 Yancheng Kong
Yancheng Kong Di Wang
Di Wang Xu Jin2†
Xu Jin2†- 1Trauma Emergency Center, Changzhou Hospital of Traditional Chinese Medicine, Changzhou, China
- 2Changzhou Hospital Affiliated to Nanjing University of Chinese Medicine, Changzhou, China
Stroke has long been a major threat to human health worldwide. Hemorrhagic stroke, including intracerebral hemorrhage and subarachnoid hemorrhage, exhibits a high incidence rate and a high mortality and disability rate, imposing a substantial burden on both public health and the economy and society. In recent years, the triggering receptor expressed on myeloid cells (TREM) family has garnered extensive attention in various pathological conditions, including hemorrhagic stroke. This review comprehensively summarizes the structure and function of TREM1/2, as well as their roles and potential mechanisms in hemorrhagic stroke, with the aim of providing guidance for the development of targeted therapeutic strategies in the future.
1 Introduction
Stroke is the second leading global cause of death, the third major contributor to disability, and a primary cause of dementia (1). Its incidence is rising, regardless of whether one resides in a high-income or low-to middle-income country, particularly among the younger population (under 55 years). Projections suggest that the total cost of stroke will increase from $891 billion annually in 2017 to a staggering $23.1 trillion by 2050. Stroke can be categorized into ischemic stroke, which has a higher incidence rate, and hemorrhagic stroke, characterized by a significantly higher mortality and disability rate, making it more threatening (2, 3). Current treatment strategies for stroke are far from satisfactory. Recently, the Triggering Receptor Expressed on Myeloid cells (TREM)-1/2 has been discovered to play a significant role in both physiological and pathological processes (4), and its involvement in stroke has attracted widespread attention, as evidenced by a series of preclinical studies (5–8). The role of TREMs in ischemic stroke has been well-summarized and, therefore, is beyond the scope of this review (9–11). In this review, we have systematically outlined the structure and function of TREM1/2, as well as their roles and potential mechanisms in hemorrhagic stroke, with the aim of providing new insights into the development of potential therapeutic options.
2 Overview of hemorrhagic stroke
Hemorrhagic stroke comprises intracerebral hemorrhage (ICH) and subarachnoid hemorrhage (SAH). ICH refers to the rupture of blood vessels within the brain parenchyma, leading to the leakage of blood into adjacent brain tissue. ICH is typically caused by the spontaneous rupture of intracranial blood vessels, called spontaneous ICH (12). Additionally, factors such as trauma, tumors, vascular malformations, and the use of anticoagulants/fibrinolytic drugs can also lead to ICH, which is beyond the scope of this article. Spontaneous ICH is the most common form of hemorrhagic stroke, typically caused by hypertension and cerebral amyloid angiopathy. It carries a high mortality and disability rate, affecting approximately 2 million people globally each year (13). Gaining a deeper understanding of the pathological mechanisms of ICH is of paramount significance, as this will aid in developing targeted treatment approaches.
The injury mechanisms of ICH encompass both primary and subsequent secondary damage (14). Primary damage occurs within the initial hours after the onset of bleeding, primarily driven by the mass effect of the hematoma (15). Secondary damage refers to a cascade of reactions resulting from primary injury and the release of blood clot components, including excitotoxicity (16, 17), oxidative stress (18, 19), neuroinflammation (16, 18, 20, 21), disruption of the blood–brain barrier (BBB) (21, 22), ultimately leading to neuronal cell death (18, 23). Of these, neuroinflammation mainly involves early activation of resident microglia, release of pro-inflammatory mediators and influx of peripheral immune cells (24, 25). Stimulation of ICH acts on different microglia receptors including Toll-like receptors (TLRs) and the receptor of advanced glycosylation endproducts (26). TLRs, including TLR4, are involved in the neuroinflammatory process after ICH (27). TLR4 is predominantly expressed in CD11b + microglia and is upregulated early after ICH, followed by upregulation of pro-inflammatory genes through the nuclear factor-κB (NF-κB) signaling pathway (28). In addition to the above, blood components such as thrombin, fibrin and hemoglobin can trigger the inflammatory process via the TLR/NF-κB pathway (29, 30). Expression of adhesion molecules on leukocytes and their ligands is increased on endothelial cells of small postcapillary veins during neuroinflammatory processes. Infiltration of blood-borne leukocytes and immune cells at the site of brain injury has been found in experimental and clinical ICH (31–33). Currently, the management of acute ICH typically involves a comprehensive treatment approach that includes conservative therapy (strict blood press control, hemostatic drug administration, and prevention of complications) and surgical interventions (for cases where the hematoma’s mass effect leads to malignant intracranial pressure elevation, and even brain herniation) (34, 35). If patients are fortunate enough to survive the acute phase, chronic phase treatment typically involves the management of cognitive impairments (if present), active rehabilitation exercises (for functions such as language and motor skills), and long-term effective home care measures (36).
SAH is another subtype of hemorrhagic stroke. While it accounts for only 3–5% of stroke incidence, it carries a high mortality rate, poor prognosis and imposes a significant burden on individuals, families, and society (37, 38). SAH occurs when blood flows into the subarachnoid space following the rupture of intracranial blood vessels, with the rupture of intracranial aneurysms being the most common cause (39). The brain damage caused by SAH can also be divided into two stages: early brain injury (EBI) and delayed cerebral injury (DCI). EBI occurs within the first 3 days after the onset of SAH and is believed to be caused by transient global cerebral ischemia, the toxic effects of subarachnoid blood, and direct damage to brain tissue due to bleeding (40). The mechanisms of EBI are still not fully understood, but it is generally believed to be associated with oxidative stress, endoplasmic reticulum stress, BBB disruption, excessive inflammatory responses, and cell apoptosis (38, 40). An increasing body of evidence suggests that EBI is the primary cause of poor prognosis in SAH patients (41, 42). DCI is one of the hallmarks of SAH and is believed to be caused by the breakdown products of blood and inflammatory responses. It commonly occurs within 3–14 days after SAH and can lead to delayed neurological deterioration. The pathological basis of DCI may involve microcirculatory disturbances due to various factors, including large artery/microvascular spasms, microthrombus formation, and impaired venous outflow (43).
In summary, current treatment approaches have not yet significantly improved the prognosis of patients with hemorrhagic stroke. The limited understanding of the pathological mechanisms of hemorrhagic stroke constrains the exploration of more targeted therapeutic interventions. There is an urgent need for in-depth research into the pathogenesis of such diseases to guide the development of effective treatment strategies.
3 Overview of the TREM family
The TREM family is a recently discovered class of pattern recognition receptors predominantly expressed on the surface of myeloid cells. They can recognize one or multiple pathogen-associated molecular patterns (4). TREM genes in both humans and mice are situated within gene clusters on human chromosome 6p21.1 and mouse chromosome 17C (44, 45). With an immunoglobulin-like folded extracellular domain, TREM is a member of the evolutionarily conserved immunoglobulin superfamily (46). The human cluster includes NCR2 (encoding NKp44), TREM1, TREML4 (encoding TREM-like 4), TREML2, TREM2 and TREML1. The mouse cluster consists of Trem5, Trem4, Trem1, Trem3, Treml4, Treml2, Treml6, Trem2, and Treml1 (47). The TREM family has been found to be expressed on the surface of various cell types, including granulocytes, monocytes, tissue macrophages, macrophages, and dendritic cells (DCs). In general, the primary function of TREMs is to modulate the threshold and duration of myeloid cell responses. Research has revealed that TREMs have both positive and negative roles in regulating the activation and differentiation of myeloid cells and are associated with various pathological and physiological conditions, including inflammation, neurodegenerative diseases, bone remodeling, metabolic syndrome, atherosclerosis, and cancer (4).
3.1 Structure and function of TREM1
The earliest discovered TREM1 has been characterized as an amplifier of immune responses, capable of enhancing monocyte/granulocyte responses to microbial products (48). There are two isoforms of TREM-1: membrane-bound (mTREM-1) and soluble (sTREM-1). mTREM-1 is a type I transmembrane glycoprotein receptor composed of an extracellular immunoglobulin domain, a transmembrane region, and a cytoplasmic domain (49). The extracellular domain is primarily responsible for ligand binding, while the transmembrane region associates with the DNAX-activating protein 12 (DAP12) adapter protein through non-covalent bonds, transmitting signals within the cell (50). sTREM-1 is a glycosylated peptide with only one immunoglobulin-like domain. The ligands of mTREM-1 initiate the activation of the TREM-1 signaling pathway. Since this receptor lacks an immunoreceptor tyrosine-based activation motif (ITAM), it requires association with the ITAM-containing adapter protein DAP12 to transmit signals. The transmembrane structural domain of TREM-1 features positively charged lysine residues that form noncovalent bonds with the negatively charged aspartate residues found in the transmembrane region of the DAP12 junction protein. When DAP12 binds, tyrosine phosphorylation in the immunoreceptor tyrosine-based activation motif (ITAM) is facilitated by Src-family kinases. This, in turn, recruits the zeta-associated protein of 70 kDa (ZAP70) and spleen tyrosine kinase (SYK) (51), initiating a cascade of downstream signaling reactions. These reactions encompass the activation of pathways including phosphoinositide phospholipase C-gamma (PLCγ), phosphoinositide 3-kinase (PI3K), Janus kinase (JAK), and mitogen-activated protein kinase (MAPK). These pathways collectively induce processes such as Ca2+ mobilization, actin cytoskeleton rearrangement, and the activation of transcription factors. These transcription factors (including the nuclear factor of activated T cells (NFAT), signal transducer and activator of transcription 3/5 (STAT3/5)) are responsible for encoding the expression of cell-surface molecules, proinflammatory cytokines, and chemokines (52, 53). Little is known about the ligands that activate TREM1. Limited evidence suggested that high mobility group box 1 (HMGB1), PGN recognition protein 1 (PGLYRP1), heat shock protein 70 (HSP70), CD177, actin, and extracellular cold-inducible RNA-binding protein (eCIRP) may be ligands for TREM-1 (50, 54, 55).
3.2 Structure and function of TREM2
TREM2 is a transmembrane receptor belonging to the immunoglobulin superfamily of lectin-like proteins. Its ligands encompass various anionic molecules, freely present or bound to the cell membrane, including bacterial products, DNA, lipoproteins, and phospholipids (56). TREM2 is a crucial innate immune receptor predominantly expressed in myeloid cells, such as immature dendritic cells, osteoclasts, tissue macrophages, and microglial cells, among other cell types, both on the cell surface and within intracellular pools (4, 56, 57). TREM2 is an activating receptor associated with DAP12 and DAP10. It consists of a single V-type immunoglobulin domain, a short extracellular domain, a transmembrane helix, and a short cytoplasmic tail lacking any signaling or transport motifs (58). Research on mouse macrophages has shown that TREM2 binds to the adapter proteins DAP12 and DAP10 through reverse-charged residues in its transmembrane domain (58). Following the interaction of TREM2 with its ligands, these co-receptors become phosphorylated, initiating intracellular signaling mechanisms. DAP12, also known as Tyrosine Kinase Binding Protein (TYROBP), mediates the activation of SYK, whereas DAP10 promotes signal propagation by recruiting PI3K (59).
4 The role of TREM1 in hemorrhagic stroke
To date, several preclinical studies have reported the role and potential mechanisms of TREM1 in hemorrhagic stroke (see Figure 1). Current evidence suggests that TREM1 mediates EBI after hemorrhagic stroke by engaging neuroinflammation and BBB destruction (summarized in Table 1).
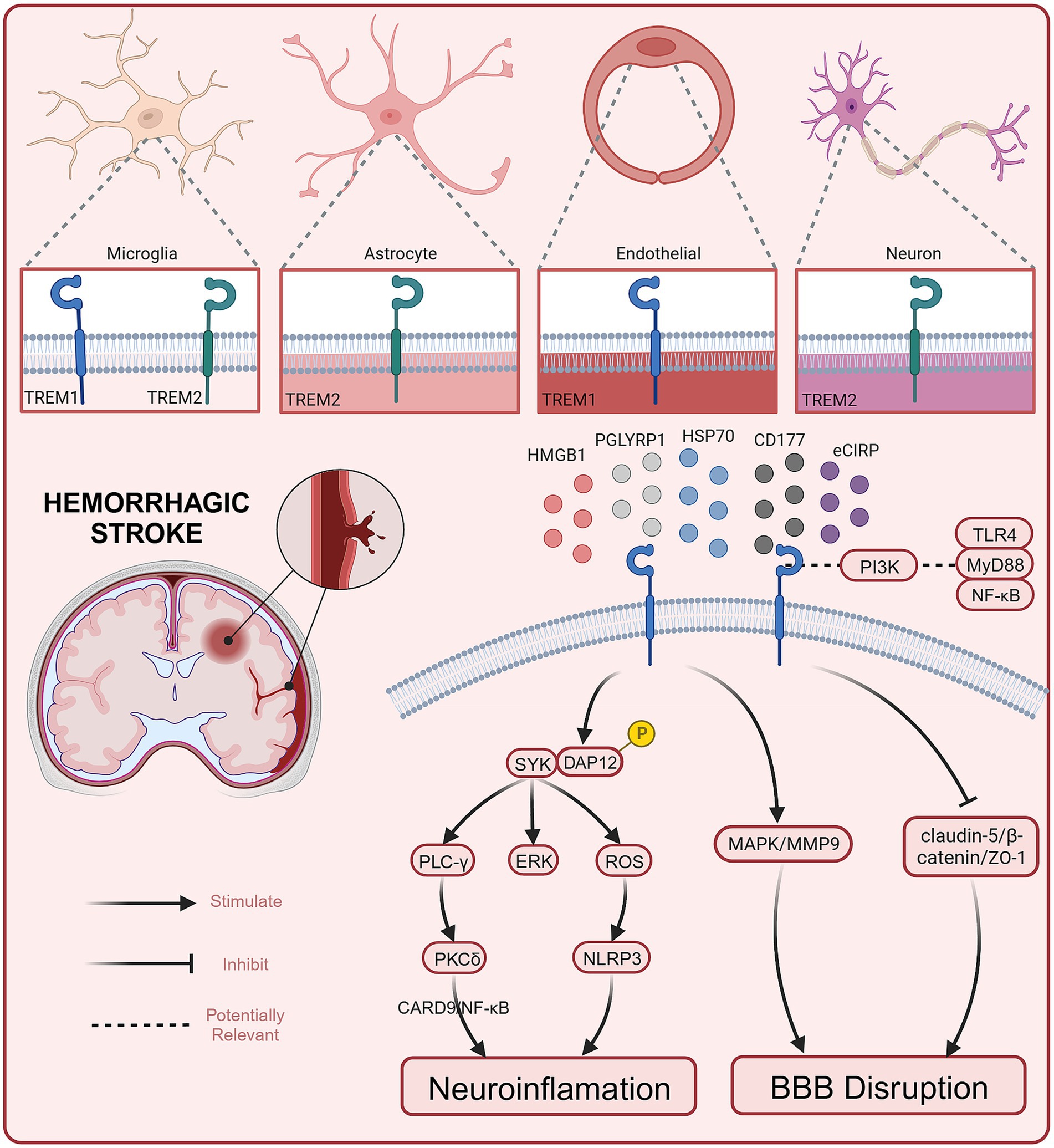
Figure 1. Role and potential mechanisms of TREM1 in hemorrhagic stroke.
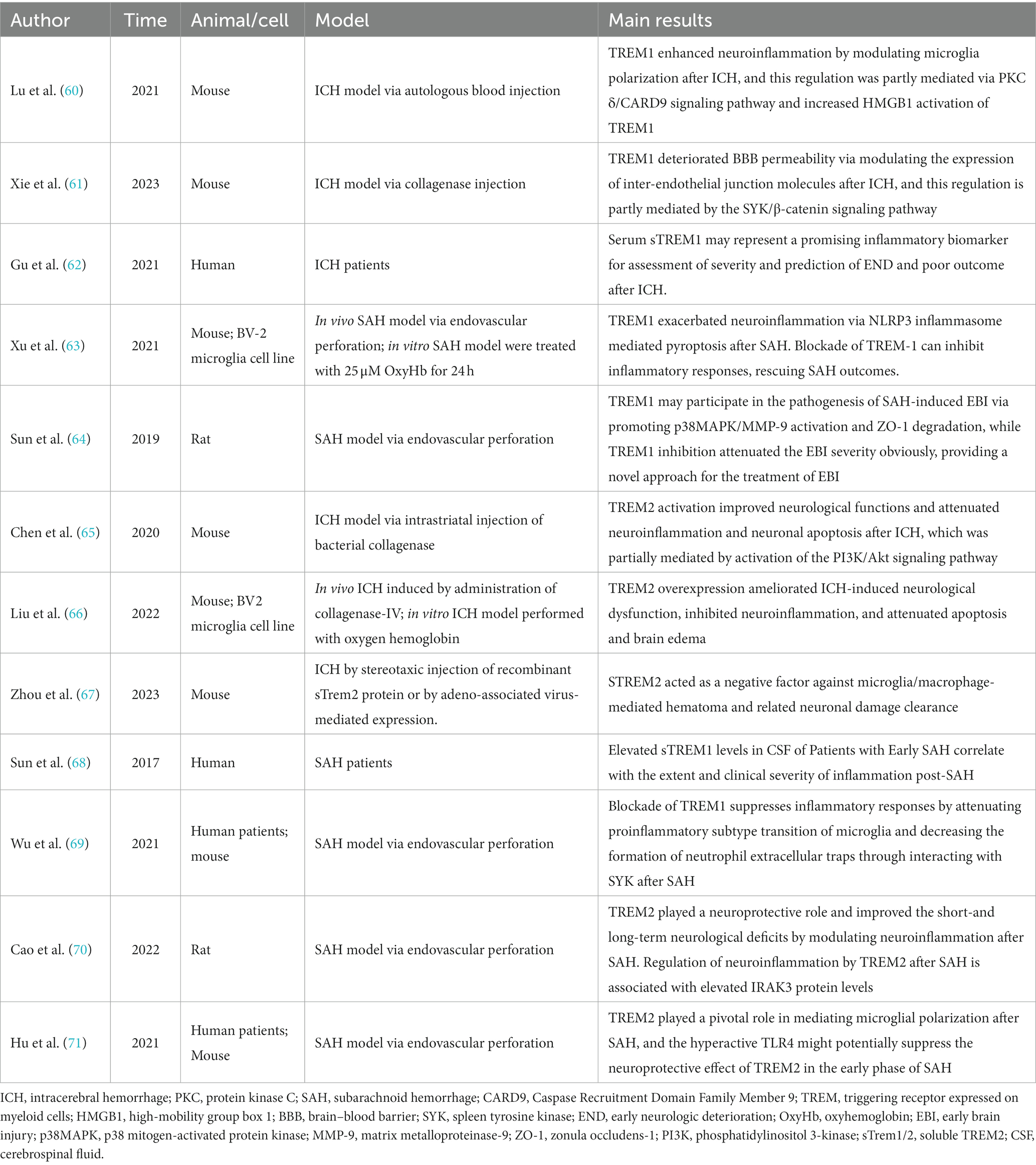
Table 1. Established reports of TREM1/2 in hemorrhagic stroke.
4.1 The role of TREM1 in ICH
TREM1 is an inflammation amplifier typically expressed on myeloid cells (72). Both in ICH and SAH, the secondary neuroinflammation is closely associated with the progression and prognosis of the diseases (20). Activated TREM-1 provides a docking site for SYK through the phosphorylation of DAP12, and SYK can activate the PLC-γ and extracellular signal-regulated kinase (ERK) pathways (73). It has been reported that PLC-γ can activate Protein Kinase C(PKC)-δ, and this activation has been demonstrated to stimulate microglia through the caspase recruitment domain family member 9 (CARD9)/NF-κB signaling pathway (74). In 2021, a study conducted by a Chinese team showed that serum sTREM1 levels were significantly higher in patients with ICH compared to healthy controls from admission to 24 h after admission. Furthermore, Gu et.al found that serum sTREM-1 concentrations were closely associated with National Institutes of Health Stroke Scale (NIHSS) scores, hematoma volume, blood leukocyte count, and serum C-reactive protein levels. By measuring serum sTREM-1 levels and three-month follow-up scores at prognosis (NIHSS score and modified Rankin scale (mRS) score), they found a significant increase in serum sTREM-1 concentration after ICH that may reflect the inflammatory response, hemorrhage severity and long-term functional prognosis. Therefore, sTREM-1 can be used as an inflammatory biomarker to assess the severity of ICH and predict early neurological function deterioration (62). TREM1 is primarily expressed in microglia/macrophages within the central nervous system (50). TREM-1 and HMGB1 expression increased after ICH, and the two proteins interacted to promote an inflammatory response. HMGB1 inhibitors alleviated neuroinflammation after ICH (60). In the EBI stage, microglia/macrophages polarize to the M1 phenotype and secrete pro-inflammatory factors involved in the process of EBI. Within the first 7 days after ICH, microglia/macrophages polarize to the M2 phenotype, aiding in hematoma clearance and brain injury repair (75). TREM1 can enhance neuroinflammatory responses by regulating the polarization of microglia/macrophages following ICH, causing them to shift toward the M1 phenotype. Selective knockout or pharmacological inhibition of TREM1 (LP17) reduces the secretion of pro-inflammatory cytokines after ICH, promoting the polarization of M2 phenotype. This, in turn, decreases brain edema and inflammatory responses after ICH and improves neurological symptoms in mice (60). In addition to microglia/macrophages, TREM1 has also been reported to be expressed in brain vascular endothelial cells (61). Another study found that early and delayed administration of LP17 significantly reduces brain edema and improves neurological behavioral performance 24 h after ICH. Specifically, TREM1 inhibition decreased BBB permeability by modulating the SYK/β-catenin signaling pathway and increasing the expression of β-catenin, claudin-5, and ZO-1 (61). In summary, TREM1 is expressed in different cell types in the CNS and plays a dual “harmful” role in promoting inflammation and exacerbating BBB disruption following ICH. Targeted interventions to modulate TREM1 may have a positive impact on improving the prognosis of ICH. Elevated serum levels of TREM1 in patients with ICH, although awaiting further confirmation in a multicenter study, suggests a basis for future clinical studies to be conducted.
4.2 The role of TREM1 in SAH
Extensive research suggests that EBI is a significant contributor to poor outcomes in SAH, and EBI is closely associated with neuroinflammation (41, 42). Sun et al. found a substantial increase in sTREM-1 levels in the cerebrospinal fluid (CSF) of SAH patients. The extent of this increase was closely related to the intensity of inflammatory responses (TNF-α, IL-6, white blood cell count, and CRP levels) and the severity of the disease (Glasgow coma scale (GCS), World Federation of Neurosurgical Societies (WFNS) scale, Hunt and Hess scale). This suggests that TREM-1 may be involved in initiating and amplifying the inflammatory cascade processes in EBI following SAH (68, 76). Sun et al. reported that TREM1 is primarily expressed in microglia/macrophages and brain vascular endothelial cells, consistent with previous reports. Additionally, they demonstrated that the expression of TREM-1, TLR4, MyD88, and NF-κB p65 increases following SAH. Inhibiting TREM-1 can alleviate EBI associated with TLR4/MyD88/NF-κB inhibition. Inhibiting TLR4 can prevent TREM-1 induction and improve EBI. Furthermore, the levels of sTREM-1 in SAH patients are positively correlated with Hunt-Hess score (76). All of this evidence suggests a potential role for TREM1 in mediating early brain injury after SAH. Previous research has also found the involvement of PI3K in both the TREM-1/DAP12 pathway and the TLR4/MyD88 pathway. PI3K seems to play a crucial role in both TREM-1 and TLR4 signal transduction, leading to the synergistic release of ROS (77). This suggests that there may be a connection between the TREM-1 pathway and the TLR4/MyD88/NF-κB signaling pathway (78). Wu et al. further explored the role of TREM1 in neuroinflammation following SAH and its potential mechanisms (69). Mechanistic studies have shown that TREM1 activates a downstream proinflammatory pathway through interaction with splenic tyrosine kinase (SYK). Pharmacological inhibition of TREM1 attenuates the proinflammatory subtype switching of microglia/macrophages and the formation of extracellular traps in neutrophils, which reduces neuroinflammation after SAH, decreases brain water content, reduces neuronal damage, and ultimately attenuates neurological deficits.
In addition, the role of TREM-1 in BBB disruption following SAH has also been explored. The BBB comprises a structural barrier formed by tight junction-associated proteins, including ZO-1. The degradation of ZO-1 disrupts tight junctions and leads to BBB breakdown (79). Previous studies have suggested that TREM-1 is involved in the pathogenesis of BBB disruption by activating MAPK and degrading ZO-1. Meanwhile, TLR4, through the MyD88 pathway, leads to NF-κB induction and MAPK activation. Subsequently, MAPKs participate in the degradation of ZO-1 and the breakdown of the BBB following SAH (64, 80). Therefore, TREM-1 and TLR4 may form a positive feedback loop by activating MAPKs, exacerbating EBI. In another study, TREM-1, the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome, cleaved caspase-1, mature IL-1β, and mature IL-18 increased expression following SAH. The NLRP3 inflammasome can be activated through active oxygen production dependent on SYK, and the TREM-1/DAP12 signaling cascade can recruit SYK. This suggests that TREM-1 may activate NLRP3, triggering an inflammatory response and leading to the death of microglia/macrophages through SYK (63, 81). Combining in vivo and in vitro research, Xu et al. found that activated TREM-1 could trigger pyroptosis in microglia and exacerbated neuroinflammation after SAH through activation of NLRP3 inflammasome (63). All evidence suggests that elevated TREM1 after SAH is indeed involved in the progression of early brain injury, at least in part by exacerbating neuroinflammation and BBB destruction. Targeting TREM1 is expected to exert a neuroprotective effect as one of the potential therapeutic options for patients with SAH. However, before that can happen, more precise mechanistic pathways and extensive and effective clinical studies must first be addressed.
5 The role of TREM2 in hemorrhagic stroke
In recent years, several teams have investigated the role of TREM2 in hemorrhagic stroke. Although there are few reports, limited evidence suggests that TREM2 exerts some neuroprotective effects in hemorrhagic stroke (Table 1).
5.1 The role of TREM2 in ICH
In animal models of neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and ischemic stroke, overexpression of TREM2 has been reported to suppress neuroinflammation and exhibit significant neuroprotective effects (9, 82–84). In recent years, several studies have explored the role of TREM2 in ICH. Chen et al.’s research suggests that TREM2 expression is significantly upregulated in microglia/macrophages, astrocytes, and neurons following ICH. Intranasal administration of the TREM2 ligand COG1410 can upregulate TREM2, PI3K, phosphorylated Akt, reduce brain edema, suppress microglia/macrophage activation and neutrophil infiltration, and inhibit neuronal apoptosis in the perihematomal region after ICH. Ultimately, this leads to improved neurological function. These conclusions are further validated by the fact that knockdown of TREM2 resulted in worsening neurologic dysfunction (65). Another study reported similar neuroprotective effects of TREM2 in ICH (66). An in vivo/ex vivo study by Liu et al. demonstrated that TREM2 attenuates neuroinflammation and neuronal apoptosis during the acute phase of ICH by negatively regulating the TLR4 signaling pathway and ultimately ameliorates ICH mice’s neurological dysfunction. The latest study delves deeper into the potential role of TREM2 in hematoma dissipation (67). Their findings suggest that soluble TREM2 (sTREM2) hinders hematoma resolution and affects the recovery of motor and sensory functions. Specifically, sTREM2 is a negative regulator inhibiting microglial/macrophage-mediated clearance of hematoma and associated neuronal damage. This may partially explain the impaired erythrophagocytosis following ICH. The evidence above suggests that TREM2 is involved in regulating neuroinflammation and hematoma clearance following ICH by modulating the activation and infiltration of immune cells, particularly microglial cells and macrophages. Further studies could address the above different roles of TREM2 in detail, for example, whether there is crosstalk between neuroinflammation and hematoma clearance mechanisms. It has been shown that TREM2 is expressed in the astrocytes, but whether it is involved in BBB destruction has not been further reported. In addition, TREM2 is also expressed in neurons, but its specific function is unknown. Extensive and in-depth studies based on in vivo and in vitro experiments with the help of multi-omics analysis, including single-cell sequencing, are needed to answer these questions.
5.2 The role of TREM2 in SAH
There is limited evidence regarding the role of TREM2 in SAH, but Hu et al. conducted a study on this (71). The results indicated that TREM2 is expressed in microglia/macrophages and increases delayed after SAH. TREM2 knockout led to activation of microglia/macrophages, increased pro-inflammatory factors and deterioration of neurological function. As the evidence suggested, TLR4 knockout increased TREM2 expression while improving neuroinflammation and neurological function. The authors speculate that the neuroprotective effect of TREM2 may be suppressed by the exaggerated TLR4 expression in the early stages of SAH, which could explain the delayed increase in expression. One year later, Cao et al. found that elevated TREM2 could regulate neuroinflammation by increasing IRAK3 expression and exert neuroprotective effects after experimental SAH in rats (70). This complements the findings of Hu et al. by clarifying the upstream and downstream regulators of TREM2 after SAH, respectively. Further in-depth studies are expected to clarify the mechanism of TREM2 pathway in regulating neuroinflammation after SAH. Limited evidence suggests that, unlike the “deleterious” role of TREM1 in SAH, TREM2 appears to be neuroprotective. It is intriguing why structurally similar TREM receptors exhibit almost opposite functions, and further in-depth studies will help us better understand the underlying mechanisms.
6 Summary and prospects
In summary, current evidence suggests that the TREM1 signaling pathway is involved in the inflammatory response and BBB disruption in hemorrhagic stroke and ultimately exacerbates secondary brain damage. Targeting the inhibition of the TREM1 signaling pathway, such as using the TREM1 inhibitor LP17, can mitigate the inflammatory response and blood–brain barrier disruption, improving neurological function in animal models of ICH and SAH. Conversely, preclinical studies suggest a neuroprotective role for TREM2. As the evidence indicates, TREM2 can inhibit microglial/macrophage activation and neutrophil infiltration, reduce brain edema, and inhibit neuronal apoptosis in the perihematomal region. TREM1 and TREM2 have similar structures but different functions. This difference is well summarized by Zhang et al. (50). The potential roles of TREM1 and TREM2 in neuroinflammation and BBB disruption provide new insights into targeted therapies for ICH and SAH. Moreover, serum sTREM1 was elevated within 24 h after ICH (62) and positively correlated with Hunt-Hess scale in patients with SAH (76). Further studies need to explore its potential as a biomarker for hemorrhagic stroke.
It is well known that during acute stress, microglia/macrophages release a range of inflammatory mediators, including cytokines and chemokines. These acute-phase responses are often thought to favor neuronal survival, attenuate secondary damage and ultimately help restore cellular homeostasis (85, 86). During chronic inflammation, the continued release of inflammatory mediators by microglia/macrophages causes sustained oxidative stress and is therefore considered to be detrimental (87, 88). Studies have focused on TREM1/2 in the acute phase of ischemic stroke; what role TREM1/2 fills in the chronic phase is largely unknown, which is a direction for future research. Notably, most published ICH studies have been unable to distinguish between macrophages and microglia (which we refer to here as microglia/macrophages) because the two cell types are difficult to distinguish in vivo (89, 90). With the help of more specific cellular markers (e.g., Tmem119 for microglia) and multichannel flow cytometry, these two cell types are expected to be rigorously distinguished in the next explorations (91, 92). On this basis, it will be fully explored whether microglia and macrophages play different roles (if any) in ICH.
In addition, the above studies inevitably have other limitations. For instance, the exact roles of TREM1/2 in other cell types in the central nervous system (e.g., TREM2 in astrocytes and neurons) have not been fully elucidated. Additionally, the current research is primarily preclinical and mainly involves rodent animals. The roles of TREM1/2 in hemorrhagic stroke need further validation in higher primates and clinical studies. Finally, elucidating the mechanisms of action and signal regulation between TREM1/2 and their respective ligands will help us understand their biological properties and provide a solid theoretical basis for developing potential future treatment options.
Author contributions
YK: Conceptualization, Funding acquisition, Methodology, Resources, Writing – original draft, Writing – review & editing. DW: Investigation, Visualization, Writing – original draft. XJ: Formal analysis, Writing – review & editing. YL: Supervision, Writing – review & editing. HX: Data curation, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Collaborators GBDS. Global, regional, and national burden of stroke and its risk factors, 1990-2019: a systematic analysis for the global burden of disease study 2019. Lancet Neurol. (2021) 20:795–820. doi: 10.1016/S1474-4422(21)00252-0
2. Kalantri, A, and Kalantri, S. Distinguishing hemorrhagic stroke from ischemic stroke. JAMA. (2010) 304:1327. doi: 10.1001/jama.2010.1341
3. Runchey, S, and McGee, S. Does this patient have a hemorrhagic stroke?: clinical findings distinguishing hemorrhagic stroke from ischemic stroke. JAMA. (2010) 303:2280–6. doi: 10.1001/jama.2010.754
4. Colonna, M. The biology of TREM receptors. Nat Rev Immunol. (2023) 23:580–94. doi: 10.1038/s41577-023-00837-1
5. François, B, Lambden, S, Fivez, T, Gibot, S, Derive, M, Grouin, J-M, et al. Prospective evaluation of the efficacy, safety, and optimal biomarker enrichment strategy for nangibotide, a TREM-1 inhibitor, in patients with septic shock (ASTONISH): a double-blind, randomised, controlled, phase 2b trial. Lancet Respir Med. (2023) 11:894–904. doi: 10.1016/S2213-2600(23)00158-3
6. Labiano, I, Agirre-Lizaso, A, Olaizola, P, Echebarria, A, Huici-Izagirre, M, Olaizola, I, et al. TREM-2 plays a protective role in cholestasis by acting as a negative regulator of inflammation. J Hepatol. (2022) 77:991–1004. doi: 10.1016/j.jhep.2022.05.044
7. Jolly, L, Carrasco, K, Salcedo-Magguilli, M, Garaud, J-J, Lambden, S, van der Poll, T, et al. sTREM-1 is a specific biomarker of TREM-1 pathway activation. Cell Mol Immunol. (2021) 18:2054–6. doi: 10.1038/s41423-021-00733-5
8. Boufenzer, A, Carrasco, K, Jolly, L, Brustolin, B, Di-Pillo, E, Derive, M, et al. Potentiation of NETs release is novel characteristic of TREM-1 activation and the pharmacological inhibition of TREM-1 could prevent from the deleterious consequences of NETs release in sepsis. Cell Mol Immunol. (2021) 18:452–60. doi: 10.1038/s41423-020-00591-7
9. Gervois, P, and Lambrichts, I. The emerging role of triggering receptor expressed on myeloid cells 2 as a target for immunomodulation in ischemic stroke. Front Immunol. (2019) 10:1668. doi: 10.3389/fimmu.2019.01668
10. Ma, W-Y, Wang, S-S, Wu, Q-L, Zhou, X, Chu, S-F, and Chen, N-H. The versatile role of TREM2 in regulating of microglia fate in the ischemic stroke. Int Immunopharmacol. (2022) 109:108733. doi: 10.1016/j.intimp.2022.108733
11. Wang, H, Li, X, Wang, Q, Ma, J, Gao, X, and Wang, M. TREM2, microglial and ischemic stroke. J Neuroimmunol. (2023) 381:578108. doi: 10.1016/j.jneuroim.2023.578108
12. Puy, L, Parry-Jones, AR, Sandset, EC, Dowlatshahi, D, Ziai, W, and Cordonnier, C. Intracerebral haemorrhage. Nat Rev Dis Primers. (2023) 9:14. doi: 10.1038/s41572-023-00424-7
13. Cordonnier, C, Demchuk, A, Ziai, W, and Anderson, CS. Intracerebral haemorrhage: current approaches to acute management. Lancet. (2018) 392:1257–68. doi: 10.1016/S0140-6736(18)31878-6
14. Bautista, W, Adelson, PD, Bicher, N, Themistocleous, M, Tsivgoulis, G, and Chang, JJ. Secondary mechanisms of injury and viable pathophysiological targets in intracerebral hemorrhage. Ther Adv Neurol Disord. (2021) 14:175628642110492. doi: 10.1177/17562864211049208
15. Zhou, Y, Wang, Y, Wang, J, Anne Stetler, R, and Yang, Q-W. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. (2014) 115:25–44. doi: 10.1016/j.pneurobio.2013.11.003
16. Ren, R, Fang, Y, Sherchan, P, Lu, Q, Lenahan, C, Zhang, JH, et al. Kynurenine/aryl hydrocarbon receptor modulates mitochondria-mediated oxidative stress and neuronal apoptosis in experimental intracerebral hemorrhage. Antioxid Redox Signal. (2022) 37:1111–29. doi: 10.1089/ars.2021.0215
17. Li, H, Liu, S, Sun, X, Yang, J, Yang, Z, Shen, H, et al. Critical role for Annexin A7 in secondary brain injury mediated by its phosphorylation after experimental intracerebral hemorrhage in rats. Neurobiol Dis. (2018) 110:82–92. doi: 10.1016/j.nbd.2017.11.012
18. Zheng, Y, Li, R, and Fan, X. Targeting oxidative stress in intracerebral hemorrhage: prospects of the natural products approach. Antioxidants (Basel). (2022) 11:1811. doi: 10.3390/antiox11091811
19. Zhang, Y, Khan, S, Liu, Y, Wu, G, Yong, VW, and Xue, M. Oxidative stress following intracerebral hemorrhage: from molecular mechanisms to therapeutic targets. Front Immunol. (2022) 13:847246. doi: 10.3389/fimmu.2022.847246
20. Zhang, Z, Zhang, Z, Lu, H, Yang, Q, Wu, H, and Wang, J. Microglial polarization and inflammatory mediators after intracerebral hemorrhage. Mol Neurobiol. (2017) 54:1874–86. doi: 10.1007/s12035-016-9785-6
21. Lu, Q, Gao, L, Huang, L, Ruan, L, Yang, J, Huang, W, et al. Inhibition of mammalian target of rapamycin improves neurobehavioral deficit and modulates immune response after intracerebral hemorrhage in rat. J Neuroinflammation. (2014) 11:44. doi: 10.1186/1742-2094-11-44
22. Lim-Hing, K, and Rincon, F. Secondary hematoma expansion and Perihemorrhagic edema after intracerebral hemorrhage: from bench work to practical aspects. Front Neurol. (2017) 8:74. doi: 10.3389/fneur.2017.00074
23. Wan, Y, Holste, KG, Hua, Y, Keep, RF, and Xi, G. Brain edema formation and therapy after intracerebral hemorrhage. Neurobiol Dis. (2023) 176:105948. doi: 10.1016/j.nbd.2022.105948
24. Wang, J, and Dore, S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. (2007) 27:894–908. doi: 10.1038/sj.jcbfm.9600403
25. Wang, J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. (2010) 92:463–77. doi: 10.1016/j.pneurobio.2010.08.001
26. Taylor, RA, and Sansing, LH. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Dev Immunol. (2013) 2013:746068. doi: 10.1155/2013/746068
27. Fang, H, Wang, PF, Zhou, Y, Wang, YC, and Yang, QW. Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. J Neuroinflammation. (2013) 10:27. doi: 10.1186/1742-2094-10-27
28. Lin, S, Yin, Q, Zhong, Q, Lv, FL, Zhou, Y, Li, JQ, et al. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflammation. (2012) 9:46. doi: 10.1186/1742-2094-9-46
29. Loftspring, MC, Hansen, C, and Clark, JF. A novel brain injury mechanism after intracerebral hemorrhage: the interaction between heme products and the immune system. Med Hypotheses. (2010) 74:63–6. doi: 10.1016/j.mehy.2009.08.002
30. Wang, YC, Zhou, Y, Fang, H, Lin, S, Wang, PF, Xiong, RP, et al. Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann Neurol. (2014) 75:876–89. doi: 10.1002/ana.24159
31. Mayne, M, Fotheringham, J, Yan, HJ, Power, C, Del Bigio, MR, Peeling, J, et al. Adenosine A2A receptor activation reduces proinflammatory events and decreases cell death following intracerebral hemorrhage. Ann Neurol. (2001) 49:727–35. doi: 10.1002/ana.1010
32. Peeling, J, Yan, HJ, Corbett, D, Xue, M, and Del Bigio, MR. Effect of FK-506 on inflammation and behavioral outcome following intracerebral hemorrhage in rat. Exp Neurol. (2001) 167:341–7. doi: 10.1006/exnr.2000.7564
33. Wang, J, and Tsirka, SE. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain. (2005) 128:1622–33. doi: 10.1093/brain/awh489
34. Hemphill, JC, Greenberg, SM, Anderson, CS, Becker, K, Bendok, BR, Cushman, M, et al. Guidelines for the Management of Spontaneous Intracerebral Hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. (2015) 46:2032–60. doi: 10.1161/STR.0000000000000069
35. Li, Z, Khan, S, Liu, Y, Wei, R, Yong, VW, and Xue, M. Therapeutic strategies for intracerebral hemorrhage. Front Neurol. (2022) 13:1032343. doi: 10.3389/fneur.2022.1032343
36. Mahase, E. Stroke patients should be offered at least 15 hours of rehabilitation a week. NICE advises BMJ. (2023) 383:2417. doi: 10.1136/bmj.p2417
37. Muehlschlegel, S. Subarachnoid hemorrhage. Continuum (Minneap Minn). (2018) 24:1623–57. doi: 10.1212/CON.0000000000000679
38. Lidington, D, Wan, H, and Bolz, S-S. Cerebral autoregulation in subarachnoid hemorrhage. Front Neurol. (2021) 12:688362. doi: 10.3389/fneur.2021.688362
39. Wang, X-Y, Wu, F, Zhan, R-Y, and Zhou, H-J. Inflammatory role of microglia in brain injury caused by subarachnoid hemorrhage. Front Cell Neurosci. (2022) 16:956185. doi: 10.3389/fncel.2022.956185
40. Lu, D, Wang, L, Liu, G, Wang, S, Wang, Y, Wu, Y, et al. Role of hydrogen sulfide in subarachnoid hemorrhage. CNS Neurosci Ther. (2022) 28:805–17. doi: 10.1111/cns.13828
41. Rass, V, and Helbok, R. Early brain injury after poor-grade subarachnoid hemorrhage. Curr Neurol Neurosci Rep. (2019) 19:78. doi: 10.1007/s11910-019-0990-3
42. Xu, G, Guo, J, and Sun, C. Eucalyptol ameliorates early brain injury after subarachnoid haemorrhage via antioxidant and anti-inflammatory effects in a rat model. Pharm Biol. (2021) 59:114–20. doi: 10.1080/13880209.2021.1876101
43. Suzuki, H, Kanamaru, H, Kawakita, F, Asada, R, Fujimoto, M, and Shiba, M. Cerebrovascular pathophysiology of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Histol Histopathol. (2021) 36:143–58. doi: 10.14670/HH-18-253
44. Benitez, BA, Jin, SC, Guerreiro, R, Graham, R, Lord, J, Harold, D, et al. Missense variant in TREML2 protects against Alzheimer’s disease. Neurobiol Aging. (2014) 35:1510.e19–26. doi: 10.1016/j.neurobiolaging.2013.12.010
45. Washington, AV, Quigley, L, and McVicar, DW. Initial characterization of TREM-like transcript (TLT)-1: a putative inhibitory receptor within the TREM cluster. Blood. (2002) 100:3822–4. doi: 10.1182/blood-2002-02-0523
46. Biadgilign, S, Mgutshini, T, Deribew, A, Gelaye, B, and Memiah, P. Association of maternal psychological distress with children with overweight/obesity in Ethiopia. Child Care Health Dev. (2023) 49:392–9. doi: 10.1111/cch.13057
47. Kasamatsu, J, Deng, M, Azuma, M, Funami, K, Shime, H, Oshiumi, H, et al. Double-stranded RNA analog and type I interferon regulate expression of Trem paired receptors in murine myeloid cells. BMC Immunol. (2016) 17:9. doi: 10.1186/s12865-016-0147-y
48. Bouchon, A, Dietrich, J, and Colonna, M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. (2000) 164:4991–5. doi: 10.4049/jimmunol.164.10.4991
49. Leonard, CT. Childhood motor disorders. Pediatrics. (2003) 112:1462–3. doi: 10.1542/peds.112.6.1462
50. Zhang, C, Kan, X, Zhang, B, Ni, H, and Shao, J. The role of triggering receptor expressed on myeloid cells-1 (TREM-1) in central nervous system diseases. Mol Brain. (2022) 15:84. doi: 10.1186/s13041-022-00969-w
51. Billadeau, DD, and Leibson, PJ. ITAMs versus ITIMs: striking a balance during cell regulation. J Clin Invest. (2002) 109:161–8. doi: 10.1172/JCI0214843
52. Aoki, N, Kimura, S, and Xing, Z. Role of DAP12 in innate and adaptive immune responses. Curr Pharm Des. (2003) 9:7–10. doi: 10.2174/1381612033392503
53. Radsak, MP, Salih, HR, Rammensee, H-G, and Schild, H. Triggering receptor expressed on myeloid cells-1 in neutrophil inflammatory responses: differential regulation of activation and survival. J Immunol. (2004) 172:4956–63. doi: 10.4049/jimmunol.172.8.4956
54. El Mezayen, R, El Gazzar, M, Seeds, MC, McCall, CE, Dreskin, SC, and Nicolls, MR. Endogenous signals released from necrotic cells augment inflammatory responses to bacterial endotoxin. Immunol Lett. (2007) 111:36–44. doi: 10.1016/j.imlet.2007.04.011
55. Denning, NL, Aziz, M, Murao, A, Gurien, SD, Ochani, M, Prince, JM, et al. Extracellular CIRP as an endogenous TREM-1 ligand to fuel inflammation in sepsis. JCI. Insight. (2020) 5:172. doi: 10.1172/jci.insight.134172
56. Kober, DL, and Brett, TJ. TREM2-ligand interactions in health and disease. J Mol Biol. (2017) 429:1607–29. doi: 10.1016/j.jmb.2017.04.004
57. Ulrich, JD, and Holtzman, DM. TREM2 function in Alzheimer’s disease and neurodegeneration. ACS Chem Neurosci. (2016) 7:420–7. doi: 10.1021/acschemneuro.5b00313
58. Deczkowska, A, Weiner, A, and Amit, I. The physiology, pathology, and potential therapeutic applications of the TREM2 signaling pathway. Cell. (2020) 181:1207–17. doi: 10.1016/j.cell.2020.05.003
59. Peng, Q, Malhotra, S, Torchia, JA, Kerr, WG, Coggeshall, KM, and Humphrey, MB. TREM2-and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal. (2010) 3:500. doi: 10.1126/scisignal.2000500
60. Lu, Q, Liu, R, Sherchan, P, Ren, R, He, W, Fang, Y, et al. TREM (triggering receptor expressed on myeloid cells)-1 inhibition attenuates Neuroinflammation via PKC (protein kinase C) δ/CARD9 (caspase recruitment domain family member 9) signaling pathway after intracerebral hemorrhage in mice. Stroke. (2021) 52:2162–73. doi: 10.1161/STROKEAHA.120.032736
61. Xie, Y, He, W, Ma, L, Ren, R, Yang, S, and Lu, Q. Endothelial TREM-1 receptor regulates the blood-brain barrier integrity after intracerebral hemorrhage in mice via SYK/β-catenin signaling. CNS Neurosci Ther. (2023) 29:3228–38. doi: 10.1111/cns.14255
62. Gu, Y, Deng, X, Liang, C, Chen, Y, Lei, H, and Zhang, Q. Soluble triggering receptor expressed on myeloid cells-1 as a serum biomarker of early neurologic deterioration and prognosis in acute supratentorial intracerebral hemorrhage. Clinica Chimica Acta; international journal of. Clin Chem. (2021) 523:290–6. doi: 10.1016/j.cca.2021.10.010
63. Xu, P, Hong, Y, Xie, Y, Yuan, K, Li, J, Sun, R, et al. TREM-1 exacerbates Neuroinflammatory injury via NLRP3 Inflammasome-mediated Pyroptosis in experimental subarachnoid hemorrhage. Transl Stroke Res. (2021) 12:643–59. doi: 10.1007/s12975-020-00840-x
64. Sun, X-G, Duan, H, Jing, G, Wang, G, Hou, Y, and Zhang, M. Inhibition of TREM-1 attenuates early brain injury after subarachnoid hemorrhage via downregulation of p38MAPK/MMP-9 and preservation of ZO-1. Neuroscience. (2019) 406:369–75. doi: 10.1016/j.neuroscience.2019.03.032
65. Chen, S, Peng, J, Sherchan, P, Ma, Y, Xiang, S, Yan, F, et al. TREM2 activation attenuates neuroinflammation and neuronal apoptosis via PI3K/Akt pathway after intracerebral hemorrhage in mice. J Neuroinflammation. (2020) 17:168. doi: 10.1186/s12974-020-01853-x
66. Liu, S, Cao, X, Wu, Z, Deng, S, Fu, H, Wang, Y, et al. TREM2 improves neurological dysfunction and attenuates neuroinflammation, TLR signaling and neuronal apoptosis in the acute phase of intracerebral hemorrhage. Front Aging Neurosci. (2022) 14:967825. doi: 10.3389/fnagi.2022.967825
67. Zhou, H, Li, J, Hu, L, Yu, J, Fu, X, Liang, F, et al. Soluble Trem2 is a negative regulator of erythrophagocytosis after intracerebral hemorrhage in a CD36 receptor recycling manner. J Adv Res. (2023) 44:185–99. doi: 10.1016/j.jare.2022.03.011
68. Sun, X-G, Ma, Q, Jing, G, Wang, L, Hao, X-D, and Wang, G-Q. Early elevated levels of soluble triggering receptor expressed on myeloid cells-1 in subarachnoid hemorrhage patients. Neurol Sci. (2017) 38:873–7. doi: 10.1007/s10072-017-2853-5
69. Wu, X, Zeng, H, Xu, C, Chen, H, Fan, L, Zhou, H, et al. TREM1 regulates Neuroinflammatory injury by modulate Proinflammatory subtype transition of microglia and formation of neutrophil extracellular traps via interaction with SYK in experimental subarachnoid hemorrhage. Front Immunol. (2021) 12:766178. doi: 10.3389/fimmu.2021.766178
70. Cao, C, Ding, J, Cao, D, Li, B, Wu, J, Li, X, et al. TREM2 modulates neuroinflammation with elevated IRAK3 expression and plays a neuroprotective role after experimental SAH in rats. Neurobiol Dis. (2022) 171:105809. doi: 10.1016/j.nbd.2022.105809
71. Hu, Y, Li, C, Wang, X, Chen, W, Qian, Y, and Dai, X. TREM2, driving the microglial polarization, has a TLR4 sensitivity profile after subarachnoid hemorrhage. Front Cell Dev Biol. (2021) 9:693342. doi: 10.3389/fcell.2021.693342
72. Arts, RJW, Joosten, LAB, van der Meer, JWM, and Netea, MG. TREM-1: intracellular signaling pathways and interaction with pattern recognition receptors. J Leukoc Biol. (2013) 93:209–15. doi: 10.1189/jlb.0312145
73. Tammaro, A, Derive, M, Gibot, S, Leemans, JC, Florquin, S, and Dessing, MC. TREM-1 and its potential ligands in non-infectious diseases: from biology to clinical perspectives. Pharmacol Ther. (2017) 177:81–95. doi: 10.1016/j.pharmthera.2017.02.043
74. Yang, Q, Langston, JC, Tang, Y, Kiani, MF, and Kilpatrick, LE. The role of tyrosine phosphorylation of protein kinase C Delta in infection and inflammation. Int J Mol Sci. (2019) 20:498. doi: 10.3390/ijms20061498
75. Tschoe, C, Bushnell, CD, Duncan, PW, Alexander-Miller, MA, and Wolfe, SQ. Neuroinflammation after intracerebral hemorrhage and potential therapeutic targets. J Stroke. (2020) 22:29–46. doi: 10.5853/jos.2019.02236
76. Sun, X-G, Ma, Q, Jing, G, Wang, G-Q, Hao, X-D, and Wang, L. Increased levels of soluble triggering receptor expressed on myeloid cells-1 in cerebrospinal fluid of subarachnoid hemorrhage patients. J Clin Neurosci. (2017) 35:139–43. doi: 10.1016/j.jocn.2016.09.005
77. Yamamori, T, Inanami, O, Nagahata, H, Cui, Y, and Kuwabara, M. Roles of p38 MAPK, PKC and PI3-K in the signaling pathways of NADPH oxidase activation and phagocytosis in bovine polymorphonuclear leukocytes. FEBS Lett. (2000) 467:253–8. doi: 10.1016/S0014-5793(00)01167-4
78. Sun, X-G, Zhang, M-M, Liu, S-Y, Chu, X-H, Xue, G-Q, Zhang, B-C, et al. Role of TREM-1 in the development of early brain injury after subarachnoid hemorrhage. Exp Neurol. (2021) 341:113692. doi: 10.1016/j.expneurol.2021.113692
79. Liu, L, Kawakita, F, Fujimoto, M, Nakano, F, Imanaka-Yoshida, K, Yoshida, T, et al. Role of Periostin in early brain injury after subarachnoid hemorrhage in mice. Stroke. (2017) 48:1108–11. doi: 10.1161/STROKEAHA.117.016629
80. Kwon, MS, Woo, SK, Kurland, DB, Yoon, SH, Palmer, AF, Banerjee, U, et al. Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int J Mol Sci. (2015) 16:5028–46. doi: 10.3390/ijms16035028
81. Ford, JW, and McVicar, DW. TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol. (2009) 21:38–46. doi: 10.1016/j.coi.2009.01.009
82. Raha, AA, Henderson, JW, Stott, SRW, Vuono, R, Foscarin, S, Friedland, RP, et al. Neuroprotective effect of TREM-2 in aging and Alzheimer’s disease model. J Alzheimers Dis. (2017) 55:199–217. doi: 10.3233/JAD-160663
83. Takahashi, K, Prinz, M, Stagi, M, Chechneva, O, and Neumann, H. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. (2007) 4:e124. doi: 10.1371/journal.pmed.0040124
84. Zhai, Q, Li, F, Chen, X, Jia, J, Sun, S, Zhou, D, et al. Triggering receptor expressed on myeloid cells 2, a novel regulator of Immunocyte phenotypes, confers neuroprotection by relieving Neuroinflammation. Anesthesiology. (2017) 127:98–110. doi: 10.1097/ALN.0000000000001628
85. Li, Q, and Barres, BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. (2018) 18:225–42. doi: 10.1038/nri.2017.125
86. Salter, MW, and Stevens, B. Microglia emerge as central players in brain disease. Nat Med. (2017) 23:1018–27. doi: 10.1038/nm.4397
87. Keane, L, Cheray, M, Blomgren, K, and Joseph, B. Multifaceted microglia - key players in primary brain tumour heterogeneity. Nat Rev Neurol. (2021) 17:243–59. doi: 10.1038/s41582-021-00463-2
88. Prinz, M, and Priller, J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. (2014) 15:300–12. doi: 10.1038/nrn3722
89. Zarruk, JG, Greenhalgh, AD, and David, S. Microglia and macrophages differ in their inflammatory profile after permanent brain ischemia. Exp Neurol. (2018) 301:120–32. doi: 10.1016/j.expneurol.2017.08.011
90. Bennett, ML, Bennett, FC, Liddelow, SA, Ajami, B, Zamanian, JL, Fernhoff, NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA. (2016) 113:E1738–46. doi: 10.1073/pnas.1525528113
91. Chang, C-F, Goods, BA, Askenase, MH, Hammond, MD, Renfroe, SC, Steinschneider, AF, et al. Erythrocyte efferocytosis modulates macrophages towards recovery after intracerebral hemorrhage. J Clin Invest. (2018) 128:607–24. doi: 10.1172/JCI95612
Keywords: TREM, intracerebral hemorrhage, subarachnoid hemorrhage, neuroinflammation, neuroprotection
Citation: Kong Y, Wang D, Jin X, Liu Y and Xu H (2024) Unveiling the significance of TREM1/2 in hemorrhagic stroke: structure, function, and therapeutic implications. Front. Neurol. 15:1334786. doi: 10.3389/fneur.2024.1334786
Edited by:
Heike Wulff, University of California, Davis, United StatesReviewed by:
Anuska V. Andjelkovic, University of Michigan, United StatesMai Anwar, National Organization for Drug Control and Research, Egypt
Ana Beatriz DePaula-Silva, The University of Utah, United States
Copyright © 2024 Kong, Wang, Jin, Liu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yancheng Kong, MTczMTUxMjc3OTNAMTYzLmNvbQ==
†These authors have contributed equally to this work