
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 18 December 2023
Sec. Pediatric Neurology
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1310419
 Hongjun Fang
Hongjun Fang Wenjing Hu
Wenjing Hu Qingyun Kang
Qingyun Kang Xiaojun KuangLijuan WangXiao ZhangHongmei LiaoLiming Yang
Xiaojun KuangLijuan WangXiao ZhangHongmei LiaoLiming Yang Haiyan Yang
Haiyan Yang Zhi Jiang
Zhi Jiang Liwen Wu*
Liwen Wu*Objective: This study aimed to examine the clinical and gene-mutation characteristics of pediatric patients with sodium channel gene mutation-related childhood epilepsy and to provide a basis for precision treatment and genetic counseling.
Methods: The clinical data from 94 patients with sodium channel gene mutation-related childhood epilepsy who were treated at Hunan Children's Hospital from August 2012 to December 2022 were retrospectively evaluated, and the clinical characteristics, gene variants, treatment, and follow-up status were analyzed and summarized.
Results: Our 94 pediatric patients with sodium channel gene variant-related childhood epilepsy comprised 37 girls and 57 boys. The age of disease onset ranged from 1 day to 3 years. We observed seven different sodium channel gene variants, and 55, 14, 9, 6, 6, 2, and 2 patients had SCNlA, SCN2A, SCN8A, SCN9A, SCN1B, SCN11A, and SCN3A variants, respectively. We noted that 52 were reported variants and 42 were novel variants. Among all gene types, SCN1A, SCN2A, and SCN8A variants were associated with an earlier disease onset age. With the exception of the SCN1B, the other six genes were associated with clustering seizures. Except for variants SCN3A and SCN11A, some patients with other variants had status epilepticus (SE). The main diagnosis of children with SCN1A variants was Dravet syndrome (DS) (72.7%), whereas patients with SCN2A and SCN8A variants were mainly diagnosed with various types of epileptic encephalopathy, accounting for 85.7% (12 of 14) and 88.9% (8 of 9) respectively. A total of five cases of sudden unexpected death in epilepsy (SUDEP) occurred in patients with SCN1A, SCN2A, and SCN8A variants. The proportion of benign epilepsy in patients with SCN9A, SCN11A, and SCN1B variants was relatively high, and the epilepsy control rate was higher than the rate of other variant types.
Conclusion: Sodium channel gene variants involve different epileptic syndromes, and the treatment responses also vary. We herein reported 42 novel variants, and we are also the first ever to report two patients with SCN11A variants, thereby increasing the gene spectrum and phenotypic profile of sodium channel dysfunction. We provide a basis for precision treatment and prognostic assessment.
Epilepsy is a common neurologic disorder that reflects diverse causes, including genetic factors, brain disease, and generalized or systemic disease. As our knowledge of genetics gradually improves, we now recognize an abundance of findings that show that ion channel gene mutations are intimately associated with epilepsy (1). Among these mutations, those encoding for sodium channels are highly correlated with childhood epilepsy (2). The voltage-gated sodium channel (Nav) is a protein complex consisting of one α subunit and two β subunits and is primarily present in the central nervous system (CNS), peripheral nervous system (PNS), skeletal muscles, and myocardium (3). The α subunit of the Nav channel is a functional subunit of ~2,000 amino acids that is highly conserved. There are nine different Nav α subtypes (Nav1.1–Nav1.9), and mammalian tissues express all nine (4). These subtype proteins are named Nav1.1–Nav1.9 and the corresponding genes are designated SCN1A–SCN5A and SCN8A–SCN11A. Of these, Nav1.1, Nav1.2, Nav1.3, and Nav1.6 are highly expressed in the central nervous system (CNS) and two different subtypes are expressed in muscles (Nav1.4 in adult skeletal muscle and Nav1.5 in myocardium), while Nav1.7, Nav1.8, and Nav1.9 are principally expressed in the peripheral nervous system (5). There are four Nav channel β subunits that are encoded by four genes, i.e., SCN1B–SCN4B (6). Nav channel mutations are related to epilepsy, including α subunits, such as Nav1.1 (SCN1A), Nav1.2 (SCN2A), Nav1.3 (SCN3A), Nav1.6 (SCN8A), and Nav1.7 (SCN9A), and the β subunit (SCN1B) (7). In order to strengthen our understanding of the different sodium channel genotypes and clinical phenotypes, and to guide personalized treatment, determination of disease prognosis, and genetic counseling, we herein summarized the clinical and gene-mutation characteristics of 94 sodium channel gene mutation-related childhood epilepsy patients who were treated at Hunan Children's Hospital. This review, therefore, comprises the broadest analysis of sodium channel genes currently available.
This study was a retrospective summary of cases. We collected data from epileptic patients who were treated in the neurology outpatient clinic or the inpatient unit at Hunan Children's Hospital from August 2012 to December 2022, and who harbored sodium channel gene variants upon genetic testing. These patients sought medical attention for epilepsy of unknown cause and underwent whole-exome sequencing for personalized treatment, prognostic assessment, and genetic counseling. A detailed clinical data registry (including name, sex, date of birth, age at disease onset, status of mental development, seizure presentation, perinatal condition, past medical history, family history, response to anticonvulsants, genetic test results, head MRI, and electroencephalography) was established for every patient. Patients were followed up through outpatient visits, readmission, or telephone follow-ups. The clinical and genetic characteristics of these patients were retrospectively analyzed, and the guardians of all pediatric patients provided signed informed consent. The clinical data of patients and the DNA from the peripheral blood of the patients and their parents were also collected. This study was approved by the Ethics Committee of Hunan Children's Hospital.
Agilent exon chip capture + high-throughput sequencing was used for whole-exome sequencing to screen for gene mutations in pediatric patients, and first-generation sequencing was executed on the patients and their parents to validate mutation sites and determine the source of the mutation.
The response to anticonvulsants was divided into four types: controlled (no seizure for at least 3 months), reduced (seizure frequency decreased by ≥25%), ineffective (seizure frequency decreased by <25%), and worsened. Controlled and reduced responses were considered to be effective responses (8).
All children were scored for intellectual development: children aged 6 years were evaluated for intelligence using the Chinese standardized Gesell scale; children older than 6 years were assessed for intelligence using the Wechsler Intelligence Scale for Children. A score of <70 indicates intellectual disability; 50–69 indicates mild intellectual disability; 35–49 indicates moderate intellectual disability; 20–34 indicates severe intellectual disability; and <20 indicates extremely severe intellectual disability (9, 10).
We implemented the SPSS24.0 statistics suite for this analysis. Measurement data with a normal distribution are presented as the mean ± standard deviation, and measurement data with a non-normal distribution are presented as the median. In addition, qualitative data are presented as the number of patients (percentage). The independent-sample Kruskal–Wallis test was applied to compare the differences in age at onset with respect to the seven gene variants, and we conducted pairwise comparisons. The Mann–Whitney U-test was used to compare differences in the responses to these seven mutated genes.
We collected data from 94 patients with sodium channel gene variant-related childhood epilepsy, comprising 37 girls and 57 boys (see Table 1 for clinical presentations and gene-variant results). The age at onset was 1 day to 3 years, and the median age was 7.0 (4.5, 10.2) months. The children's parents were not consanguineous and the patients were not related to each other. We detected a total of seven different sodium channel gene variants in our patients, and 55, 14, 9, 6, 6, 2, and 2 patients had SCNlA, SCN2A, SCN8A, SCN9A, SCN1B, SCN11A, and SCN3A variants, respectively (Figure 1). A total of 62 patients harbored missense variants; 11 were frameshift variants, 10 were splicing variants, nine were non-sense variants, 1 was a deletion variant, and 1 was a chimeric duplication variant. A total of 52 cases were reported to be pathogenic variants, 42 were novel variants, 22 were inherited variants, and 72 were de novo variants. There were more male patients with sodium channel-related epilepsy than female patients. The most common variant was SCN1A, accounting for 58.5% of cases, and missense variants constituted the most common type, accounting for 66.0% of cases. Pediatric patients mainly had de novo variants (accounting for 76.6%) and some inherited-mutation patients presented with genetic epilepsy with febrile seizures plus (GEFS+) and febrile seizures plus (FS+) that showed good prognoses (Table 2).
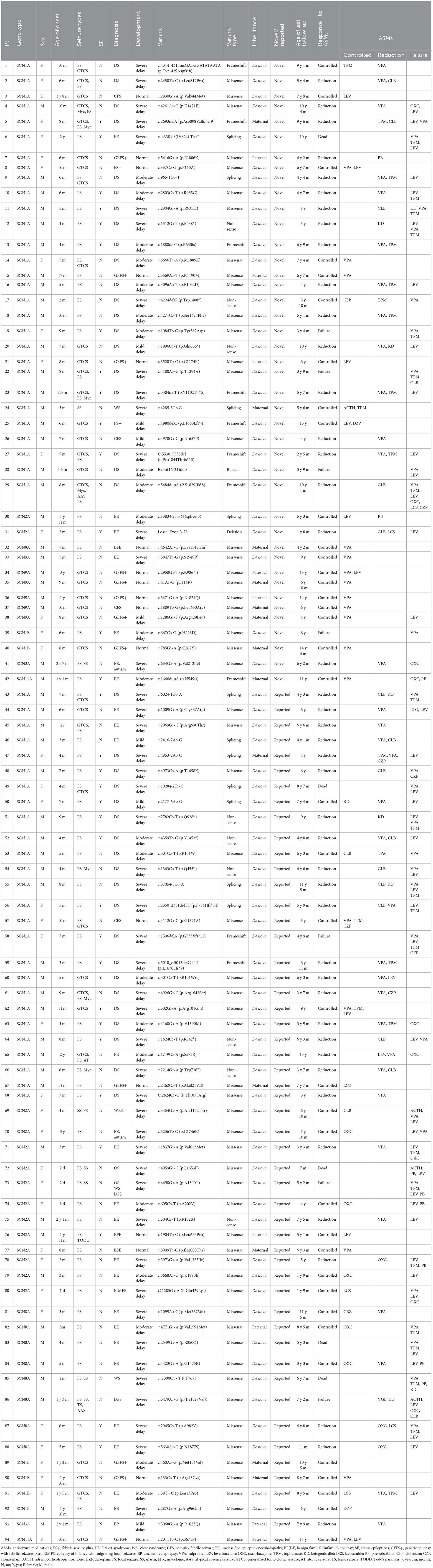
Table 1. Clinical findings of novel and reported sodium channel gene variants.
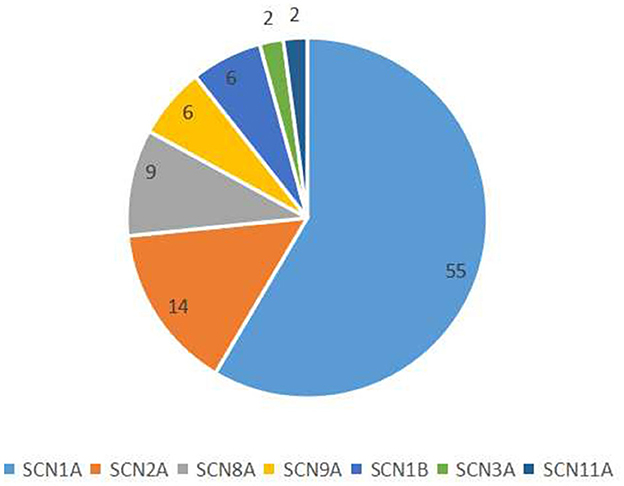
Figure 1. Classification of sodium channel gene variants.
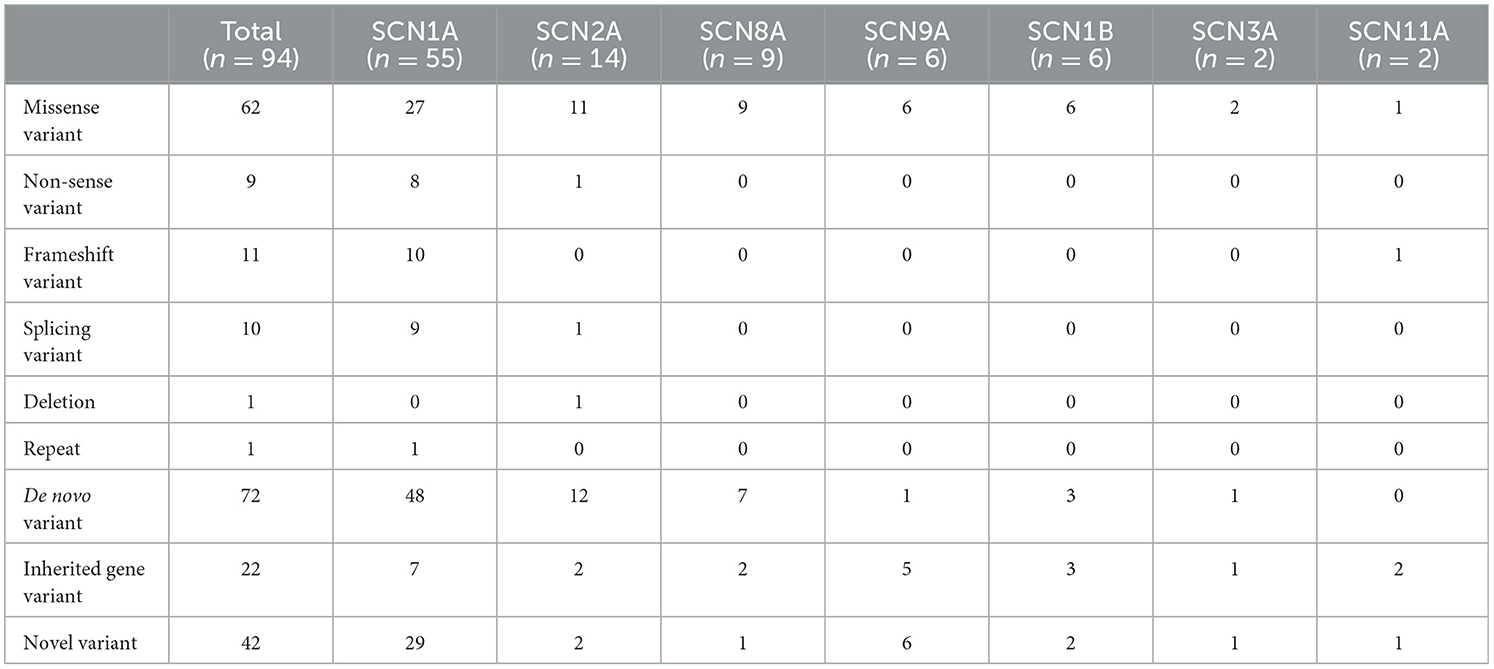
Table 2. Distributional characteristics of sodium channel gene variants.
Out of the seven genes we evaluated, the genotypes with early disease onset were SCN1A, SCN2A, and SCN8A, and the median age at onset was 7.0 (5.0, 9.0), 3.0 (0.1, 23.0), and 5.0 (3.5, 7.5) months, respectively. The age at onset for SCN1A ranged from 2 months to 2 years. There was one patient with disease onset within 3 months, 19 patients within 3–6 months, 31 patients between 6 and 12 months, and five patients manifested the disease onset after 1 year of age. The age at onset of patients with the SCN2A ranged from day 1 of birth to 3 years of age. There were eight patients with disease onset within 3 months (accounting for 57.1%), of whom four exhibited disease onset within 1–2 days after birth. There were six patients with disease onset after 3 months (accounting for 42.9%), of whom four patients developed the disease after 1 year of age. The age at disease onset of patients with the SCN8A ranged from 1 month to 1 year and 3 months, of whom one patient showed disease onset within 3 months (accounting for 11.1%), and eight patients after 3 months (accounting for 88.9%), of whom one patient developed the disease after 1 year of age. Disease onset was later for SCN9A, SCN1B, SCN3A, and SCN11A variant patients. With the exception of the SCN1B, the other six genes were associated with clustering seizures; these were ranked in descending order of patient percentage as SCN1A (78.2%), SCN2A (64.3%), SCN8A (55.6%), SCN3A (50%), SCN11A (50%), and SCN9A (33.3%). With the exception of the SCN3A, the genes had a tendency to correlate with fever sensitivity. Fever sensitivity occurred in all patients with the SCN9A, SCN11A, and SCN1B variants, while the proportions of individuals with SCN1A, SCN2A, and SCN8A variants who developed fever sensitivity were 92.7, 21.4, and 11.1%, respectively. With the exception of SCN3A and SCN11A variants, several patients with the other gene variants experienced SE, and the SE duration was the longest in SCN1A variant patients, ranging from 20 min to 4 h. There were varying proportions of the seven gene variants reflected by patients who showed delayed mental development. The seizures of SCN1A variant patients were predominantly focal seizures, followed by generalized tonic-clonic seizure (GTCS), while several patients exhibited comorbid myoclonus, spasms, absence seizures, and atonic seizures. SCN2A variant patients developed focal seizures, of whom three patients showed comorbid spasms. All SCN8A variant patients manifested focal seizures, of whom one patient had comorbid spasms; another patient exhibited comorbid spasms, absence seizures, and clonic seizures. Two SCN3A variant patients had focal seizures, of whom one showed comorbid spasms. SCN9A, SCN11A, and SCN1B variant patients experienced focal seizures and GTCS but did not experience any other types of epileptic seizures. With regard to epilepsy diagnosis, SCN1A variant patients were chiefly diagnosed with DS (72.7%, 40 of 55), and the remaining patients were diagnosed with unclassified epileptic encephalopathy (EE), GEFS+, complex febrile seizure (CFS), FS+, and West syndrome. Eight patients demonstrated comorbid ataxia, and two GEFS+ patients developed immune-related encephalitis during the course of the disease. Of 14 SCN2A variant patients, two were diagnosed with OS, one with West syndrome, one had epilepsy of infancy with migrating focal seizures (EIMFS), two had benign familial epilepsy, and eight had non-specific EE. One patient developed Todd's paralysis after convulsions and one patient acquired aphasia after convulsions; the latter was considered to be caused by speech-center involvement generated by the convulsions. One patient had autism spectrum disorder (ASD) and one showed bilateral hearing loss. Of nine SCN8A variant patients, one was diagnosed with West syndrome, one with Lennox–Gastaut syndrome (LGS), one with benign familial infantile epilepsy (BFIE), and six with non-specific EE. One patient manifested comorbid congenital joint dysplasia and two exhibited prolonged postpartum jaundice. Of the six SCN9A variant patients, four were diagnosed with GEFS+, one with CFS, one with non-specific EE, and one patient had comorbid cryptorchidism. Of the six SCN1B variant patients, three were diagnosed with GEFS+ and three with non-specific EE. Of the two SCN3A variant patients, one was diagnosed with non-specific focal seizures. This latter patient was diagnosed with ASD at 1 year and 9 months of age, had focal seizures at 2 years and 7 months of age, and exhibited spasms at 3 years of age. Of two SCN11A variant patients, one was diagnosed with EE and one with GEFS+ (Table 3).
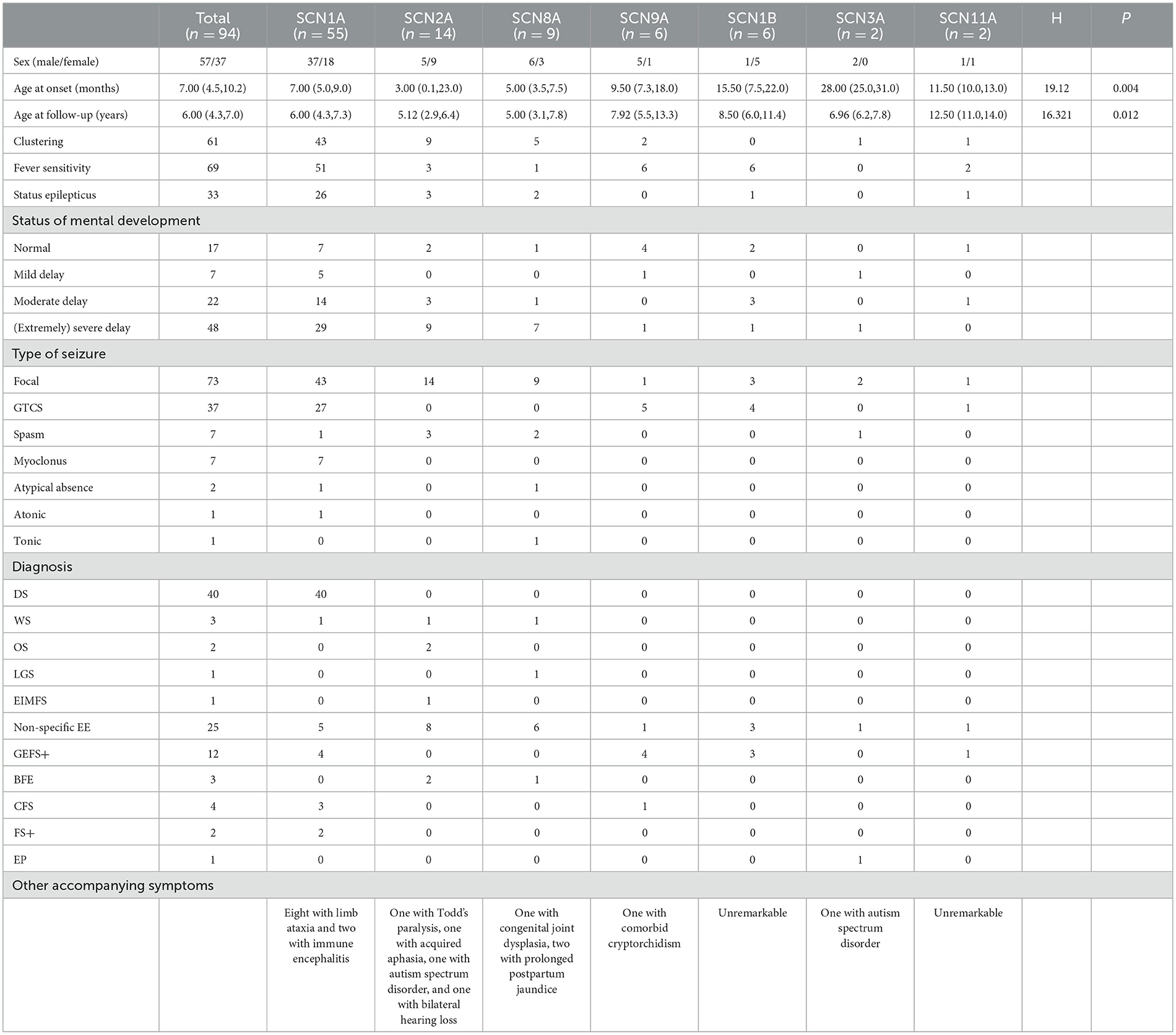
Table 3. Clinical phenotypic characteristics of sodium channel gene variants.
All 94 pediatric patients underwent cranial magnetic resonance imaging (MRI) and the majority showed a normal head MRI (74.5%, 70 of 94). A minority of patients exhibited brain dysplasia or non-specific structural abnormalities. Eleven SCN1A variant patients produced an abnormal head MRI, of whom four had dilated cerebral hemispheric gyri, three had slightly intense signals in the frontal, parietal, and temporal white matter, two showed corpus callosum dysplasia, one had diminished hippocampal volume, and one manifested cerebral dysplasia. Seven SCN2A variant patients exhibited abnormal head MRIs, of whom five had a dilated extracerebral space and two possessed ependymal cysts. Five SCN8A variant patients produced abnormal head MRIs, of whom one showed cerebellar atrophy, two had a dilated extracerebral space, one had abnormal frontal cerebral blood vessel development, and one had slightly thicker and enhanced meninges in the right frontal lobe. Of the six SCN9A variant patients, one manifested brain white matter dysplasia in head MRIs and the remaining patients generated normal head MRIs. All SCN1B, SCN3A, and SCN11A patients showed normal MRIs. All pediatric patients underwent at least one video electroencephalography, and the majority showed abnormal electroencephalograms (76.6%, 72 of 94) that mainly indicated slow waves, focal discharge, generalized discharge, multifocal discharge, hypsarrhythmia, and burst suppression. Patients with SCN1A, SCN2A, SCN8A, and SCN3A variants were prone to background slowing. In addition, the first electroencephalograms were normal in 29 SCN1A variant patients, but electroencephalograms were abnormal upon 1-year follow-up in 18 patients (Table 4).
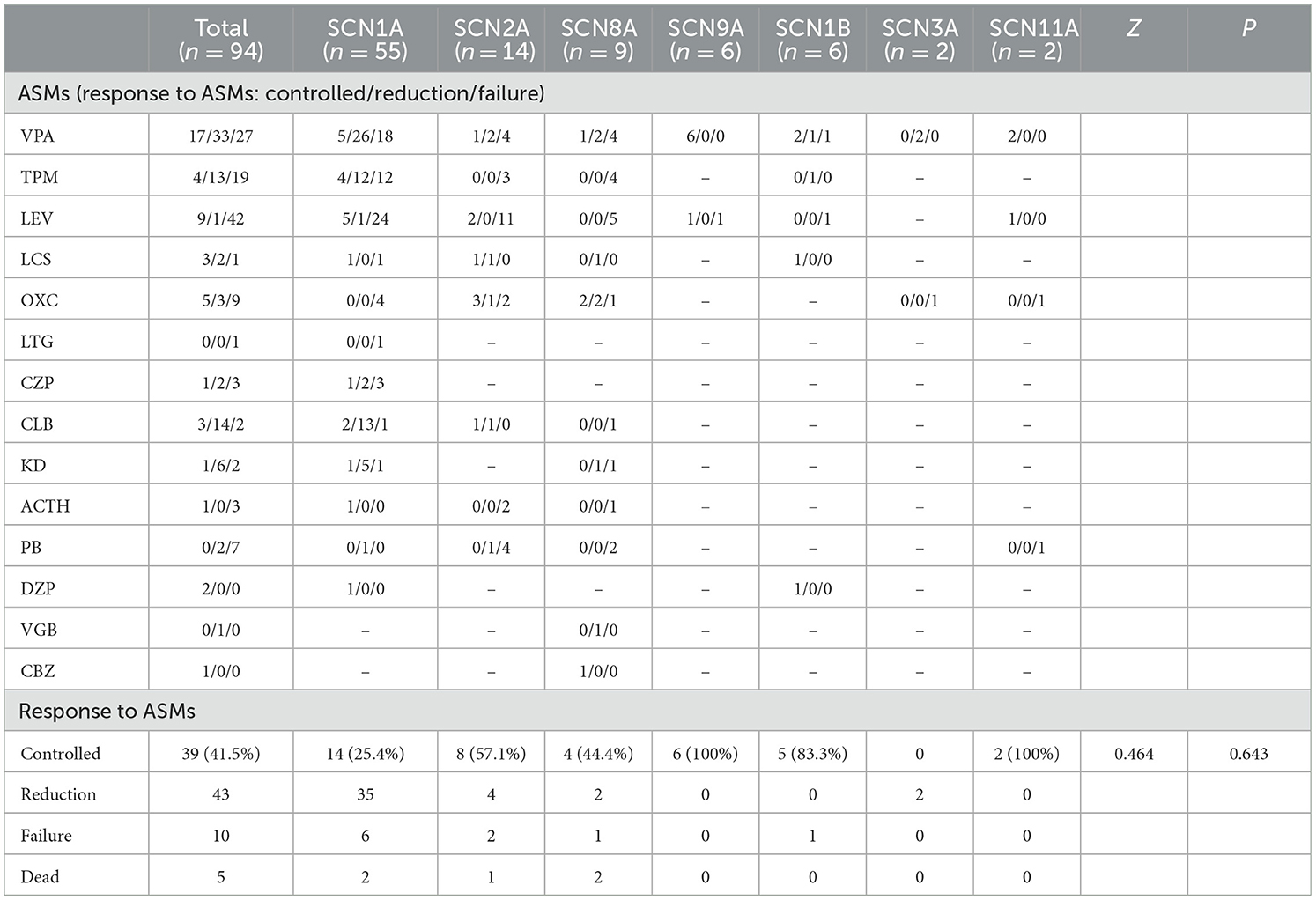
Table 4. Treatment and follow-up of epileptic patients with sodium channel gene variants.
In the present study, one patient did not receive anticonvulsant drugs, while the remaining 93 received one or more anticonvulsant drugs. Patients were followed up to a median age of 6.0 (4.3, 7.0) years, and epilepsy symptoms were controlled (41.5%, 39 of 94) or reduced (45.7%, 43 of 94) in most patients. In SCN1A variant patients, anticonvulsants were effective in 89.1% (49 of 55) of patients, epilepsy was controlled in 25.5% (14 of 55), and the seizure frequency decreased in 63.9% (35 of 55). Drug efficacies in descending order were clobazam (CLB) at 93.8% (15 of 16), ketogenic diet (KD) at 85.7% (6 of 7), valproic acid (VPA) at 63.3% (31 of 49), topiramate (TPM) at 57.1% (16 of 28), diazepam (DZP) at 50.0% (3 of 6), lacosamide (LCS) at 50.0% (1 of 2), and levetiracetam (LEV) at 20.0% (6 of 30). Adrenocorticotropic hormone (ACTH) was effective in one patient, oxcarbazepine (OXC) did not show any improvement in four patients, and lamotrigine (LTG) was ineffective in one patient. In SCN2A variant patients, anticonvulsants were effective in 85.7% (12 of 14) of patients, epilepsy was controlled in 57.1% (8 of 14), and the seizure frequency decreased in 16.3% (4 of 14). Drug efficacies in descending order were LCS at 100% (2 of 2), CLB at 100% (2 of 2), OXC at 66.7% (4 of 6), VPA at 42.9% (3 of 7), and LEV at 15.4% (2 of 13). TPM was ineffective in three patients and ACTH was ineffective in two others. In total, six of the 14 patients with SCN2A variant epilepsy had early onset EE, of whom four received sodium channel blockers, seizures were controlled in 1 patient after OXC addition, and seizures decreased in one patient after OXC addition. Seizures diminished in one patient after LCS addition, and OXC addition was ineffective in one patient, but seizures completely resolved after LCS addition. This latter pediatric patient was an SCN2A variant (c.1285G>A)-related EIMFS patient who experienced convulsions on the first day after birth (Figures 2, 3). The neonate experienced over 50 seizures daily, and convulsions could not be controlled even after multiple anticonvulsants were administered. Seizures stopped when LCS was administered at 11 weeks, and convulsions were absent at one-and-a-half years of outpatient follow-up. In the present study, three patients with late-onset SCN2A-related epilepsy received OXC: OXC addition was ineffective in one patient, and seizures were completely controlled after OXC addition in the two others. In SCN8A variant patients, anticonvulsants were effective in 66.7% (six of nine) of patients, epilepsy was controlled in 25.5% (four of nine), and the seizure frequency declined in 22.2% (two of nine). Drug efficacies in descending order were carbamazepine (CBZ) at 100% (one of one), LCS at 100% (one of one), vigabatrin at 100% (one of one), OXC at 80% (four of five), KD at 50% (one of two), and VPA at 42.9% (three of seven). TPM was ineffective in all four patients treated, LEV was ineffective in all five patients treated, and ACTH was ineffective in the one patient treated. Epileptic seizures were controlled in six SCN9A patients (100%, six of six) and drug efficacies were 100% (six of six) with VPA and 50% with LEV (one of two). Epilepsy was controlled in 83.3% (five of six) of SCN1B variant patients. Three of four patients experienced efficacy with VPA (75%, three of four), and seizures were controlled in one patient treated with LCS. Seizures declined in two SCN3A patients and VPA was effective, whereas OXC was ineffective. Epilepsy symptoms were controlled in two SCN11A variant patients, in whom VPA and LEV were effective and where OXC was ineffective. Of all patients, five developed SUDEP, of whom two harbored an SCN1A variant; one patient was diagnosed with non-specific EE, one patient with DS, and two patients with a splicing variant. The age at death was 5 years of age in both patients, and one patient had a younger monozygotic twin sister who died of SE at 4 years of age. In SCN1A variant patients, one OS patient developed SUDEP at 7 months. Among the SCN8A variant patients, two developed SUDEP, one of whom with non-specific EE died at 3 years of age, and the other patient with West syndrome died at 5 years of age (Table 5).
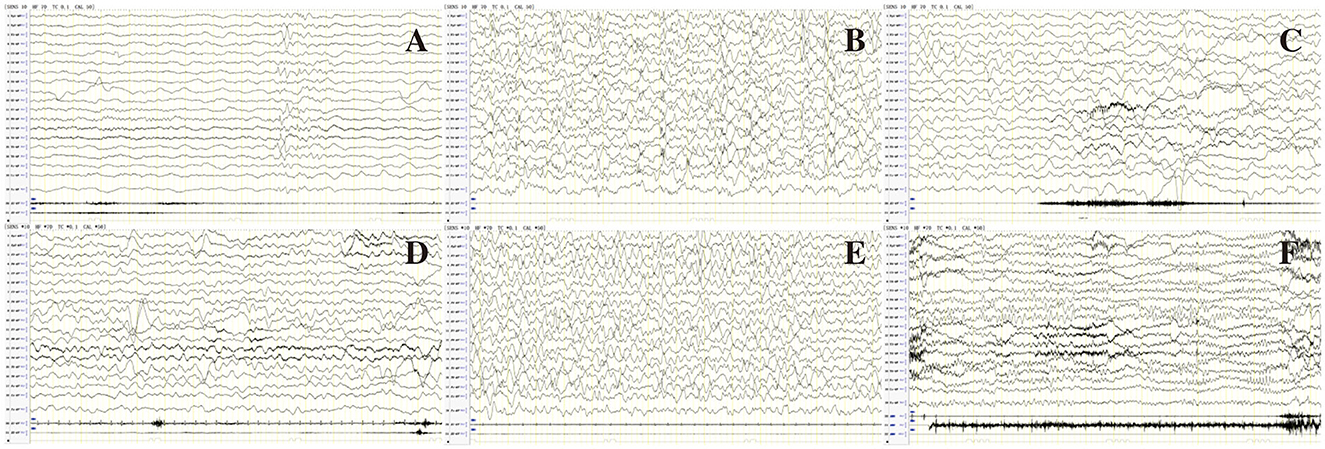
Figure 2. EEG of patient 80 in Table 1 before (A–C) and after (D–F) treatment of LCS. (A) Interictal EEG showed slow background, multiple focal discharges during wakefulness; (B) Atypical hypsarrhythmia during sleep; (C) Ictal EEG showed focal discharge associated with focal seizures. (D) Normal background activity during wakefulness after treatment with LCS; (E) Rare spikes during sleep; (F) Follow-up EEG showed normal background activity, rare spikes during wakefulness at age 1 year and 5 months.
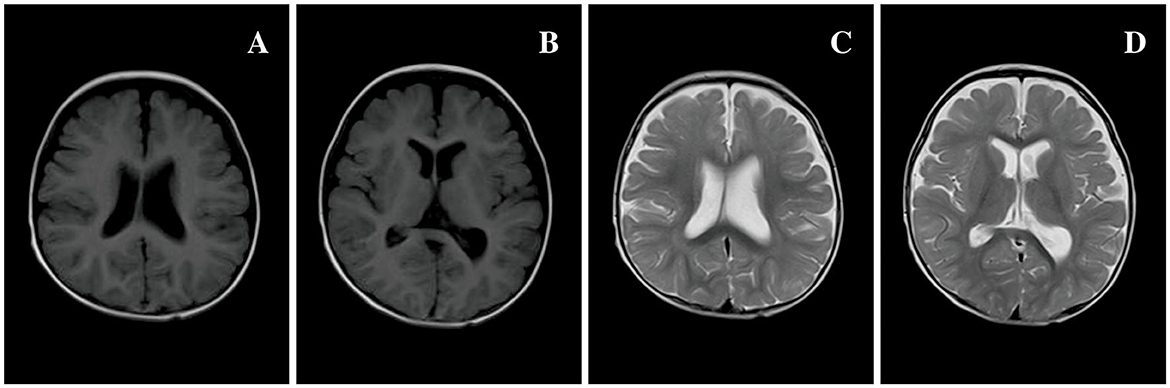
Figure 3. Brain MRI of 80 patients in Table 1 at 7 months of age. (A, B) T1 MRI and (C, D) T2 MRI: bilateral extracerebral spaces of frontal and temporal lobes widened, and the left ventricle was slightly dilated compared to the right side.
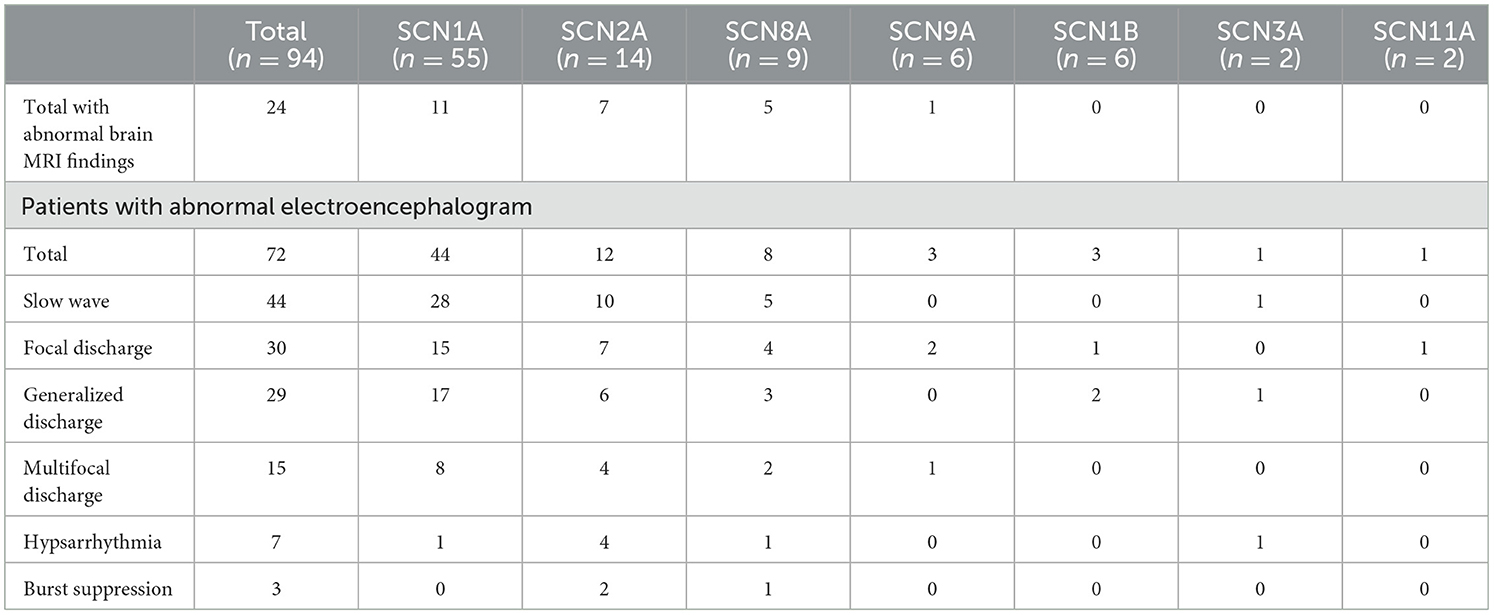
Table 5. Auxiliary examinations of epileptic patients with sodium channel gene variants.
We observed significant differences in the age at onset among the aforementioned seven subtypes of sodium channel gene variant-related epilepsy patients (P = 0.004, Table 3), revealing that age at onset may portend genetic heterogeneity. SCN1A/SCN2A/SCN8A variant-related epilepsy was generally manifested at an earlier age, while the SCN3A/SCN9A/SCN1B variant-related subtype showed a later age at onset. We noted no significant differences in treatment responses among the different subtypes (P = 0.643, Table 4).
Ion channels constitute the material basis for electrical activity in neurons, and abnormalities in ion channel structure or function can result in electrophysiologic changes that cause abnormal epileptiform discharge. Most of the currently confirmed pathogenic mutations in epilepsy genes encode for ion channels or their auxiliary subunits; of these, gene variants in ion channels comprise the most common cause of genetic epilepsy and sodium channel-related epilepsy and reflect intense areas of research in recent years (11). Sodium channels include nine α subunits and four β subunits, with the SCN1A, SCN2A, SCN3A, SCN8A, SCN9A, SCN11A, and SCN1B encoding human Nav α1, α2, α3, α6, α7, α9, and β1, respectively. Different gene mutations then produce epileptic seizures of varying severity and types.
The SCN1A is located on chromosome 2q 24.3; it contains 31 exons and encodes the Nav1.1 α subunit. As our understanding of the SCN1A improves, we note an increasing number of reports describing SCN1A mutations. The diseases caused by various mutation sites and types are different and range from mild diseases such as GEFS+ and partial epilepsy with febrile seizures plus (PEFS+) to severe diseases such as DS (2). In our study, the 55 SCN1A variant patients encompassed those with relatively mild diseases (four with GEFS+, three with CFS, and two with FS+). With the exception of one patient with a frameshift variant, the remaining patients harbored missense variants. One study showed that the SCN1A mutation was localized to the pedigrees of 5–10% of GEFS+ patients. The GEFS+-related SCN1A mutation is principally concentrated in the intracellular domain of Nav1.1 (12), and almost all mutations are missense mutations, which is consistent with the results from the current study. A previous study indicated that truncation mutations and missense mutations resulted in the most severe clinical symptoms of all SCN1A mutations (13). We herein demonstrated that de novo splicing variants in the SCN1A were present in two patients and in the monozygotic younger sister of one patient, and that these patients died due to status epilepticus. Four patients who failed treatment were DS patients, of whom two harbored missense variants, one had a frameshift variant, and one had a duplication variant.; this, however, was different from previous studies and may have been due to our smaller sample size. A previous study showed that the most common gene mutations in SCN1A-related DS were frameshift mutations, missense mutations, and non-sense mutations; however, splice-site, duplication, and deletion mutations were rarely observed (14). Our study revealed that 40 SCN1A variant patients manifested DS, of whom 16 had missense variants, nine showed frameshift variants, eight had non-sense variants, six possessed splicing variants, and one exhibited a duplication variant; this was generally consistent with previous investigations. Analysis of the treatment responses in 55 patients revealed that CLB, KD, VPA, and TPM produced better efficacy in SCN1A variant-related epilepsy patients, LEV showed slight efficacy, and OXC and LTG were ineffective in controlling convulsions. Symptoms worsened in two of our GEFS+ patients as they experienced recurrent febrile seizures and were ultimately diagnosed with immune-related encephalitis. This suggested that the observed worsening of symptoms during treatment of patients with gene mutation-related epilepsy was not attributable solely to gene mutations, but that other factors such as immune factors and infection should be considered in order to adopt appropriate treatment.
Ever since the first case of SCN2A mutation-induced benign (familial) neonatal/infantile seizure was discovered and confirmed in 2001, the epilepsy spectrum as provoked by SCN2A mutations has continuously expanded to include OS syndrome, infantile spasms, EIMFS, DS, myoclonic-atonic epilepsy, LGS, GEFS+, and other unclassified severe EE (15, 16). Mild epilepsy is primarily related to inherited missense mutations and severe epilepsy is principally precipitated by de novo mutations, with the majority of patients showing delayed mental and motor development (17). In our study, 12 of 14 EE patients harbored de novo variants and manifested moderately severe mental developmental delay. Two children had benign familial epilepsy due to inherited variants, exhibited normal mental and motor development, and responded well to treatment, all of which was consistent with the literature (17). Studies revealed that patients with SCN2A mutation-related EE primarily showed neonatal or infantile onset, and over half showed a neonatal onset (8). In the present study, four EE patients experienced a neonatal onset and two had disease onset before 1–3 months of age. Six patients had a disease onset of 3 months or later, a situation completely different from that described in other reports. Studies showed that the SCN2A mutation generated two Nav1.2 functional changes: gain-of-function (GoF) and loss-of-function (LoF). Early-onset (age at onset < 3 months of age) Nav1.2 is mainly due to GoF and responds well to SCBs. Late-onset (onset ≥ 3 months of age) or non-epilepsy intellectual disability/autism spectrum disorder (ID/ASD) Nav1.2 is mainly due to LoF, responds poorly to SCBs, and may even augur for worsening seizures (8, 18). In our study, we applied sodium channel blockers to four of six patients with early-onset SCN2A-related epilepsy, and OXC and LCS were effective in two patients each. Although sodium channel blockers exhibit some efficacy in early-onset SCN2A-related epilepsy, OXC is not effective in all cases, and LCS and other sodium channel blockers can be used when OXC shows a poor response. We implemented OXC to treat three late-onset SCN2A-related epilepsy patients, and OXC was ineffective in one patient while seizures were completely controlled in the other two. It is obvious, then, that Nav1.2 electrophysiologic changes noted in SCN2A mutation-related EE are more complex. Studies have shown that many patients with epilepsy-related SCN2A mutations present with complex GoF or LoF characteristics, and some rare mutations share both characteristics (19, 20). Hence, it is difficult to apply a simple scheme with respect to classification. Additional experience in future diagnoses and treatments is certainly necessitated to establish more effective methods that can determine GoF or LoF characteristics, so as to produce suitable precision-therapy pharmaceuticals.
SCN8A encodes for the Nav1.6 α subunit and is highly expressed in the central nervous system. Since it was identified as an epilepsy-related gene in 2012, investigators have ascertained that SCN8A mutation-related epileptic seizures are diverse, as they can be manifested as systemic tonic-clonic seizures, spasms, absence, and focal seizures, and can also be present as epileptic syndromes that include BFIE or epileptic encephalopathies, such as Lennox-Gastaut syndrome and West syndrome. In addition to causing epileptic seizures, SCN8A mutations can affect speech, intelligence, and development in pediatric patients (21, 22). In our study, there were nine SCN8A variant-related epilepsy patients, of whom eight showed moderately severe mental and motor impairment. We observed diverse seizure forms but discerned that all patients showed focal seizures. Studies on the functionality of SCN8A mutations revealed that the electrophysiologic changes in the Nav1.6 channel included a persistent increase in Na+ current (INaP), complete channel inactivation, and steady-state rapid inactivation voltage-dependent depolarization transition (23). These three electrophysiologic changes are all GoF alterations of the Nav1.6 channel that increase neuronal excitation due to mutated channels, and correcting the GoF of this channel may therefore constitute a therapeutic target. A related study showed that high-dose sodium channel blockers such as carbamazepine, OXC, and phenytoin inhibited epileptic seizures in patients with the SCN8A mutation (24). In our study, six patients were treated with sodium channel blockers, and convulsions were controlled in one patient after CBZ was added; seizures were controlled in two patients and reduced in one patient; and treatment was ineffective in one patient after OXC treatment. In addition, although seizures decreased in one patient after OXC addition, they still occurred but diminished after LCS was added. This case shows that sodium channel blockers such as CBZ, OXC, and LCS are highly or partially effective in SCN8A variant-related epilepsy. In the present study, the seizure frequency did not decrease or increase in five patients after LEV was applied. VPA was used in seven patients and was effective in three patients and ineffective in four patients. This showed that LEV was ineffective in SCN8A-related epilepsy or that it may have even worsened epilepsy, while VPA exhibited some efficacy consistent with previous studies (25). One study depicted SCN8A-related EE as exhibiting a high mortality rate, reflecting the cause of death as SUDEP primarily. More than 10% of SCN8A mutation patients will develop SUDEP (26, 27), and two of nine patients (22%) died during epileptic seizures in our analysis.
SCN9A is located on chromosome 2q24.3, contains 27 exons, encodes for a 1977- amino-acid Nav1.7 α subunit, and was initially classified as a peripheral nervous system channel gene. It was subsequently reported that Nav1.7 is expressed in the cerebrum, particularly in the fetal hippocampus, showing that it plays a role in the central nervous system (28). Recent reports show that SCN9A mutations are mostly associated with febrile seizures and DS (29). One study showed that SCN9A mutations coexist with SCN1A mutations, that this mutation is a genetic modification factor in SCN1A mutation-related FS+, and that it may worsen the symptoms of mild FS+ (30). As another study showed that the SCN9A variant caused febrile seizures in families without the SCN1A variant (29, 31), it is possible that some SCN9A mutations may portray a modifying role in the presence of more potent mutations, or that it may itself induce mild epileptic seizures (32). There are very few extant reports on epilepsy caused by SCN9A mutations, and the pathogenesis of GEFS+ without an SCN1A mutation remains arcane. In our study, there were six SCN9A variant patients and all manifested fever sensitivity consistent with previous studies. All six patients had unreported missense variants and did not possess SCN1A variants. Of these, four patients were diagnosed with GEFS+, and one patient was diagnosed with CFS; we also reported a patient with non-specific EE. This latter patient began developing recurrent febrile convulsions at 5 months and received VPA as an anticonvulsant treatment. At 7 years and 6 months, seizures were controlled, although the patient exhibited severe developmental delay since infancy. In our study, seizures were controlled after one to two anticonvulsants were applied to six patients, and the response rate with VPA was 100% (six of six). This shows that SCN9A variant-related epilepsy predominantly consists of heterozygous missense variants. Most of the patients reflected fever sensitivity, prominently generalized seizures, and relatively mild symptoms; and anticonvulsants showed good efficacy in these patients.
SCN1B was the first sodium channel gene found to be associated with epilepsy, as Wallace et al. ascertained a point mutation in chromosome 19q13.1 in a GEFS+ pedigree in 1998 (33). Studies showed that heterozygous mutations in this gene were manifested as FS+ and others as non-EE (34), while patients with homozygous recessive mutations presented with DS-like EE symptoms and were at high risk of premature death (35). Compared with classical DS, the clinical symptoms of patients with homozygous SCN1B mutations are more consistent with early infantile developmental and epileptic encephalopathy (DEE), and the latter is more severe than DS (36). We found six SCN1B variant-related epilepsy patients. Three were diagnosed with GEFS+ and three with non-specific EE, and all six patients showed fever sensitivity; the three patients with non-specific EE had moderately mild DS-like symptoms. However, these six patients harbored heterozygous missense variants that were different from those described in previous studies (34, 35). Some investigators demonstrated that there were no differences in surface sodium channel β1 subunit expression in SCN1B mutant (p.Arg 85 Cys) cells compared with wild-type cells. However, the β1 subunit regulates Nav1.1 sodium ion current disappearance, revealing that this SCN1B mutation is a LoF mutation (34), thus providing a basis for the treatment of SCN1B-related epilepsy. With the exception of one patient in our study who did not take anticonvulsants regularly and who exhibited recurrent seizures, seizures were controlled in the other five patients. One patient did not self-administer any anticonvulsant drugs and convulsions did not occur after 6 years of age. Two patients received VPA monotherapy, one patient received diazepam tablets due to convulsion prophylaxis during fever, and another had a poor response to LEV—with seizures decreasing after the patient was switched to VPA and TPM, and controlled after adding LCS. Seizures were ultimately controlled in this patient after the sodium channel blocker LCS was applied, which was different from previous studies that showed that most SCN1B mutations were LoF mutations. Further evaluations of the specific biological mechanisms involved are therefore required.
The SCN3A is located on chromosome 2q24 and encodes for the Nav1.3 channel. Nav1.3 appears to be expressed at different sites at various times. For example, during embryonic and fetal development, Nav1.3 is principally expressed in the central nervous system, with its expression extremely low after birth. During infancy, Nav1.3 is gradually replaced by Nav1.1. SCN3A is also considered to be related to neurodevelopmental disorders and epilepsy, and SCN3A pathogenic mutations reflect many phenotypes. In mild cases, there is a mild cognitive disability, while in severe cases, there is a severe developmental delay due to cerebellar dysfunction (30). We herein noted two SCN3A variant patients, both with cognitive disabilities. The head MRI of these two patients was normal and there were no apparent structural abnormalities. These two SCN3A patients harbored missense variants. However, as our sample size was small, we only had a few SCN3A variant-induced epilepsy cases, and additional in-depth study is thus required.
The SCN11A codes for the sodium channel Nav1.9 is expressed in peripheral pan-sensing neurons and visceral afferent nerves and has been proven to be a threshold channel that is associated with peripheral neuropathy. There is no extant report of SCN11A and epilepsy. One study depicted the SCN11A variant as present in SUDEP patients, but it is unclear whether this variant causes epilepsy or SUDEP (37). We herein found two patients with epilepsy carrying gene variants in SCN11A: one patient was diagnosed with EE, with an unreported c.1646dupA frameshift variant in SCN11A from the patient's mother. The other patient was diagnosed with GEFS+ and had a reported c.2011T>C missense variant in SCN11A from the patient's father, who had a history of febrile seizures when he was young. Nav1.9 is specifically expressed in peripheral nociceptors, and animal models and clinical genetic studies showed that the channel protein plays an important role in inflammatory pain, neurogenic pain, and cold pain but its underlying mechanisms of action in epilepsy remain unelucidated. In our study, we enrolled two cases with SCN11A variants. However, more cases and further genetic functional verification are needed to confirm the relationship between SCN11A and epilepsy.
SCN1A, SCN2A, SCN3A, SCN8A, SCN9A, SCN11A, and SCN1B sodium channel gene mutations were intimately associated with epilepsy and displayed significant heterogeneity in pathogenesis and clinical symptoms. For example, SCN1A mutation-related epilepsy had a disease onset of several months, its clinical presentation was mainly DS, and few patients showed febrile seizures; while sodium channel blockers generally caused symptoms to worsen. SCN2A can cause early-onset encephalopathy within several days after birth or delayed epileptic encephalopathy after 3 months. A majority of seizures exhibited clustering and most sodium channel blockers provided favorable efficacy. Additionally, SCN1A, SCN2A, and SCN8A were associated with SUDEP. The treatment response to antiseizure medications (ASMs) varies depending on the type of Nav channel mutation. Therefore, it is of great significance to study the relationship between genotype, clinical phenotype, and efficacy of ASMs in children with epilepsy. Such studies should provide not only a reference for precise and individualized treatment of epilepsy but also a basis for genetic counseling and prenatal testing to maximize the benefits for children.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the Ethics Committee of Hunan Children's Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
HF: Data curation, Formal analysis, Funding acquisition, Methodology, Supervision, Writing–original draft, Writing–review & editing. WH: Formal analysis, Writing–review & editing. QK: Investigation, Writing–review & editing. XK: Resources, Writing–review & editing. LWa: Writing–review & editing. XZ: Writing– review & editing. HL: Writing–review & editing. LY: Writing–review & editing. HY: Writing–review & editing. ZJ: Methodology, Writing–review & editing. LWu: Funding acquisition, Project administration, Resources, Writing–review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by grants from the Natural Science Foundation of Hunan Province (Nos. 2022JJ70087 and 2021JJ30393), the National Natural Science Foundation of China (No. 82171453), the Scientific Research Projects of Hunan Health Committee (No. 2021SK50515), the Huxiang Youth Talent Support Program (No. 2021RC3117), and the Key Projects Supported by Hunan Provincial Health Commission (No. 202203074966).
The authors appreciate the patients' participation in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bartolini E, Campostrini R, Kiferle L, Pradella S, Rosati E, Chinthapalli K, et al. Epilepsy and brain channelopathies from infancy to adulthood. Neurol Sci. (2020) 41:749–61. doi: 10.1007/s10072-019-04190-x
2. Feng Y, Zhang S, Zhang Z, Guo J, Tan Z, Zhu Y, et al. Understanding genotypes and phenotypes of the mutations in voltage-gated sodium channel α subunits in epilepsy. CNS Neurol Disord Drug Targets. (2019) 18:266–72. doi: 10.2174/1871527317666181026164825
3. Ahern CA, Payandeh J, Bosmans F, Chanda B. The hitchhiker's guide to the voltage-gated sodium channel galaxy. Gen Physiol. (2016) 147:1–24. doi: 10.1085/jgp.201511492
4. Catterall WA, Kalume F, Oakley JC. NaV1.1 channels and epilepsy. Physiology. (2010) 588 (Pt 11):1849–59. doi: 10.1113/jphysiol.2010.187484
5. Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev. (2008) 88:1407–47. doi: 10.1152/physrev.00002.2008
6. Deng H, Xiu X, Song Z. The molecular biology of genetic based epilepsies. Mol Neurobiol. (2014) 49:352–67. doi: 10.1007/s12035-013-8523-6
7. Kaplan DI, Isom LL, Petrou S. Role of sodium channels in epilepsy. Cold Spring Harb Perspect. (2016) 6:1. doi: 10.1101/cshperspect.a022814
8. Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. (2017) 140:1316–36. doi: 10.1093/brain/awx054
9. Sun Y-Y, Wang L, Peng J-L, Huang Y-J, Qiao F-Q, Wang P. Effects of repetitive transcranial magnetic stimulation on motor function and language ability in cerebral palsy: a systematic review and meta-analysis. Front Pediatr. (2023) 11:835472. doi: 10.3389/fped.2023.835472
10. Shi Y, Feng Y, Zhang Y, Liu H, Luo H, Shang L, et al. Assessment of event-related evoked potentials and China-Wechsler intelligence scale for cognitive dysfunction in children with obstructive sleep apnea. Int J Pediatr Otorhinolaryngol. (2021) 150:110901. doi: 10.1016/j.ijporl.2021.110901
11. Du J, Simmons S, Brunklaus A, Adiconis X, Hession CC, Fu Z, et al. Differential excitatory vs inhibitory SCN expression at single cell level regulates brain sodium channel function in neurodevelopmental disorders. Eur J Paediatr Neurol. (2020) 24:129–33. doi: 10.1016/j.ejpn.2019.12.019
12. Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA. SCN1A mutations and epilepsy. Hum Mutat. (2005) 25:535–42. doi: 10.1002/humu.20178
13. Lange IM, Mulder F, Slot R, Sonsma ACM, Kempen MJA, Nijman IJ, et al. Modifier genes in SCN1A-related epilepsy syndromes. Mol Genet Genomic Med. (2020) 8:e1103. doi: 10.1002/mgg3.1103
14. Guerrini R. Dravet syndrome: The main issues. Eur J Paediatr Neurol. (2012) 16 (Suppl. 1): S1–4. doi: 10.1016/j.ejpn.2012.04.006
15. Passi GR, Mohammad SS. Dominant SCN2A mutation with variable phenotype in two generations. Brain Dev. (2021) 43:166–9. doi: 10.1016/j.braindev.2020.08.009
16. Schwarz N, Hahn A, Bast T, Müller S, Löffler H, Maljevic S, et al. Mutations in the Sodium Channel gene SCN2A cause neonatal epilepsywithlate-onset episodic ataxia. J Neurol. (2016) 263:334–43. doi: 10.1007/s00415-015-7984-0
17. Carroll LS, Woolf R, Ibrahim Y, Williams HJ, Dwyer S, Walters J, et al. Mutation screening of SCN2A in schizophrenia and identification of a novel loss-of-function mutation. Psychiatr Genet. (2016) 26:60–5. doi: 10.1097/YPG.0000000000000110
18. Berecki G, Howell KB, Heighway J, Olivier N, Rodda J, Overmars I, et al. Functional correlates of clinical phenotype and severity in recurrent SCN2A variants. Commun Biol. (2022) 5:515. doi: 10.1038/s42003-022-03454-1
19. Yang X-R, Ginjupalli VKM, Theriault O, Poulin H, Appendino JP, Au PYB, et al. SCN2A-related epilepsy of infancy with migrating focal seizures: report of a variant with apparent gain- and loss-of-function effects. J Neurophysiol. (2022) 127:1388–97. doi: 10.1152/jn.00309.2021
20. Thompson CH, Potet F, Abramova TV, DeKeyser JM, Ghabra NF, Vanoye CG, et al. Epilepsy-associated SCN2A (NaV1.2) variants exhibit diverse and complex functional properties. J Gen Physiol. (2023) 155:e202313375. doi: 10.1085/jgp.202313375
21. Wengert ER, Tronhjem CE, Wagnon JL, Johannesen KM, Petit H, Krey I, et al. Biallelic inherited SCN8A variants, a rarecause of SCN8A-related developmental and epileptic encephalopathy. Epilepsia. (2019) 60:2277–85. doi: 10.1111/epi.16371
22. Solazzi R, Castellotti B, Canafoglia L, Messina G, Magri S, Freri E, et al. Paroxysmal tonic upgaze in a child with SCN8A-related encephalopathy. Epileptic Disord. (2021) 23:643–7. doi: 10.1684/epd.2021.1305
23. Veeramah KR, O'Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. (2012) 90:502–10. doi: 10.1016/j.ajhg.2012.01.006
24. Talwar D, Hammer FM. SCN8A epilepsy, developmental encephalopathy, and related disorders. Pediatr Neurol. (2021) 122:76–83. doi: 10.1016/j.pediatrneurol.2021.06.011
25. Schreiber JM, Tochen L, Brown M, Evans S, Ball LJ, Bumbut A, et al. A multi-disciplinary clinic for SCN8A related epilepsy. Epilepsy Res. (2020) 159:106261. doi: 10.1016/j.eplepsyres.2019.106261
26. Johannesen KM, Gardella E, Scheffer I, Howell K, Smith DM, Helbig I, et al. Early mortality in SCN8A-related epilepsies. Epilepsy Res. (2018) 143:79–81. doi: 10.1016/j.eplepsyres.2018.04.008
27. Donnan AM, Schneider AL, Russ-Hall S, Churilov L, Scheffer IE. Rates of status epilepticus and sudden unexplained death in epilepsy in people with genetic developmental and epileptic encephalopathies. Neurology. (2023) 100:e1712–22. doi: 10.1212/WNL.0000000000207080
28. Mechaly I, Scamps F, Chabbert C, Sans A, Valmier J. Molecular diversity of voltage-gated sodium channel alpha subunits expressed in neuronal and non-neuronal excitable cells. Neuroscience. (2005) 130:389–96. doi: 10.1016/j.neuroscience.2004.09.034
29. Zhang T, Chen M, Zhu A, Zhang X, Fang T. Novel mutation of SCN9A gene causing generalized epilepsy with febrile seizures plus in a Chinese family. Neurol Sci. (2020) 41:1913–7. doi: 10.1007/s10072-020-04284-x
30. Ma H, Guo Y, Chen Z, Wang L, Tang Z, Zhang J, et al. Mutations in the sodium channel genes SCN1A, SCN3A, and SCN9A in children with epilepsy with febrile seizures plus(EFS+). Seizure. (2021) 88:146–52. doi: 10.1016/j.seizure.2021.04.006
31. Yang C, Hua Y, Zhang W, Xu J, Xu L, Gao F, et al. Variable epilepsy phenotypes associated with heterozygous mutation in the SCN9A gene: report of two cases. Neurol Sci. (2018) 39:1113–5. doi: 10.1007/s10072-018-3300-y
32. Zhang S, Zhang Z, Shen Y, Zhu Y, Du K, Guo J, et al. SCN9A epileptic encephalopathy mutations display a gain-of-function phenotype and distinct sensitivity to oxcarbazepine. Neurosci Bull. (2020) 36:11–24. doi: 10.1007/s12264-019-00413-5
33. Wallace RH, Wang DW, Singh R, Scheffer IE, George AL, Phillips HA, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet. (1998) 19:366–70. doi: 10.1038/1252
34. Aeby A, Sculier C, Bouza AA, Askar B, Lederer D, Schoonjans A-S, et al. SCN1B-linked early infantile developmental and epileptic encephalopathy. Ann Clin Transl Neurol. (2019) 6:2354–67. doi: 10.1002/acn3.50921
35. Darras N, Ha TK, Rego S, Martin P-M, Barroso E, Slavotinek AM, et al. Developmental and epileptic encephalopathy in two siblings with a novel, homozygous missense variant in SCN1B. Am J Med Ggenet A. (2019) 179:2190–95. doi: 10.1002/ajmg.a.61344
36. Gong J-E, Liao H-M, Long H-Y, Li X-M, Long L-L, Zhou L, et al. SCN1B and SCN2B gene variants analysis in dravet syndrome patients: analysis of 22 cases. Medicine. (2019) 98:e14974. doi: 10.1097/MD.0000000000014974
Keywords: epilepsy, sodium channel, gene, mutation, pediatrics
Citation: Fang H, Hu W, Kang Q, Kuang X, Wang L, Zhang X, Liao H, Yang L, Yang H, Jiang Z and Wu L (2023) Clinical characteristics and genetic analysis of pediatric patients with sodium channel gene mutation-related childhood epilepsy: a review of 94 patients. Front. Neurol. 14:1310419. doi: 10.3389/fneur.2023.1310419
Received: 09 October 2023; Accepted: 27 November 2023;
Published: 18 December 2023.
Edited by:
Carl E. Stafstrom, Johns Hopkins University, United StatesReviewed by:
Yang Tian, Guangzhou Medical University, ChinaCopyright © 2023 Fang, Hu, Kang, Kuang, Wang, Zhang, Liao, Yang, Yang, Jiang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liwen Wu, d3VsaXdlbjIwMjBAc2luYS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.