
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Neurol., 29 February 2024
Sec. Pediatric Neurology
Volume 15 - 2024 | https://doi.org/10.3389/fneur.2024.1308296
 Claudia Dosi
Claudia Dosi Riccardo Masson*
Riccardo Masson*Objective: To review the clinical characteristics and effect of treatment in patients with spinal muscular atrophy (SMA) and three copies of the SMN2 gene.
Methods: We conducted a literature search in October 2022 to identify English-language clinical research on SMA that included SMN2 copy number according to PRISMA guidelines.
Results: Our search identified 44 studies examining the impact of three SMN2 copies on clinical characteristics (21 on phenotype, 13 on natural history, and 15 on functional status and other signs/symptoms). In children with type I SMA or presymptomatic infants with an SMN1 deletion, three SMN2 copies was associated with later symptom onset, slower decline in motor function and longer survival compared with two SMN2 copies. In patients with SMA type II or III, three SMN2 copies is associated with earlier symptom onset, loss of ambulation, and ventilator dependence compared with four SMN2 copies. Eleven studies examined treatment effects with nusinersen (nine studies), onasemnogene abeparvovec (one study), and a range of treatments (one study) in patients with three SMN2 copies. In presymptomatic infants, early treatment delayed the onset of symptoms and maintained motor function in those with three SMN2 copies. The impact of copy number on treatment response in symptomatic patients is still unclear.
Conclusion: SMN2 copy number is strongly correlated with SMA phenotype in patients with SMN1 deletion, while no correlation was found in patients with an SMN1 mutation. Patients with three SMN2 copies show a highly variable clinical phenotype. Early initiation of treatment is highly effective in presymptomatic patients with three SMN2 copies.
Spinal muscular atrophy (SMA) is a rare autosomal recessive condition, with an incidence of approximately 1:15,000 live births (1, 2) and greater prevalence in regions with high consanguinity rates (3). Affected individuals develop progressive muscle weakness and atrophy as a result of degeneration of motor neurons in the anterior horn of the spinal cord and brain stem nuclei (3). Muscle weakness is generally symmetrical and worse in proximal than distal muscle groups.
The severity of clinical presentation differs markedly between individuals, and may be classified into one of five clinical phenotypes depending on the maximal level of motor function achieved and age of onset (Supplementary Table S1), with type 0 being the worst (usually resulting in death shortly after birth) and type IV the mildest (onset in adulthood) (3). Almost all patients with SMA have a homozygous deletion of the SMN1 gene, which codes for the survival motor neuron (SMN) protein (4). However, there are multiple other genetic factors that influence the phenotypic expression of SMA, including SMN2 gene variants and how many copies of the SMN2 gene the patient has (4).
A higher SMN2 copy number is generally associated with a milder disease phenotype, but there is considerable variability in the clinical presentation of SMA within subgroups of patients based on SMN2 copy number, and discordance between the expected phenotype based on SMN2 copy number (5). Variability in clinical presentation is most marked in patients with three SMN2 copies (6).
In the past 5–7 years, disease-modifying therapies have become available for the treatment of SMA, including nusinersen, risdiplam, and onasemnogene abeparvovec, which are affecting the expected disease course of SMA patients. These treatments are expensive (7), and healthcare payers may use selection criteria to reimburse treatment for those most likely to achieve benefit, including SMN2 copy number (8). Such decisions are likely to be especially difficult in patients with three SMN2 copies, who have a more variable clinical presentation. Therefore, the aim of the current systematic literature review is to comprehensively map the clinical characteristics of patients with three SMN2 copies and to identify the effect of disease-modifying treatment in this group.
We conducted a systematic literature review and report according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) standards (9). A literature search was undertaken in October 2022 of the PubMed, Medline, Web of Science, and Cochrane databases using a combination of MeSH terms and free text items to identify articles on SMA that included SMN2 copy number (see Supplementary Methods for full search strategies). No date limits were included. The search results were de-duplicated, and then articles were manually reviewed for inclusion based on the following criteria: (1) English language, and (2) clinical studies reporting SMN2 copy number, which allows identification/definition of subgroups of patients with three SMN2 copies.
Articles were excluded if they were published in languages other than English, or if they were animal or in vitro studies, conference presentations or proceedings, case reports or case series in which fewer than five patients had three or more SMN2 copy numbers, review articles, articles about diseases other than SMA, studies focused on the methodologic detection of SMN2 copies, or studies in which SMN2 copy number was not reported.
From the remaining papers, the authors closely assessed the articles and chose those that specifically examined the impact of three SMN2 copies on disease characteristics or treatment effect.
Studies examining the relationship between SMN2 copy and clinical parameters were assessed for bias using the Appraisal tool for Cross-sectional Studies (AXIS) (10), or the Newcastle-Ottawa Scale (NOS) for cohort or case–control studies (11). The AXIS tool poses 20 questions (one relating to the study introduction, 10 about methods, five about the presentation of the results, two about the discussion of study findings and limitations, one about funding, and one about ethics). Each question is answered with “yes,” “no,” or “do not know” (10). The AXIS tool does not provide an overall rating of bias. The NOS tools assess eight study design elements across three key domains in case–control and cohort studies (11). In case–control studies, the domains are selection of cases and controls (four elements – definition and representativeness of cases and controls), comparability of groups (one element – matching or controlling for confounders) and exposure (three elements – ascertainment of exposure and non-responder rate) (11). In cohort studies, the domains are similar: selection of cohort (four elements – definition and representativeness of cohort, selection of non-exposed cohort, and ascertainment of exposure), comparability of exposed and non-exposed groups (one element – matching or controlling for confounders), and outcome (three elements – ascertainment of outcome, follow-up duration and adequacy of cohort follow-up). Assessors of bias using NOS can apply star ratings of between one and nine stars for each study, with 0–4 stars for selection, 1 or 2 for comparability, and 0–3 for exposure (case–control) or outcomes (cohort) (11). We took account of the rarity of SMA and disease characteristics when applying the NOS. For example, for the assessment of outcomes, NOS gives one star for blinded assessment, but we gave the study one star if outcomes were assessed by trained evaluators using well-established rating scales, even if assessors were not blinded, because blinding is likely not possible in SMA cohort or case–control studies.
Non-randomized studies assessing the impact of SMN2 copy number on treatment outcomes were assessed for bias using the Risk Of Bias In Non-randomized Studies—of Interventions (ROBINS-I) tool (12). This tool is designed to examine how much the study deviates from the ‘gold standard’ randomized, double-blind study. Seven potential sources of bias (domains) are assessed, and each is graded as having a low, moderate, serious, or critical risk of bias. These domains are: (1) confounding; (2) selection of participants into the study; (3) classification of interventions; (4) deviations from intended interventions; (5) missing data; (6) measurement of outcomes; and (7) selection of reported results (12). From these assessments, an overall risk of bias is estimated as low if the study is at low risk of bias in all domains, moderate if the study is at low or moderate risk of risk in all domains, serious if there is a serious risk of bias in at least one domain, and critical if there is a critical risk of bias in at least one domain. Studies with a critical risk of bias were not included in the current analysis, as these studies are considered too problematic to provide any useful evidence (12).
Randomized studies were assessed for bias using the revised Cochrane Risk-of-Bias tool for randomized trials (RoB 2) (13). This tool ranks five potential sources of bias (domains) as having low risk of bias, some concerns, or high risk of bias. The domains are: (1) bias arising from the randomization process; (2) bias due to deviations from the intended interventions; (3) bias due to missing outcome data; (4) bias in the measurement of the outcome; and (5) bias in the selection of the reported results. The overall risk is based on the following criteria: low if the study is judged to be at low risk of bias for all domains; some concerns if there are some concerns in at least one domain; and high if the study has a high risk of bias in at least one domain (14).
Data extraction and bias assessments were undertaken by CR and reviewed/confirmed by CD and RM.
The search identified 392 studies. Of these, 103 were excluded because of the article type and 213 were excluded because of the focus of the content (Figure 1). Overall, 76 articles were reviewed for inclusion, of which 53 assessed disease characteristics and 23 assessed treatment effect.
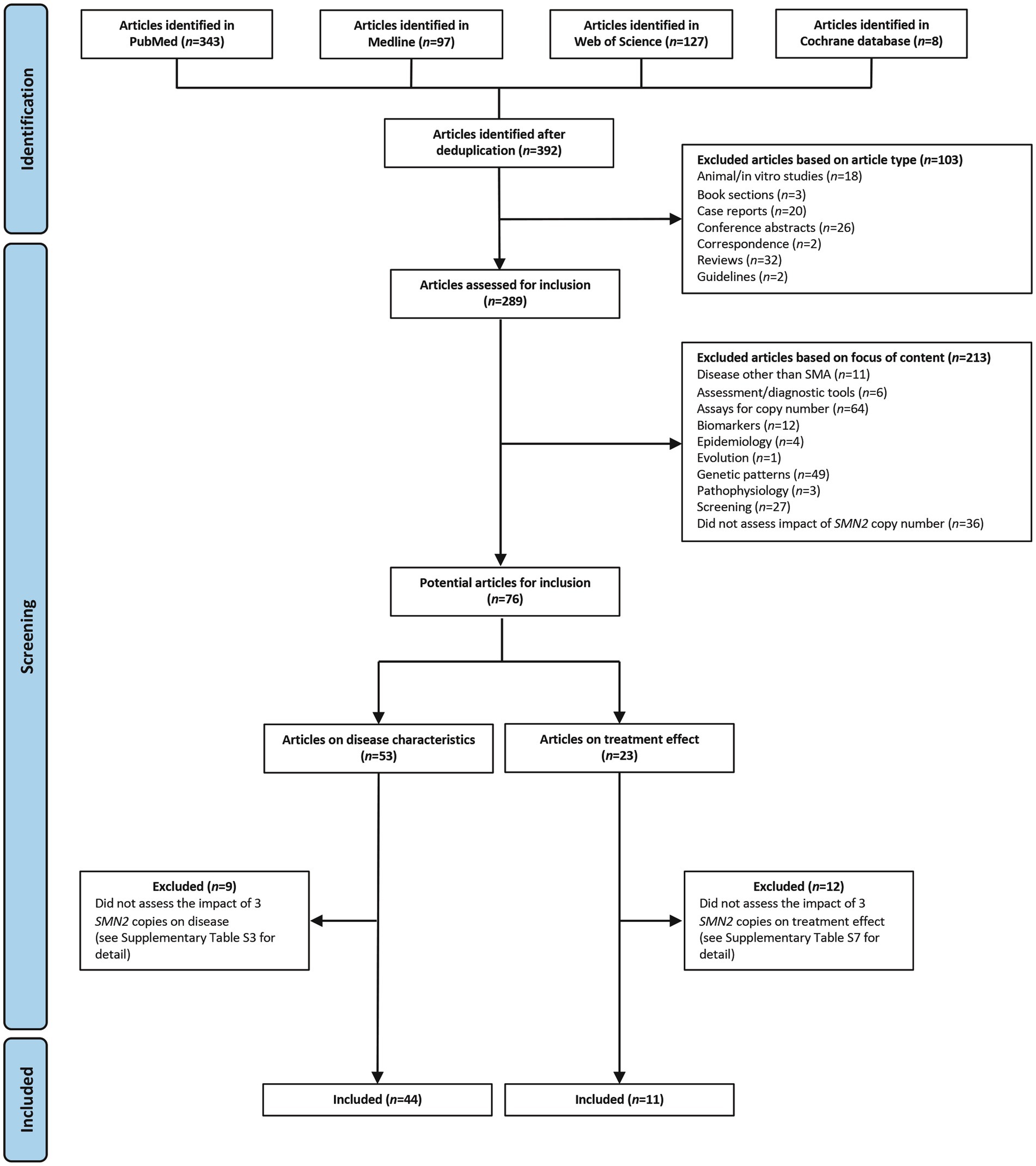
Figure 1. Search results and inclusion of studies. SMA, spinal muscular atrophy; SMN, survival motor neuron.
Of the 53 articles assessing disease characteristics, 44 described the impact of three SMN2 copies and are included in this review (Supplementary Table S2) (6, 15–57); nine articles were excluded because they did not assess the clinical impact of having three SMN2 copies (Supplementary Table S3) (58–66). Of the 44 included studies on disease characteristics, 25 were prospective, 13 were retrospective, two were case–control studies, one was a post-hoc analysis of a cohort of patients from a randomized controlled trial (RCT), and three provided insufficient information on study design to make a determination (Supplementary Table S2). Where the site of the study could be determined (43 studies), two were multinational studies, five were conducted in Italy, five in Japan, three in China, and three in the USA (Supplementary Table S2).
Of the 44 studies, 43 reported SMA types. Overall, 23 included patients with a mix of SMA phenotypes ranging from type 0/I to type III or IV; the others included type 0/I only (six studies), type I or II only (three studies), type II only (two studies), type II or III only (two studies), type II to IV (two studies), type III only (three studies), type III or IV (one study), and one study compared type I and type III.
The cross-sectional studies assessing the relationship between SMN2 copy number and clinical features were all consistent in not including a justification of sample size, and most studies did not discuss limitations (Supplementary Table S4). The studies were generally consistent in a number of features assessed by the AXIS tool, specifically clear study aims, an appropriate design, definition of the sample population, complete presentation of results, complete funding information/no evidence of potential conflicts of interest, and description of ethics (Supplementary Table S4). None of the studies in this analysis included a non-responder group, so three AXIS domains were consistently not relevant (questions 7, 13, and 14). Variability between studies was seen in AXIS domains related to the reporting of study population representativeness (questions 5 and 6).
Most studies scored highly using the NOS cohort or case–control tool, obtaining overall ratings of five (15, 21, 50, 57), six (26, 28, 29, 43, 54), or seven (32, 34, 42, 44, 45, 48, 51–53, 55) stars (Supplementary Table S5). One study obtained four stars (17); bias may be present in this study because there was limited description of how the study population was derived and 25% of patients were not followed up. The major sources of bias in the cohort and case–control studies were the proportion of patients lost to follow-up and the variability and/or adequacy of follow-up duration (Supplementary Table S5).
Of the 44 studies, 21 assessed the relationship between SMN2 copy number and SMA phenotype, and 19/21 studies reported the number of patients with three SMN2 copies within each phenotype group (Table 1) (6, 15, 16, 18, 19, 22, 24, 30, 33, 35, 36, 38, 40–42, 46, 50, 52, 55).
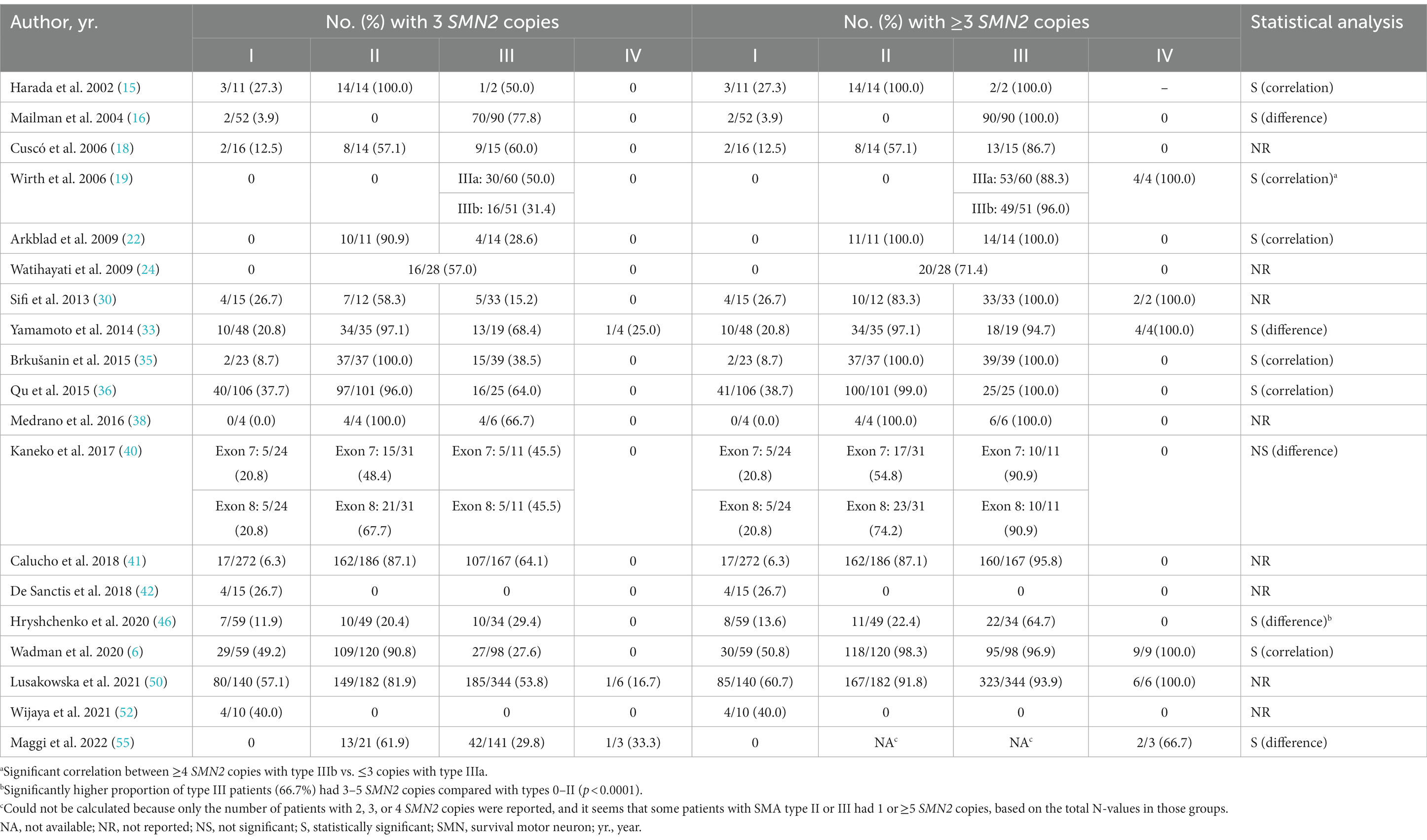
Table 1. Studies evaluating the impact of three SMN2 copies on clinical phenotype.
Wijaya and colleagues (52) reported results that were somewhat anomalous to the usual pattern of milder phenotype with increasing SMN2 copy number. This study examined only SMA patients with an intragenic variant of SMN1 (excluding those with SMN1 deletion), and found that 4/10 patients with SMA type I, but none of those with SMA type II or III, had three copies of SMN2 (52).
A study by De Sanctis and colleagues included only patients with SMA type I, and reported that 4/15 infants had three SMN2 copies; three with a milder phenotype and late onset and one with a typical type I phenotype (42).
A statistically significant effect of SMN2 copy number on SMA phenotype was reported in all but one of the 12 studies that undertook statistical analysis (6, 15, 16, 19, 22, 33, 35–37, 40, 46, 55). Excluding the study by Wijaya and colleagues (52), the studies reported that three SMN2 copies were present in between 0 and 57.1% of patients with SMA type I, 20.4%–100.0% of patients with SMA type II, 15.2%–77.8% of those with SMA type III, and 0%–33.3% of those with SMA type IV (Table 1). While the proportion of type II SMA patients with three or more SMN2 copies was quite variable (22.4%–100.0%), 65%–100% of patients with type III or type IV SMA had three or more SMN2 copies (Table 1).
Two other studies examined the relationship between SMA phenotype and SMN2 copy number but did not report the number of patients with three SMN2 copies in different phenotype groups (37, 47). Mendonça and colleagues compared patients who were homozygous for an SMN1 deletion with those who were compound heterozygotes (47). They reported that, in homozygous SMA patients, 65% of those with SMA type II and 69.6% of those with SMA type III had three SMN2 copies, and there was a clear relationship between phenotype and SMN2 copy number. However, no correlation was found between SMN2 copy number and disease phenotype in patients with a compound heterozygous genotype (47). This is consistent with the findings by Wijaya and colleagues in patients with SMN1 intragenic variants described above (52).
Zarkov and colleagues reported the mean number of SMN2 copies in patients with SMA type II, III, or IV, and found that the mean number of copies was higher in milder phenotypes (mean 3.1 in type II, 3.7 in type III and 4.2 in type IV; p < 0.05) (37).
Several studies, including the one by De Sanctis and colleagues described above, noted discordance between SMN2 copy number and expected clinical phenotype. Lusakowska and colleagues reported that 85/140 patients with type I SMA (60.7%) in the Polish SMA registry had three or more SMN2 copies (50); the cohort of 285 Dutch patients reported by Wadman and colleagues contained 30 patients with type I SMA who had three or more copies of SMN2 (6); and Hryshchenko and colleagues found three SMN2 copies in two patients with the most severe form of SMA (type 0) (46). In fact, Wadman and colleagues noted that phenotype was most variable among patients with three SMN2 copies, ranging from those unable to sit independently (type Ic) to ambulant patients (type IIIa) (6).
Of the 44 studies, 13 reported on the impact of SMN2 copy number on clinical course/prognosis (17, 21, 23, 32, 36, 37, 39, 42, 49–51, 55, 57), of which eight provided data on age at onset and/or mortality (Table 2) (21, 23, 36, 49–51, 55, 57).
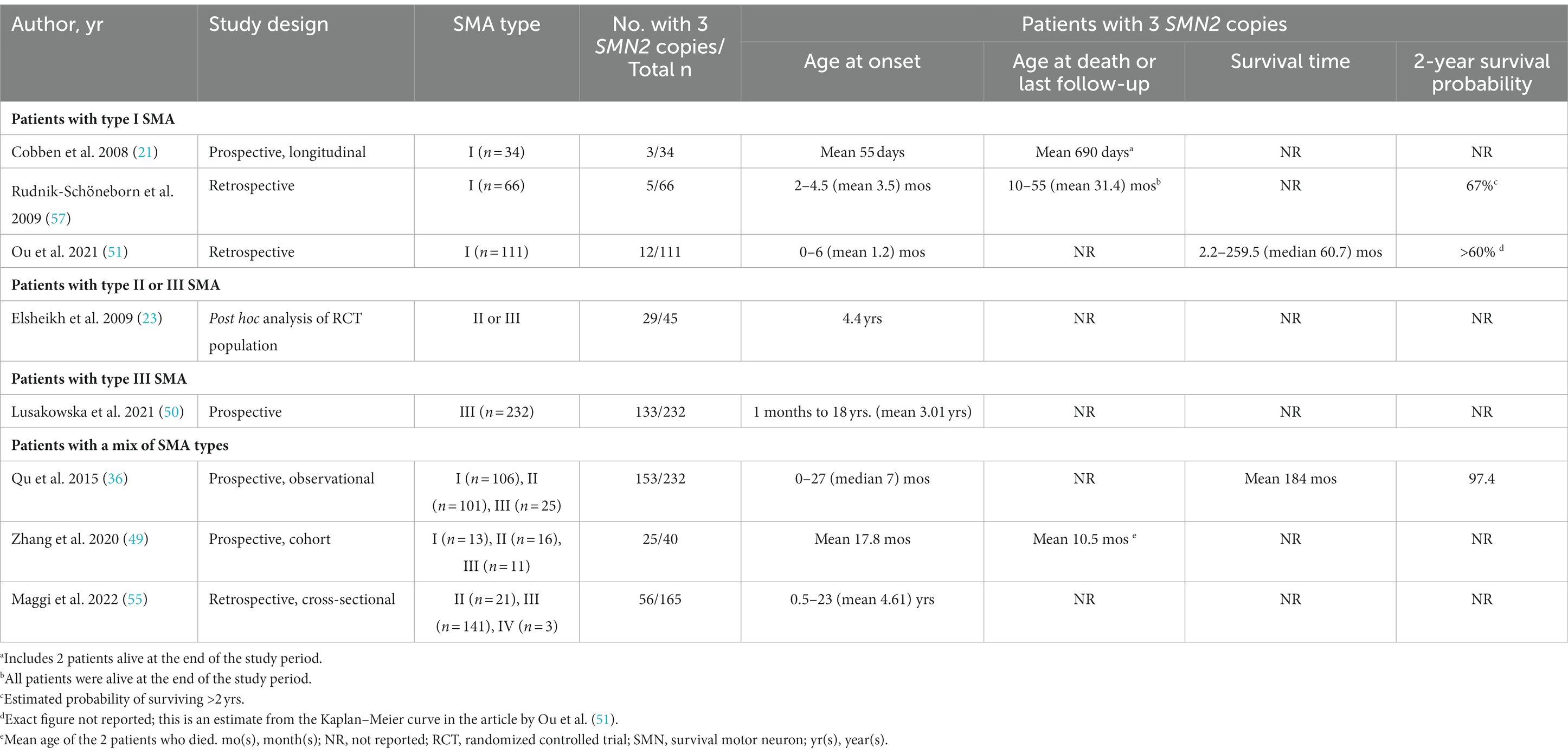
Table 2. Studies examining the relationship between SMN2 copy number and natural history.
Three studies reported the age at onset in children with type I SMA and three SMN2 copies; the mean age was 1–2 months in two studies (21, 51), and 3.5 months in the other (57). One study reported a similar age at onset in children with two or three SMN2 copies (51), but the other two studies reported a later age at onset in type I SMA patients with three SMN2 copies than in those with up to two SMN2 copies (21, 57).
Prognosis tended to be better in type I patients with three vs. two SMN2 copies in two studies (51, 57), but there were too few patients with three SMN2 copies in the study by Cobben and colleagues to draw any conclusions about the relationship between survival and SMN2 copy number (21). Survival was longer in type I SMA patients with three vs. two SMN2 copies (median 60.7 vs. 9.2 months) in a Taiwanese cohort study, but the survival probability between the groups did not reach statistical significance (p = 0.0687) (51). Two of the studies in type I SMA patients reported the estimated 2-year probability of death in those with three SMN2 copies, and the survival was 60%–67% in both studies (51, 57).
A small Italian study in patients with type I SMA (15 had SMN2 copy number available) found consistently more rapid deterioration in motor function in patients with two SMN2 copies than in those with three SMN2 copies (42), although motor function decline was variable.
In patients with type II or III SMA, the mean age at clinical onset was significantly lower in patients with three vs. four SMN2 copies [mean 4.4 vs. 7.5 years in one study (23) and mean 3.01 vs. 6.71 years in the other (50)].
Unsurprisingly, the studies in patients with a range of SMA phenotypes (I–IV) reported a wider range of ages at onset (Table 2) (36, 49, 55). Qu and colleagues reported that Chinese patients with three SMN2 copies had a median age of onset of 7 months, compared with 1.15 months in those with two SMN2 copies and 18 months in those with four SMN2 copies; age at onset was strongly correlated with SMN2 copy number (r = 0.72, p < 0.0001) (36). These researchers also found a significant association between SMN2 copy number and mortality; patients with two SMN2 copies had a much higher risk of mortality compared with those who had three or four SMN2 copies [odds ratio (OR) 186; 95% confidence interval (CI) 91.1–1812.7; p < 0.0001] (36). Similar results were reported by Zhang and colleagues in their study of 40 Chinese patients with SMA type I (n = 13), II (n = 16), or III (n = 11) (49): mean age at onset was 6.1, 17.8, and 31.4 months, respectively, in those with two, three, and four SMN2 copies. None of the five patients with four SMN2 copies had died, compared with 2/25 with three SMN2 copies (8.0%) and 5/10 (50.0%) with two SMN2 copies; mean age at death was 10.5 months in the two patients with three SMN2 copies vs. 5.2 months in the five patients with two SMN2 copies (49).
In contrast to the two Chinese studies described above (36, 49), a retrospective cross-sectional study from Italy including patients with SMA II–IV reported a similar age at onset in patients with two or three SMN2 copies (median of 2 years in both groups; mean of 7.47 years in those with two copies vs. 4.61 years in those with three copies), although it should be noted that only seven patients in this study had two SMN2 copies whereas 54 had three copies and 74 had four copies (55).
A US study examined the impact of SMN2 copy number on age at death or at the combined endpoint of death and requirement for ventilation for ≥16 h/day in 79 patients with SMA type I or II (32). Among those with SMA type I, the median age at the combined endpoint was 10.5 months for those with two SMN2 copies but not reached for those with three SMN2 copies (25th percentile age in this group was 22 months) (32).
Six studies noted a relationship between SMN2 copy number and loss/retention of ambulation (17, 23, 37, 39, 50, 55). These populations also included patients with SMA II, who by definition never achieve ambulation, but generally showed a linear relationship between SMN2 copy number and proportion of ambulatory patients in cohort studies (Figure 2). In their cohort of patients with SMA type II, III, or IV, Zarkov and colleagues noted that patients with two SMN2 copies lost ambulation earlier than patients with three SMN2 copies (after a mean of 23.7 vs. 37.4 years, respectively; p < 0.05), but found no significant correlation between SMN2 copy number and achievement of motor milestones (37). SMN2 copy number was also significantly associated with the probability of remaining ambulant over time in patients with type III SMA, with 70% of those with three SMN2 copies still walking after 10 years and 60% after 20 years, compared with 91 and 82%, respectively, of those with four SMN2 copies (p < 0.0001) in the study by Lusakowska and colleagues (50). Similarly, SMN2 copy predicted the age at loss of ambulation in an Italian cohort of patients with SMA type II, III, or IV, although ambulation was lost at a younger age in this cohort than in the study by Zarkov and colleagues (12 years in those with two SMN2 copies and 16.5 years in those with three SMN2 copies) (55).
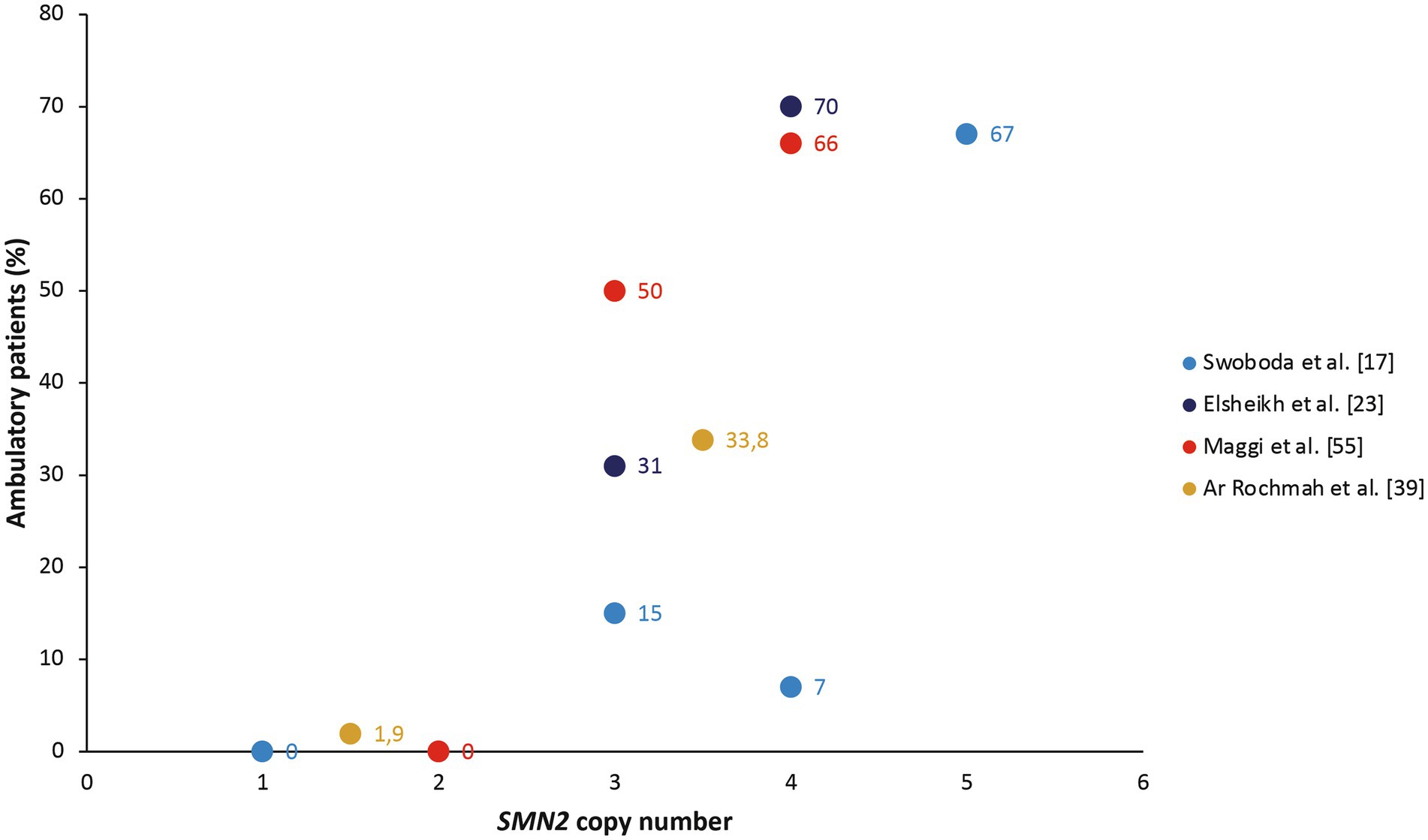
Figure 2. The proportion of ambulatory patients in cohort studies, stratified by SMN2 copy number (17, 23, 39, 55). The study by Ar Rochmah et al. grouped patients into those with one or two SMN2 copies compared with three or four SMN2 copies (39).
Of 44 studies, 16 reported on the relationship between SMN2 copy number and muscle strength, functional status (motor, neurologic, or pulmonary), and other signs/symptoms (Table 3) (17, 20, 23, 26, 28, 29, 31, 37, 42, 44, 45, 48, 53–56).

Table 3. Studies evaluating the impact of three SMN2 copies on functional parameters.
Five studies investigated the relationship between neurophysiologic parameters and SMN2 copy number (17, 23, 26, 29, 31).
Swoboda and colleagues found a greater deterioration in motor unit number estimation (MUNE) and compound muscle action potential (CMAP) over time among SMA patients with up to two SMN2 copies compared with three or more copies (17), and Farrar et al. reported a significant correlation between SMN2 number and CMAP (29). These studies were in a mixed population of patients with type I, II, or III SMA; however, the two studies that used myometry in patients with type II or III SMA found no significant relationship between muscle strength and SMN2 copy number (26, 31).
Elsheikh and colleagues found no difference in maximum voluntary isometric contraction (MVIC) between patients with three vs. four SMN2 copies, but did find a significant correlation between MVIC and SMN2 copy number (23).
One study examined neurologic function in infants identified by newborn screening (n = 17), and found that a significantly higher proportion of those with three or more SMN2 copies had optimal scores on the Hammersmith Neonatal Neurological Examination (HNNE) scale compared with infants who had up to two SMN2 copies (66.7% vs. 14.3%; p = 0.036) (56).
Eight studies examined the impact of SMN2 copy number on motor function using the Hammersmith Functional Motor Scale (HFMS) or HFMS Expanded (HFMSE; seven studies) (20, 26, 28, 29, 44, 45, 55), gross motor function measure (GMFM; two studies) (26, 28), revised version of the upper limb module (RULM; one study) (55), and Children’s Hospital of Philadelphia infant test of neuromuscular disorders (CHOP INTEND; one study in children with type I SMA) (42).
The only study in children with SMA type I reported a more rapid decline in CHOP INTEND scores in patients with two vs. three SMN2 copies, but SMN2 copy number data were available for only 15/20 patients (75%), and no statistical analysis was undertaken on these findings (42).
In cross-sectional analyses in patients with SMA type II or III, HFMSE scores were significantly lower in patients with two vs. three SMN2 copies (20), or those with three vs. four SMN2 copies (28), and there was a significant correlation between SMN2 copy number and HFMSE scores in patients with types I, II, or III (29).
On the other hand, the results of longitudinal assessments appeared to show no strong association between SMN2 copy number and changes in motor function. Kaufmann and colleagues found no significant association between SMN2 copy number and change in HFMSE or GMFM over time (26, 28). Similarly, Coratti and colleagues reported no significant association between SMN2 copy number and gain/loss of functional ability over time in patients with type II SMA (44), or between SMN2 copy number and HFMSE scores over time in patients with type III SMA (45). Farrar and colleagues reported an age-dependent decline in HFMSE scores among patients with two SMN2 copies, but relatively stable HFMSE scores in those with three copies (29).
Two studies examined ambulatory function in adult patients with type III SMA using the North Star Ambulatory Assessment (NSAA) and 6-min walk test (6MWT) (31), or the 10-min walk test (10MWT) (54).
Tiziano and colleagues found no relationship between NSAA or 6MWT and SMN2 copy number (31), whereas Krosschell and colleagues reported a 45% slower 10MWT among patients with three vs. four copies (p = 0.013) (54), in line with expectations. According to Townsend and colleagues (48), children with three SMN2 copies were eight times more likely to consistently use a stander than patients with two copies (p < 0.001).
Two studies examined the relationship between other signs/symptoms and SMN2 copy number (37, 53).
Zarkov and colleagues reported no significant correlation between SMN2 copy number and spine deformities or limb contractures in patients with type II, III, or IV SMA (37). Hanna and colleagues reported a higher incidence of hip pain among patients with type I or II SMA and three SMN2 copies (53%) compared with two SMN2 copies (17%). Only two patients in this study had four SMN2 copies and one-half of them had hip pain (53).
Four articles reported the relationship between forced vital capacity (FVC) and SMN2 copy number in patients with SMA type II or III (26, 28), type III (31), or type II, III or IV (55).
The largest of these studies (in 165 patients with type II, III, or IV) found a significantly lower FVC in patients with three vs. four SMN2 copies (74.49% vs. 92.5% predicted; p = 0.018) (55). However, in a cross-sectional study in type III patients, there was no significant association between FVC and SMN2 copy number (31), and in a prospective longitudinal study in patients with type II or III SMA, there was no significant association between SMN2 and change in FVC over time (26, 28).
Two studies examined the relationship between SMN2 copy number and plastin 3 (PLS3) expression in patients with type I, II, or III SMA (25, 34), and one examined the impact of SMN2 copy number on SMN protein levels in patients with a range of SMA types (I–IV), including nine presymptomatic patients (Table 4) (43).
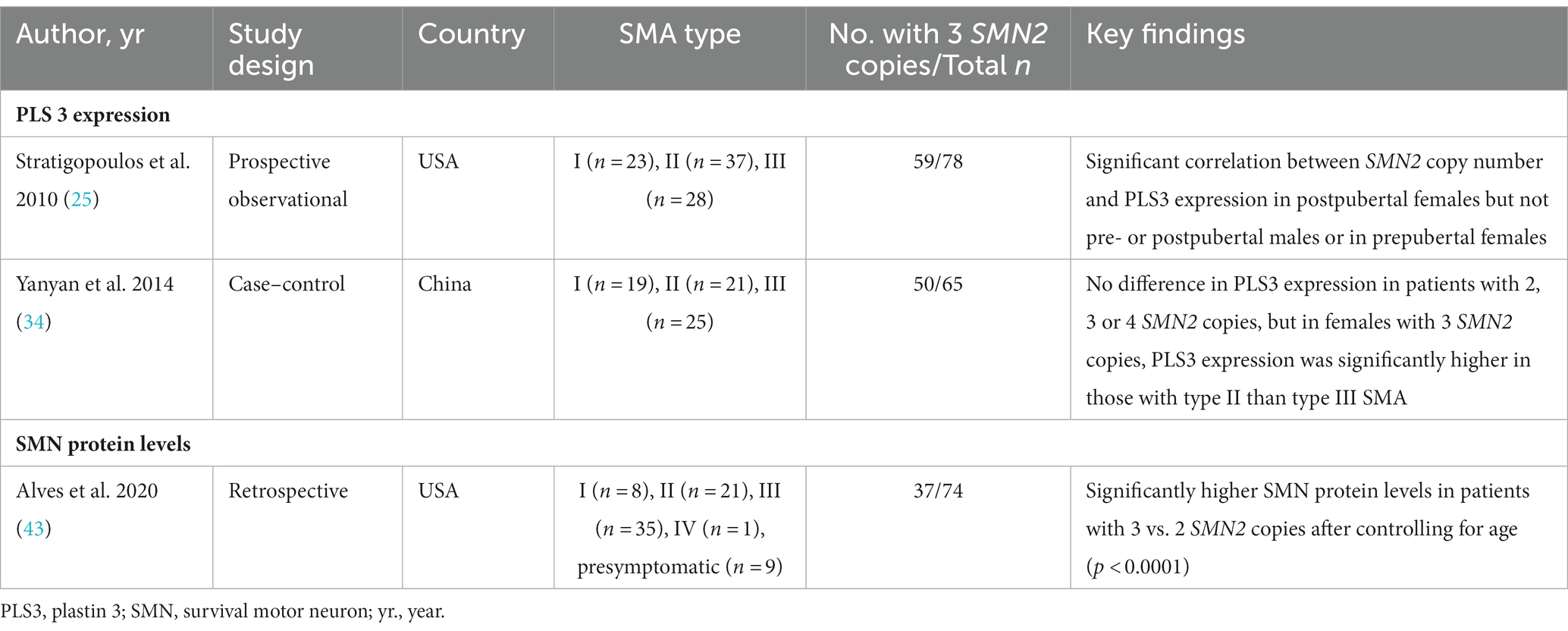
Table 4. Studies evaluating the impact of three SMN2 copies on biomarkers.
The two studies of PLS expression suggest that PLS expression may modify the SMA phenotype in an age- and/or sex-specific manner. Stratigopolous and colleagues found a significant correlation between SMN2 copy number and PLS3 transcript levels in postpubertal females, but not in prepubertal females or in males in either age group (25). In postpubertal females, PLS3 expression was also significantly correlated with motor function (assessed using GMFM) (25). A Chinese case–control study found no significant difference in PLS3 expression between patients with two, three, or four SMN2 copies (34). However, in the subgroup of female patients with three SMN2 copies, PLS expression was significantly higher in those with SMA type II vs. type III (34).
A US study found that SMN protein levels in whole blood were significantly higher in patients with three vs. two SMN2 copies (p < 0.0001), and between patients with type II, III, or IV SMA (p < 0.0001), after controlling for age (43).
Overall, 23 studies on SMA treatment included SMN2 copy number (67–89), but only 11 of these studies examined treatment effects in patients with three SMN2 copies (Supplementary Table S6) (68, 69, 71, 74, 79, 80, 83, 85, 87–89). The 12 excluded studies did not allow analysis of treatment effects in patients with three SMN2 copies (Supplementary Table S7) (67, 70, 71, 73, 75–78, 81, 82, 84, 86).
Among the 11 included studies, nine were in patients receiving nusinersen (68, 69, 71, 74, 80, 83, 85, 88, 89), one was in patients receiving onasemogene abeparvovec (87), and one was in patients receiving a range of treatments, including nusinersen, onasemnogene abeparvovec, and risdiplam (79). Study design was not clearly defined in one of the nusinersen studies (68); the study of patients receiving a mix of treatments was retrospective (79), but all other studies were prospective (69, 71, 74, 80, 83, 85, 87–89). One nusinersen study was a subanalysis of the phase 2 NURTURE study (74), one was the phase 3 randomized CHERISH study (89), and the study with onasemnogene abeparvovec was a subanalysis of the SPR1NT study (87). The other prospective studies were observational cohort studies.
The non-randomized studies in this analysis had an overall ROBINS-I rating of moderate (six studies) or serious (three studies; Supplementary Table S8). In all non-randomized studies, the risk of bias from participant selection and missing data was low (Supplementary Table S8). However, all non-randomized studies had at least a moderate risk of confounding, which resulted in none of the studies having a low overall risk of bias.
The randomized study (CHERISH) had a RoB2 overall rating of ‘some concerns’. There was a low risk of bias due to deviations from intended interventions, missing outcomes data, outcome measurement or selection of reported results (Supplementary Table S8). However, there were some concerns about bias due to randomization because nusinersen or control was administered by personnel who were aware of the group assignments.
The nine studies in patients receiving nusinersen included a total of 506 patients, of whom 230 had three SMN2 copies. Where SMA type was specified, 68 patients were presymptomatic infants; the others had type I (n = 296), type II (n = 14), or type III (n = 2) SMA. The SMA type was not specified in the CHERISH study cohort (n = 126), but these patients were considered to have type II or III SMA based on the inclusion criteria (89).
Seven studies – one subanalysis of the phase 2 NURTURE study in presymptomatic infants (74), one real-world study in patients diagnosed by newborn screening (88), and five prospective cohort studies in patients with type I SMA (68, 69, 80, 83, 85)—compared outcomes in patients with two vs. three or more SMN2 copies. Of these seven studies, four reported baseline and follow-up data in groups with two vs. three SMN2 copies, and are summarized in Table 5. There was an improvement in functional scores (CHOP INTEND, HINE-2, and MFM 20) during nusinersen treatment in patients with two or three or more SMN2 copies in five studies (69, 74, 80, 83, 85). Only one study statistically compared patients with two vs. three SMN2 copies, and found no significant difference in functional scores (HINE-2, CHOP INTEND, MFM-20 or − 32) between groups (69). However, another observational study reported that SMN2 copy number was a significant predictor of outcomes among nusinersen-treated patients with type I SMA (83).

Table 5. Studies examining the effects of nusinersen in presymptomatic or type I SMA patients with three vs. two SMN2 copies.
A study by Aragon-Gawinska and colleagues found that SMN2 copy number had no significant effect on whether children with type I SMA achieved sitting status during nusinersen treatment (68). However, this study had a serious risk of bias, because patients were assigned to groups for analysis based on sitting status achieved after treatment, and there were differences between these groups with regard to cointerventions (68).
A Polish study reported that SMA type I patients with three or more SMN2 copies had higher CHOP INTEND scores than those with two copies (p = 0.013), and tended to show a greater improvement during treatment, although this did not reach statistical significance (80).
The NURTURE subanalysis in presymptomatic patients showed that patients with three SMN2 copies receiving nusinersen were less likely to develop SMA symptoms, and more consistently achieved motor milestones and at a younger median age compared with the group with two SMN2 copies (Table 5), although the between-group difference was not statistically tested (74). This study had a moderate risk of bias, but was one of the few non-randomized studies to have a low risk of bias in outcome measurement because it was part of a clinical trial.
A separate study in infants identified by newborn screening found that presymptomatic patients treated with nusinersen remained asymptomatic and achieved normal motor milestones during follow-up, and this was true of eight patients with two SMN2 copies and six with three SMN2 copies (88). In contrast, all untreated infants with two or three SMN2 copies developed symptoms (88). However, the study authors did not adjust for confounding, so this study had a serious risk of bias.
The randomized phase 3 CHERISH study compared nusinersen and sham control in patients with type II SMA or milder (later onset SMA), and found that nusinersen was significantly more effective than control at improving HFMSE score (the primary endpoint; p < 0.001) (89). A significant improvement was seen in all subgroups of patients based on SMN2 copy number, but most patients in this study (111/126; 88%) had three SMN2 copies (89). While there are some concerns regarding the risk of bias associated with potential differences in the baseline characteristics between the nusinersen and control groups (lower mean HFMSE score at baseline, longer median disease duration and higher proportion of patients who could walk with support in the control group than the nusinersen group), the randomized controlled nature of the study means that results can be considered robust.
A small US study investigated nusinersen in patients with type I, II or III SMA, but focused on the feasibility of using an indwelling subcutaneous catheter to administer nusinersen in patients with complex spinal anatomy (71). The study reported an improvement in sum total force (an aggregate of 11 dynamometry maneuvers) after starting nusinersen in one patient aged 6 years with four SMN2 copies and in three patients aged 5–10 years with three SMN2 copies, but not in six patients with three SMN2 copies aged 13–30 years (71). Risk of bias in this study was considered to be moderate.
Strauss and colleagues reported a subgroup analysis from the phase 3 SPR1NT study in presymptomatic infants at risk of developing SMA who had three SMN2 copies (n = 15) (87). They did not directly compare outcomes in treated patients with three vs. two SMN2 copies, because the primary study endpoint was different between groups (independent sitting at any visit up to 18 months of age in those with two SMN2 copies vs. independent standing within 24 months of age in those with three SMN2 copies) (90). All 15 infants with three SMN2 copies achieved the primary endpoint (independent standing for ≥3 s at any visit up to 24 months of age); the median time to this milestone was 377 days, and 14/15 (93%) achieved independent standing within the normal developmental window, compared with 24% in the Pediatric Neuromuscular Clinical Research (PNCR) natural history study (p < 0.0001). Overall, 14/15 infants (93%) were able to walk independently for ≥5 steps at any visit up to 24 months of age, with a median age of independent walking of 422.0 days, and 11/15 (73%) within the normal developmental window (87).
Similar primary endpoint results were seen in the patients with two SMN2 copies, with 14/14 of these patients (100%) achieving independent sitting at any visit up to 18 months of age (90). However, while 93% of patients with three SMN2 copies achieved independent standing within the World Health Organization (WHO) developmental window (87), this endpoint was achieved by 50% of patients with two SMN2 copies (90), and the median time to independent walking was 422 days in those with three copies vs. 493 days in those with two copies (87, 90).
All 15 of the patients with three SMN2 copies treated with onasemnogene abeparvovec were alive and free from permanent ventilation at the end of the study (87). Risk of bias in this study was considered to be moderate.
Lee and colleagues retrospectively assessed treatment patterns and outcomes in infants identified through the newborn screening program implemented in New York state in 2018 (79). Of the 34 infants identified in the screening program, 32 received treatment, of whom 11 had three SMN2 copies. Treatments received by screened infants were onasemnogene abeparvovec alone (n = 23), nusinersen alone (n = 1), risdiplam alone (n = 1), nusinersen as a bridge to onasemnogene abeparvovec (n = 5), or risdiplam after initial treatment with onasemnogene abeparvovec (n = 2).
Ten of the 11 patients with three SMN2 copies had received onasemnogene abeparvovec alone and the other received nusinersen as a bridge to onasemnogene. The 11 infants with three SMN2 copies were all asymptomatic at the time of treatment (age 11–94 [median 34] days) and remained asymptomatic at last follow-up (age at last follow-up 1.5–26 months), when they had met age-appropriate developmental milestones and had normal neurologic function. All seven children with three SMN2 copies aged ≥ 12 months at follow-up were walking independently. None of the treated patients with three SMN2 copies required ventilatory or feeding support (79). Risk of bias in this study was considered to be serious based on the potential for confounding.
This systematic review confirms that SMN2 copy number is strongly correlated with SMA phenotype in patients with SMN1 deletion. It also confirms that patients with three copies of the SMN2 gene have a more variable clinical presentation/phenotype than patients with up to two copies or four or more copies, and that patients with three SMN2 copies may have SMA ranging from severe (type I) to mild (type IIIc) (91). These data support the contention that SMN2 copy number is not sufficient to explain the variability of clinical presentation in patients with SMA. The largest analysis of data on this issue to date indicates that three SMN2 copies are present in 20% of type I patients, 78% of type II patients, and 49% of type III patients (41), in line with the published data in our analysis (Table 1).
Our analysis found no relationship between SMN2 copy number and phenotype in SMA patients who carry an SMN1 mutation, although this was examined in only two studies in a small number of patients (47, 52). Newborn screening identifies only patients with SMN1 deletion (92), so patients with SMN1 mutation will not be identified until symptoms develop. The lack of relationship between SMN2 copy number and phenotype in these patients with SMN1 mutation makes it difficult to provide prognostic information to parents of these children.
In relation to life expectancy in SMA type I, three studies consistently reported better odds of survival and/or longer survival duration in type I SMA patients with three copies than in those with two copies (21, 51, 57). Another study reported that SMN2 copy number was a significant protective factor for survival with early death in 4/4 patients with only one SMN2 copy (mean survival 4 months) (66). The mean SMN2 copy number was significantly higher in type I patients who survived >2 years (mean 2.89 copies) than in those who died before the age of 2 years (mean 2 copies) (66). The lack of information on the impact of SMN2 copy on survival duration in patients with types II, III, or IV SMA is not surprising, given the life expectancy of these patients (93), and the need for large cohorts and long follow-up to develop accurate data.
In addition, the data indicate that patients with three SMN2 copies have an age at onset and time to ventilator dependence or loss of ambulation that is intermediate between patients with up to two copies or four or more copies. Cross-sectional studies using tests of ambulatory function (e.g., 6MWT) did not find a significant difference between groups based on SMN2 copy number, which is consistent with other reports that these parameters show too much interindividual variation and overlap between phenotype groups to provide valuable information on their own (94). This indicates that, within the same class of motor milestones (e.g., walking), the number of SMN2 copies is not sufficient to define clinical classification, even if there is an overall correlation with prognosis.
Our findings indicate that the relationship between SMN2 copy number and neurophysiologic parameters depends on what is measured; CMAP was significantly related to SMN2 copy number (17, 29), but myometric measures were not (26, 31). A relationship between CMAP amplitude and SMN2 copy number has even been detected in presymptomatic patients identified by newborn screening, and CMAP may be a sensitive measure of motor function impairment in infants before overt symptoms develop (95). Our analysis also suggests that motor function (measured using standard scales such as HFMSE or CHOP INTEND) was better in patients with three vs. two SMN2 copies, at least in cross-sectional studies, but the evidence from longitudinal studies did not consistently show a slower decline in motor function among patients with three vs. two SMN2 copies.
While these data provide additional information on the natural history of SMA which may assist in the counseling of patients/parents about what to expect, SMN2 copy number is only one factor moderating the clinical severity/phenotype of SMA patients. Therefore, the identification of other biomarkers is needed to guide phenotypic or prognostic estimations. NAIP copy number also shows a relationship with the clinical severity of SMA (6, 24, 27, 36, 49). Two groups of Chinese researchers used combined genotype information from SMN1-SMN2-NAIP as a prognostic marker, and both noted that patients with a 0–3-1 genotype were significantly less likely to develop type I SMA and to have significantly better survival compared with patients harboring the 0–2-0 genotype (36, 49). Within groups of patients with two or three SMN2 copies, the presence of NAIP copies modified the risk of survival and disease progression (36, 49), suggesting that more nuanced genotyping will become part of the SMA prognostic algorithm in future. Some of the other biomarkers being considered in SMA include PLS3 and/or coronin 1C expression, SMN protein levels in blood, microRNA, neurofilament proteins, creatine kinase or creatinine levels, and Tau levels in the CSF (43, 94, 96–98). We identified only three papers evaluating the relationship between SMN2 copy number and PLS3 or SMN protein levels (25, 34, 43). The authors of the study on SMN protein levels in whole blood speculated that this biomarker could provide adjunctive information (in addition to SMN2 copy number) about the likely phenotype of young patients with SMA and help to inform treatment decisions (43). Further research is needed to identify biomarkers relevant to the natural history and treatment response of patients with SMA.
Our review identified 11 studies examining the impact of SMN2 copy number on treatment effects, but like previous systematic reviews (99), we did not identify any studies that assessed the impact of copy number on survival in treated patients. To date, the largest number of publications are about nusinersen. Based on the available data, nusinersen appears to be as effective in patients with three SMN2 copies as in those with up to two copies (68, 69, 71, 74, 80, 83, 85, 88, 89). However, there is potential for bias in observational assessments, particularly when SMN2 copy number is related to disease phenotype, and therefore to decisions about treatment. In the studies that presented baseline clinical characteristics in the groups with two vs. three SMN2 copies, age-related motor function at baseline tended to be better and age at first nusinersen dose tended to be higher in those with three vs. two copies (69, 74). Even if age at first dose is comparable, the expected slower decline in motor function in untreated patients with three vs. two SMN2 copies complicates the comparison of treatment response between copy number groups (17, 29, 42).
The randomized phase 3 CHERISH study was the only study to include a control group, and the data showed similar motor function improvement during nusinersen treatment in the patients with three vs. two SMN2 copies; however, only 9% of patients in this analysis had two SMN2 copies compared with 87% with three SMN2 copies (89), so the data should be confirmed in larger studies.
The SPR1NT study examined the effect of SMN2 copy number on gene therapy (onasemnogene abeparvovec) efficacy in presymptomatic patients, and found that outcomes were significantly better in treated patients with three SMN2 copies than in a historical control group of untreated patients with two or three SMN2 copies (87). However, the lack of a direct comparator hampered interpretation of these findings.
As newborn screening becomes more widespread, a growing proportion of the candidates for treatment are presymptomatic infants, and questions arise about the cost-effective application of treatment-modifying therapies, particularly in infants with a milder phenotype. The European ad hoc guidelines for the use of gene therapy in SMA recommend that, in presymptomatic infants, SMN2 copy number should be used to select patients for treatment, because it is currently the most accurate predictor of age at onset and clinical severity (100). In all other patients, age at onset, disease duration, and motor function status are the most important factors that predict response to pharmacologic treatments.
Our review identified three studies on the use of nusinersen in this setting [the open-label phase 2 NURTURE study (74), a prospective cohort study (88), and a retrospective study (79)] and two on onasemnogene abeparvovec [a retrospective study (79) and the SPR1NT study (87, 90)], but no published studies on risdiplam. These data showed that early use of disease-modifying treatment delayed the onset of symptoms and maintained motor function in patients with three SMN2 copies. A recent systematic review of clinical trials and real-world studies examining the impact of treatment in presymptomatic infants seems to confirm these findings, although a longer follow-up will be necessary to verify the clinical outcome of these patients (1). Real-world data show that almost all infants with three SMN2 copies who began treatment with disease-modifying therapy within the first 6 weeks of life had normal motor development and reached age-appropriate milestones; infants with two SMN2 copies also derive considerable benefit but the proportion of patients achieving normal motor development was smaller in this group than in those with three SMN2 copies (1).
Quantification of SMN2 copy number is not straightforward and a number of different techniques may be used (5). The most common technique is multiplex ligation-dependent probe amplification (MLPA), but results are not always concordant between MLPA assay kits or between laboratories using the same assays (88, 101). European guidelines recommend that SMN2 copy number analysis is undertaken by an expert laboratory with effective quality control measures in place (100).
SMN2 copy number may not align with expectations based on the patient phenotype (5), and our review suggests that this is most likely in patients with three SMN2 copies. A number of specific mutations in SMN2 may modify disease severity, and there is evidence of structural differences in the SMN2 gene copies within the same patient (6). Patients with two or three SMN2 copies and an unexpected phenotype may have rare positive variants of SMN2 associated with a milder phenotype (e.g., c.859G>C or c.835-44A>G) (5, 6). However, if patients with one or four or more SMN2 copies show a discordant phenotype, physicians should consider retesting for SMN2 copy number with a new sample and/or a different laboratory or technique (5).
Because of the difficulties inherent in conducting clinical trials in patients with rare diseases (102), this systematic review included a small number of randomized trials, as only a few trials are conducted in patients with SMA. Our search identified only one RCT, so there was no opportunity for meta-analysis. However, this is to be expected in rare disease research where there are few patients and ethical concerns often preclude the use of placebo (102). Most studies were small, and many were excluded because they did not specifically assess the impact of three SMN2 copies on outcomes. Differences in the study designs and endpoints limit the conclusions that could be drawn. Several pharmacologic studies have recently been presented at conferences, but were excluded from this analysis because the data have not been peer-reviewed.
Some studies in this review lacked sufficient detail in reporting study design to conduct a detailed assessment of bias. Moreover, almost all studies in our analysis were observational and potentially affected by bias. This may be particularly true in observational treatment studies where decisions around the use and timing of treatment are likely to be related to disease phenotype, which (as we have shown) is affected by SMN2 copy number. For example, we used the ROBINS-I tool for the assessment of bias in non-randomized intervention studies (12); most non-randomized trials will be assessed as having at least moderate risk of bias using this tool (103).
This review of the available literature indicates that SMA patients with three SMN2 copies show a more variable clinical presentation than those with one, two, four, or five copies. In infants and children with type I SMA or in presymptomatic infants with an SMN1 deletion, having three SMN2 copies is associated with a later onset of symptoms, a slower decline in motor function and longer survival compared with having two copies of SMN2. In patients with SMA type II or III, having three SMN2 copies is associated with earlier symptom onset, loss of ambulation and ventilator dependence compared with having four SMN2 copies. Early disease-modifying treatment (with nusinersen or onasemnogene abeparvovec) delays the onset of symptoms in presymptomatic patients, and may help these patients with three SMN2 copies to remain asymptomatic and meet normal motor milestones, but the studies were mostly small and uncontrolled. In the RCT (CHERISH), nusinersen was as effective at improving motor function in patients with three SMN2 copies as it was in those with two SMN2 copies. Given the variable clinical phenotype of SMA patients with three SMN2 copies, more research is needed in additional biomarkers to help the prognosis and response to treatment of patients with three SMN2 copies.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
CD: Conceptualization, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing. RM: Conceptualization, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors declare that this study received funding from Novartis Pharma Italy. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
The authors would like to thank Catherine Rees who, under the guidance of the authors, undertook data extraction, wrote the outline and first draft of this manuscript and conducted the bias assessments, on behalf of Springer Healthcare Communications. This medical writing assistance was funded by Novartis Pharma Italy.
The authors report personal fees from Biogen S.R.L., Roche, Avexis, and Novartis Gene Therapies outside the submitted work.
The reviewer SD declared a shared parent affiliation with the authors to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2024.1308296/full#supplementary-material
1. Aragon-Gawinska, K, Mouraux, C, Dangouloff, T, and Servais, L. Spinal muscular atrophy treatment in patients identified by Newborn screening-a systematic review. Genes. (2023) 14:1377. doi: 10.3390/genes14071377
2. CureSMA. State of SMA 2022 Report (2023). Available at: https://www.curesma.org/wp-content/uploads/2023/06/9062023_State-of-SMA_vWeb.pdf
3. Prior, TW, Leach, ME, and Finanger, E. Spinal muscular atrophy In: MP Adam, DB Everman, GM Mirzaa, RA Pagon, SE Wallace, and B LJH, et al., editors. GeneReviews®. Seattle (WA): University of Washington (2000)
4. Farrar, MA, and Kiernan, MC. The genetics of spinal muscular atrophy: progress and challenges. Neurotherapeutics. (2015) 12:290–302. doi: 10.1007/s13311-014-0314-x
5. Cuscó, I, Bernal, S, Blasco-Pérez, L, Calucho, M, Alias, L, Fuentes-Prior, P, et al. Practical guidelines to manage discordant situations of SMN2 copy number in patients with spinal muscular atrophy. Neurol Genet. (2020) 6:e530. doi: 10.1212/nxg.0000000000000530
6. Wadman, RI, Jansen, MD, Stam, M, Wijngaarde, CA, Curial, CAD, Medic, J, et al. Intragenic and structural variation in the SMN locus and clinical variability in spinal muscular atrophy. Brain Commun. (2020) 2:fcaa075. doi: 10.1093/braincomms/fcaa075
7. Dangouloff, T, Boemer, F, Dideberg, V, Caberg, JH, and Servais, L. Reader response: discrepancy in redetermination of SMN2 copy numbers in children with SMA. Neurology. (2020) 95:144–5. doi: 10.1212/wnl.0000000000009907
8. Medical Services Advisory Committee (Australia). Public Summary Document: Application No. 1589—Prognostic value of the number of copies of the survival of motor neurone 2 (SMN2) gene for the severity of spinal muscular atrophy to determine eligibility for nusinersen in pre-symptomatic patients. (2019). Available at: http://www.msac.gov.au/internet/msac/publishing.nsf/Content/552F338BE4BEBA0FCA2583D000051738/$File/1589%20-%20Final%20PSD.pdf.
9. Moher, D, Liberati, A, Tetzlaff, J, and Altman, DGPRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. (2009) 6:e1000097. doi: 10.1371/journal.pmed.1000097
10. Downes, MJ, Brennan, ML, Williams, HC, and Dean, RS. Development of a critical appraisal tool to assess the quality of cross-sectional studies (AXIS). BMJ Open. (2016) 6:e011458. doi: 10.1136/bmjopen-2016-011458
11. Wells, GA, Shea, B, O'Connell, D, Peterson, J, Welch, V, Losos, M, et al. The Newcastle-Ottawa scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses: The Ottawa Hospital Research Institute (2021). Available at: https://www.ohri.ca/programs/clinical_epidemiology/oxford.asp.
12. Sterne, JA, Hernán, MA, Reeves, BC, Savović, J, Berkman, ND, Viswanathan, M, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ. (2016) 355:i4919. doi: 10.1136/bmj.i4919
13. Sterne, JAC, Savović, J, Page, MJ, Elbers, RG, Blencowe, NS, Boutron, I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. (2019) 366:l4898. doi: 10.1136/bmj.l4898
14. Higgins, JP, Savovic, J, Page, MJ, and Sterne, JA. Revised Cochrane risk-of-bias tool for randomized trials (RoB 2) (2019). Available at: https://www.riskofbias.info/welcome/rob-2-0-tool/current-version-of-rob-2.
15. Harada, Y, Sutomo, R, Sadewa, AH, Akutsu, T, Takeshima, Y, Wada, H, et al. Correlation between SMN2 copy number and clinical phenotype of spinal muscular atrophy: three SMN2 copies fail to rescue some patients from the disease severity. J Neurol. (2002) 249:1211–9. doi: 10.1007/s00415-002-0811-4
16. Mailman, MD, Heinz, JW, Papp, AC, Snyder, PJ, Sedra, MS, Wirth, B, et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med. (2002) 4:20–6. doi: 10.1097/00125817-200201000-00004
17. Swoboda, KJ, Prior, TW, Scott, CB, McNaught, TP, Wride, MC, Reyna, SP, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. (2005) 57:704–12. doi: 10.1002/ana.20473
18. Cuscó, I, Barceló, MJ, Rojas-García, R, Illa, I, Gámez, J, Cervera, C, et al. SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J Neurol. (2006) 253:21–5. doi: 10.1007/s00415-005-0912-y
19. Wirth, B, Brichta, L, Schrank, B, Lochmüller, H, Blick, S, Baasner, A, et al. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet. (2006) 119:422–8. doi: 10.1007/s00439-006-0156-7
20. Tiziano, FD, Bertini, E, Messina, S, Angelozzi, C, Pane, M, D'Amico, A, et al. The hammersmith functional score correlates with the SMN2 copy number: a multicentric study. Neuromuscul Disord. (2007) 17:400–3. doi: 10.1016/j.nmd.2007.02.006
21. Cobben, JM, Lemmink, HH, Snoeck, I, Barth, PA, van der Lee, JH, and de Visser, M. Survival in SMA type I: a prospective analysis of 34 consecutive cases. Neuromuscul Disord. (2008) 18:541–4. doi: 10.1016/j.nmd.2008.05.008
22. Arkblad, E, Tulinius, M, Kroksmark, AK, Henricsson, M, and Darin, N. A population-based study of genotypic and phenotypic variability in children with spinal muscular atrophy. Acta Paediatr. (2009) 98:865–72. doi: 10.1111/j.1651-2227.2008.01201.x
23. Elsheikh, B, Prior, T, Zhang, X, Miller, R, Kolb, SJ, Moore, D, et al. An analysis of disease severity based on SMN2 copy number in adults with spinal muscular atrophy. Muscle Nerve. (2009) 40:652–6. doi: 10.1002/mus.21350
24. Watihayati, MS, Fatemeh, H, Marini, M, Atif, AB, Zahiruddin, WM, Sasongko, TH, et al. Combination of SMN2 copy number and NAIP deletion predicts disease severity in spinal muscular atrophy. Brain and Development. (2009) 31:42–5. doi: 10.1016/j.braindev.2008.08.012
25. Stratigopoulos, G, Lanzano, P, Deng, L, Guo, J, Kaufmann, P, Darras, B, et al. Association of plastin 3 expression with disease severity in spinal muscular atrophy only in postpubertal females. Arch Neurol. (2010) 67:1252–6. doi: 10.1001/archneurol.2010.239
26. Kaufmann, P, McDermott, MP, Darras, BT, Finkel, R, Kang, P, Oskoui, M, et al. Observational study of spinal muscular atrophy type 2 and 3: functional outcomes over 1 year. Arch Neurol. (2011) 68:779–86. doi: 10.1001/archneurol.2010.373
27. Amara, A, Adala, L, Ben Charfeddine, I, Mamaï, O, Mili, A, Lazreg, TB, et al. Correlation of SMN2, NAIP, p44, H4F5 and Occludin genes copy number with spinal muscular atrophy phenotype in Tunisian patients. Eur J Paediatr Neurol. (2012) 16:167–74. doi: 10.1016/j.ejpn.2011.07.007
28. Kaufmann, P, McDermott, MP, Darras, BT, Finkel, RS, Sproule, DM, Kang, PB, et al. Prospective cohort study of spinal muscular atrophy types 2 and 3. Neurology. (2012) 79:1889–97. doi: 10.1212/WNL.0b013e318271f7e4
29. Farrar, MA, Vucic, S, Johnston, HM, du Sart, D, and Kiernan, MC. Pathophysiological insights derived by natural history and motor function of spinal muscular atrophy. J Pediatr. (2013) 162:155–9. doi: 10.1016/j.jpeds.2012.05.067
30. Sifi, Y, Sifi, K, Boulefkhad, A, Abadi, N, Bouderda, Z, Cheriet, R, et al. Clinical and genetic study of Algerian patients with spinal muscular atrophy. J Neurodegener Dis. (2013) 2013:903875. doi: 10.1155/2013/903875
31. Tiziano, FD, Lomastro, R, Di Pietro, L, Barbara Pasanisi, M, Fiori, S, Angelozzi, C, et al. Clinical and molecular cross-sectional study of a cohort of adult type III spinal muscular atrophy patients: clues from a biomarker study. Eur J Hum Genet. (2013) 21:630–6. doi: 10.1038/ejhg.2012.233
32. Finkel, RS, McDermott, MP, Kaufmann, P, Darras, BT, Chung, WK, Sproule, DM, et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. (2014) 83:810–7. doi: 10.1212/wnl.0000000000000741
33. Yamamoto, T, Sato, H, Lai, PS, Nurputra, DK, Harahap, NI, Morikawa, S, et al. Intragenic mutations in SMN1 may contribute more significantly to clinical severity than SMN2 copy numbers in some spinal muscular atrophy (SMA) patients. Brain Dev. (2014) 36:914–20. doi: 10.1016/j.braindev.2013.11.009
34. Yanyan, C, Yujin, Q, Jinli, B, Yuwei, J, Hong, W, and Fang, S. Correlation of PLS3 expression with disease severity in children with spinal muscular atrophy. J Hum Genet. (2014) 59:24–7. doi: 10.1038/jhg.2013.111
35. Brkušanin, M, Kosać, A, Jovanović, V, Pešović, J, Brajušković, G, Dimitrijević, N, et al. Joint effect of the SMN2 and SERF1A genes on childhood-onset types of spinal muscular atrophy in Serbian patients. J Hum Genet. (2015) 60:723–8. doi: 10.1038/jhg.2015.104
36. Qu, YJ, Ge, XS, Bai, JL, Wang, LW, Cao, YY, Lu, YY, et al. Association of copy numbers of survival motor neuron gene 2 and neuronal apoptosis inhibitory protein gene with the natural history in a Chinese spinal muscular atrophy cohort. J Child Neurol. (2015) 30:429–36. doi: 10.1177/0883073814553271
37. Zarkov, M, Stojadinović, A, Sekulić, S, Barjaktarović, I, Perić, S, Keković, G, et al. Association between the SMN2 gene copy number and clinical characteristics of patients with spinal muscular atrophy with homozygous deletion of exon 7 of the SMN1 gene. Vojnosanit Pregl. (2015) 72:859–63. doi: 10.2298/vsp140328072z
38. Medrano, S, Monges, S, Gravina, LP, Alías, L, Mozzoni, J, Aráoz, HV, et al. Genotype-phenotype correlation of SMN locus genes in spinal muscular atrophy children from Argentina. Eur J Paediatr Neurol. (2016) 20:910–7. doi: 10.1016/j.ejpn.2016.07.017
39. Ar Rochmah, M, Shima, A, Harahap, NIF, Niba, ETE, Morisada, N, Yanagisawa, S, et al. Gender effects on the clinical phenotype in Japanese patients with spinal muscular atrophy. Kobe J Med Sci. (2017) 63:E41–4.
40. Kaneko, K, Arakawa, R, Urano, M, Aoki, R, and Saito, K. Relationships between long-term observations of motor milestones and genotype analysis results in childhood-onset Japanese spinal muscular atrophy patients. Brain Dev. (2017) 39:763–73. doi: 10.1016/j.braindev.2017.04.018
41. Calucho, M, Bernal, S, Alías, L, March, F, Venceslá, A, Rodríguez-Álvarez, FJ, et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord. (2018) 28:208–15. doi: 10.1016/j.nmd.2018.01.003
42. De Sanctis, R, Pane, M, Coratti, G, Palermo, C, Leone, D, Pera, MC, et al. Clinical phenotypes and trajectories of disease progression in type 1 spinal muscular atrophy. Neuromuscul Disord. (2018) 28:24–8. doi: 10.1016/j.nmd.2017.09.015
43. Alves, CRR, Zhang, R, Johnstone, AJ, Garner, R, Eichelberger, EJ, Lepez, S, et al. Whole blood survival motor neuron protein levels correlate with severity of denervation in spinal muscular atrophy. Muscle Nerve. (2020) 62:351–7. doi: 10.1002/mus.26995
44. Coratti, G, Lucibello, S, Pera, MC, Duong, T, Muni Lofra, R, Civitello, M, et al. Gain and loss of abilities in type II SMA: a 12-month natural history study. Neuromuscul Disord. (2020) 30:765–71. doi: 10.1016/j.nmd.2020.07.004
45. Coratti, G, Messina, S, Lucibello, S, Pera, MC, Montes, J, Pasternak, A, et al. Clinical variability in spinal muscular atrophy type III. Ann Neurol. (2020) 88:1109–17. doi: 10.1002/ana.25900
46. Hryshchenko, NV, Yurchenko, AA, Karaman, HS, and Livshits, LA. Genetic modifiers of the spinal muscular atrophy phenotype. Cytol Genet. (2020) 54:130–6. doi: 10.3103/s0095452720020073
47. Mendonça, RH, Matsui, C Jr, Polido, GJ, Silva, AMS, Kulikowski, L, Torchio Dias, A, et al. Intragenic variants in the SMN1 gene determine the clinical phenotype in 5q spinal muscular atrophy. Neurol Genet. (2020) 6:e505. doi: 10.1212/nxg.0000000000000505
48. Townsend, EL, Simeone, SD, Krosschell, KJ, Zhang, RZ, and Swoboda, KJ. Project Cure SMA Investigator's network. Stander use in spinal muscular atrophy: results from a large natural history database. Pediatr Phys Ther. (2020) 32:235–41. doi: 10.1097/pep.0000000000000713
49. Zhang, Y, He, J, Zhang, Y, Li, L, Tang, X, Wang, L, et al. The analysis of the association between the copy numbers of survival motor neuron gene 2 and neuronal apoptosis inhibitory protein genes and the clinical phenotypes in 40 patients with spinal muscular atrophy: observational study. Medicine (Baltimore). (2020) 99:e18809. doi: 10.1097/md.0000000000018809
50. Lusakowska, A, Jedrzejowska, M, Kaminska, A, Janiszewska, K, Grochowski, P, Zimowski, J, et al. Observation of the natural course of type 3 spinal muscular atrophy: data from the polish registry of spinal muscular atrophy. Orphanet J Rare Dis. (2021) 16:150. doi: 10.1186/s13023-021-01771-y
51. Ou, SF, Ho, CS, Lee, WT, Lin, KL, Jones, CC, and Jong, YJ. Natural history in spinal muscular atrophy type I in Taiwanese population: a longitudinal study. Brain and Development. (2021) 43:127–34. doi: 10.1016/j.braindev.2020.07.012
52. Wijaya, YOS, Ar Rohmah, M, Niba, ETE, Morisada, N, Noguchi, Y, Hidaka, Y, et al. Phenotypes of SMA patients retaining SMN1 with intragenic mutation. Brain and Development. (2021) 43:745–58. doi: 10.1016/j.braindev.2021.03.006
53. Hanna, RB, Nahm, N, Bent, MA, Sund, S, Patterson, K, Schroth, MK, et al. Hip pain in nonambulatory children with type-I or II spinal muscular atrophy. JB JS Open Access. (2022) 7:e22.00011. doi: 10.2106/jbjs.Oa.22.00011
54. Krosschell, KJ, Townsend, EL, Kiefer, M, Simeone, SD, Zumpf, K, Welty, L, et al. Natural history of 10-meter walk/run test performance in spinal muscular atrophy: a longitudinal analysis. Neuromuscul Disord. (2022) 32:125–34. doi: 10.1016/j.nmd.2021.08.010
55. Maggi, L, Bello, L, Bonanno, S, Govoni, A, Caponnetto, C, Passamano, L, et al. Adults with spinal muscular atrophy: a large-scale natural history study shows gender effect on disease. J Neurol Neurosurg Psychiatry. (2022) 93:jnnp-2022-329320–1261. doi: 10.1136/jnnp-2022-329320
56. Pane, M, Donati, MA, Cutrona, C, De Sanctis, R, Pirinu, M, Coratti, G, et al. Neurological assessment of newborns with spinal muscular atrophy identified through neonatal screening. Eur J Pediatr. (2022) 181:2821–9. doi: 10.1007/s00431-022-04470-3
57. Rudnik-Schöneborn, S, Berg, C, Zerres, K, Betzler, C, Grimm, T, Eggermann, T, et al. Genotype-phenotype studies in infantile spinal muscular atrophy (SMA) type I in Germany: implications for clinical trials and genetic counselling. Clin Genet. (2009) 76:168–78. doi: 10.1111/j.1399-0004.2009.01200.x
58. Belter, L, Jarecki, J, Reyna, SP, Cruz, R, Jones, CC, Schroth, M, et al. The Cure SMA membership surveys: highlights of key demographic and clinical characteristics of individuals with spinal muscular atrophy. J Neuromuscul Dis. (2021) 8:109–23. doi: 10.3233/jnd-200563
59. Carson, VJ, Puffenberger, EG, Bowser, LE, Brigatti, KW, Young, M, Korulczyk, D, et al. Spinal muscular atrophy within Amish and Mennonite populations: ancestral haplotypes and natural history. PLoS ONE. (2018) 13:e0202104. doi: 10.1371/journal.pone.0202104
60. Kesari, A, Idris, MM, Chandak, GR, and Mittal, B. Genotype-phenotype correlation of SMN locus genes in spinal muscular atrophy patients from India. Exp Mol Med. (2005) 37:147–54. doi: 10.1038/emm.2005.20
61. Rudnik-Schöneborn, S, Heller, R, Berg, C, Betzler, C, Grimm, T, Eggermann, T, et al. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J Med Genet. (2008) 45:635–8. doi: 10.1136/jmg.2008.057950
62. Souza, PVS, Pinto, W, Ricarte, A, Badia, BML, Seneor, DD, Teixeira, DT, et al. Clinical and radiological profile of patients with spinal muscular atrophy type 4. Eur J Neurol. (2021) 28:609–19. doi: 10.1111/ene.14587
63. Taylor, JE, Thomas, NH, Lewis, CM, Abbs, SJ, Rodrigues, NR, Davies, KE, et al. Correlation of SMNt and SMNc gene copy number with age of onset and survival in spinal muscular atrophy. Eur J Hum Genet. (1998) 6:467–74. doi: 10.1038/sj.ejhg.5200210
64. Bowen, BM, Truty, R, Aradhya, S, Bristow, SL, Johnson, BA, Morales, A, et al. SMA identified: clinical and molecular findings from a sponsored testing program for spinal muscular atrophy in more than 2,000 individuals. Front Neurol. (2021) 12:663911. doi: 10.3389/fneur.2021.663911
65. Alvarez, K, Suarez, B, Palomino, MA, Hervias, C, Calcagno, G, Martínez-Jalilie, M, et al. Observations from a nationwide vigilance program in medical care for spinal muscular atrophy patients in Chile. Arq Neuropsiquiatr. (2019) 77:470–7. doi: 10.1590/0004-282x20190073
66. Petit, F, Cuisset, JM, Rouaix-Emery, N, Cancés, C, Sablonnière, B, Bieth, E, et al. Insights into genotype-phenotype correlations in spinal muscular atrophy: a retrospective study of 103 patients. Muscle Nerve. (2011) 43:26–30. doi: 10.1002/mus.21832
67. Aharoni, S, Bistritzer, J, Levine, H, Sagi, L, Fattal-Valevski, A, Ginzberg, M, et al. Adeno-associated virus serotype 9 antibody titers in patients with SMA pre-screened for treatment with onasemnogene abeparvovec -routine care evidence. Gene Ther. (2023) 30:101–6. doi: 10.1038/s41434-022-00339-0
68. Aragon-Gawinska, K, Daron, A, Ulinici, A, Vanden Brande, L, Seferian, A, Gidaro, T, et al. Sitting in patients with spinal muscular atrophy type 1 treated with nusinersen. Dev Med Child Neurol. (2020) 62:310–4. doi: 10.1111/dmcn.14412
69. Aragon-Gawinska, K, Seferian, AM, Daron, A, Gargaun, E, Vuillerot, C, Cances, C, et al. Nusinersen in patients older than 7 months with spinal muscular atrophy type 1: a cohort study. Neurology. (2018) 91:e1312–8. doi: 10.1212/wnl.0000000000006281
70. Audic, F, de la Banda, MGG, Bernoux, D, Ramirez-Garcia, P, Durigneux, J, Barnerias, C, et al. Effects of nusinersen after one year of treatment in 123 children with SMA type 1 or 2: a French real-life observational study. Orphanet J Rare Dis. (2020) 15:148. doi: 10.1186/s13023-020-01414-8
71. Carson, VJ, Young, M, Brigatti, KW, Robinson, DL, Reed, RM, Sohn, J, et al. Nusinersen by subcutaneous intrathecal catheter for symptomatic spinal muscular atrophy patients with complex spine anatomy. Muscle Nerve. (2022) 65:51–9. doi: 10.1002/mus.27425
72. Coratti, G, Pane, M, Lucibello, S, Pera, MC, Pasternak, A, Montes, J, et al. Age related treatment effect in type II spinal muscular atrophy pediatric patients treated with nusinersen. Neuromuscul Disord. (2021) 31:596–602. doi: 10.1016/j.nmd.2021.03.012
73. Darbar, IA, Plaggert, PG, Resende, MB, Zanoteli, E, and Reed, UC. Evaluation of muscle strength and motor abilities in children with type II and III spinal muscle atrophy treated with valproic acid. BMC Neurol. (2011) 11:36. doi: 10.1186/1471-2377-11-36
74. De Vivo, DC, Bertini, E, Swoboda, KJ, Hwu, WL, Crawford TOFinkel, RS, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. (2019) 29:842–56. doi: 10.1016/j.nmd.2019.09.007
75. Elsheikh, B, Severyn, S, Zhao, S, Kline, D, Linsenmayer, M, Kelly, K, et al. Safety, tolerability, and effect of nusinersen treatment in ambulatory adults with 5q-SMA. Front Neurol. (2021) 12:650535. doi: 10.3389/fneur.2021.650535
76. Finkel, RS, Chiriboga, CA, Vajsar, J, Day, JW, Montes, J, De Vivo, DC, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: final report of a phase 2, open-label, multicentre, dose-escalation study. Lancet Child Adolesc Health. (2021) 5:491–500. doi: 10.1016/S2352-4642(21)00100-0
77. Friese, J, Geitmann, S, Holzwarth, D, Müller, N, Sassen, R, Baur, U, et al. Safety monitoring of gene therapy for spinal muscular atrophy with onasemnogene abeparvovec—a single Centre experience. J Neuromuscul Dis. (2021) 8:209–16. doi: 10.3233/jnd-200593
78. Hahn, A, Gunther, R, Ludolph, A, Schwartz, O, Trollmann, R, Weydt, P, et al. Short-term safety results from compassionate use of risdiplam in patients with spinal muscular atrophy in Germany. Orphanet J Rare Dis. (2022) 17:276. doi: 10.1186/s13023-022-02420-8
79. Lee, BH, Deng, S, Chiriboga, CA, Kay, DM, Irumudomon, O, Laureta, E, et al. Newborn screening for spinal muscular atrophy in New York state: clinical outcomes from the first 3 years. Neurology. (2022) 99:e1527–37. doi: 10.1212/wnl.0000000000200986
80. Modrzejewska, S, Kotulska, K, Kopyta, I, Grędowska, E, Emich-Widera, E, Tomaszek, K, et al. Nusinersen treatment of spinal muscular atrophy type 1—results of expanded access programme in Poland. Neurol Neurochir Pol. (2021) 55:289–94. doi: 10.5603/PJNNS.a2021.0020
81. Orbach, R, Sagi, L, Sadot, E, Tokatly Latzer, I, Shtamler, A, Zisberg, T, et al. Cerebrospinal fluid characteristics of patients treated with intrathecal nusinersen for spinal muscular atrophy. Muscle Nerve. (2022) 66:762–6. doi: 10.1002/mus.27731
82. Osredkar, D, Jílková, M, Butenko, T, Loboda, T, Golli, T, Fuchsová, P, et al. Children and young adults with spinal muscular atrophy treated with nusinersen. Eur J Paediatr Neurol. (2021) 30:1–8. doi: 10.1016/j.ejpn.2020.11.004
83. Pane, M, Coratti, G, Sansone, VA, Messina, S, Bruno, C, Catteruccia, M, et al. Nusinersen in type 1 spinal muscular atrophy: twelve-month real-world data. Ann Neurol. (2019) 86:443–51. doi: 10.1002/ana.25533
84. Pane, M, Coratti, G, Sansone, VA, Messina, S, Catteruccia, M, Bruno, C, et al. Type I SMA "new natural history": long-term data in nusinersen-treated patients. Ann Clin Transl Neurol. (2021) 8:548–57. doi: 10.1002/acn3.51276
85. Pane, M, Palermo, C, Messina, S, Sansone, VA, Bruno, C, Catteruccia, M, et al. Nusinersen in type 1 SMA infants, children and young adults: preliminary results on motor function. Neuromuscul Disord. (2018) 28:582–5. doi: 10.1016/j.nmd.2018.05.010
86. Strauss, KA, Carson, VJ, Brigatti, KW, Young, M, Robinson, DL, Hendrickson, C, et al. Preliminary safety and tolerability of a novel subcutaneous intrathecal catheter system for repeated outpatient dosing of nusinersen to children and adults with spinal muscular atrophy. J Pediatr Orthop. (2018) 38:e610–7. doi: 10.1097/bpo.0000000000001247
87. Strauss, KA, Farrar, MA, Muntoni, F, Saito, K, Mendell, JR, Servais, L, et al. Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: the phase III SPR1NT trial. Nat Med. (2022) 28:1390–7. doi: 10.1038/s41591-022-01867-3
88. Vill, K, Schwartz, O, Blaschek, A, Gläser, D, Nennstiel, U, Wirth, B, et al. Newborn screening for spinal muscular atrophy in Germany: clinical results after 2 years. Orphanet J Rare Dis. (2021) 16:153. doi: 10.1186/s13023-021-01783-8
89. Mercuri, E, Darras, BT, Chiriboga, CA, Day, JW, Campbell, C, Connolly, AM, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. (2018) 378:625–35. doi: 10.1056/NEJMoa1710504
90. Strauss, KA, Farrar, MA, Muntoni, F, Saito, K, Mendell, JR, Servais, L, et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: the phase III SPR1NT trial. Nat Med. (2022) 28:1381–9. doi: 10.1038/s41591-022-01866-4
91. Butchbach, ME. Copy number variations in the survival motor neuron genes: implications for spinal muscular atrophy and other neurodegenerative diseases. Front Mol Biosci. (2016) 3:7. doi: 10.3389/fmolb.2016.00007
92. Chien, YH, Chiang, SC, Weng, WC, Lee, NC, Lin, CJ, Hsieh, WS, et al. Presymptomatic diagnosis of spinal muscular atrophy through newborn screening. J Pediatr. (2017) 190:124–129.e1. doi: 10.1016/j.jpeds.2017.06.042
93. Bodamer, OA. Spinal muscular atrophy: UpToDate, Inc. (2023). Available at: https://www.uptodate.com/contents/spinal-muscular-atrophy.
94. Smeriglio, P, Langard, P, Querin, G, and Biferi, MG. The identification of novel biomarkers is required to improve adult SMA patient stratification, diagnosis and treatment. J Pers Med. (2020) 10:75. doi: 10.3390/jpm10030075
95. Weng, WC, Hsu, YK, Chang, FM, Lin, CY, Hwu, WL, Lee, WT, et al. CMAP changes upon symptom onset and during treatment in spinal muscular atrophy patients: lessons learned from newborn screening. Genet Med. (2021) 23:415–20. doi: 10.1038/s41436-020-00987-w
96. Pino, MG, Rich, KA, and Kolb, SJ. Update on biomarkers in spinal muscular atrophy. Biomark Insights. (2021) 16:11772719211035643. doi: 10.1177/11772719211035643
97. Heesen, L, Peitz, M, Torres-Benito, L, Hölker, I, Hupperich, K, Dobrindt, K, et al. Plastin 3 is upregulated in iPSC-derived motoneurons from asymptomatic SMN1-deleted individuals. Cell Mol Life Sci. (2016) 73:2089–104. doi: 10.1007/s00018-015-2084-y
98. Kariyawasam, DST, D'Silva, A, Lin, C, Ryan, MM, and Farrar, MA. Biomarkers and the development of a personalized medicine approach in spinal muscular atrophy. Front Neurol. (2019) 10:898. doi: 10.3389/fneur.2019.00898
99. Baranello, G, Gorni, K, Daigl, M, Kotzeva, A, Evans, R, Hawkins, N, et al. Prognostic factors and treatment-effect modifiers in spinal muscular atrophy. Clin Pharmacol Ther. (2021) 110:1435–54. doi: 10.1002/cpt.2247
100. Kirschner, J, Butoianu, N, Goemans, N, Haberlova, J, Kostera-Pruszczyk, A, Mercuri, E, et al. European ad-hoc consensus statement on gene replacement therapy for spinal muscular atrophy. Eur J Paediatr Neurol. (2020) 28:38–43. doi: 10.1016/j.ejpn.2020.07.001
101. Schorling, DC, Becker, J, Pechmann, A, Langer, T, Wirth, B, and Kirschner, J. Discrepancy in redetermination of SMN2 copy numbers in children with SMA. Neurology. (2019) 93:267–9. doi: 10.1212/wnl.0000000000007836
102. Kempf, L, Goldsmith, JC, and Temple, R. Challenges of developing and conducting clinical trials in rare disorders. Am J Med Genet A. (2018) 176:773–83. doi: 10.1002/ajmg.a.38413
103. Sterne, JAC, Higgins, JPT, Elbers, RG, and Reeves, BC. Risk of Bias in Non-randomized Studies of Interventions (ROBINS-I): detailed guidance (2016). Available at: https://www.riskofbias.info/welcome/home/current-version-of-robins-i/robins-i-detailed-guidance-2016
Keywords: nusinersen, onasemnogene abeparvovec, prognosis, SMN2 gene copy, spinal muscular atrophy
Citation: Dosi C and Masson R (2024) The impact of three SMN2 gene copies on clinical characteristics and effect of disease-modifying treatment in patients with spinal muscular atrophy: a systematic literature review. Front. Neurol. 15:1308296. doi: 10.3389/fneur.2024.1308296
Edited by:
Alessandro Simonati, University of Verona, ItalyReviewed by:
Stefania Della Vecchia, Stella Maris Foundation (IRCCS), ItalyCopyright © 2024 Dosi and Masson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Riccardo Masson, cmljY2FyZG8ubWFzc29uQGlzdGl0dXRvLWJlc3RhLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.