and P2 [c.9232_9247delCAGCGATCAGAACCGT (p.Gln3078fs*13)], P3 [c.5068dupA (p.Arg1690fs*10)], P4 [c.10815_10819delAAGAA/p.Lys3605fs*7], and P5 [c.6911_6912insA (p.Met2304fs*8)] had frameshift variants.
Patient 1 was a 3-year and 12-month-old boy, the second child of healthy, unrelated Chinese parents. He was born full-term with a birth weight of 3,250 g and a body length of 48.5 cm. At 3 years and 10 months, he was admitted to the Pediatric Intensive Care Unit of the Guangxi Zhuang Autonomous Region Maternal and Child Health Hospital due to hypoxic acute respiratory failure caused by pneumonia. His developmental milestones were globally delayed, with independent sitting at 9 months, standing with support at 20 months, and walking without support at 25 months. He began speaking single words by the age of 2 years and started forming sentences by the age of 3 years. He had moderate ID and behavioral abnormality, including stereotypies such as body rocking, clapping and hand flapping, spinning, hitting objects, and self-talking. Physical examination revealed that the patient was severely underweight (weight: 9 kg, BMI: 10.9 kg/m2) and had a short stature (height: 91 cm, −3 SD). Unfortunately, due to economic factors and an unfavorable prognosis, the patient discontinued treatment and passed away a month after discharge.
Patient 2 was a 1-year-old male child, born at full term but small for gestational age, with a birth weight of 2,300 g (< 10th centile), a body length of 44 cm, and a head circumference of 32.4 cm (< 10th centile). He presented with severe ID and global developmental delay. At 11 months, his Full Scale IQ was 43, according to the Wechsler Intelligence Scale for Children. He was unable to stand or walk independently and showed no signs of speech development. Additionally, he experienced postnatal growth retardation with short stature (−2 SD), weight loss (−2.1 SD), and microcephaly (−1.9 SD). The child also exhibited midface hypoplasia and strabismus.
Patient 3 was a female child born at 38 weeks of gestation with normal weight, length, and head circumference. At 13 months of age, she was initially assessed at the Child Healthcare Department of the Guangxi Zhuang Autonomous Region Maternal and Child Health Hospital for feeding difficulty, comprehensive developmental delay, and failure to thrive. She exhibited mild developmental delay, began walking at 15 months, and started talking at 1 year and 8 months. She continued to display short stature (−2.1 SD), mild development delay, speech delays, and learning disabilities. At the age of 8 years, she was readmitted for learning difficulty and hyperactivity and was diagnosed with attention-deficit-hyperactivity disorder (ADHD) and weight loss (BMI: 10.8 kg/m2). Treatment with an amino acid-based high-energy formula, high-energy high-protein diet, and aripiprazole (1.25 mg/day) resulted in a 6-kg weight gain after 1 year. However, her ADHD symptoms persisted. At the age of 9 years and 1 month, she began taking 18 mg/day of methylphenidate accompanied by behavioral interventions implemented by her parents, leading to a significant alleviation of her ADHD symptoms. In the most recent examination at the age of 9 years and 5 months, she exhibited mild intellectual disability (Full Scale IQ, 69; Verbal Comprehension Index, 70; Perceptual Reasoning Index, 64; Working Memory Index, 68; and Processing Speed Index, 65). Minor facial dysmorphisms, including prominent eyebrows, a thick lower lip, and a short nose, were observed.
Patient 4 was a 14-year-old boy, the first child of healthy non-consanguineous Chinese parents, who presented with severe short stature (< -4.1 SD). An arginine clonidine GH stimulation test yielded an IGF-1 level of 113 ng/ml (reference range 49–283), an IGF-BP3 level of 2.89 μg/ml (reference range 1.0–4.7), and a peak GH level of 12.65 ng/ml, ruling out the GH deficiency. He demonstrated normal gross and fine motor skills, with a bone age of 12 years. His general performance on the language scale was 80 and below the average range (86%). Expressive language skills were within the normal range, while receptive language skills were impaired with scaled scores of 10 and 5, respectively. The patient also experienced sleep disturbances, including insomnia, difficulty falling asleep, abnormal sleep duration, excessive daytime sleepiness, and disrupted circadian rhythms.
Patient 5, the first child of healthy non-consanguineous Chinese parents, was delivered at 34 weeks plus 2 days of gestation due to premature rupture of membranes. He was 8 years old at the time of the last physical examination. He began walking at 18 months and exhibited hypotonia and talking at 40 months. He had moderate intellectual disability (Wechsler Intelligence Scale for Children-IV, Full Scale IQ = 48). Behaviorally, he exhibited a typical ASD phenotype with speech impairment, social interaction impairment, poor response when called, communication difficulties, behavioral problems, and hyperactivity. He had some dysmorphic features, including a flattened midface, everted lower lip, and esotropia. It is noteworthy that his mother exhibits a mild cognitive impairment, with no other apparent dysmorphic features detected in the family history report.
Using WES, we identified five heterozygous variants in the KMT2C gene in patients 1–5 as follows: c.9166C>T (p.Gln3056Ter) in patient 1, c.9232_9247delCAGCGATCAGAACCGT (p.Gln3078fs*13) in patient 2, c.5068dupA (p.Arg1690fs*10) in patient 3, c.10815_10819delAAGAA(p.Lys3605fs*7) in patient 4, and c.6911_6912insA (p.Met2304fs*8) in patient 5 (Figure 1). These five variants were validated by Sanger sequencing. Additionally, it was shown that the c.6911_6912insA (p.Met2304fs*8) variant was inherited from the mother of patient 5, while the other four variants were not present in the samples of the four parents in the study. Specifically, c.9232_9247delCAGCGATCAGAACCGT (p.Gln3078fs*13), c.5068dupA (p.Arg1690fs*10), c.10815_10819delAAGAA (p.Lys3605fs*7), c.6911_6912insA (p.Met2304fs*8), and c.2519_2520dup (p.Thr841fs*37) were novel variants, which were not reported in gnomAD, Human Gene Mutation database, 1,000 Genomes Project, Exome Sequencing Project, ExAC, ClinVar, and the Single Nucleotide Polymorphism database. The c.9166C>T (p.GLn3056*) non-sense variant is reported in the gnomAD database with a low allele frequency (1 mutated allele out of 833,108 alleles). While this variant has not been previously associated with a disease, multiple computational pieces of evidence predict that it is damaging. These variants were predicted to be deleterious by in silico tools. The pathogenicity prediction analysis and ACMG/AMP rating of the five KMT2C variants are shown in Table 2.
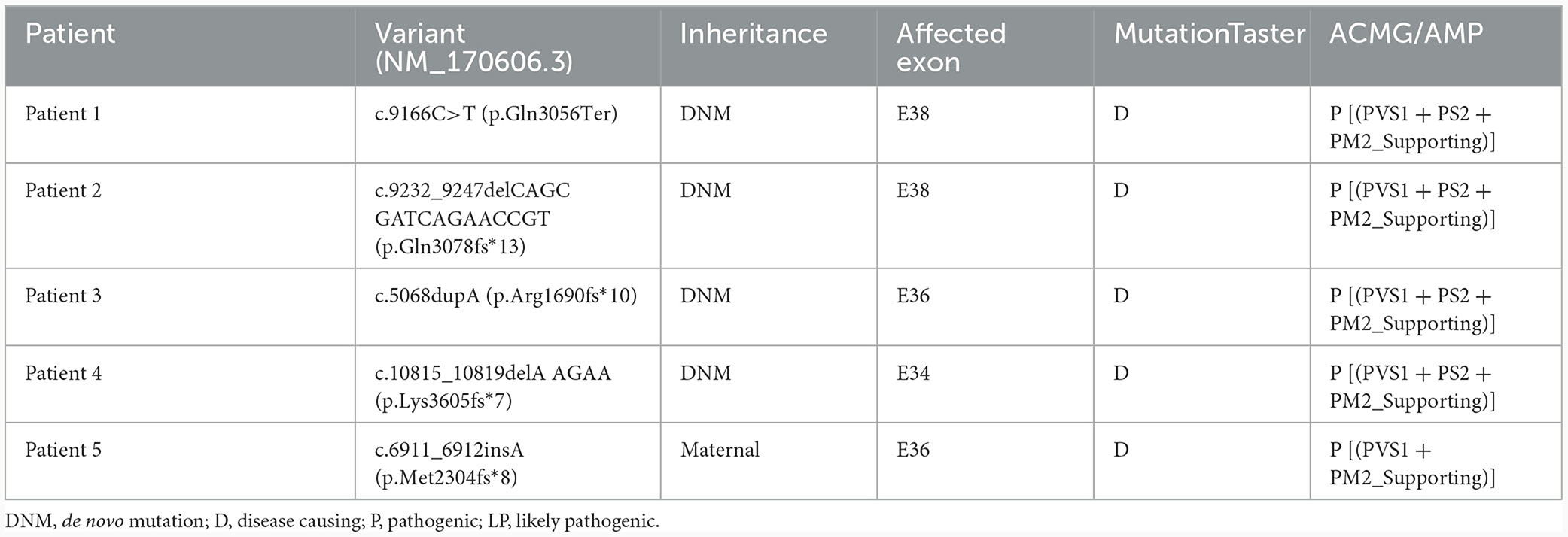
Table 2. Predicted pathogenicity of novel or de novo KMT2C variants.
KMT2C, also known as MLL3, is a histone lysine methyltransferase enzyme that plays an important role in modifying histone proteins (6, 8, 20, 21). It is involved in H3K4me1 and H3K4me3 marks related to active enhancers or transcriptionally active regions, respectively (22, 23). KMT2C is part of the protein complex associated with Set1 (COMPASS complex), which plays a vital role in eukaryotic developmental signaling pathways (24–26). KMT2C knockout mice display ASD-like repetitive behaviors, social deficits, and ID (27). In human, variants in KMT2C have been reported to be associated with ASD-like behaviors and Kleefstra syndrome-2 (18, 21, 25, 28–32). Notably, only 13 patients have been reported with variants in KMT2C. Among these patients, only one long-term report showed a long-term persistent phenotype. Therefore, the phenotypes associated with KMT2C mutations remain incompletely characterized. In this study, we found five KMT2C variants related to neurodevelopmental abnormalities. The patients exhibited common disease phenotypes, including mild-to-moderate ID and DD, failure to thrive, facial dysmorphism, speech delay, hypotonia, feeding difficulties, short stature, weight loss, microcephaly, and behavioral abnormalities.
In the patients described in this study, five variants were identified in KMT2C, including one non-sense variant and four frameshift variants. In silico, these null variants may cause a premature termination codon or a translational frameshift, leading to significantly reduced truncated proteins and markedly reduced mRNA levels due to NMD degradation. These variants were assessed as likely pathogenic or pathogenic according to the ACMG/AMP guidelines (Table 2).
Patient 5 harbors a potentially pathogenic heterozygous variant in the KMT2C gene inherited from his mother. Inconsistent phenotypes were observed in patient 5 and his mother. Patient 5 manifested a more extensive and severe phenotype, including delayed motor development, severe language impairment, moderate intellectual disability, autistic behaviors, ADHD, and dysmorphic facial features, while his mother exhibited only a mild cognitive impairment. Consequently, these findings suggest a high degree of variability in the clinical features among individuals with heterozygous mutations in the KMT2C gene.
In the current study, we describe the genotype and clinical phenotype of five patients with variants in KMT2C, bringing the total number of reported individuals to 18. The list of reported variants of KMT2C and the clinical phenotype of our patients and the other reported patients are summarized in Table 3 and Supplementary Table S1 (8, 14–18). Except for one missense variant and one in-frame deletion, all reported variants were loss-of-function variants (LoF; including non-sense variants, splicing variants, frameshifts, in-frame, and microdeletions). Some common phenotypes were observed in patients with these variants. All 18 patients exhibited a range of intellectual disability from mild to severe; 88% of the patients (15/17) displayed diverse minor facial dysmorphisms, including midface hypoplasia, thick and everted lower lip, abnormal eyebrows (arched or prominent), nose abnormalities (saddle bridge, bulbous tip nose, and short nose), and eye abnormalities (deep set eyes, strabismus, ptosis, down-slanting palpebral fissures, and hypertelorism). Speech delay was observed in all patients (17/17), ranging from delayed speech to remaining non-verbal after the age of 15 years. Nearly all patients (14/15) presented with motor delay or dyskinesia. All patients learned to walk, but some did not learn to walk until after the age of 4 years. Behavioral abnormalities were also observed in most patients (13/16) and became more characteristic with age. These abnormalities included autism, ADHD, sleeping disorder, aggressiveness, hyperactivity, automutilation, and elective mutism. Hypotonia was common (11/16), with one patient exhibiting a broad gait and another showing developmental regression. Short stature (12/17) and weight loss (11/17) were the prevalent features, both falling 2–4 standard deviations below the mean for the general population. Aripiprazole medication effectively improved weight in patients with low body weight; two patients, including ours, showed significant weight gain after aripiprazole medication, particularly in abdominal fat. Relative microcephaly was observed in nine out of 18 patients, while macrocephaly was noted in two patients. Additionally, various other variable dysmorphic features, such as brain abnormalities, seizures, phenylketonuria, recurrent respiratory infections, cryptorchidism, bifid uvula, bilateral inguinal hernia, delayed puberty, hearing impairment, insensitivity to pain, constipation, slight telelia, ligamentous hyperlaxity, and eczema, were also observed.
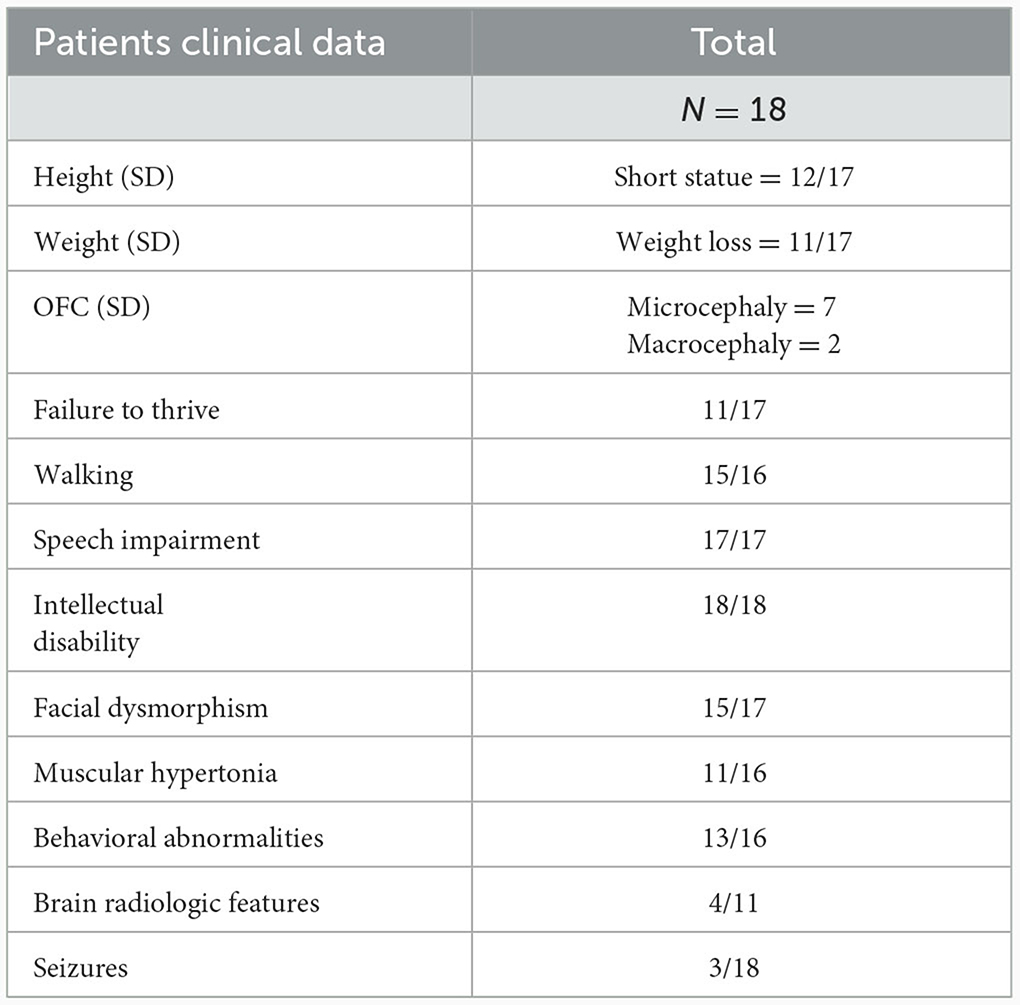
Table 3. Clinical features of patients with KMT2C variants.
Neurodevelopmental disorders caused by chromatin-regulated protein-encoding genes often exhibit overlapping clinical manifestations (33–35). Comparing the clinical manifestations of these different related disorders can enhance our understanding of the broad clinical spectrum of these diseases and the significance of WES in the diagnostic process (36, 37). Although some consistent clinical manifestations of KLEFS2 have been observed, we currently cannot establish a clear correlation between the genotype and the phenotype, which is attributed to the complexity and heterogeneity of the KLEFS2 phenotypes. The diverse phenotype observation indicates that variants occurring at different positions may have site-specific effects on the phenotypes. Moreover, in this cohort of 18 patients, the male-to-female ratio was 1:1. Some severe phenotypes are more likely to be observed in women, including severe ID (4/9), severe speech impairment (3/9), brain abnormalities (3/9), and macrocephaly (2/9), suggesting that sex may influence the outcome. The underrepresentation of patients in this cohort may reflect ascertainment bias, and further confirmation of the observations is expected as the number of patients increases.
The exact mechanisms underlying how these variants cause ID, facial dysmorphisms, language and motor delays, behavioral abnormalities, hypotonia, short stature and weight loss, and additional clinical symptoms remain unclear. The KMT2C gene is highly conserved from archaea to eukaryotes and is widely expressed in human tissues with high levels in the cerebellum of the developing and adult human brain (38, 39). This gene encodes an enzyme that monomethylates lysine 4 of histone H3 (H3K4me1) and epigenetically regulates many gene expressions in the brain through chromatin modification (40). In 2017, Koemans et al. found that KMT2C knockdown drosophila exhibited severe defects in memory formation (8). Moreover, they found that KMT2C binds to the promoters of many genes that play important roles in neuronal processes (6). In vitro experiments show that reduced KMT2C expression leads to a decrease in the overall level of histone tail modifications H3K4me1 and H3K4me3 (6, 41). Furthermore, rare KMT2C variants have been associated with Kleefstra syndrome, ID, ASD, schizophrenia, non-syndromic primary failure of tooth eruption, and a family with colorectal cancer and acute myeloid leukemia (8, 14–18, 42). These findings suggest that the KMT2C variant has pleiotropic effects. Functional studies of these variants are needed to fully understand KLEFS2 and its mechanisms of action.
In conclusion, we present detailed clinical and phenotypic information on five additional unrelated Chinese patients with five novel heterozygous KMT2C variants, expanding the mutational spectrum of KLEFS2. This is the first report of the c.9232_9247delCAGCGATCAGAACCGT (p.Gln3078fs*13), c.5068dupA (p.Arg1690fs*10), c.10815_10819delAAGAA (p.Lys3605fs*7), and c.6911_6912insA (p.Met2304fs*8) variants in the KMT2C gene. Our study also indicates that there is currently no significant correlation between the genotype and phenotype, providing a foundation for clinical diagnosis and genetic counseling for KLEFS2.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
The studies involving humans were approved by the Institutional Review Board and Ethics Committee of Guangxi Maternal and Child Health Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
QY: Conceptualization, Data curation, Formal analysis, Funding acquisition, Software, Writing—original draft, Writing—review & editing. QZ: Data curation, Methodology, Validation, Writing—review & editing. SheY: Writing—review & editing. SZ: Writing—review & editing. ShaY: Writing—review & editing. XZ: Writing—review & editing. ZQ: Writing—review & editing. BC: Writing—review & editing. JL: Writing—review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by the Health Department of Guangxi Province (Grant Nos. Z-A20220256 and Z-2016702), the Guangxi Medical and Health Appropriate Technology Development and Promotion Application (S2020060), and the Guangxi Clinical Research Center for Pediatric Diseases (Guike AD22035121).
The authors are grateful to the patients and the parents who participated in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2024.1340458/full#supplementary-material
1. Kleefstra T, Smidt M, Banning MJG, Oudakker AR, Van Esch H, de Brouwer APM, et al. Disruption of the gene euchromatin histone methyl transferase1 (Eu-HMTase1) is associated with the 9q34 subtelomeric deletion syndrome. J Med Genet. (2005) 42:299–306. doi: 10.1136/jmg.2004.028464
2. Knight SJ, Regan R, Nicod A, Horsley SW, Kearney L, Homfray T, et al. Subtle chromosomal rearrangements in children with unexplained mental retardation. Lancet. (1999) 354:1676–81. doi: 10.1016/S0140-6736(99)03070-6
3. Kleefstra T, de Leeuw N. Kleefstra syndrome. In:Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, , editors. GeneReviews® 1993–2023. Seattle, WA: University of Washington (2010).
4. Willemsen MH, Vulto-van Silfhout AT, Nillesen WM, Wissink-Lindhout WM, van Bokhoven H, Philip N, et al. Update on Kleefstra Syndrome. Mol Syndromol. (2012) 2:202–12. doi: 10.1159/000335648
5. Shen E, Shulha H, Weng Z, Akbarian S. Regulation of histone H3K4 methylation in brain development and disease. Philos Trans R Soc Lond B Biol Sci. (2014) 369:20130514. doi: 10.1098/rstb.2013.0514
6. Faundes V, Newman WG, Bernardini L, Canham N, Clayton-Smith J, Dallapiccola B, et al. Clinical Assessment of the Utility of Sequencing and Evaluation as a Service (CAUSES) Study; Deciphering Developmental Disorders (DDD) Study. Am J Hum Genet. (2018) 102:175–87. doi: 10.1016/j.ajhg.2017.11.013
7. Vallianatos CN, Iwase S. Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics. (2015) 7:503–19. doi: 10.2217/epi.15.1
8. Koemans TS, Kleefstra T, Chubak MC, Stone MH, Reijnders MRF, de Munnik S, et al. Functional convergence of histone methyltransferases EHMT1 and KMT2C involved in intellectual disability and autism spectrum disorder. PLoS Genet. (2017) 13:e1006864. doi: 10.1371/journal.pgen.1006864
9. Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. (2014) 515:216–21. doi: 10.1038/nature13908
10. De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. (2014) 515:209–15. doi: 10.1038/nature13772
11. Freed D, Pevsner J. The contribution of mosaic variants to autism spectrum disorder. PLoS Genet. (2016) 12:e1006245. doi: 10.1371/journal.pgen.1006245
12. Senormanci O, Karakas Celik S, Valipour E, Valipour E, Dogan V, Senormanci G, et al. Determination of candidate genes involved in schizophrenia using the whole-exome sequencing. Bratisl Lek Listy. (2018) 119:572–6. doi: 10.4149/BLL_2018_104
13. Assiry AA, Albalawi AM, Zafar MS, Khan SD, Ullah A, Almatrafi A, et al. KMT2C, a histone methyltransferase, is mutated in a family segregating non-syndromic primary failure of tooth eruption. Sci Rep. (2019) 9:16469. doi: 10.1038/s41598-019-52935-7
14. Kleefstra T, Kramer JM, Neveling K, Willemsen MH, Koemans TS, Vissers LELM, et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet. (2012) 91:73–82. doi: 10.1016/j.ajhg.2012.05.003
15. Wu D, Li R. Case report: long-term treatment and follow-up of Kleefstra Syndrome-2. Front Pediatr. (2022) 10:881838. doi: 10.3389/fped.2022.881838
16. Chen CH, Huang A, Huang YS, Fang TH. Identification of a rare novel KMT2C mutation that presents with schizophrenia in a multiplex family. J Pers Med. (2021) 11:1254. doi: 10.3390/jpm11121254
17. Schoch K, Tan QK-G, Stong N, Deak KL, McConkie-Rosell A, McDonald MT, et al. Alternative transcripts in variant interpretation: the potential for missed diagnoses and misdiagnoses. Genet Med. (2020) 22:1269–75. doi: 10.1038/s41436-020-0781-x
18. Siano MA, De Maggio I, Petillo R, Cocciadiferro D, Agolini E, Majolo M, et al. De novo mutation in KMT2C manifesting as Kleefstra syndrome 2: case report and literature review. Pediatr Rep. (2022) 14:131–9. doi: 10.3390/pediatric14010019
19. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
20. Eshraghi AA, Liu G, Kay S-IS, Eshraghi RS, Mittal J, Moshiree B, et al. Epigenetics and autism spectrum disorder: is there a correlation? Front Cell Neurosci. (2018) 12:78. doi: 10.3389/fncel.2018.00078
21. Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. (2020) 180:568–84.e23. doi: 10.1016/j.cell.2019.12.036
22. Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. (2007) 25:15–30. doi: 10.1016/j.molcel.2006.12.014
23. Jozwik KM, Chernukhin I, Serandour AA, Nagarajan S, Carroll JS. FOXA1 directs H3K4 monomethylation at enhancers via recruitment of the methyltransferase MLL3. Cell Rep. (2016) 17:2715–23. doi: 10.1016/j.celrep.2016.11.028
24. Fagan RJ, Dingwall AK. COMPASS ascending: emerging clues regarding the roles of MLL3/KMT2C and MLL2/KMT2D proteins in cancer. Cancer Lett. (2019) 458:56–65. doi: 10.1016/j.canlet.2019.05.024
25. Lavery WJ, Barski A, Wiley S, Schorry EK, Lindsley AW. KMT2C/D COMPASS complex-associated diseases [KCDCOM-ADs]: an emerging class of congenital regulopathies. Clin Epigenet. (2020) 12:10. doi: 10.1186/s13148-019-0802-2
26. Cenik BK, Shilatifard A. COMPASS and SWI/SNF complexes in development and disease. Nat Rev Genet. (2021) 22:38–58. doi: 10.1038/s41576-020-0278-0
27. Brauer B, Merino-Veliz N, Ahumada-Marchant C, Arriagada G, Bustos FJ. KMT2C knockout generates ASD-like behaviors in mice. Front Cell Dev Biol. (2023) 11:1227723. doi: 10.3389/fcell.2023.1227723
28. Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. (2012) 74:285–99. doi: 10.1016/j.neuron.2012.04.009
29. Stessman HAF, Xiong B, Coe BP, Wang T, Hoekzema K, Fenckova M, et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet. (2017) 49:515–26. doi: 10.1038/ng.3792
30. Dhaliwal J, Qiao Y, Calli K, Martell S, Race S, Chijiwa C, et al. Contribution of multiple inherited variants to autism spectrum disorder (ASD) in a family with 3 affected siblings. Genes. (2021) 12:1053. doi: 10.3390/genes12071053
31. Tuncay IO, Parmalee NL, Khalil R, Kaur K, Kumar A, Jimale M, et al. Analysis of recent shared ancestry in a familial cohort identifies coding and noncoding autism spectrum disorder variants. NPJ Genom Med. (2022) 7:13. doi: 10.1038/s41525-022-00284-2
32. Zhou X, Feliciano P, Shu C, Wang T, Astrovskaya I, Hall JB, et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat Genet. (2022) 54:1305–19. doi: 10.1038/s41588-022-01148-2
33. Kochinke K, Zweier C, Nijhof B, Fenckova M, Cizek P, Honti F, et al. Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am J Hum Genet. (2016) 98:149–64. doi: 10.1016/j.ajhg.2015.11.024
34. Bjornsson HT. The Mendelian disorders of the epigenetic machinery. Genome Res. (2015) 25:1473–81. doi: 10.1101/gr.190629.115
35. Kleefstra T, Schenck A, Kramer JM, van Bokhoven H. The genetics of cognitive epigenetics. Neuropharmacology. (2014) 80:83–94. doi: 10.1016/j.neuropharm.2013.12.025
36. Tan WH, Bird LM, Thibert RL, Williams CA. If not Angelman, what is it? A review of Angelman-like syndromes. Am J Med Genet A. (2014) 164A:975–92. doi: 10.1002/ajmg.a.36416
37. Mullegama SV, Alaimo JT, Chen L, Elsea SH. Phenotypic and molecular convergence of 2q231 deletion syndrome with other neurodevelopmental syndromes associated with autism spectrum disorder. Int J Mol Sci. (2015) 16:7627–43. doi: 10.3390/ijms16047627
38. GTEx Consortium Human Genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science. (2015) 348:648−60. doi: 10.1126/science.1262110
39. Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, et al. Transcriptional landscape of the prenatal human brain. Nature. (2014) 508:199–206. doi: 10.1038/nature13185
40. Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. (2012) 48:491–507. doi: 10.1016/j.molcel.2012.11.006
41. Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. (2012) 81:65–95. doi: 10.1146/annurev-biochem-051710-134100
Keywords: Kleefstra syndrome-2, KMT2C, novel variants, whole-exome sequencing, genotype-phenotype correlation
Citation: Yang Q, Zhang Q, Yi S, Zhang S, Yi S, Zhou X, Qin Z, Chen B and Luo J (2024) Novel germline variants in KMT2C in Chinese patients with Kleefstra syndrome-2. Front. Neurol. 15:1340458. doi: 10.3389/fneur.2024.1340458
Received: 18 November 2023; Accepted: 10 January 2024;
Published: 31 January 2024.
Edited by:
Suzanne Lesage, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Muhammad Ilyas, International Islamic University, Islamabad, PakistanCopyright © 2024 Yang, Zhang, Yi, Zhang, Yi, Zhou, Qin, Chen and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingsi Luo, eWFuZ3Fpc2tsbWcxMjZAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.