 Mickael Bonnan
Mickael Bonnan Stephane Debeugny
Stephane Debeugny- 1Service de Neurologie, Hôpital Delafontaine, Saint-Denis, France
- 2Département d'Information Médicale, Centre Hospitalier de Pau, Pau, France
Stroke is a rare and severe complication of giant cell arteritis (GCA). Although early diagnosis and treatment initiation are essential, the mechanism of stroke is often related to vasculitis complicated by arterial stenosis and occlusion. Its recurrence is often attributed to early steroid resistance or late GCA relapse, so immunosuppressive treatment is often reinforced. However, many questions concerning the mechanisms of stroke remain elusive, and no review to date has examined the whole data set concerning GCA-related stroke. We therefore undertook this scoping review. GCA-related stroke does not necessarily display general signs and inflammatory parameters are sometimes normal, so clinicians should observe caution. Ischemic lesions often show patterns predating watershed areas and are associated with stenosis or thrombosis of the respective arteries, which are often bilateral. Lesions predominate in the siphon in the internal carotid arteries, whereas all the vertebral arteries may be involved with a predominance in the V3-V4 segments. Ultrasonography of the cervical arteries may reveal edema of the intima (halo sign), which is highly sensitive and specific of GCA, and precedes stenosis. The brain arteries are spared although very proximal arteritis may rarely occur, if the patient has microstructural anatomical variants. Temporal artery biopsy reveals the combination of mechanisms leading to slit-like stenosis, which involves granulomatous inflammation and intimal hyperplasia. The lumen is sometimes occluded by thrombi (<15%), suggesting that embolic lesions may also occur, although imaging studies have not provided strong evidence for this. Moreover, persistence of intimal hyperplasia might explain persisting arterial stenosis, which may account for delayed stroke occurring in watershed areas. Other possible mechanisms of stroke are also discussed. Overall, GCA-related stroke mainly involves hemodynamic mechanisms. Besides early diagnosis and treatment initiation, future studies could seek to establish specific preventive or curative treatments using angioplasty or targeting intimal proliferation.
1 Introduction
Giant cell arteritis (GCA), formerly called granulomatous arteritis, is a form of vasculitis involving the large and medium-sized arteries branching to the aorta. GCA is the most frequent form in Western countries. Its incidence increases sharply after the fifth decade to 10.9 per 100.000 inhabitants per year (1), and the lifetime risk prevalence is about 1% in women and 0.5% in men (2). Clinical severity is driven by ischemic complications occurring in the cranial arteries due to active recurrence, sequalae and increased risk of death [up to 28% compared with non-stroke GCA patients (3–7)].
Ophthalmic ischemia is the most common complication. In historical series, bilateral blindness occurred in up to 38% of GCA patients (8–10), whereas its incidence dramatically decreased in the era of steroid treatment down to <3% of monocular blindness (11–13). On the other hand, the incidence of GCA-related stroke at onset has always been low in the range of 2–6% (4, 14–17), and mainly involves the vertebrobasilar territories (4, 15).
A metanalysis confirmed a slightly higher risk of stroke (odds ratio ~1.4) in GCA patients than in age-matched controls (18), and the vasculitis process itself is considered to be the main cause of stroke (15). However, comorbid conditions are frequently found in this age group, although their prevalence is roughly similar with age-matched stroke-free GCA (4, 19, 20), even though this point remains controversial (15, 16). The mechanisms involved in GCA-related stroke are sometimes difficult to ascertain and should not be confused with events occurring frequently in aged patients (3, 21, 22).
Series and reviews of intracranial GCA focused on the main clinical and predictive features of intracranial lesions (4, 5, 17, 23–30), but none of them examined stroke mechanisms in detail, and most data about GCA-related stroke derive from unrelated or heterogeneous case reports that were never systematically examined. Pathological examination of arteries involved in GCA reveals an extended range of transmural inflammation, with granuloma reducing the inner caliber of the artery. The endothelium is mostly spared by inflammation but clots are sometimes observed inside the reduced lumen. Cervical arteritis may extend to the internal carotid and vertebral terminations, the basilar artery and rarely to the very proximal M1 branches, whereas the distal branches are always spared. However, it remains unclear how GCA is related with brain symptoms, resistance to steroid, and stroke relapse. This paper examines questions concerning GCA-related stroke that have received little attention until now:
1) Is there a pattern of stroke associated with GCA? Since extracranial involvement is associated with stenosis/occlusion, stroke involving watershed areas and hemodynamic mechanisms should be over-represented.
2) What is the most distal boundary of GCA-related arterial lesions, and what is the link with internal elastic limitans or the vasa vasorum?
3) Response to steroid initiation is often described as paradoxical since the frequency of stroke relapse may increase within the first days of drug initiation. Delayed stroke suggests resistance to drug treatment, clinical relapse requiring reinforcement of treatment, or unrelated events. Briefly, can the natural history of critical arterial stenosis persisting from GCA relapse be characterized?
4) The diagnosis of cranial involvement in GCA is challenged by the fact that some arteries are spared, and by the lower frequency of general signs or normal inflammatory parameters, especially in ophthalmology. What is the proportion of these 'cold' or misleading cases among patients with GCA-related stroke?
5) What is the kinetics of stenosis, and in turn the kinetics of involution of the arterial inflammation? What is the frequency of persisting stenosis and occlusion?
6) Since a hemodynamical mechanism is the most plausible, can stenting prevent dramatic stenosis and stroke?
We do not deal with the general signs and management of GCA [see Mollan et al. (31)], the mechanisms and clinical management of ophthalmic lesions [see Mollan et al. (32)], nor temporal artery biopsy (TAB) sensitivity. However, data obtained from ophthalmologic studies were used to throw light on the mechanisms of GCA-associated stroke.
2 Methods
PubMed request was: (temporal arteritis OR giant-cell arteritis) AND (stroke OR brain OR cerebral OR cerebellar OR carotid OR basilar OR vertebral OR watershed). All abstracts of selected articles were read. All articles dealing with the questions raised in the introduction were carefully read and possibly used in the literature review. References of articles were also examined for unnoticed related articles. For statistical methods, see Supplementary material 2.
3 Results
3.1 Establishing GCA diagnosis in presenting strokes
3.1.1 Definitions
GCA is called temporal arteritis, or cranial arteritis (as opposed to extracranial involvement), which hampers the distinction with brain involvement. Involvement of the cervical non-temporal or intracranial arteries is often not clearly described among admitted GCA extensions (solitary cranial or extracranial, mixed, see Supplementary Table S1). Various terms were used to define this involvement: intracranial involvement is based on the location of the ischemic consequence (includes brain stroke but remains unclear about ophthalmic lesions), whereas other authors preferred to use intra- or extra-dural artery lesions based on the location of putative mechanisms (33). We use the term cervical arteries to define arteries abutting to the brain (e.g., carotid or vertebral). Otherwise, strict artery nomenclature was preferred to avoid confusion.
3.1.2 Limitations of ACR criteria applied early
GCA diagnosis is based on the ACR 1990 criteria (34) which were revised in 2022 (35). Cranial ischemic signs were included (jaw/tongue claudication, sudden visual loss) but stroke was excluded (Supplementary Tables 1, 2). Patients presenting at onset with GCA-related stroke before the GCA diagnosis has been made may not meet the criteria due to their lower sensitivity.
In series of patients with ischemic complications (Table 1), new onset localized headache was observed in 74% of patients and temporal artery signs in 56%. The prevalence of abnormal inflammatory parameters may be lower than in non-ischemic GCA patients but the issue is still elusive (often expressed as median compared to non-ischemic patients). When the diagnosis is evoked, TAB gives definitive positive arguments for the diagnosis of GCA. However, the sensitivity of TAB is lower in patients recruited due to ischemic complications in fast-track series (46%−56%) (12, 41). The prevalence of the ACR 1990 criteria cannot be assessed owing to a circularity bias, and no data is available about the 2022 criteria. Moreover, since the sensitivity of fulfilled ACR criteria is high but incomplete (~93%), they are required for clinical research but not for clinical diagnosis (e.g., typical GCA may be diagnosed in a 48-year-old patient). Clinicians faced with stroke should observe caution, although the incidence of GCA-related stroke remains low. The absence of typical GCA signs should not rule out the hypothesis and FDG-PET and/or US of the cranial arteries should be performed owing to their diagnostic accuracy in this setting.
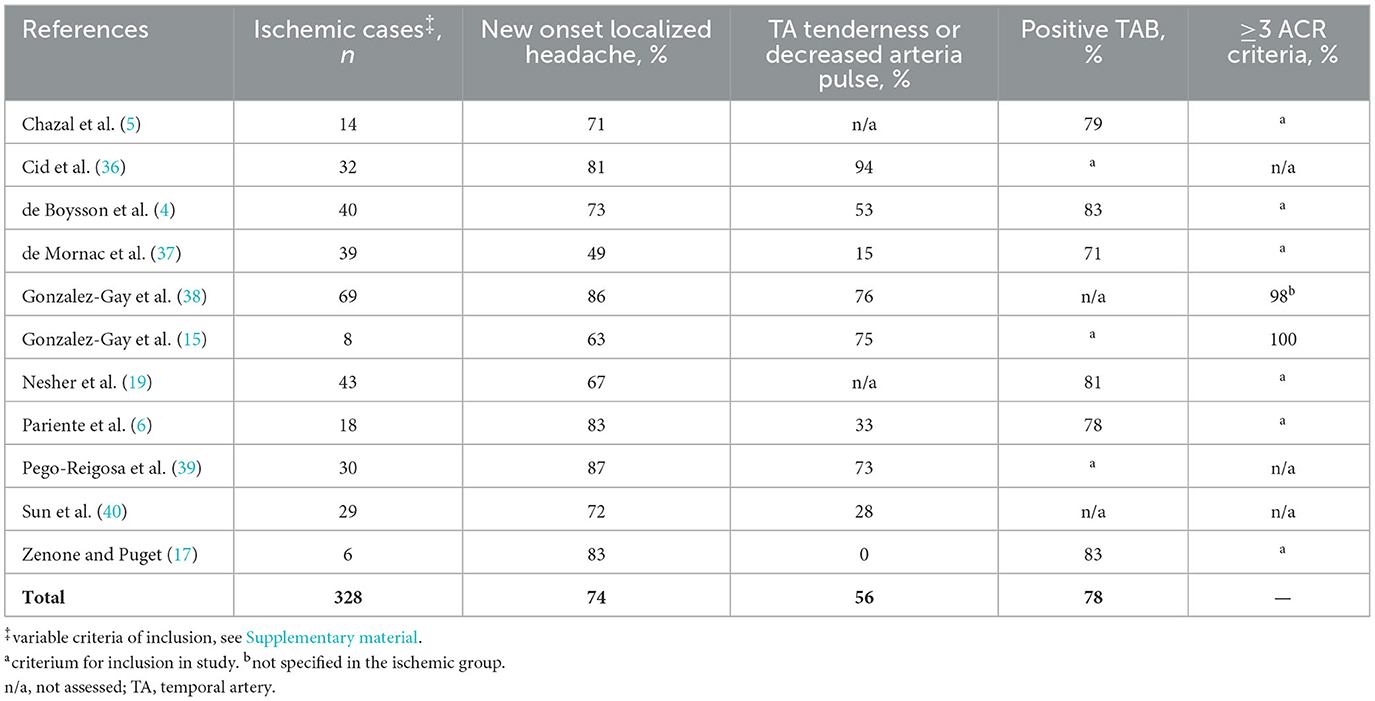
Table 1. Prevalence of ACR criteria 1990 in series of ischemic case.
3.2 Frequency of stroke and cervical artery lesions
GCA is a rare cause of stroke, accounting for 0.15% to 0.4% of patients admitted for stroke and systematically checked by ultrasonography for “halo sign” (1, 42–44) (Table 2). The incidence of GCA-related stroke over the age of 50 is 0.76 per 100.000 inhabitants per year (1). Stroke is also admitted to be a rare complication of GCA, estimated at below 16%, and congruent for the range of 2–3% of GCA over the series defined in the period extending from GCA onset to the first month following steroid initiation (4, 6, 14–17, 28, 37).

Table 2. Red flags for the diagnosis of GCA in stroke.
Cervical arteries are very often involved in GCA, either in isolation (37%), or in association with large vessels [42%, in De Mornac et al. (37)], which contrasts with the rarity of observed stroke and suggests that the cervical arteries involved are not so prone to distal complications. However, ischemic complications were possibly underestimated in older series in which stroke was diagnosed on clinical grounds without systematic MRI screening, whereas systematic screening for silent stroke in recent series led to an increased prevalence in non-selected GCA patients [up to 15% in Siemonsen et al. (47)].
A systematic autopsy series found 0.4% of (unsuspected) aortic GCA in the general population (48), suggesting that GCA could be more prevalent than previously thought. However, no data supports the hypothesis of serendipitous cranial GCA underlying stroke.
3.3 Lessons from non-brain arteries
We briefly examine mechanism of ischemic manifestations in the eyes and limbs and show that low flow is the main problem whereas embolic events are mostly absent.
3.3.1 Lessons from optic ischemia
Our study did not focus upon ophthalmic signs, but the frequency of optic involvement is higher than stroke and probably shares similar mechanisms. Ophthalmic, post-ciliary and circles of Zinn arteries are often involved in GCA arteritis (9). In angiography, massive delayed choroidal filling is characteristic of GCA, but various patterns of ischemia are possible: choroidal ischemia, arteritic anterior ischemic optic neuropathy (AAION), posterior ischemic optic neuropathy, or central retinal artery occlusion. In series of GCA patients, 15% developed permanent visual loss, either unilateral (51%), bilateral simultaneous or sequential (30%), and 17% had a visual field defect (36, 49). Premonitory visual symptoms were present in a quarter to three quarters of patients (19, 24, 49).
A third of involved eyes deteriorated within the first 6 days after steroid initiation, 11% of uninvolved eyes deteriorated within a day, whereas only 15% recovered more than two lines of visual acuity, although remaining deeply impaired (50). Overall, the duration of GCA symptoms was shorter in patients with ischemic events (mean 5 weeks from onset) (36). These data suggest that although most patients benefit from early treatment, some could still be at high risk of cranial complications (36). The precise nature of the risk factors remains unclear in these patients (random lesion of critical arteries? persisting inflammation or thrombosis of stenosed artery?).
Amaurosis fugax revealing GCA is a major sign of compromised ophthalmic perfusion: fluorescein fundus angiography confirms sluggish circulation of blood in relation with low arterial pressure. Moreover, relapse or aggravation of visual signs could be related to a fall of prefusion pressure secondary to hypertensive drugs or to a rise in intraocular pressure (51). MRI may demonstrate wall enhancement of ophthalmic arteries (28) and peri-phlebitis (52). Amaurosis fugax rarely relapses after treatment initiation (24).
Doppler exploration of the ophthalmic artery demonstrates various hemodynamic alterations: reversal of flow (without carotid change), turbulent flow between two focal stenoses, increase in vascular resistance (due to vascular bed stenosis), reduced ocular pulse amplitude, or loss of flow. Interestingly, high flow velocity and vascular resistance obtained at onset are associated with clinical progression and visual deterioration after steroid initiation (53). These data suggest that patients who undergo clinical deterioration have more advanced and critical vasculitis lesions at onset than those who 'responded' to steroids (53). During treatment, vascular resistance still increases whereas blood flow decreases, suggesting a residual intimal thickening and wall fibrosis resistant to steroids (or lag before efficacy), even in unaffected eyes (53). An improvement on fluorescein angiography is indicative of clinical recovery (54, 55), suggesting that retinal penumbra may explain early optic impairment at some point. Finally, lessons from optic ischemia point to the major role of hemodynamical mechanisms downstream arterial stenosis/occlusion.
3.3.2 Lessons from GCA-associated limb arteriopathy
Limb claudication is one of the most frequent symptoms, although sometimes remaining poorly symptomatic (56). Vascular limb manifestations accompany or often reveal general symptoms in ~30–40% of patients (56–59). In half of cases, vascular symptoms appear after treatment initiation (56). Local vascular relapses (stenosis) may occur at the end of the treatment (56). Except in rare cases showing dramatic improvement of stenosis, the resolution of symptoms does not generally accompany arteriographic improvement of stenosis and results from the development of artery collaterals (56, 57). Unlike other types of vasculitis, gangrene is not a common feature of GCA (7), which suggests that lesions of the distal bed or emboli are not involved.
3.4 Clinical cranial signs and symptoms
Cranial ischemic events in GCA could be defined as transient or irreversible signs. Transient signs are typical of reversible ischemia or insufficient arterial flow: jaw claudication (painful muscles during chewing), amaurosis fugax or blurred vision, and transient ischemic attack (TIA) (Table 3). Irreversible signs are mainly tongue or scalp necrosis, diverse ischemic ophthalmic signs, and stroke. The incidence of ischemic events occurring within 2 weeks after GCA diagnosis, defined as transient and irreversible events, was 73%, whereas severe irreversible events (AION and stroke) occurred in 34% of GCA (60). Carotid and orbital bruits related with flow turbulence and disappearing a few days after steroid initiation were noted early (14, 61, 62).
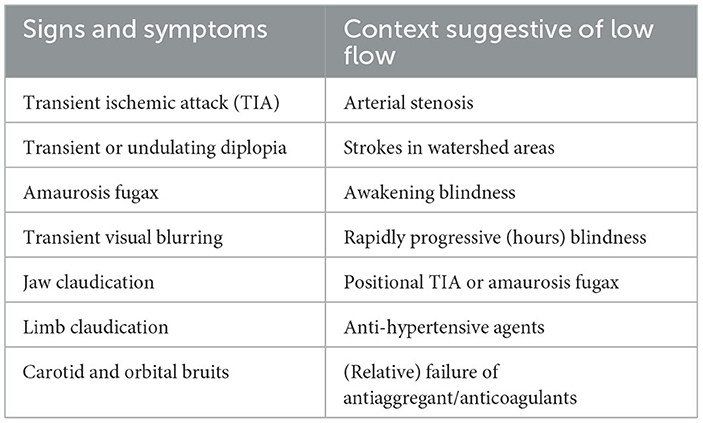
Table 3. Signs and symptoms related with hemodynamic impairment.
Headache is the most frequent extracranial symptom (72%). Clinical symptoms of intracranial GCA are the following: unilateral blindness (26%), cerebellar signs (72%), motor impairment (62%) and impaired level of consciousness (49%), and about half of patients died (26). Dementia is always the consequence of relapsing and extensive infarction (63).
Transient or undulating diplopia from third or sixth nerve palsy may occur in more than 10% of GCA cases in relation with oculomotor muscle or nerve ischemia (32, 64), and may be complicated by oculomotor synkinesis or aberrant nerve regeneration, suggesting peripheral nerve involvement [see Reich et al. (64)]. MRI may reveal third nerve enhancement (“check mark sign”) (65).
3.5 Meta-analysis of signs associated with ischemic events
Several authors compared groups of ischemic events among GCA patients to delineate a high-risk clinical profile and select patients who might benefit from aggressive treatments (19). Although the available data are highly heterogeneous and mix brain and extra-brain ischemic events, we provide a metanalysis of clinical and paraclinical signs associated with cranial ischemic events including stroke (Table 4, all the results below are findings from the metanalysis, detailed methods and results are provided in Supplementary material 2).
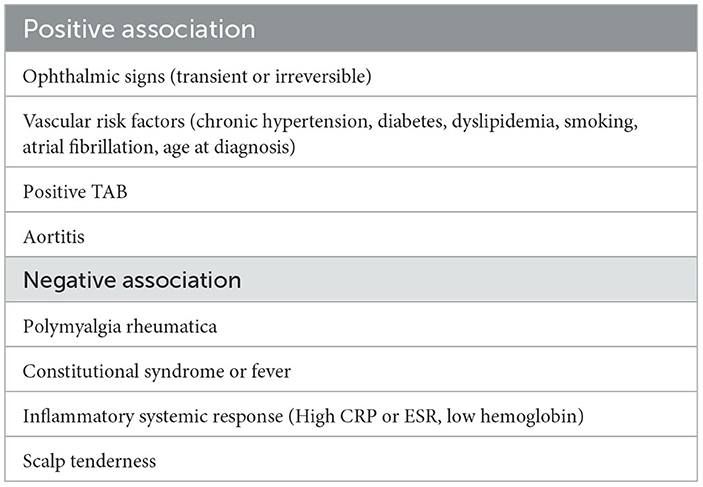
Table 4. Signs predicting stroke events.
Transient signs (OR 5.03 [CI 95% 2.81, 9.01], p < 10−5), mixing amaurosis fugax and TIA, signs of aortic branch lesions, like aortitis (OR 3.09 [1.39, 6.85], p = 0.005) and positive TAB (OR 1.69 [1.11, 2.56], p = 0.01), are strongly associated with ischemic events. Other signs of cranial involvement, like abnormal TA on physical examination, jaw claudication, and headache, were not significantly associated.
General signs, including constitutional syndrome (OR 0.64 [0.47, 0.88], p = 0.005) and fever (OR 0.60 [0.40, 0.91], p = 0.01), PMR (OR 0.66 [0.51, 0.85], p = 0.001) were all negatively associated. Inflammatory systemic response was also lower in patients with ischemia: CRP (mean difference, MD −29.01 [−42.61, −15.41], p < 0.001), ESR (MD −5.70 [−9.71, −1.69], p = 0.005), whereas hemoglobin was higher (MD 0.49 [0.19, 0.79], p = 0.001). Platelet and albumin levels were not significantly different.
Among vascular risk factors, chronic hypertension was the strongest predictive factor (1.82 [1.38, 2.41], 10−4). Unfortunately, specific data (complications, pressure control or drugs administered) were not available, especially at the time of ischemic events. One series found that treatment by anti-hypertensive agents, especially β-adrenergic inhibitors, was associated with a risk of optic ischemic events but not hypertension (66). Since hypertension is a vascular risk factor, it is often suspected that the risk associated with antihypertensive drugs is a confounding effect (13, 32). Nevertheless, we think that antihypertensive drugs may have facilitated hemodynamic mechanisms that, in turn, triggered ischemic events.
Other vascular risk factors were also predictive of ischemic events: diabetes (OR 1.65 [1.15, 2.36], p = 0.006), smoking (OR 1.69 [1.06, 2.68], p = 0.003), hypercholesterolemia (OR 1.37 [1.02, 1.84], p = 0.04), and atrial fibrillation (OR 2.23 [1.20, 4.13], p = 0.01). Among vascular risk factors, diabetes mellitus was the only other significant vascular risk factor (OR 1.84 [1.15, 2.94], p = 0.01), whereas a history of ischemic heart diseases was not significant. Antiaggregation or anticoagulation before the diagnosis did not provide significant protection, but an interaction with vascular risk factors may mask a protective effect. Although socio-economic deprivation is probably a proxy for higher tobacco exposure and atherosclerotic risk, a study adjusting for common vascular risks found that deprivation could be a major risk factor of ischemic events, and patients who took aspirin before GCA were still at higher risk of ischemic events (67). Finally, GCA in itself is probably an additional vascular risk factor since the cumulative incidence of myocardial infarction and peripheral vascular disease are higher in GCA patients (68).
Ischemic events at onset are strong predictors of future events. Transient cranial ischemic events like jaw claudication, amaurosis fugax, and irreversible visual loss or stroke are strongly associated with the risk of future strokes (15, 19, 36, 38, 54, 60, 69, 70). Ischemic events developing after GCA diagnosis are often recurrences since 21% of them are relapses, but only 4% of GCA without ischemic events developed a recurrence after steroid initiation (19, 50).
3.6 Systemic inflammatory response (SIR)
It is widely thought that the risk of cranial ischemic symptoms is inversely proportional to SIR (36, 71). However, the strict definition of inflammation severity remains unclear. If 'strong' SIR is defined by ≥3 parameters in: fever, weight loss, ESR>85 mm/h, or hemoglobin <11 g/dL (36, 71–73), no patients with an ischemic event fulfilled all four positive criteria (36). However, replication is lacking, and this stratification is not used in common practice. Inflammatory parameters (e.g., ESR, CRP, fibrinogen, anemia) could be lower in GCA patients with stroke (4, 36, 74), although mean ESR remains similar in others (5). A series with short delays from GCA onset to diagnosis found similar levels of inflammation in patients with stroke (75). This suggests that ischemic patients may be referred early before the full-blown systemic response (36, 72, 76). Indeed, GCA patients recruited in fast-track clinics displayed lower mean CRP levels, although higher levels were observed in cases with visual loss (77). Multivariate analysis did not confirm a difference in CRP levels between GCA subgroups with stroke or amaurosis (23, 78). Although a promoting role of neo-angiogenesis was proposed (79), inflammatory syndrome probably does not exert any protective role on ischemia (24). Finally, although it is possible that ischemic complications occur in patients with lower inflammatory parameters, the minor biological difference does not add any clinical value at individual level (80). Of note, CRP levels are variably expressed in mg/dL or in mg/L, leading to erroneous underestimation of CRP levels. Normal ESR at onset is possible in <10% of GCA (81), but CRP is far more sensitive to systemic inflammation. At GCA onset, CRP is very rarely normal [below ≤ 2.5% (81, 82)]. It is typically associated with optic signs and rises rapidly within a few days (83). Combined normal CRP and ESR occurs in <1.2% of cases (84). Series of ischemic cases reported low (but abnormal) median CRP at diagnosis (6 mg/L, range 2–28 mg/L)(5), but normal CRP levels were extremely rare in published cases of stroke (85, 86). In our review of GCA-related stroke, 22% cases had CRP below 20 mg/L, but only one patient displayed double-negative CRP/ESR [none in Hayreh et al. (81) and Bonnan and Balley1, and 0.8% in Parikh et al. (82)]. In another series, double-negative CRP/ESR was present in 36% (9/25) of patients with stroke and 18% (3/16) of other GCA patients (87). Therefore, although SIR is not always dramatic, completely false negative biology for inflammation is not an uncommon finding at onset in GCA-related stroke.
Severe ischemic GCA patients were found to display a higher prevalence of VEGF polymorphism associated with a reduced circulating level (88), but this finding has not been confirmed until now.
3.7 Does pathology give clues to ischemic mechanisms?
GCA is characterized by inflammatory infiltrates of the medium and large vessels, with variable abundance of giant cells and internal elastic lamina (IEL) fragmentation (89) (Figure 1). Random lesions occurring along the arteries are separated by normal tissue (“skip lesions”). Large transmural inflammatory infiltrates correlate (90) or not (91) with higher levels of biological inflammation. The influence of steroids on the main parameters of inflammatory lesions remains modest during the first month (91), whereas healing lesions are observed on biopsies performed more than a year later (92).
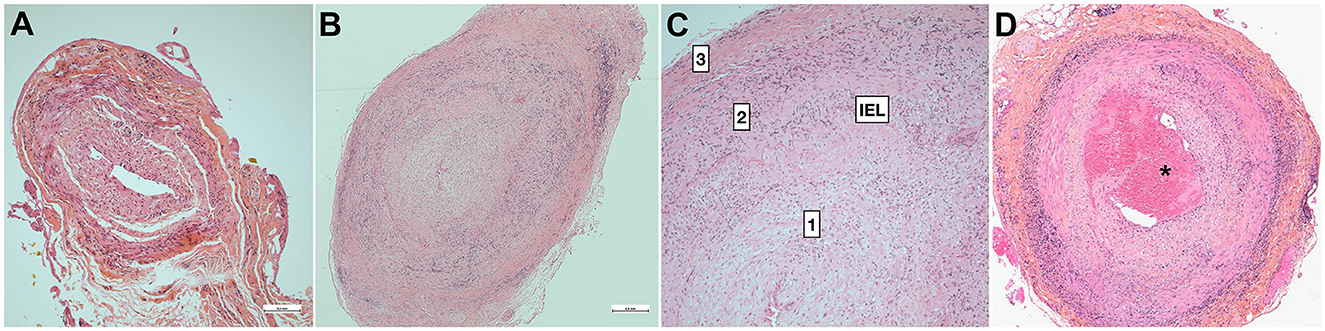
Figure 1. Pathology of temporal artery biopsy. (A) False-negative temporal artery biopsy. Minimal non-specific intimal proliferation. (B) Positive temporal artery biopsy with full-thickness granulomatous inflammation and slit-like luminal stenosis related with major hyperplasia of the intima [magnified in (C)]. (D) Positive TAB with intraluminal thrombus (*). 1: intima, 2: media, 3: adventitia. H&E staining. Bar is 0.5mm (Dr Badaro).
Ophthalmic involvement is generally associated with intensity of inflammation, giant cells, and plasmocytes, or with the severity of intimal hyperplasia (93–95). However, the correlation is mostly absent in stroke patients, since none of the histological parameters obtained from TAB (especially intimal proliferation) differed in ischemic groups (90). Luminal thrombosis is observed at the same frequency (95) (Figure 1D). A major limitation is that the temporal arteries do not irrigate the brain or ophthalmic tissues, so focal thrombi found on TAB do not convey any risk of brain or eye embolization. Since the culprit arteries cannot be evidenced except by autopsy, it is uncertain whether the lesions of temporal artery lesions mirror those of the culprit arteries.
One study found a higher ischemic risk in the presence of vascular calcifications (96), but stenosis combining atherosclerotic and arteritis lesions has been rarely described (97). Age-related intimal thickening is aggravated by atherosclerosis (98), but remains moderate and non-occlusive. GCA does not promote atherosclerosis (99), and ischemic events in GCA are not related to complicated atherosclerotic lesions.
Fibrinoid degeneration is always absent in large GCA series (96, 100, 101) and would point to antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides (102). Autopsied cases from long ago reported thrombosis of the coronary, vertebral or basilar arteries infiltrated by arteritis (103, 104). However, detailed pathology was not provided, so it remains unclear whether the mural thrombosis developed on endothelial lesions as arteritis, atheromatous plaque, or an artery stump. Luminal thrombus, which is generally subtle, is observed in <15% of TAB (90, 101, 105), irrespective of steroid treatment (96), and may participate only in luminal occlusion, which is mainly driven by intimal proliferation (95, 101, 106–108) (Figures 1B, C). Reduction of the arterial lumen to slit-like is mostly the consequence of the major intimal proliferation observed in half of the cases (101), and developing until occlusion (101, 109). Moreover, the long-term evolution of intimal hyperplasia after treatment is unclear considering the frequency, extension and degree of involution, although persistence may be variable (110). One case showed a minute focal hemorrhage within the thickened intima (33).
Intimal hyperplasia, which may drive ischemic events, is variable among GCA patients. Delayed treatment, which may lead to an increased burden of lesions, is associated with an increased risk of ischemic events. However, the degree of intimal hyperplasia (on TAB) may be independent from the lag between GCA onset and TAB (91, 94). Since intimal hyperplasia is assessed on a different artery (e.g., TAB) than those involved in ischemic events, its implication may be difficult to confirm (96). We believe that the risk of ischemic events may be related with both the duration of the natural history before treatment, and with each patient's kinetics of reactive intimal hyperplasia. Moreover, persisting intimal hyperplasia in healed patients may contribute to the delayed risk of hemodynamic ischemic events. Intimal hyperplasia is significantly associated with T-cell synthesis of IFNγ and IL1β, and with PDGF-A/B, VEGF and GM-CSF secreted by macrophages and multinucleated giant cells (71, 93), which are high value targets for future treatments.
3.8 Explaining GCA selectivity to extradural arteries: putative role of basic structural variations in cranial arteries
It is commonly admitted that GCA lesions target elastic fibers and especially IEL, which is fragmented in the lesions. Why are the intradural arteries spared by GCA? Some authors posit that IEL could be absent in the intracranial arteries, which is erroneous, or that the lower content of elastic fibers might play a protective role, which is contradictory with the persistence of a thick IEL.
3.8.1 Elastic fibers and laminae in extra- or intra-dural arteries
The walls of the cranial arteries are thinner than those of other arteries of the body, owing to thinner adventitial and medial layers (111). Elastic fibers in the tunica media and external elastic lamina (EEL) progressively vanish along the horizontal segment of the cavernous portion of the internal carotid, and within the more distal 5 mm of the intradural vertebral arteries (111–114). Predominance of elastic fibers in the vertebral arteries rather than in the carotids may explain predominance of VA inflammation (115). The discontinuation of EEL is heterogenous (Figure 2), while continuous and even more fragmented EEL may be observed in the intracranial arteries at low frequency (below 20%) beyond the dural entry point (116). In the posterior circulation, continuous EEL is commonly observed in VA and more rarely in the distal arteries (i.e., basilar and anterior inferior/superior cerebellar arteries) (116). EEL disappears in the ophthalmic arteries after penetrating the optic nerve and globe, which correlates with observations of GCA-spared retinal arteries (115).

Figure 2. Basic structural variation of intracranial arteries. (A) Intradural arteries are characterized by the presence of large internal elastic lamina (IEL), whereas EEL disappear from the entry point into the dura mater. Extradural arteries have prominent EEL and IEL, and elastic fibers in the tunica media and adventitia. In the adventitia, vasa vasorum (VV) surround extradural arteries including the vertebral and carotid arteries, whereas VV are usually absent from the intradural arteries. (B) Classically, the transition from extra- to intra-dural artery type occurs in the cavernous portion of the carotid between the posterior (EEL always present) and anterior knees (EEL mostly absent) (113), and within the 5mm of vertebral artery around the dural entry point (111). Although EEL is considered absent from intracranial arteries, structural variations may occur in healthy subjects, and EEL may persist as a continuous or fragmented structure in the intracranial arteries [from Denswil et al. (116)]. ACA, anterior cerebral; ACoA, anterior communicating; AICA, anterior inferior cerebellar; BA, basilar; ICA, internal carotid; MCA, middle cerebral; PCA, posterior cerebral; PCoA, posterior communicating; PICA, posterior inferior cerebellar; SCA, uperior cerebellar; VA, vertebral.
On the other hand, the thickness of the IEL is mostly unchanged between the extra and intradural segments (111, 113), or even thicker in the brain arteries (114).
3.8.2 Vasa vasorum
Others posited that the absence of intradural vasa vasorum (VV), which might be key structures for initiating inflammation, could be protective (32, 33). At birth, VV are present in the peripheral arteries but not in the brain arteries. During aging, VV develop in the continuity of extradural VV toward the proximal segments of the intradural arteries and are mostly associated with intracranial atherosclerosis (117). In GCA, VV might be the entry point of autoreactive CD4 lymphocytes and VV vasculitis occurring in the adventitia. Consequently, the risk of intracranial GCA lesions may correlate with pre-existing VV (89, 91, 117), which is age-dependent, variable and limited to the proximal segments of brain arteries. Moreover, VV might be more frequent in the intradural vertebral and basilar arteries. Finally, there is probably an individual and age-dependent vulnerability to the proximal intracranial extension of GCA lesions, while the distal intracranial arteries are always spared.
3.9 Imaging techniques in GCA-related stroke
Imaging techniques are briefly examined for red flags, tips and pitfalls in the clinical context of GCA-related strokes.
3.9.1 Computed tomography (CT) and CT angiography (CTA)
Thoracic CTA may show aortic dilatation or stenosis of the branches. Severe stenosis and occlusion of the vertebral and internal carotid arteries, but also lesions of branches of the external carotid, are easily observed. Occlusion and inflammation of the temporal arteries appear as a blurred artery wall with perivascular enhancement (cigar smoke sign) in 71% of GCA (118).
Increased arterial wall thickness is highly typical of aortitis and may be observed in the smaller branches (see cut-off in Supplementary Table 3). However, CT examination of the cervical artery wall (internal carotid or vertebral arteries) is not as sensitive as other techniques.
3.9.2 Brain MRI
Ischemic brain lesions are not specific and their patterns are discussed elsewhere. Involvement of the frontal branch of the temporal artery is so common that it gave the disease its former name. The cranial arteries emerging from the external carotid, occipital arteries, and the frontal and parietal branches of the temporal arteries are commonly involved. Cranial MRI acquired with protocols dedicated to vessel-wall imaging (i.e., postcontrast fat-saturated 3D-T1-weighted spin-echo acquired on 3T scanner) demonstrate a high proportion of multifocal arterial involvement (119). Arterial lesions display bright contrast enhancement and mural thickening extending to the surrounding tissue of these medium arteries (119), and MRI sensitivity is close to 90% vs ACR criteria, and 100% vs TAB (119). T2-weighted sequences are poorly sensitive to vessel-wall lesions, except in the event of massive inflammation (120). High field 7T MRI confirms the findings and improves the visibility of the small branches of the temporal artery (121).
3.9.3 Ultrasonography (US)
US is an easy-access cost-effective technique which reveals typical signs of GCA, described as an hypoechogenic 'halo' of the arterial wall secondary to the increased intima-media thickness (IMT) that persists during artery compression (122). Cut-off values for IMT may be highly specific and sensitive (~100%) (123) (Supplementary Table 3). Halo sign may be observed in all arteries involved by GCA, especially the vertebral and temporal arteries, and it is rarely influenced by vascular risk factors. US is more sensitive than conventional CT or MRI of the cervical arteries to detect changes. Cases with limited evidence of GCA, e.g., absence of headache and inflammation, can be revealed by this sign, and fast-track management is now based on US screening (12, 41). In patients presenting with a cranial ischemic event, positivity of the temporal artery may be low (29) whereas the vertebral arteries are more frequently involved (124).
3.9.4 PET scan
FDG-PET/CT explores most of the cranial and extracranial arteries involved but is slightly less sensitive than clinical diagnosis (Se 71%, CI 95% [48–89]) or TAB (Se 92%, [62–100]) (125). Positivity correlates with inflammatory parameters (126). Examples are given in Supplementary Figure 3.
It is worthy to note the lower risk of systemic involvement of the large arteries in FDG-PET, especially aortitis in GCA patients suffering from ischemic lesions of the brain and eyes (127–129). The cranial arteries are rarely examined by FDG-PET since assessment of small-diameter arteries was hampered by low resolution in earlier studies. Nowadays, combined FDG-PET/CT allows accurate assessment of the external carotid, temporal, mandibular, vertebral, occipital arteries (130) and of the lower internal carotids, but not their terminations which are too close to the brain. Positivity of one of these arteries is highly indicative of GCA (130). The predictive value of artery FDG-PET positivity for actual or future stenosis or stroke remains unknown.
3.10 Drug treatment of GCA-related strokes
In this section, we do not review strategies for treating GCA but focus rather on treatment strategies in the context of GCA-related ischemic events.
3.10.1 Treatment of GCA
High-dose steroid treatment should be started as soon as the diagnosis is made. Fast-track management in specialized units improves delays in diagnosis and earlier treatment decreases the rate of blindness (12, 41, 77). Guidelines for preventing stroke complications are mostly based on retrospective data, but no controlled trial has yet been performed.
Patients with complicated GCA are commonly administered high-dose steroids, either oral (1 mg/kg/d) or IV (15 mg/kg for 1 to 3 days) (4, 131). Megadose or standard oral dose steroids prevented an attack in the contralateral eye but slightly influenced visual outcome in the already affected eye (51). Data obtained in the treatment of visual loss suggest that delayed initiation of steroids (≥24 h after visual loss) dramatically decreases the expected rate of sight improvement (38). Although not fully analyzed, this observation suggests that ophthalmic ischemia could be at least partly reversible (albeit with minimal clinical gain), as a penumbra caused by reduced blood flow may be reversed by an acute effect of steroids on vasculitis. Tapering steroids may trigger GCA relapse, and the patient should receive careful and comprehensive monitoring of potential relapses.
Immunosuppressive drugs are sometimes used early (132). The influence of methotrexate on the risk of future visual loss remains uncertain [no stroke occurred in Hoffman et al. (133)]. An incident stroke was observed in an infliximab-treated group but not in the placebo group (134). Tocilizumab (TCZ), which targets IL6-receptors, can be given in association or as monotherapy to avoid steroid treatment. However, various issues are to be considered in the context of GCA-associated strokes:
a) no patient developed early stroke in the GiACTA trial, so the preventive effect against stroke could not be compared with the effect of steroids (only one stroke and visual loss occurred at day 254) (135), whereas in an observational study, 40% (2 of 5) of TCZ-treated patients (136);
b) worsening of arterial stenosis occurred during TCZ treatment so arterial follow-up should not be neglected (137);
c) although no head-to-head comparison is available, the rate of vision loss occurring after initiation of treatment was low but roughly similar in TCZ- or steroid-treated patients [see Amsler et al. (129)]. The lower incidence of visual complications in patients receiving late add-on TCZ may also illustrate the spontaneous decrease in relapse risk (138);
d) TCZ does not improve acquired vision loss any more than steroids.
Consequently, TCZ is an interesting treatment for GCA, but its benefits (steroid-sparing) related with stroke prevention have not yet been proven.
3.10.2 The so-called resistance to steroids during treatment initiation
Non-compliance with steroid therapy and premature discontinuation due to side-effects occurred in 3 of the 8 patients who suffered early ischemic complications (19). However, stroke developing within the first days after steroids are puzzling and authors discussed several putative causes: arteritis refractory to steroids (139, 140); steroids triggering ischemic events (15, 22, 51, 103, 141–148); insufficient steroid doses (103, 149, 150); or promotion of thrombosis by steroids via effective inhibition of prostacyclin but defective inhibition of platelet thromboxane (151).
Several days of steroid therapy may be needed to halt the inflammatory process, whatever the steroid dose (24, 51). This simple explanation for this is that most of the ischemic events occurring after steroid initiation occur within 5–7 days, especially optic events (51), but are rare thereafter, even though the dose is still high. This pleads against the triggering role of steroids. Consequently, remission (i.e. no subsequent ischemic event) obtained by immunosuppressive drugs given as add-on therapy after a within-a-week relapse is probably more due to natural history than to steroid resistance being overcome by add-on drugs (129, 140).
3.10.3 Treatment of GCA-related stroke
Use of antiplatelets and heparin is proposed empirically at GCA diagnosis (17, 19, 20), and has been strongly recommended since the seminal work of Nesher et al., who reported a 5-fold decreased risk of ischemic events (19, 152). In another study, taking low-dose aspirin and warfarin before GCA was the main difference in patients remaining free of ischemic events (20). However, the protective effect of treatments remains controversial and patients with severe ischemic complications more often receive anti-aggregants or anticoagulants (15, 19, 29, 60). Other studies failed to demonstrate any difference in the incidence of ischemic events between GCA patients previously treated by anti-aggregants or not (4, 16, 29, 153, 154), suggesting a lack of protection, although a protective effect may have been counterbalanced by a higher prevalence of vascular risk factors. Moreover, recurrent TIAs, which are probably related to stenosis, seem resistant to heparin (155). The mechanisms involved in the preventive action of aspirin on GCA-induced stroke are not clearly understood: prevention of embolization from artery to artery (in keeping with the efficiency of warfarin), or the preventive effect of aspirin on intimal hyperplasia via the NF-κB pathway (156), like the action of steroids, or the repression of IFNγ transcription (with no effect on the transcription of IL1β and IL6).
Heparin, warfarin or anticoagulants were used to prevent and treat ischemic complications on the hypothetical basis that thrombosis is promoted by anti-cardiolipin (aCL) antibodies (157). However, aCL are recovered in up to a third of GCA patients, involute within months after steroid treatment, and are not significantly associated with acute ischemic or stenotic events in GCA (19, 158–160).
Anticoagulants might hypothetically prevent thrombo-embolic lesions promoted by severe stenosis or occlusion, although more evidence is still required. Future therapies would ideally combine suppression of inflammation, prevention of thrombosis, and above all should target (both prevent and reverse) luminal occlusion [e.g., imatinib decreases outgrowth of myointimal cells by targeting PDGF (161)].
Since stenosis and occlusion of the cervical arteries are common, these patients are also at high risk of hemodynamic stroke. Flat bed rest is recommended in patients suffering from optic ischemic events and is mandatory to prevent hemodynamic TIA and stroke in high-risk patients.
3.11 Transient ischemic attacks (TIA)
TIA and amaurosis fugax are common warning symptoms of GCA. Around 25–50% of patients with amaurosis fugax may develop definitive vision loss, usually within days (38, 69), whereas amaurosis fugax preceded acute vision loss in 30% to 65% of cases (19, 24, 49, 54). TIA may also involve uncommon territories, e.g., lingual artery leading to paroxysmal tongue numbness (14). TIA preceded stroke in only 6% to 23% of GCA-related strokes (19, 36), and also occurred in 2% of uncomplicated GCA (36). However, the frequency of unspecific symptoms like dizziness observed at GCA onset suggests that the incidence of TIA is underestimated. TIA commonly occurs in GCA patients suffering from vertebral or internal carotid artery stenosis (cavernous segment), and may occur (and recur) months before diagnosis (162). TIA commonly disappears following initiation of steroids but may rarely recur (163, 164), probably depending on the reversion or compensation of arterial stenosis (165).
The relationship between TIA in the territory of the stenotic carotid and low blood pressure is rarely questioned. In one case of GCA-related stroke where angioplasty was ruled out, stenosis persisted despite IS therapy and strokes recurred over several months (166), while TIA never recurred in others treated by angioplasty (167). Transient symptoms, especially orthostatic complaints (including dizziness, syncope), commonly occur in patients suffering from severe bilateral vertebral stenosis (168); positional or pressure changes dramatically modify symptoms and perfusion (169–172); persisting decrease in perfusion may reverse after steroids (173); and bright-light amaurosis fugax is a symptom of severe carotid stenosis (171). The GCA literature is mainly focused on irreversible lesions, whereas the influence of hemodynamic triggers on amaurosis fugax, TIA and strokes in watershed areas (WA) has not been systematically examined.
3.12 Patterns of lesions: stroke and cervical arteries
3.12.1 Patterns of stroke
Data were collected from a previous review of the literature which analyzed 75 documented CGA-related cases (see text footnote 1).
3.12.1.1 Ischemic lesions predate watershed areas
Given the high rate of stenosis and occlusion involving the carotid or vertebral arteries, WA are expected to be the most common targets of ischemic lesions occurring at onset (Table 5). As expected, all cases of ischemic lesions occurring in WA also present severe stenosis or occlusion of the afferent arteries. In the carotid territory, WA are probably involved in up to 89% of strokes, both sides being involved in half of them (Supplementary Figures 1, 2). Moreover, the difference between transient symptoms and minute ischemic lesions gives strong clues to the hemodynamical mechanisms involved [example in Dimancea et al. (174)]. Recurring perfusion impairment may precede imminent hemodynamic stroke (175).
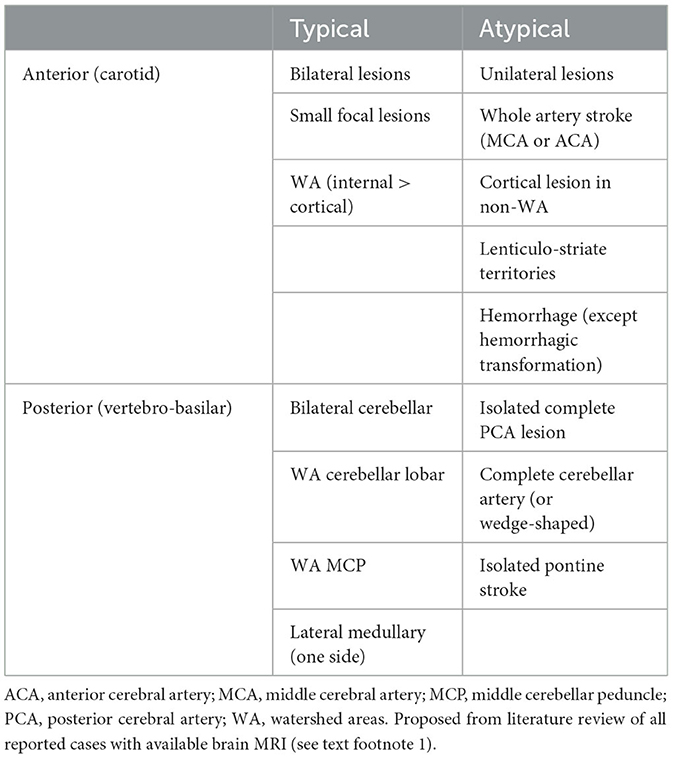
Table 5. Typical ischemic MRI patterns in GCA-related strokes.
WA are more difficult to ascertain in the posterior circulation but could be involved in up to 67% of patients. A typical posterior WA involves one or two sides of the middle cerebellar peduncles (MCP), as was found in a study in which a quarter of patients had a posterior GCA-associated stroke [(43), see text footnote 1]. MCP are very rarely involved in non-GCA strokes (below 0.12% of strokes (176), and among 13 bilateral cases of MCP lesions found in the literature, one suffered from GCA (177, 178). Other WA in cerebellum, pons, or brainstem are difficult to evaluate. The hypothesis of hemodynamic stroke in WA is rarely evoked by authors, so its prevalence remains uncertain.
3.12.1.2 Non-WA are also involved
Ischemic lesions extending to all the arterial territories seem exceptional and mostly involve complete cervical occlusion of the cervical arteries in late or recurring cases. Except in these severe cases, strokes involving an entire arterial territory (e.g., complete MCA, PICA, or pontine paramedian artery) were almost never observed at onset, and stroke involving cerebellar cortex almost never appeared as typical wedge-shaped ischemia. Instead, territory lesions were almost always incomplete, small, multiple, or ill-defined, and often associated with typical WA lesions (179). In fact, a publication bias may have excluded typical territorial stroke patterns from the selected figures. Non-specific small lesions involving the pontine or cerebral peduncle were described, but the mechanism and territories could not be ascertained.
Lesions of the perforating arteries (lenticulostriate) seem very rare (180–182). Curiously, the anterior choroidal artery (AChA) seems always to be spared, since GCA lesions are limited upstream to the extradural carotid, whereas the AChA is a branch of the internal carotid arising downstream near the posterior communicating artery.
Lesions of the lateral medullary may occur and are probably due to the abutting of the artery within the first 5 mm of the intradural part of the vertebral arteries, which are heavily involved by GCA (115). In cases with artery abutment above the end of the vertebral arteritis process, a thrombus may extend upwards from the arteritic segment into the proximal part of the PICA (115). Rare cases of lateral medullary lesions may also occur in patients apparently free of vertebral stenosis, although TOF sequences may reveal an irregular wall (183) and contrast enhancement of the wall provides obvious evidence in some cases (28). These lesions do not have the 'comma-sign' suggestive of medulla WA, which is a linear ischemic lesion located at the medial border of the lateral medullary territory (184). Cases previously treated by aspirin-dipyridamole and coumadin (due to vascular risk factors) may develop stroke in the lateral medullary area (183), thus suggesting an elective arteritis location.
3.12.2 Patterns of involvement of brain-afferent arteries
The common carotid arteries (CCA) were long considered to be spared by GCA (115), and may be used to implant a vascular by-pass (185). However, up to 30% of CCA were involved in a recent series (35). The ophthalmic and ciliary arteries are always involved outside the eyeball and the optic nerve. Classically, the vertebral arteries are severely involved two to three times more often, whereas the internal carotids are mostly spared by GCA (9, 14, 87, 115, 186). Autopsy series highlighted the discrepancy of the less severely affected internal carotid lesions, even in the most involved cavernous segment (9, 97, 115). However, older series based on angiography probably underestimated the prevalence of siphon stenosis (62). In a classical series of 166 TBA-positive GCA collected in 1988, 31% failed to demonstrate involvement of the large arteries, although 19% had permanent or transient ischemic complications (14), which is a very unusual finding in view of the recent literature.
In a series of 62 consecutive GCA (non-selected for stroke), the frequency of arteriographic lesions (with ischemic event or not) was 17–21% in each carotid artery and 11–17% in each vertebral artery (187). The prevalence was higher with optimized brain MRI sensitive to mural inflammation: ICA 40%, MCA 3.5%, VA 68%, and BA in none (47). In this systematic brain assessment study, up to 15% of GCA patients presented with stroke, all of them displaying cervical artery involvement (47), which is far higher than the usual incidence of clinical stroke. In a series of GCA-related stroke, the frequency of artery lesions is even higher: ICA 48%, MCA 19%, VA 84% (stenosis V3 and V4 segments), BA 16% (87) (Figure 3). Therefore, the frequency of cervical artery involvement was probably largely underestimated in earlier studies.
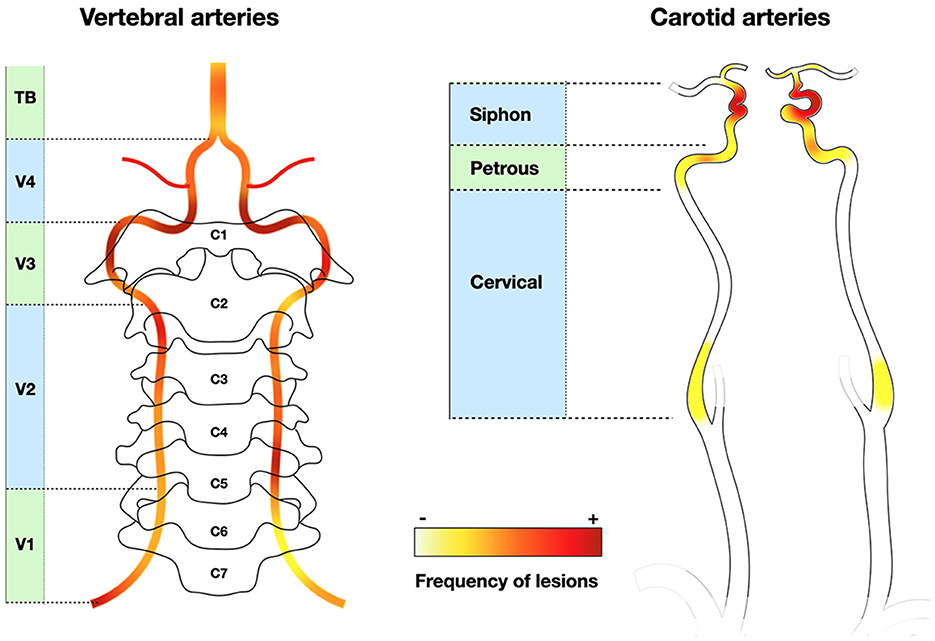
Figure 3. Frequency of stenotic lesions in cervical arteries. Vertebral arteries are often involved, mainly in V3 and V4, whereas internal carotid lesions are located at the level of the siphon. External carotid branches are not depicted. Classically, lesions are localized in V1 and V4 for vertebral arteries (9, 64), or in the entire vertebral and terminal carotid arteries (not represented) (115). Heatmap based on Beuker et al. (87) and Ruegg et al. (188).
The modality of artery assessment strongly influences the association with the risk of ischemic complications: with FDG-PET, the prevalence of carotid or vertebral artery involvement was similar between groups, whereas exams sensitive to luminal stenosis/occlusion demonstrated more frequent lesions in ischemic patients (124). The discrepancy between apparently moderate carotid stenosis and perfusion deficit with bilateral stroke in WA is sometimes surprising (189), again underlining the risk of underestimation due to short stenosis of the ill-depicted tortuous carotid siphon.
3.12.3 Carotid and vertebral artery lesions cluster separately
A tree dendrogram algorithm classifies carotid and vertebral involvement in very different subgroups of patients: the former with mesenteric and aortic lesions, the latter with renal and iliofemoral lesions (187). Our review of stroke patients also points to a separation between anterior and posterior stroke, since fewer than 15% of cases involved both anterior and posterior circulation (at least at onset) (see text footnote 1), but data concerning the other arteries were lacking. Tandem lesions of the arteries (e.g., both carotids, or both vertebral arteries) are common, and bilateral infarction occurred in half of the cases (87). No explanation yet accounts for these associations. There might also be a clustering of patients prone to ischemic complications (190). This hypothesis was proposed by Font et al. (49) to explain visual loss after steroid initiation by exclusive determinism, although alternative explanations are possible.
3.12.4 GCA-related stroke without apparent artery lesions
Sometimes, no stenosis is reported yet it cannot be ruled out (28, 145, 191) (#8). Nevertheless, cases of strokes with parietal inflammation but no apparent stenosis of the vertebral arteries have been reported. For example, a case of minute stroke in the area postrema was associated only with a vertebral halo sign but no stenosis (192). Other reports describe cases of transient vertebrobasilar attacks or Wallenberg syndrome with vertebral artery inflammation and no stenosis (79, 131#3, 178).
Although rarely reported, cases of GCA-related stroke might occur without apparent stenosis at the time of the stroke. In these cases, one may suppose the selective ostial involvement of small arteries (perforating or PICA) abutting to lesioned vertebral/basilar arteries.
3.12.5 GCA is not a form of brain angiitis, except in rare cases
Cases involving the vertebral segment V4 below the basilar artery [e.g. (33)] or the few millimeters of the intradural carotid siphon were excluded from the definition of brain angiitis. A diagnosis of brain angiitis was sometimes offered to explain stroke occurring in patients with stable extra-cranial lesions and intra-dural lesions of the vertebral/carotid arteries (132). Moreover, the causal role of GCA-related brain angiitis is often put forward as a possible cause of stroke, but without providing any reference to unequivocal cases. The term should be avoided since it supposes the implicit and unconfirmed expectation of vasculitis of the medium and small arteries of the brain. No proof of small-vessel arteritis is found in autopsy cases (9, 115), and cuffing of rare peri-venous lymphocytes may reflect a reactive inflammation, as observed in ischemic brain tissue (obtained at autopsy), rather than arteritis (193). Only very rare cases definitively involved proximal MCA stenosis up to the M2 bifurcation (175, 194–197), in which reversiblity of luminal stenosis was in favor of the hypothesis of GCA. However, in the absence of pathological examination, other reversible or non-reversible mechanisms (vasospasms or atheroma) could also be evoked. At pathological examination, the basilar arteries are sometimes involved (198, 199), as well as the leptomeningeal arteries (198). To our knowledge, the cerebral arteries are exceptionally involved in their proximal segment (9) and are always spared distally from their first segment, so stenosis of the cerebral arteries merely reflects associated intracranial atheroma. Severe stroke does not necessarily mean cerebral arteritis but rather reflects proximal cervical occlusion. Autopsy of a steroid-resistant case with progressive occlusion of the vertebral and carotid arteries and diffuse brain stroke failed to demonstrate any thrombosis in the brain and cerebellar arteries (200).
Although non-GCA granulomatous arteritis of the small brain arteries (i.e., primary arteritis of the CNS or PACNS, or Aβ-related amyloid angiitis) is now clearly documented, it was long confounded by the old nosology, and it may still mask stroke mechanisms associated with GCA (33). Interestingly, rare patients with atypical signs suggesting granulomatous brain angiitis were also those younger than 50, and/or having granulomatous lesions restricted to the intracranial vessels or meningitis (see Supplementary Table 5), which are all red flags against GCA. Rarely, GCA may reveal ANCA-associated vasculitides although ischemic brain lesions are not observed in these cases (102).
Pachymeningitis is rare and dural biopsy may reveal typical inflammation in the small dural arteries, since the dura is vascularized by branches of the external carotid (201).
Cerebrospinal fluid (CSF) is usually normal in GCA (202), and CSF WBC count never exceeds 20 cells/mm3 (33, 132). In a series of 7 GCA-related stroke, one displayed pleocytosis and leukocyte subsets were normal in all of them (87). Finally, meningitis is mostly associated with a differential diagnosis, especially PACNS (203, 204).
3.13 Follow-up and endovascular treatment of arterial lesions
3.13.1 Follow-up
The wall thickness of the aorta and its branches is often increased by inflammation, and contrast enhancement is observed. Follow-up evaluation at 1 year after treatment demonstrates persistence of abnormal wall thickness: the ascending aorta is abnormal in 69% at onset and 43% at year one, while the carotid arteries in 31% at onset and 11% after 1 year (91). Since contrast enhancement is systematically cleared, persistent mural thickening probably involves residual fibrosis and intimal proliferation (91).
At the cervical level, stenosis apparent on angiography may mask the full extent of arterial lesions, whereas mural enhancement may reveal GCA lesions in arteries of apparently normal caliber (26, 166). Inflammatory lesions progressively abate over time. Artery wall enhancement decreases as soon as day two of treatment (205, 206), and MRI sensitivity decreases after day 10 (207). In the cranial arteries, most of the inflammatory signs had disappeared at follow-up several months later (208). On the other hand, a third of the aortic branches (including the common carotid) still show unchanged enhancement, even in TCZ-treated patients (209). Wall enhancement is not always associated with stenosis at onset (166), and its value in predicting future arterial stenosis is unknown. Late persistence of wall enhancement in a segment undergoing stenosis suggests the incomplete control of GCA (33, 210, 211). On ultrasonography, arterial halo persists at day 7 but disappears before week 3 (212, 213). However, an unexpected pattern of change may be observed in the temporal arteries but not in the axillary or subclavian arteries: a sharp decrease in intima-media thickness occurred first in TCZ-treated patients, followed by a transient but small increase to baseline around week 4, then a decrease to a lower thickness (147, 214). If the pattern of change were to be similar in the brain or ocular arteries, this paradoxical transient small change in volume occurring in treated patients might dramatically impede an already reduced blood flow and trigger hemodynamic stroke. To our knowledge, the hypothesis that treatment may transiently expand lesion burden and trigger ischemic events has never been investigated.
The late outcome of stenosis has never been systematically followed up, and uncertainties exist about rates of outcome. Stenosis or occlusion may persist after treatment initiation (132, 166, 167, 215–217) or variably regress, from partial change (30, 46, 168, 218), to complete re-permeabilization (173, 194, 219–223). However, stenosis sometimes appears or worsens after steroid and/or TCZ initiation (44, 86, 137, 166, 200).
Moreover, stroke occurring late after initiation of treatment may not be directly due to GCA but rather to severe arterial stenosis (22, 167). Beyond a persisting stenotic scar, it is unclear whether stenosis can also exacerbate due, its own dynamics in the absence of focal small-grade GCA recurrence. Persistent occlusion may be associated with pre-existing confirmed atherosclerosis (103, 215, 224). One case of GCA-related stroke treated only by antiplatelets demonstrated spontaneous re-opening of an occluded vertebral artery during follow-up in the absence of steroid treatment (27). In brief, systematic follow-up of a known stenosis is very important if a perfusion deficit remains in its territory (189).
3.13.2 Endovascular treatment of stenotic cranial arteries
Since the action of steroids may be delayed or incomplete, surgical endarterectomy (185) followed by endovascular therapy was proposed to overcome threatening stenosis. In a short series of limb artery stenosis receiving balloon angioplasty, immediate success was obtained although moderate artery dissection occurred in up to 40% of procedures, and late restenosis was limited to previously affected areas in association with the biological recurrence of GCA (59). Balloon dilatation was performed in a few patients, sometimes on multiple arteries and/or several times in the same subjects (166, 175, 225). Dilatation of severe stenosis immediately improved the clinical signs (200, 217). Post-catheter dissection occurred in two cases (225).
To our knowledge, stents are rarely used to relieve GCA-related ischemia in the limbs, bowels or heart (58, 190, 226, 227), and since the coronaries are mostly spared by GCA, most procedures involve the cranial arteries. They were mostly used to treat severe relapsing stroke and had a variable outcome: one patient suffering from a rapid progressive course was offered a stent and partial remission was obtained, whereas two others died due to procedural complications (87). Patients with recurrent hemodynamic stroke in WA received distal ICA or VA stenting and improved without relapse [(26, 139, 164, 167, 177, 218, 228, 229)#2]. Another received a superficial temporal artery to MCA bypass and encephalo-myosynangiosis to improve MCA blood flow in the setting of persisting severe carotid stenosis (165). A carotid stent was also used to treat a putative atherothrombotic stenosis re-occluded within months in a patient with occult GCA that had gone untreated (110). Stroke recurred in another patient despite steroids at day 7 due severe carotid stenosis: although the patient received balloon angioplasty, another relapse occurred in the WA within weeks due to restenosis and poor MCA perfusion (175).
Finally, procedures are increasingly being used to treat severe symptomatic stenosis. Considering the high risk of procedural complications due to the frequent location of the stenosis in the clinoid segment, the indications for intracranial carotid stent placement to target high-risk patients with hemodynamic stroke are restricted (Table 6).

3.14 Non-stenotic mechanisms of stroke
3.14.1 No GCA-induced microemboli?
Microemboli generated by artery wall lesions or a hypercoagulable state were postulated to account for brain and eye GCA-related ischemic lesions, although the evidence is insufficient. Acute central retinal artery occlusion (CRAO) due to an embolic mechanism is classically associated with sonographic visualization of emboli in the retinal circulation (the 'spot sign'), although this sign was not observed in GCA-related CRAO (232), thus ruling out an embolic mechanism. Cotton-wool spots on the retina are usually associated with vasculitis but small retinal vessels are spared by GCA. The authors hypothesized that microemboli may have given rise to these lesions (233), but direct proof was lacking.
A single study detected microembolic signals by transcranial doppler in the ophthalmic and cranial arteries in association with GCA (234). Systematic investigation of microemboli in GCA has not yet been undertaken, so unless the finding is reproduced, their causal role in WA infarction or multifocal brain lesions seems unlikely, since their location and size are not characteristic of microembolic infarction in the distal arterial bed (235). Moreover, microemboli related with atheroma seems to a play only a minor role, if any, in downstream ischemic watershed brain lesions, which are mostly related with the degree of stenosis.
3.14.2 Arguments supporting artery-to-artery macro-emboli
Thrombosis is common in PACNS since the intima is involved by inflammation, but it differs from GCA pathology in which luminal thrombosis is not a common feature (236). Although no proof is yet available, the release of thrombi from inflamed vessels and an embolic mechanism very likely account for GCA-related stroke, probably due to the high causal expectation (4, 21, 191).
Unlike stenosis, mural thrombi are uncommon radiological features of GCA and rare cases have been reported. There was a unique case of a cervical artery lined by multiple mural thrombi that disappeared within days after anticoagulation (237). In another case of a probable GCA, multiple thrombi were observed in the ascending aorta (238), and a third case was observed during autopsy (106). Cases of coronary thrombosis may involve multiple mechanisms (239, 240). Cases of GCA-related limb ischemia are related with large artery occlusion, but embolization is not a common mechanism in this setting. One acute case explored by a Fogarty catheter was finally explained by humeral stenosis relieved by angioplasty, but no thrombotic material was evidenced (58). Such a rarity of reported mural thrombi is even more surprising in the era of widespread CTA imaging.
Thrombosis of the brain arteries has rarely been observed during autopsy (198, 241), sometimes extending from an occluded vertebral artery downwards to PICA (141, 144, 242), and is thought to cause distal embolic stroke in PCA (106, 115). Data from autopsy cases may be biased toward advanced thrombotic states, which may not reflect the mechanisms involved in early ischemic lesions. Thrombi were also found in the retinal artery of a GCA patient with visual loss but no apparent local arteritis (97, 243). The structure and composition of thrombi have never been the subject of scrutiny, except for fibrinous thrombi observed in a unique autopsy case (198). Of note, although they were hypothesized long ago (10), hemodynamic mechanisms were ruled out by authors who considered that the cause was embolic and did not envision alternative mechanisms (115).
Finally, thrombi may occur in GCA as infrequent findings. Apart from typical expected changes related with GCA, post-mortem findings in severely affected patients revealed an organizing mural thrombus in the stump of the carotid arteries (115, 244, 245). Clotting probably reflects blood stasis downstream arterial occlusion, and two hypothetical mechanisms can be proposed: (a) migration of stasis thrombus located downstream a thrombosed carotid or vertebral artery (stump syndrome); (b) migration of stasis thrombus located upstream the arterial occlusion, which may migrate after the blood flow restoration due to the steroid-induced resolution of wall inflammation. This hypothesis may account for stroke occurring early after steroid initiation in patients without anticoagulation (246), or for bilateral PCA thrombosis in bilateral occlusion of VA (106). It would be appealing to monitor the natural history of changes occurring in occluded arteries (kinetics of repermeabilization, presence of mural/luminal thrombi, etc.).
3.14.3 Improbable stroke mechanisms and brain vessel complications, especially vasospasm
Unlike Takayasu arteritis, steal phenomenon remains rather elusive. To our knowledge, no systematic doppler study of Willis circle circulation has been undertaken in GCA, the perfusion consequences of severe stenosis remain to be established in these cases, and steal phenomenon in GCA is still undemonstrated, although advocated (21, 193, 247).
Venous phlebitis and pulmonary embolism are common complications of GCA that occur more frequently than in the general population (248), but cerebral venous thrombosis does not occur in association with GCA.
Vasospasm has been posited to account for transient lesions and AION, although convincing data were lacking (249). A patient with normal brain MRI 2 days before demonstrated new severe carotid and MCA stenosis, but incomplete resolution of the symptoms suggested an explanation other than vasospasm (195). Two patients suffering from amaurosis fugax presented transient focal stenosis of the retinal arteries (195), but the diagnosis of stenosis may be explained by orthostatic factors (250). Others attributed amaurosis fugax or unexpected visual recovery after AION to vasospasm (51, 249), although hemodynamic factors may have been involved. Headache associated with GCA is described as dull and continuous, but icepick headache compatible with vasospasm has sometimes been proposed despite the absence of imaging evidence (251, 252). We did not find any definitive evidence that vasospasm can trigger stenosis or strokes in GCA, although the issue remains to be investigated thoroughly.
Apart from secondary hemorrhage in the supra-tentorial ischemic areas, spontaneous brain hemorrhage is very exceptional and its cumulative incidence is similar in GCA with paired controls (253). A complete review in 1988 found only two cases (14), and we found none in our review. This is an important point distinguishing GCA from other types of brain arteritis.
Unlike the aorta, the cervical and brain arteries are not prone to GCA-related aneurysm, except for an isolated case of intracranial aneurysm in the distal carotid (231).
3.14.4 GCA-induced dissection of the cervical arteries: fact or pitfall?
While aortic dissection may complicate GCA, dissection of the cervical arteries requires caution. Segmental wall involvement may progressively lead to a transition from normal to stenotic segments, i.e. the slope sign, extending over a long arterial segment and mimicking artery dissection (87, 254). Wall thickening with or without stenosis should lead the clinician to suspect artery “dissection” (28, 45, 255–260), but general signs, old age, and the absence of high signal or the presence of gadolinium enhancement on T1-weighted images are in favor of a diagnosis of GCA. Images showing dissection of the cervical arteries are not typical and specific high-signal wall lesions in fat-saturated T1-weighted are absent, so this purported dissection may finally be explained by inflammatory wall infiltration. Severe ptosis related to third cranial nerve palsy homolateral to a carotid lesion may suggest Horner syndrome (260). Pathological autopsy confirmation of vertebral dissection was obtained twice, but one patient had already undergone salvage angioplasty before (257), which is also a risk factor for dissection. However, a cleavage plane cut appeared devoid of erythrocytes (104, 257), which may suggest an artifact commonly observed with GCA lesions [e.g., (33, 105)]. Finally, although GCA-related cervical dissection may occur, GCA may mimic cervical dissection.
3.15 Early and late stroke recurrence are GCA-related and -unrelated
3.15.1 Early recurrence
Stroke recurrence may indicate incomplete control of GCA shortly after treatment initiation, a relapse of GCA during steroid tapering, the natural history of persisting severe stenosis, or an unrelated event in elderly patients (3, 189) (Table 7). Two periods are vulnerable to stroke recurrence (19): within days after steroid initiation, and months later during steroid tapering. Patients should be clearly briefed before steroid tapering so that they immediately report any general or even minor sign, in order to avoid delayed treatment and GCA-related stroke relapse.
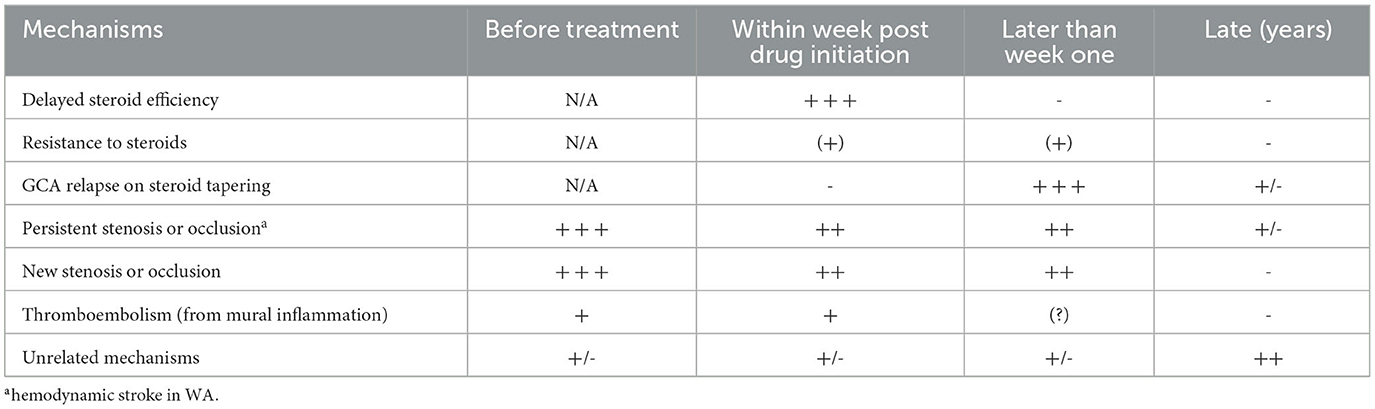
Table 7. Possible mechanisms of GCA-related TIA and stroke.
Recurrent strokes were observed in 8% to 38% of patients in some series (19, 87), suggesting that there is a subset of patients with a rapidly progressive course (87). However, most strokes occur before therapy onset or immediately after, and they occur much more frequently in the vertebrobasilar territories than in the carotid ones (15). Stroke recurrence may occur after treatment initiation although stenotic lesions apparently improve on control exams (193), raising the question of a focal low-grade relapse or unrelated cause. Early initiation of drugs (including cyclophosphamide or TCZ) failed to suppress stroke recurrence (87, 136), reinforcing the hypothesis that GCA relapses do not completely account for ischemic relapses.
3.15.2 Late recurrence
Stroke occurring later than the first month after treatment initiation raises specific problems and should be carefully scrutinized: failure of steroid tapering requiring treatment intensification, decompensation of persistent, worsening or resistant stenosis, or expression of unrelated vascular risk factors? Once treated, GCA patients have only a slightly higher risk of stroke than controls, but patients with stroke have a higher risk of GCA relapse (6). The prevalence of GCA relapses among cases of late stroke is unknown, but many authors consider the latter to be co-extensive of GCA relapse, although biological parameters of inflammation may remain low and precise exploration of arteries (i.e., relapse of local parietal inflammation on MRI or FDG-Pet) is mostly unreported in the literature. A woeful diagnosis of 'brain vasculitis' mechanically leads to treatment intensification, even though the mechanisms triggering stroke remain uncertain (132).
Recurrent carotid stroke in WA has been reported in association with persisting stenosis of the terminal carotid despite aggressive treatment (110). Autopsy demonstrated that the stenotic segment involved in these late relapsing cases of stroke displayed only fibrosis, re-permeabilization, and fragmentation of the IEL, but no active arteritis. This rare finding supports the hypothesis of a “burned-out vasculitis”, i.e., a sequel of former arteritis as observed in Takayasu arteritis. Therefore, the real challenge is to distinguish the persistence of stenotic arterial scars from active arteritis resistant to steroids and/or the required intensification of immunosuppression. In practical terms, confirmation of active (resistant) GCA or residual intimal fibrosis is technically undetermined, although wall enhancement on MRI may be of major interest. Stroke relapsing in the same arterial territory vascularized by a sub-occlusive artery merely suggests hemodynamic impairment, and immediate improvement is expected after revascularization (217).
Reduction of the lumen caliber of the cervical arteries by intimal proliferation mostly persists after treatment. Early GCA and its relapses may produce a stepwise reduction of “luminal reserve”until the hemodynamic consequences trigger stroke. Therefore, early treatment of GCA may prevent the occurrence of acute events, preserve the luminal reserve and reduce the risk of late ischemic events. Luminal reserve may also account for the predominance of stroke in territories vascularized by the vertebral arteries rather than the carotid ones.
3.15.3 Criteria for GCA-related and -unrelated stroke: late strokes are often unrelated
Diagnosing GCA-unrelated stroke poses problem since the definitions of GCA-related stroke are subjective: (a) “occurrence close to GCA onset”; (b) up to 2 or 4 weeks after steroid initiation; (c) in the absence of obvious vascular risk factor or atrial fibrillation (Supplementary material 2, Table 1). Some cases occurring around GCA onset and during follow-up are unrelated with GCA. Although difficult to ascertain especially around GCA onset, up to a quarter of incident cases could be GCA-unrelated during the first year (262), and a similar cumulative incidence of stroke with paired controls suggests that subsequent cases are mainly unrelated (253). We found no data (e.g., ischemic pattern or clinical signs) that could help to identify GCA-unrelated strokes occurring during follow-up. It is noteworthy that only one case of stroke (196) reported in the literature was considered eligible for thrombectomy, suggesting that the real percentage of GCA-related stroke at onset could be underestimated in the presence of obvious risk factors like atrial fibrillation or atheromatosis. Moreover, a GCA-related general inflammatory state may increase the risk of an embolic event, and a mural thrombus due to cardiac stroke may occur (263).
GCA patients also share the age-risk of stroke (Figure 4). Long-term follow-up confirmed new ischemic events in up to 40% of GCA patients at month 160 (39). At 10 years follow-up, studies based on large series of control-matched GCA confirmed a similar risk of combined ischemic events, TIA, and ischemic or hemorrhagic stroke, although amaurosis fugax was more frequent in former GCA patients (68, 253), suggesting persisting compromised vascularization. The rate of atherosclerotic change does not seem to be increased in former GCA patients (99).
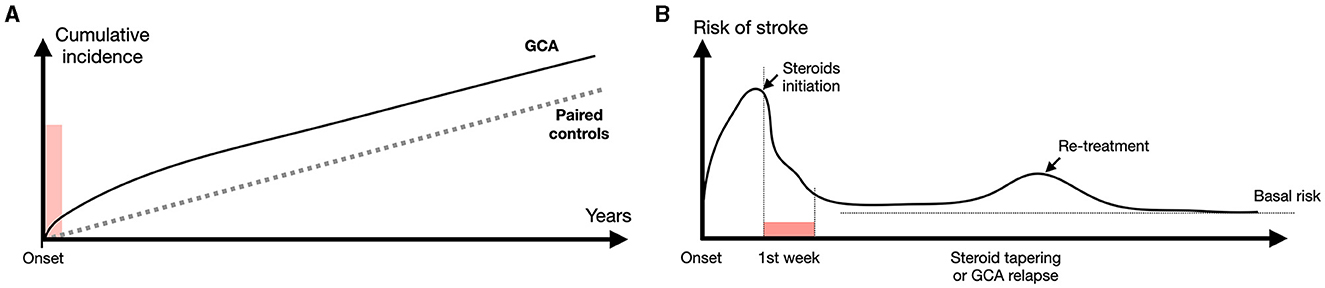
Figure 4. Risk of stroke during follow-up of GCA patients. (A) Overall risk of stroke in GCA patients compared with paired controls [from Tomasson et al. (68), Lo Gullo et al. (253) and Amiri et al. (261)]. A slightly higher incidence of stroke is observed within weeks after treatment initiation: thereafter the overall incidence remains roughly similar with paired controls. (B) The higher risk of ischemic event precedes treatment initiation, which is followed by a drop close to basal level within a week. Risk slightly increases during treatment tapering and/or GCA relapses. However, this incidence remains low compared with unrelated strokes.
Recurrence of inflammation may be observed during relapses, but GCA is frequently self-limiting thereafter for over a year (7). Therefore, the recurrence of GCA or a new arterial lesion is highly improbable after 1 or 2 years. Extended follow-up over several years demonstrates that late strokes predominate in the carotid territory in the absence of GCA relapse (15, 39), suggesting that most cases are unrelated with previous GCA.
4 Conclusion
Ischemic complications of GCA are often attributed to arteritis without any further thought, yet this is a shortcut avoiding a real investigation into the various mechanisms of stroke. This review demonstrates that this simplistic explanation is inadequate and that specific mechanisms may be involved (Figure 5). First, proof of clotting or an arterial embolism is mentioned in passing more often than it is confirmed. On the other hand, the typical clinical features of GCA-related ischemic complications, i.e. amaurosis fugax, rapidly progressive blindness, TIA and strokes in WA, fit well with the hemodynamic mechanisms related to stenosis/occlusion. The rule of thumb for treatment should be to restore the inner arterial caliber as soon as possible. Steroids should be initiated as early as possible, although they are fully efficient on the inner arterial caliber probably only after several days. Aspirin is recommended to prevent stroke although its preventive efficiency remains modest. Drugs are efficient to control general symptoms and biological parameters in GCA, but their results do not always match with imaging features, especially stenosis. In a context of severe arterial stenosis, the best preventive strategy should be to attenuate the local inflammation and to release the obstacle to blood flow.
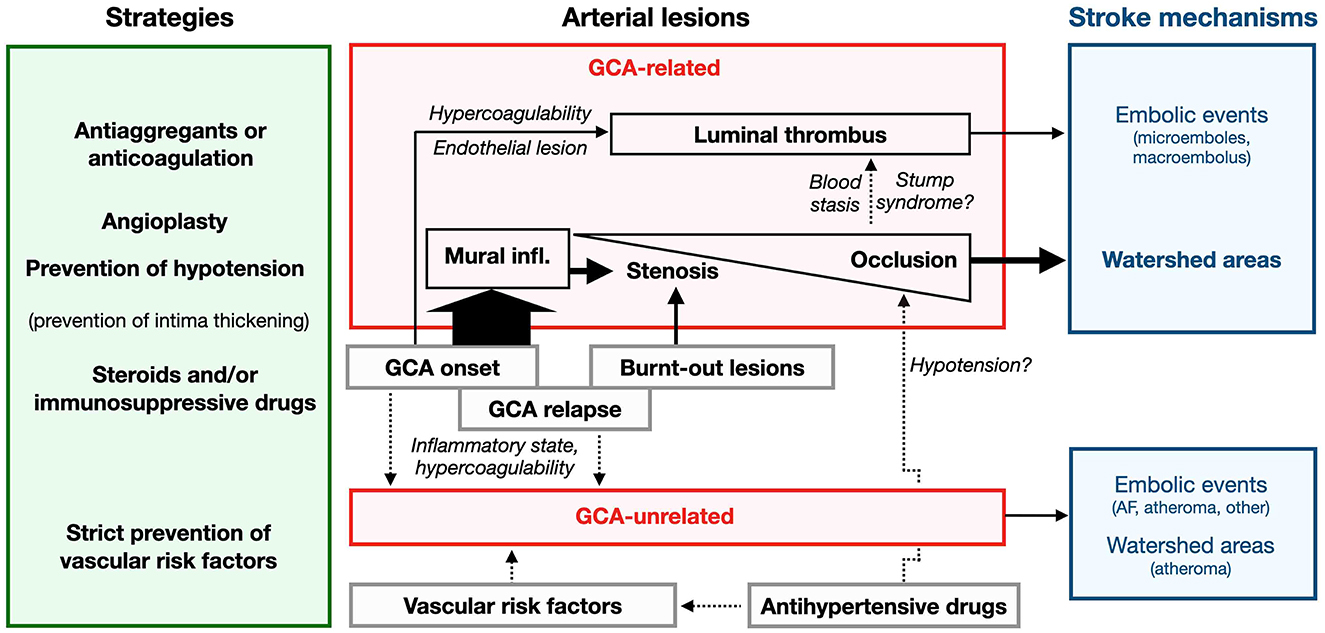
Figure 5. Proposed GCA-related stroke mechanisms and treatment strategies.
Finally, GCA-related strokes are most due to a hemodynamic mechanism restricted to patients with major wall lesions. Nowadays, strokes have become rare events and the early diagnosis of GCA is probably the best way to prevent these severe complications, as is already the case in the prevention of ophthalmic ischemia.
Author contributions
MB: Conceptualization, Methodology, Writing—original draft, Writing—review & editing. SD: Formal analysis, Methodology, Software, Writing—review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank Dr. D. Badaro for providing figures of TABs.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1305093/full#supplementary-material
Footnotes
1. ^Bonnan M, Balley G. Stroke Pattern in Giant-Cell Arteritis Mostly Involves Watershed Areas. (2022).
References
1. Samson M, Jacquin A, Audia S, Daubail B, Devilliers H, Petrella T, et al. Stroke associated with giant cell arteritis: a population-based study. J Neurol Neurosurg Psychiatry. (2015) 86:216–21. doi: 10.1136/jnnp-2014-307614
2. Crowson CS, Matteson EL. Contemporary prevalence estimates for giant cell arteritis and polymyalgia rheumatica, 2015. Semin Arthritis Rheum. (2017) 47:253–6. doi: 10.1016/j.semarthrit.2017.04.001
3. Graham E, Holland A, Avery A, Russell RW. Prognosis in giant-cell arteritis. Br Med J (Clin Res Ed). (1981) 282:269–71. doi: 10.1136/bmj.282.6260.269
4. De Boysson H, Liozon E, Lariviere D, Samson M, Parienti JJ, Boutemy J, et al. Giant cell arteritis-related stroke: a retrospective multicenter case-control study. J Rheumatol. (2017) 44:297–303. doi: 10.3899/jrheum.161033
5. Chazal T, Couture P, Rosso C, Haroche J, Leger A, Hervier B, et al. Cerebrovascular events are associated with lower survival in giant cell arteritis: a case-controlled multicenter study. Joint Bone Spine. (2018) 85:383–5. doi: 10.1016/j.jbspin.2017.05.017
6. Pariente A, Guedon A, Alamowitch S, Thietart S, Carrat F, Delorme S, et al. Ischemic stroke in giant-cell arteritis: French retrospective study. J Autoimmun. (2019) 99:48–51. doi: 10.1016/j.jaut.2019.01.009
7. Cooke WT, Cloake PC. Temporal arteritis; a generalized vascular disease. Q J Med. (1946) 15:47–75. doi: 10.1093/qjmed/15.57.47
8. Bruce GM. Temporal arteritis as a cause of blindness, review of the literature and report of a case. Am J Ophthalmol. (1950) 33:1568–73. doi: 10.1016/0002-9394(50)91218-9
9. Crompton MR. The visual changes in temporal (giant-cell) arteritis. Report of a case with autopsy findings. Brain. (1959) 82:377–90. doi: 10.1093/brain/82.3.377
10. Whitfield AG, Bateman M, Cooke WT. Temporal arteritis. Br J Ophthalmol. (1963) 47:555–66. doi: 10.1136/bjo.47.9.555
11. Singh AG, Kermani TA, Crowson CS, Weyand CM, Matteson EL, Warrington KJ. Visual manifestations in giant cell arteritis: trend over 5 decades in a population-based cohort. J Rheumatol. (2015) 42:309–15. doi: 10.3899/jrheum.140188
12. Diamantopoulos AP, Haugeberg G, Lindland A, Myklebust G. The fast-track ultrasound clinic for early diagnosis of giant cell arteritis significantly reduces permanent visual impairment: towards a more effective strategy to improve clinical outcome in giant cell arteritis? Rheumatology (Oxford). (2016) 55:66–70. doi: 10.1093/rheumatology/kev289
13. Gonzalez-Gay MA, Castaneda S, Llorca J. Giant cell arteritis: visual loss is our major concern. J Rheumatol. (2016) 43:1458–61. doi: 10.3899/jrheum.160466
14. Caselli RJ, Hunder GG, Whisnant JP. Neurologic disease in biopsy-proven giant cell (temporal) arteritis. Neurology. (1988) 38:352–9. doi: 10.1212/WNL.38.3.352
15. Gonzalez-Gay MA, Vazquez-Rodriguez TR, Gomez-Acebo I, Pego-Reigosa R, Lopez-Diaz MJ, Vazquez-Trinanes MC, et al. Strokes at time of disease diagnosis in a series of 287 patients with biopsy-proven giant cell arteritis. Medicine (Baltimore). (2009) 88:227–35. doi: 10.1097/MD.0b013e3181af4518
16. Salvarani C, Della Bella C, Cimino L, Macchioni P, Formisano D, Bajocchi G, et al. Risk factors for severe cranial ischaemic events in an Italian population-based cohort of patients with giant cell arteritis. Rheumatology (Oxford). (2009) 48:250–3. doi: 10.1093/rheumatology/ken465
17. Zenone T, Puget M. Characteristics of cerebrovascular accidents at time of diagnosis in a series of 98 patients with giant cell arteritis. Rheumatol Int. (2013) 33:3017–23. doi: 10.1007/s00296-013-2814-0
18. Ungprasert P, Wijarnpreecha K, Koster MJ, Thongprayoon C, Warrington KJ. Cerebrovascular accident in patients with giant cell arteritis: a systematic review and meta-analysis of cohort studies. Semin Arthritis Rheum. (2016) 46:361–6. doi: 10.1016/j.semarthrit.2016.07.005
19. Nesher G, Berkun Y, Mates M, Baras M, Rubinow A, Sonnenblick M. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum. (2004) 50:1332–7. doi: 10.1002/art.20171
20. Lee MS, Smith SD, Galor A, Hoffman GS. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum. (2006) 54:3306–9. doi: 10.1002/art.22141
21. Solans-Laque R, Bosch-Gil JA, Molina-Catenario CA, Ortega-Aznar A, Alvarez-Sabin J, Vilardell-Tarres M. Stroke and multi-infarct dementia as presenting symptoms of giant cell arteritis: report of 7 cases and review of the literature. Medicine (Baltimore). (2008) 87:335–44. doi: 10.1097/MD.0b013e3181908e96
22. Fitzgerald AJ, Ironside JW, Summers DM, Dennis MS, Mcrorie ER. Two cases of recurrent stroke in treated giant cell arteritis: diagnostic and therapeutic dilemmas. J Clin Rheumatol. (2010) 16:225–8. doi: 10.1097/RHU.0b013e3181e9a338
23. Liozon E, Jauberteau MO, Loustaud V, Vidal E. Association between the inflammatory response and the risk of developing irreversible cranial ischemic complications: comment on the article by Cid et al and the letter by Nesher and Sonnenblick. Arthritis Rheum. (1999) 42:2256–8.3
24. Liozon E, Herrmann FR, Ly K, Jauberteau MO, Loustaud V, Soria P, et al. [Risk factors for irreversible cerebral ischemia complications from Horton's disease: prospective study of 178 patients]. Rev Med Interne. (2001) 22:30–41. doi: 10.1016/S0248-8663(00)00283-6
25. Belenguer-Benavides A, Vilar-Cambies C, Geffner-Sclarsky D. Stroke as the first manifestation of temporal arteritis: three case reports and a review of its pathogenesis and treatment. Rev Neurol. (2004) 39:227–32. doi: 10.33588/rn.3903.2004096
26. Alsolaimani RS, Bhavsar SV, Khalidi NA, Pagnoux C, Mandzia JL, Tay K, et al. Severe intracranial involvement in giant cell arteritis: 5 cases and literature review. J Rheumatol. (2016) 43:648–56. doi: 10.3899/jrheum.150143
27. Elhfnawy AM, Bieber M, Schliesser M, Kraft P. Atypical presentation of giant cell arteritis in a patient with vertebrobasilar stroke: a case report. Medicine (Baltimore). (2019) 98:e16737. doi: 10.1097/MD.0000000000016737
28. Donaldson L, Nanji K, Rebello R, Khalidi NA, Rodriguez AR. Involvement of the intracranial circulation in giant cell arteritis. Can J Ophthalmol. (2020) 55:391–400. doi: 10.1016/j.jcjo.2020.04.002
29. Hocevar A, Jese R, Tomsic M, Rotar Z. Risk factors for severe cranial ischaemic complications in giant cell arteritis. Rheumatology (Oxford). (2020) 59:2953–9. doi: 10.1093/rheumatology/keaa058
30. Conticini E, Falsetti P, Bardelli M, Cantarini L, Frediani B. Giant cell arteritis presenting as a stroke in the internal carotid artery territory: a case-based review. Reumatologia. (2021) 59:121–5. doi: 10.5114/reum.2021.105414
31. Mollan SP, Paemeleire K, Versijpt J, Luqmani R, Sinclair AJ. European Headache Federation recommendations for neurologists managing giant cell arteritis. J Headache Pain. (2020) 21:28. doi: 10.1186/s10194-020-01093-7
32. Soriano A, Muratore F, Pipitone N, Boiardi L, Cimino L, Salvarani C. Visual loss and other cranial ischaemic complications in giant cell arteritis. Nat Rev Rheumatol. (2017) 13:476–84. doi: 10.1038/nrrheum.2017.98
33. Salvarani C, Giannini C, Miller DV, Hunder G. Giant cell arteritis: Involvement of intracranial arteries. Arthritis Rheum. (2006) 55:985–9. doi: 10.1002/art.22359
34. Hunder GG, Bloch DA, Michel BA, Stevens MB, Arend WP, Calabrese LH, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. (1990) 33:1122–8. doi: 10.1002/art.1780330810
35. Ponte C, Grayson PC, Robson JC, Suppiah R, Gribbons KB, Judge A, et al. 2022 American College of Rheumatology/EULAR classification criteria for giant cell arteritis. Ann Rheum Dis. (2022) 81:1647–53. doi: 10.1136/ard-2022-223480
36. Cid MC, Font C, Oristrell J, De La Sierra A, Coll-Vinent B, Lopez-Soto A, et al. Association between strong inflammatory response and low risk of developing visual loss and other cranial ischemic complications in giant cell (temporal) arteritis. Arthritis Rheum. (1998) 41:26–32.
37. De Mornac D, Espitia O, Neel A, Connault J, Masseau A, Espitia-Thibault A, et al. Large-vessel involvement is predictive of multiple relapses in giant cell arteritis. Ther Adv Musculoskelet Dis. (2021) 13:1759720X211009029. doi: 10.1177/1759720X211009029
38. Gonzalez-Gay MA, Blanco R, Rodriguez-Valverde V, Martinez-Taboada VM, Delgado-Rodriguez M, Figueroa M, et al. Permanent visual loss and cerebrovascular accidents in giant cell arteritis: predictors and response to treatment. Arthritis Rheum. (1998) 41:1497–504.
39. Pego-Reigosa R, Garcia-Porrua C, Pineiro A, Dierssen T, Llorca J, Gonzalez-Gay MA. Predictors of cerebrovascular accidents in giant cell arteritis in a defined population. Clin Exp Rheumatol. (2004) 22:S13−17.
40. Sun F, Ma S, Zheng W, Tian X, Zeng X. A Retrospective Study of Chinese Patients With Giant Cell Arteritis (GCA): Clinical Features and Factors Associated With Severe Ischemic Manifestations. Medicine (Baltimore). (2016) 95:e3213. doi: 10.1097/MD.0000000000003213
41. Monti S, Bartoletti A, Bellis E, Delvino P, Montecucco C. Fast-Track Ultrasound Clinic for the Diagnosis of Giant Cell Arteritis Changes the Prognosis of the Disease but Not the Risk of Future Relapse. Front Med (Lausanne). (2020) 7:589794. doi: 10.3389/fmed.2020.589794
42. Wiszniewska M, Devuyst G, Bogousslavsky J. Giant cell arteritis as a cause of first-ever stroke. Cerebrovasc Dis. (2007) 24:226–30. doi: 10.1159/000104482
43. Garcia-Garcia J, Ayo-Martin O, Argandona-Palacios L, Segura T. Vertebral artery halo sign in patients with stroke: a key clue for the prompt diagnosis of giant cell arteritis. Stroke. (2011) 42:3287–90. doi: 10.1161/STROKEAHA.111.625152
44. Guisado-Alonso D, Edo MC, Estrada Alarcon PV, Garcia-Sanchez SM, Font MA, Mena Romo L, et al. Progression of large vessel disease in patients with giant cell arteritis-associated ischemic stroke: the role of vascular imaging: a case series. J Clin Rheumatol. (2021) 27:e418–24. doi: 10.1097/RHU.0000000000001498
45. Zwicker J, Atkins EJ, Lum C, Sharma M. An atypical presentation of giant cell arteritis. CMAJ. (2011) 183:E301–305. doi: 10.1503/cmaj.100380
46. Kong KK, Mackinnon AD, Bridges LR, Cloud GC. A giant cause of stroke. Acute Med. (2014) 13:68–71. doi: 10.52964/AMJA.0346
47. Siemonsen S, Brekenfeld C, Holst B, Kaufmann-Buehler AK, Fiehler J, Bley TA. 3T MRI reveals extra- and intracranial involvement in giant cell arteritis. AJNR Am J Neuroradiol. (2015) 36:91–7. doi: 10.3174/ajnr.A4086
48. Ostberg G. An arteritis with special reference to polymyalgia arteritica. Acta Pathol Microbiol Scand Suppl. (1973) 237:231–259.
49. Font C, Cid MC, Coll-Vinent B, Lopez-Soto A, Grau JM. Clinical features in patients with permanent visual loss due to biopsy-proven giant cell arteritis. Br J Rheumatol. (1997) 36:251–4. doi: 10.1093/rheumatology/36.2.251
50. Danesh-Meyer H, Savino PJ, Gamble GG. Poor prognosis of visual outcome after visual loss from giant cell arteritis. Ophthalmology. (2005) 112:1098–103. doi: 10.1016/j.ophtha.2005.01.036
51. Hayreh SS, Zimmerman B. Visual deterioration in giant cell arteritis patients while on high doses of corticosteroid therapy. Ophthalmology. (2003) 110:1204–15. doi: 10.1016/S0161-6420(03)00228-8
52. Gospe SM, Amrhein TJ, Malinzak MD, Bhatti MT, Mettu P, El-Dairi MA. Magnetic resonance imaging abnormalities of the optic nerve sheath and intracranial internal carotid artery in giant cell arteritis. J Neuroophthalmol. (2021) 41:54–59. doi: 10.1097/WNO.0000000000000860
53. Ho AC, Sergott RC, Regillo CD, Savino PJ, Lieb WE, Flaharty PM, et al. Color Doppler hemodynamics of giant cell arteritis. Arch Ophthalmol. (1994) 112:938–45. doi: 10.1001/archopht.1994.01090190086026
54. Liu GT, Glaser JS, Schatz NJ, Smith JL. Visual morbidity in giant cell arteritis. Clinical characteristics and prognosis for vision. Ophthalmology. (1994) 101:1779–85. doi: 10.1016/S0161-6420(94)31102-X
55. Thurtell MJ, Kardon RH. Recovery of vision from no light perception in giant cell arteritis. Arch Ophthalmol. (2012) 130:1080–2. doi: 10.1001/archophthalmol.2012.308
56. Ninet JP, Bachet P, Dumontet CM, Du Colombier PB, Stewart MD, Pasquier JH. Subclavian and axillary involvement in temporal arteritis and polymyalgia rheumatica. Am J Med. (1990) 88:13–20. doi: 10.1016/0002-9343(90)90121-S
57. Rischmueller M, Davies RP, Smith MD. Three year follow-up of a case of giant cell arteritis presenting with a chronic cough and upper limb ischaemic symptoms. Br J Rheumatol. (1996) 35:800–2. doi: 10.1093/rheumatology/35.8.800
58. Monte R, Gonzalez-Gay MA, Garcia-Porrua C, Lopez-Alvarez MJ, Pulpeiro JR. Successful response to angioplasty in a patient with upper limb ischaemia secondary to giant cell arteritis. Br J Rheumatol. (1998) 37:344. doi: 10.1093/rheumatology/37.3.344a
59. Both M, Aries PM, Muller-Hulsbeck S, Jahnke T, Schafer PJ, Gross WL, et al. Balloon angioplasty of arteries of the upper extremities in patients with extracranial giant-cell arteritis. Ann Rheum Dis. (2006) 65:1124–30. doi: 10.1136/ard.2005.048470
60. Berger CT, Wolbers M, Meyer P, Daikeler T, Hess C. High incidence of severe ischaemic complications in patients with giant cell arteritis irrespective of platelet count and size, and platelet inhibition. Rheumatology (Oxford). (2009) 48:258–61. doi: 10.1093/rheumatology/ken480
62. Vincent FM, Vincent T. Bilateral carotid siphon involvement in giant cell arteritis. Neurosurgery. (1986) 18:773–6. doi: 10.1227/00006123-198606000-00016
63. Ely GM. Giant cell arteritis complicated by multi-infarct dementia. J Clin Rheumatol. (1998) 4:209–15. doi: 10.1097/00124743-199808000-00011
64. Reich KA, Giansiracusa DF, Strongwater SL. Neurologic manifestations of giant cell arteritis. Am J Med. (1990) 89:67–72. doi: 10.1016/0002-9343(90)90100-R
65. Mournet S, Sene T, Charbonneau F, Poillon G, Vignal C, Clavel G, et al. High-resolution MRI demonstrates signal abnormalities of the 3rd cranial nerve in giant cell arteritis patients with 3rd cranial nerve impairment. Eur Radiol. (2021) 31:4472–80. doi: 10.1007/s00330-020-07595-x
66. Saleh M, Turesson C, Englund M, Merkel PA, Mohammad AJ. Visual Complications in Patients with Biopsy-proven Giant Cell Arteritis: A Population-based Study. J Rheumatol. (2016) 43:1559–65. doi: 10.3899/jrheum.151033
67. Mackie SL, Dasgupta B, Hordon L, Gough A, Green M, Hollywood J, et al. Ischaemic manifestations in giant cell arteritis are associated with area level socio-economic deprivation, but not cardiovascular risk factors. Rheumatology (Oxford). (2011) 50:2014–22. doi: 10.1093/rheumatology/ker265
68. Tomasson G, Peloquin C, Mohammad A, Love TJ, Zhang Y, Choi HK, et al. Risk for cardiovascular disease early and late after a diagnosis of giant-cell arteritis: a cohort study. Ann Intern Med. (2014) 160:73–80. doi: 10.7326/M12-3046
69. Aiello PD, Trautmann JC, Mcphee TJ, Kunselman AR, Hunder GG. Visual prognosis in giant cell arteritis. Ophthalmology. (1993) 100:550–5. doi: 10.1016/S0161-6420(93)31608-8
70. Gonzalez-Gay MA, Garcia-Porrua C, Llorca J, Hajeer AH, Branas F, Dababneh A, et al. Visual manifestations of giant cell arteritis. Trends and clinical spectrum in 161 patients. Medicine (Baltimore) 79. (2000) 283–92. doi: 10.1097/00005792-200009000-00001
71. Van Der Geest KSM, Sandovici M, Van Sleen Y, Sanders JS, Bos NA, Abdulahad WH, et al. Review: what is the current evidence for disease subsets in giant cell arteritis? Arthritis Rheumatol. (2018) 70:1366–76. doi: 10.1002/art.40520
72. Hernandez-Rodriguez J, Garcia-Martinez A, Casademont J, Filella X, Esteban MJ, Lopez-Soto A, et al. A strong initial systemic inflammatory response is associated with higher corticosteroid requirements and longer duration of therapy in patients with giant-cell arteritis. Arthritis Rheum. (2002) 47:29–35. doi: 10.1002/art1.10161
73. Nesher G, Nesher R, Mates M, Sonnenblick M, Breuer GS. Giant cell arteritis: intensity of the initial systemic inflammatory response and the course of the disease. Clin Exp Rheumatol. (2008) 26:S30–34.
74. Zhang Y, Wang D, Yin Y, Fan H, Zhang W, Zeng X. Clinical comparisons of patients with giant cell arteritis with versus without fever at onset. J Int Med Res. (2019) 47:5613–22. doi: 10.1177/0300060519875379
75. Nesher G, Sonnenblick M. No association between the inflammatory response and the risk of developing irreversible cranial ischemic complications: comment on the article by Cid et al. Arthritis Rheum. (1998) 41:2088–9.
76. Liozon E, Dalmay F, Lalloue F, Gondran G, Bezanahary H, Fauchais AL, et al. Risk factors for permanent visual loss in biopsy-proven giant cell arteritis: a study of 339 patients. J Rheumatol. (2016) 43:1393–9. doi: 10.3899/jrheum.151135
77. Patil P, Williams M, Maw WW, Achilleos K, Elsideeg S, Dejaco C, et al. Fast track pathway reduces sight loss in giant cell arteritis: results of a longitudinal observational cohort study. Clin Exp Rheumatol. (2015) 33:103–6.
78. Hayreh SS, Podhajsky PA, Zimmerman B. Ocular manifestations of giant cell arteritis. Am J Ophthalmol. (1998) 125:509–20. doi: 10.1016/S0002-9394(99)80192-5
79. Cid MC, Hernandez-Rodriguez J, Esteban MJ, Cebrian M, Gho YS, Font C, et al. Tissue and serum angiogenic activity is associated with low prevalence of ischemic complications in patients with giant-cell arteritis. Circulation. (2002) 106:1664–71. doi: 10.1161/01.CIR.0000030185.67510.C0
80. Lehmann T, Seitz M. Acute-phase response and risk of developing ischemic complications in giant cell arteritis: comment on the article by Cid et al. Arthritis Rheum. (1999) 42:190. doi: 10.1002/1529-0131(199901)42:1<190::aid-anr27>3.0.co;2-p
81. Hayreh SS, Podhajsky PA, Raman R, Zimmerman B. Giant cell arteritis: validity and reliability of various diagnostic criteria. Am J Ophthalmol. (1997) 123:285–96. doi: 10.1016/S0002-9394(14)70123-0
82. Parikh M, Miller NR, Lee AG, Savino PJ, Vacarezza MN, Cornblath W, et al. Prevalence of a normal C-reactive protein with an elevated erythrocyte sedimentation rate in biopsy-proven giant cell arteritis. Ophthalmology. (2006) 113:1842–5. doi: 10.1016/j.ophtha.2006.05.020
83. Mackay DD, Huesmann GR, Wu RI, Stone JR, Pless ML. Giant cell arteritis causing symmetric bilateral posterior circulation infarcts. J Clin Rheumatol. (2013) 19:393–6. doi: 10.1097/RHU.0b013e3182a6ffc1
84. Myklebust G, Gran JT. A prospective study of 287 patients with polymyalgia rheumatica and temporal arteritis: clinical and laboratory manifestations at onset of disease and at the time of diagnosis. Br J Rheumatol. (1996) 35:1161–8. doi: 10.1093/rheumatology/35.11.1161
85. Stengl KL, Buchert R, Bauknecht H, Sobesky J. A hidden giant: Wallenberg syndrome and aortal wall thickening as an atypical presentation of a giant cell arteritis. BMJ Case Rep. (2013) 2013:bcr2012006994. doi: 10.1136/bcr-2012-006994
86. Hiyama R, Oiwa H, Kanou Y, Nishibe S, Kono T, Nomura E. A case of giant cell arteritis lacking typical symptoms presenting with recurrent cerebellar infarctions: A case report and case-based review. Mod Rheumatol Case Rep. (2021). doi: 10.1093/mrcr/rxab030
87. Beuker C, Wankner MC, Thomas C, Strecker JK, Schmidt-Pogoda A, Schwindt W, et al. Characterization of extracranial giant cell arteritis with intracranial involvement and its rapidly progressive subtype. Ann Neurol. (2021) 90:118–29. doi: 10.1002/ana.26101
88. Rueda B, Lopez-Nevot MA, Lopez-Diaz MJ, Garcia-Porrua C, Martin J, Gonzalez-Gay MA. A functional variant of vascular endothelial growth factor is associated with severe ischemic complications in giant cell arteritis. J Rheumatol. (2005) 32:1737–41.
89. Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med. (2003) 349:160–9. doi: 10.1056/NEJMra022694
90. Breuer GS, Nesher R, Reinus K, Nesher G. Association between histological features in temporal artery biopsies and clinical features of patients with giant cell arteritis. Isr Med Assoc J. (2013) 15:271–4.
91. Prieto-Gonzalez S, Garcia-Martinez A, Tavera-Bahillo I, Hernandez-Rodriguez J, Gutierrez-Chacoff J, Alba MA, et al. Effect of glucocorticoid treatment on computed tomography angiography detected large-vessel inflammation in giant-cell arteritis. A prospective, longitudinal study. Medicine (Baltimore). (2015) 94:e486. doi: 10.1097/MD.0000000000000486
92. Visvanathan S, Rahman MU, Hoffman GS, Xu S, Garcia-Martinez A, Segarra M, et al. Tissue and serum markers of inflammation during the follow-up of patients with giant-cell arteritis–a prospective longitudinal study. Rheumatology (Oxford). (2011) 50:2061–70. doi: 10.1093/rheumatology/ker163
93. Kaiser M, Weyand CM, Bjornsson J, Goronzy JJ. Platelet-derived growth factor, intimal hyperplasia, and ischemic complications in giant cell arteritis. Arthritis Rheum. (1998) 41:623–33.
94. Makkuni D, Bharadwaj A, Wolfe K, Payne S, Hutchings A, Dasgupta B. Is intimal hyperplasia a marker of neuro-ophthalmic complications of giant cell arteritis? Rheumatology (Oxford). (2008) 47:488–90. doi: 10.1093/rheumatology/ken012
95. Chatelain D, Duhaut P, Schmidt J, Loire R, Bosshard S, Guernou M, et al. Pathological features of temporal arteries in patients with giant cell arteritis presenting with permanent visual loss. Ann Rheum Dis. (2009) 68:84–8. doi: 10.1136/ard.2007.084947
96. Muratore F, Boiardi L, Cavazza A, Aldigeri R, Pipitone N, Restuccia G, et al. Correlations between histopathological findings and clinical manifestations in biopsy-proven giant cell arteritis. J Autoimmun. (2016) 69:94–101. doi: 10.1016/j.jaut.2016.03.005
97. Cardell BS, Hanley T. A fatal case of giant-cell or temporal arteritis. J Pathol Bacteriol. (1951) 63:587–97. doi: 10.1002/path.1700630405
98. Allsop CJ, Gallagher PJ. Temporal artery biopsy in giant-cell arteritis. A reappraisal Am J Surg Pathol. (1981) 5:317–23. doi: 10.1097/00000478-198106000-00001
99. Gonzalez-Juanatey C, Lopez-Diaz MJ, Martin J, Llorca J, Gonzalez-Gay MA. Atherosclerosis in patients with biopsy-proven giant cell arteritis. Arthritis Rheum. (2007) 57:1481–6. doi: 10.1002/art.23114
100. Esteban MJ, Font C, Hernandez-Rodriguez J, Valls-Sole J, Sanmarti R, Cardellach F, et al. Small-vessel vasculitis surrounding a spared temporal artery: clinical and pathological findings in a series of twenty-eight patients. Arthritis Rheum. (2001) 44:1387–95.
101. Cavazza A, Muratore F, Boiardi L, Restuccia G, Pipitone N, Pazzola G, et al. Inflamed temporal artery: histologic findings in 354 biopsies, with clinical correlations. Am J Surg Pathol. (2014) 38:1360–70. doi: 10.1097/PAS.0000000000000244
102. Delaval L, Samson M, Schein F, Agard C, Trefond L, Deroux A, et al. Temporal arteritis revealing antineutrophil cytoplasmic antibody-associated vasculitides: a case-control study. Arthritis Rheumatol. (2021) 73:286–94. doi: 10.1002/art.41527
103. Save-Soderbergh J, Malmvall BE, Andersson R, Bengtsson BA. Giant cell arteritis as a cause of death. Report of nine cases. JAMA. (1986) 255:493–6. doi: 10.1001/jama.1986.03370040067025
104. Sheehan MM, Keohane C, Twomey C. Fatal vertebral giant cell arteritis. J Clin Pathol. (1993) 46:1129–31. doi: 10.1136/jcp.46.12.1129
105. Restuccia G, Boiardi L, Cavazza A, Catanoso M, Macchioni P, Muratore F, et al. Flares in Biopsy-Proven Giant Cell Arteritis in Northern Italy: Characteristics and Predictors in a Long-Term Follow-Up Study. Medicine (Baltimore). (2016) 95:e3524. doi: 10.1097/MD.0000000000003524
106. Heptinstall RH, Porter KA, Barkley H. Giant-cell (temporal) arteritis. J Pathol Bacteriol. (1954) 67:507–19. doi: 10.1002/path.1700670224
108. Bharadwaj A, Dasgupta B, Wolfe K, Nordborg C, Nordborg E. Difficulties in the development of histological scoring of the inflamed temporal arteries in giant cell arteritis. Rheumatology (Oxford). (2005) 44:1579–80. doi: 10.1093/rheumatology/kei180
109. Stone J. Diseases of small and medium-sized blood vessels. In:Buja L, Butany J, , editor. Cardiovascular Pathology. (Oxford, UK: Elsevier). (2016).
110. Lu-Emerson C, Walker M, Huber BR, Ghodke B, Longstreth WTJr, Khot SP. Lethal giant cell arteritis with multiple ischemic strokes despite aggressive immunosuppressive therapy. J Neurol Sci. (2010) 295:120–4. doi: 10.1016/j.jns.2010.05.008
111. Wilkinson IM. The vertebral artery. Extracranial and intracranial structure. Arch Neurol. (1972) 27:392–6. doi: 10.1001/archneur.1972.00490170024004
112. Hassler O, Larsson SE. The external elastic layer of the cerebral arteries in different age-groups. Acta Anat (Basel). (1962) 48:1–6. doi: 10.1159/000141824
113. Masuoka T, Hayashi N, Hori E, Kuwayama N, Ohtani O, Endo S. Distribution of internal elastic lamina and external elastic lamina in the internal carotid artery: possible relationship with atherosclerosis. Neurol Med Chir (Tokyo). (2010) 50:179–82. doi: 10.2176/nmc.50.179
114. Qin G, Wang L, Hua Y, Hou H, Zou Q, Wang D, et al. Comparative morphology of the internal elastic lamina of cerebral and peripheral arteries. Int J Clin Exp Pathol. (2020) 13:764–70.
115. Wilkinson IM, Russell RW. Arteries of the head and neck in giant cell arteritis. A pathological study to show the pattern of arterial involvement. Arch Neurol. (1972) 27:378–91. doi: 10.1001/archneur.1972.00490170010003
116. Denswil NP, Van Der Wal AC, Ritz K, De Boer OJ, Aronica E, Troost D, et al. Atherosclerosis in the circle of Willis: spatial differences in composition and in distribution of plaques. Atherosclerosis. (2016) 251:78–84. doi: 10.1016/j.atherosclerosis.2016.05.047
117. Portanova A, Hakakian N, Mikulis DJ, Virmani R, Abdalla WM, Wasserman BA. Intracranial vasa vasorum: insights and implications for imaging. Radiology. (2013) 267:667–79. doi: 10.1148/radiol.13112310
118. Conway R, Smyth AE, Kavanagh RG, O'donohoe RL, Purcell Y, Heffernan EJ, et al. Diagnostic utility of computed tomographic angiography in giant-cell arteritis. Stroke. (2018) 49:2233–6. doi: 10.1161/STROKEAHA.118.021995
119. Bley TA, Weiben O, Uhl M, Vaith P, Schmidt D, Warnatz K, et al. Assessment of the cranial involvement pattern of giant cell arteritis with 3T magnetic resonance imaging. Arthritis Rheum. (2005) 52:2470–7. doi: 10.1002/art.21226
120. Geiger J, Bley T, Uhl M, Frydrychowicz A, Langer M, Markl M. Diagnostic value of T2-weighted imaging for the detection of superficial cranial artery inflammation in giant cell arteritis. J Magn Reson Imaging. (2010) 31:470–4. doi: 10.1002/jmri.22047
121. Goll C, Thormann M, Hofmuller W, Friebe B, Behrens-Baumann W, Bley TA, et al. Feasibility study: 7 T MRI in giant cell arteritis. Graefes Arch Clin Exp Ophthalmol. (2016) 254:1111–6. doi: 10.1007/s00417-016-3337-7
122. Kishk NA, Alsayyad A, Alsayyad E, Hatem G. Isolated vertebral arteritis: a challenging cause of stroke. Acta Neurol Belg. (2021) 121:283–6. doi: 10.1007/s13760-020-01283-9
123. Schafer VS, Juche A, Ramiro S, Krause A, Schmidt WA. Ultrasound cut-off values for intima-media thickness of temporal, facial and axillary arteries in giant cell arteritis. Rheumatology (Oxford). (2017) 56:1632. doi: 10.1093/rheumatology/kex289
124. Penet T, Lambert M, Baillet C, Outteryck O, Henon H, Morell-Dubois S, et al. Giant cell arteritis-related cerebrovascular ischemic events: a French retrospective study of 271 patients, systematic review of the literature and meta-analysis. Arthritis Res Ther. (2023) 25:116. doi: 10.1186/s13075-023-03091-x
125. Sammel AM, Hsiao E, Schembri G, Nguyen K, Brewer J, Schrieber L, et al. Diagnostic accuracy of positron emission tomography/computed tomography of the head, neck, and chest for giant cell arteritis: a prospective, double-blind, cross-sectional study. Arthritis Rheumatol. (2019) 71:1319–28. doi: 10.1002/art.40864
126. Walter MA, Melzer RA, Schindler C, Muller-Brand J, Tyndall A, Nitzsche EU. The value of [18F]FDG-PET in the diagnosis of large-vessel vasculitis and the assessment of activity and extent of disease. Eur J Nucl Med Mol Imaging. (2005) 32:674–81. doi: 10.1007/s00259-004-1757-9
127. Prieto-Gonzalez S, Arguis P, Garcia-Martinez A, Espigol-Frigole G, Tavera-Bahillo I, Butjosa M, et al. Large vessel involvement in biopsy-proven giant cell arteritis: prospective study in 40 newly diagnosed patients using CT angiography. Ann Rheum Dis. (2012) 71:1170–6. doi: 10.1136/annrheumdis-2011-200865
128. Mestre-Torres J, Simo-Perdigo M, Martinez-Valle F, Navales I, Loureiro-Amigo J, Solans-Laque R. Risk of ischaemic events at giant cell arteritis diagnosis according to PET/CT findings. Eur J Nucl Med Mol Imaging. (2019) 46:1626–32. doi: 10.1007/s00259-019-04339-y
129. Amsler J, Kysela I, Tappeiner C, Seitz L, Christ L, Scholz G, et al. Vision loss in patients with giant cell arteritis treated with tocilizumab. Arthritis Res Ther. (2021) 23:92. doi: 10.1186/s13075-021-02480-4
130. Nielsen BD, Hansen IT, Kramer S, Haraldsen A, Hjorthaug K, Bogsrud TV, et al. Simple dichotomous assessment of cranial artery inflammation by conventional 18F-FDG PET/CT shows high accuracy for the diagnosis of giant cell arteritis: a case-control study. Eur J Nucl Med Mol Imaging. (2019) 46:184–93. doi: 10.1007/s00259-018-4106-0
131. Bienvenu B, Ly KH, Lambert M, Agard C, Andre M, Benhamou Y, et al. Management of giant cell arteritis: recommendations of the French Study Group for Large Vessel Vasculitis (GEFA). Rev Med Interne. (2016) 37:154–65. doi: 10.1016/j.revmed.2015.12.015
132. Lariviere D, Sacre K, Klein I, Hyafil F, Choudat L, Chauveheid MP, et al. Extra- and intracranial cerebral vasculitis in giant cell arteritis: an observational study. Medicine (Baltimore). (2014) 93:e265. doi: 10.1097/MD.0000000000000265
133. Hoffman GS, Cid MC, Hellmann DB, Guillevin L, Stone JH, Schousboe J, et al. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum. (2002) 46:1309–18. doi: 10.1002/art.10262
134. Hoffman GS, Cid MC, Rendt-Zagar KE, Merkel PA, Weyand CM, Stone JH, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. (2007) 146:621–30. doi: 10.7326/0003-4819-146-9-200705010-00004
135. Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med. (2017) 377:317–28. doi: 10.1056/NEJMoa1613849
136. Prunte MKR, Naumann A, Christ M, Naumann M, Bayas A. Giant cell arteritis with vertebral artery involvement-baseline characteristics and follow-up of a monocentric patient cohort. Front Neurol. (2023) 14:1188073. doi: 10.3389/fneur.2023.1188073
137. Naderi N. Development of intracranial vasculitis in giant cell arteritis during tocilizumab treatment. Clin Exp Rheumatol. (2020) 38:207–9.
138. Unizony S, Mcculley TJ, Spiera R, Pei J, Sidiropoulos PN, Best JH, et al. Clinical outcomes of patients with giant cell arteritis treated with tocilizumab in real-world clinical practice: decreased incidence of new visual manifestations. Arthritis Res Ther. (2021) 23:8. doi: 10.1186/s13075-020-02377-8
139. Dementovych N, Mishra R, Shah QA. Angioplasty and stent placement for complete occlusion of the vertebral artery secondary to giant cell arteritis. J Neurointerv Surg. (2012) 4:110–3. doi: 10.1136/jnis.2011.004689
140. Vionnet J, Buss G, Mayer C, Sokolov AA, Borruat FX, Spertini F. Tocilizumab for giant cell arteritis with corticosteroid-resistant progressive anterior ischemic optic neuropathy. Joint Bone Spine. (2017) 84:615–9. doi: 10.1016/j.jbspin.2017.04.009
141. Gibb WR, Urry PA, Lees AJ. Giant cell arteritis with spinal cord infarction and basilar artery thrombosis. J Neurol Neurosurg Psychiatry. (1985) 48:945–8. doi: 10.1136/jnnp.48.9.945
142. Chadenas D, Picon L, Roux C, Gauvain JB, Maitre F, Buzacoux J, et al. Flaccid paraplegia of abrupt onset. Complication of corticoid treatment of Horton's disease? Presse Med. (1986) 15:845.
144. Sendino A, Barbado FJ, Gonzalez-Anglada I, Anton E, Lopez-Barea F, Vazquez JJ. Temporal arteritis: a form of systemic panarteritis. Ann Rheum Dis. (1992) 51:1082–4. doi: 10.1136/ard.51.9.1082
145. Collazos J, Garcia-Monco C, Martin A, Rodriguez J, Gomez MA. Multiple strokes after initiation of steroid therapy in giant cell arteritis. Postgrad Med J. (1994) 70:228–30. doi: 10.1136/pgmj.70.821.228
146. Khalaf K, Pages M, Blard J, Labauge P. La Presse Medicale. Deux accidents ischémiques cérébraux au cours d'une maladie de Horton traitée: responsabilité de la corticothérapie?. (1994) 23:449.
147. Peyrade F, Boscagli A, Roa M, Taillan B, Dujardin P. Early cerebral ischemic complication following corticotherapy for Horton disease. Rev Med Interne. (1997) 18:585–6. doi: 10.1016/S0248-8663(97)80815-6
148. Staunton H, Stafford F, Leader M, O'riordain D. Deterioration of giant cell arteritis with corticosteroid therapy. Arch Neurol. (2000) 57:581–4. doi: 10.1001/archneur.57.4.581
149. Caselli RJ. Giant cell (temporal) arteritis: a treatable cause of multi-infarct dementia. Neurology. (1990) 40:753–5. doi: 10.1212/WNL.40.5.753
150. Buttner T, Heye N, Przuntek H. Temporal arteritis with cerebral complications: report of four cases. Eur Neurol. (1994) 34:162–7. doi: 10.1159/000117031
151. Conn DL, Tompkins RB, Nichols WL. Glucocorticoids in the management of vasculitis–a double edged sword? J Rheumatol. (1988) 15:1181–3.
152. Hellmann DB. Low-dose aspirin in the treatment of giant cell arteritis. Arthritis Rheum. (2004) 50:1026–7. doi: 10.1002/art.20172
153. Gonzalez-Gay MA, Pineiro A, Gomez-Gigirey A, Garcia-Porrua C, Pego-Reigosa R, Dierssen-Sotos T, et al. Influence of traditional risk factors of atherosclerosis in the development of severe ischemic complications in giant cell arteritis. Medicine (Baltimore). (2004) 83:342–7. doi: 10.1097/01.md.0000145369.25558.b5
154. Narvaez J, Bernad B, Gomez-Vaquero C, Garcia-Gomez C, Roig-Vilaseca D, Juanola X, et al. Impact of antiplatelet therapy in the development of severe ischemic complications and in the outcome of patients with giant cell arteritis. Clin Exp Rheumatol. (2008) 26:S57–62.
155. Lipton RB, Rosenbaum D, Mehler MF. Giant cell arteritis causes recurrent posterior circulation transient ischemic attacks which respond to corticosteroid. Eur Neurol. (1987) 27:97–100. doi: 10.1159/000116139
156. Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum. (2002) 46:457–66. doi: 10.1002/art.10071
157. Buono LM, Foroozan R, De Virgiliis M, Savino PJ. Heparin therapy in giant cell arteritis. Br J Ophthalmol. (2004) 88:298–301. doi: 10.1136/bjo.2003.021592
158. Cid MC, Cervera R, Font J, Lopez-Soto A, Pallares L, Navarro M, et al. Late thrombotic events in patients with temporal arteritis and anticardiolipin antibodies. Clin Exp Rheumatol. (1990) 8:359–63.
159. Liozon, E, Roussel V., Roblot P., Liozon F., Preud'homme J. L., Loustaud V., et al. (1998). Absence of anti-beta2 glycoprotein I antibodies in giant cell arteritis: a study of 45 biopsy-proven cases. Br J Rheumatol 37, 1129–1131. doi: 10.1093/rheumatology/37.10.1129
160. Liozon E, Roblot P, Paire D, Loustaud V, Liozon F, Vidal E, et al. Anticardiolipin antibody levels predict flares and relapses in patients with giant-cell (temporal) arteritis. A longitudinal study of 58 biopsy-proven cases. Rheumatology (Oxford) 39. (2000) 1089–94. doi: 10.1093/rheumatology/39.10.1089
161. Lozano E, Segarra M, Garcia-Martinez A, Hernandez-Rodriguez J, Cid MC. Imatinib mesylate inhibits in vitro and ex vivo biological responses related to vascular occlusion in giant cell arteritis. Ann Rheum Dis. (2008) 67:1581–8. doi: 10.1136/ard.2007.070805
162. Thielen KR, Wijdicks EF, Nichols DA. Giant cell (temporal) arteritis: involvement of the vertebral and internal carotid arteries. Mayo Clin Proc. (1998) 73:444–6. doi: 10.1016/S0025-6196(11)63727-0
163. Garcia-Porrua C, Pego-Reigosa R, Martinez-Vazquez F, Armesto V, Gonzalez-Gay MA. Bilateral vertebral artery occlusion in giant cell arteritis. Clin Exp Rheumatol. (2006) 24:S101.
164. Guerrero AM, Sierra-Hidalgo F, Calleja P, Navia P, Campollo J, Diaz-Guzman J. Intracranial internal carotid artery angioplasthy and stenting in giant cell arteritis. J Neuroimaging. (2015) 25:307–9. doi: 10.1111/jon.12116
165. Chowdhary V, Kallmes DF, Fulgham J, Meyer F, Matteson EL. Recurrent spells and bilateral internal carotid artery stenosis in a diabetic male: a test of patien-ce(-ts). Arthritis Rheum. (2007) 57:1098–101. doi: 10.1002/art.22905
166. Oeinck M, Rozeik C, Wattchow J, Meckel S, Schlageter M, Beeskow C, et al. Why a standard contrast-enhanced MRI might be useful in intracranial internal carotid artery stenosis. Neuroradiol J. (2016) 29:208–12. doi: 10.1177/1971400916638354
167. Neutel D, Biscoito L, Campos JE, Melo TP, Albuquerque L. Giant cell arteritis with symptomatic intracranial stenosis and endovascular treatment. Neurol Sci. (2014) 35:609–10. doi: 10.1007/s10072-013-1596-1
168. Sutter R, Renaud S, Bonati L, Lyrer P, Tolnay M, Wetzel S, et al. Bilateral vertebral giant cell arteritis–favourable outcome in two cases. J Neurol. (2008) 255:133–4. doi: 10.1007/s00415-008-0632-1
169. Hollenhorst RW. Effect of posture on retinal ischemia from temporal arteritis. Trans Am Ophthalmol Soc. (1967) 65:94–105. doi: 10.1001/archopht.1967.00980030571002
170. Wykes WN, Cullen JF. Headache and temporal arteritis. Scott Med J. (1985) 30:42. doi: 10.1177/003693308503000110
171. Galetta SL, Balcer LJ, Liu GT. Giant cell arteritis with unusual flow-related neuro-ophthalmologic manifestations. Neurology. (1997) 49:1463–5. doi: 10.1212/WNL.49.5.1463
172. Diego M, Margo CE. Postural vision loss in giant cell arteritis. J Neuroophthalmol. (1998) 18:124–6. doi: 10.1097/00041327-199806000-00011
173. Yahyavi-Firouz-Abadi N, Wynn BL, Rybicki FJ, Steigner ML, Hussain AZ, Mather R, et al. Steroid-responsive large vessel vasculitis: application of whole-brain 320-detector row dynamic volume CT angiography and perfusion. AJNR Am J Neuroradiol. (2009) 30:1409–11. doi: 10.3174/ajnr.A1532
174. Dimancea A, Guidoux C, Amarenco P. Minor ischemic stroke and a smoldering case of giant-cell arteritis: a case report. Stroke. (2021) 52:e749–52. doi: 10.1161/STROKEAHA.121.035432
175. Takatsuki K, Kojima Y, Ikeuchi Y, Kitayama J, Tanaka A, Inoue Y. Intracranial vascular stenosis in giant cell arteritis successfully treated by two balloon angioplasty procedures. Mod Rheumatol Case Rep. (2023) 7:166–71. doi: 10.1093/mrcr/rxac080
176. John S, Hegazy M, Cheng Ching E, Katzan I. Isolated bilateral middle cerebellar peduncle infarcts. J Stroke Cerebrovasc Dis. (2013) 22:e645–646. doi: 10.1016/j.jstrokecerebrovasdis.2013.03.035
177. Dong Q, Jing G, Han J. The endovascular treatment of bilateral infarction of middle cerebellar peduncles. Etiology and endovascular treatment analysis. Neurosciences (Riyadh). (2017) 22:56–61. doi: 10.17712/nsj.2017.1.20160169
178. Zhou C, Fan H, Chen H, Wang H, Li Z, Xu N, et al. Evaluation of clinical features and stroke etiology in patients with bilateral middle cerebellar peduncle infarction. Eur Neurol. (2020) 83:271–8. doi: 10.1159/000508835
179. Shelley BP, Harishchandra P, Devadas AK. Selective hand motor cortex lesions masquerading as “pseudoperipheral nerve palsy”. Ann Indian Acad Neurol. (2020) 23:688–93. doi: 10.4103/aian.AIAN_9_19
180. Murr N, Thaisetthawatkul P, Helvey J, Fayad P. Selective infarction of the anterior genu fornices associated with giant cell arteritis. J Stroke Cerebrovasc Dis. (2012) 21:327–9. doi: 10.1016/j.jstrokecerebrovasdis.2010.08.006
181. Shanklin A, Cox A, Ahmed HMA, Lieberman A, Busza A. Clinical reasoning: a 77-year-old man presenting with episodic expressive aphasia. Neurology. (2018) 90:e1822–6. doi: 10.1212/WNL.0000000000005525
182. Ketz F, Monti A, Velentza A, Breining A, Leger A, Pautas E. Strokes and Giant cell arteritis: imaging characteristics. Soins Gerontol. (2019) 24:17–9. doi: 10.1016/j.sger.2019.04.008
183. Aslam F, Brown HH, Pandey T, Russell EB. Giant cell arteritis presenting with a lateral medullary stroke in a patient with multiple atherosclerotic risk factors. Scand J Rheumatol. (2013) 42:82–3. doi: 10.3109/03009742.2012.709273
184. Merli E, Romoli M, Gentile M, Forlivesi S, Borghi AM, Zaniboni A, et al. Bulbar watershed ischemic stroke: the comma-shaped sign-a case series. Neuroradiology. (2021) 63:1947–50. doi: 10.1007/s00234-021-02754-3
185. Soury P, Brisset D, Saliou C, Sraieb T, Angel F, Fiessinger J, et al. Lésions artérielles des membres supérieurs dans la maladie de Horton. Presse Med. (1997) 26:1478–80.
186. Missen G. Involvement of the vertebro-carotid arterial system in giant-cell arteritis. J Pathol. (1972) 106:2–3.
187. Grayson PC, Maksimowicz-Mckinnon K, Clark TM, Tomasson G, Cuthbertson D, Carette S, et al. Distribution of arterial lesions in Takayasu's arteritis and giant cell arteritis. Ann Rheum Dis. (2012) 71:1329–34. doi: 10.1136/annrheumdis-2011-200795
188. Ruegg S, Engelter S, Jeanneret C, Hetzel A, Probst A, Steck AJ, et al. Bilateral vertebral artery occlusion resulting from giant cell arteritis: report of 3 cases and review of the literature. Medicine (Baltimore). (2003) 82:1–12. doi: 10.1097/00005792-200301000-00001
189. Oerding C, Kaden I, Wohlfarth K. Isolated bilateral internal carotid artery stenosis and recurrent ischemic strokes in a patient with suspected giant cell arteritis. Case Rep Neurol. (2020) 12:84–91. doi: 10.1159/000504018
190. Cid, M. C., Prieto-Gonzalez S., Arguis P., Espigol-Frigole G., Butjosa M., Hernandez-Rodriguez J., et al. (2009). The spectrum of vascular involvement in giant-cell arteritis: clinical consequences of detrimental vascular remodelling at different sites. APMIS Suppl 10–20. doi: 10.1111/j.1600-0463.2009.02471.x
191. Endres K, Anjum O, Costain N. Arterial-embolic strokes and painless vision loss due to phase ii aortitis and giant cell arteritis: a case report. Clin Pract Cases Emerg Med. (2021) 5:174–7. doi: 10.5811/cpcem.2021.2.51143
192. Silva M, Carvalho BM, Queiroz ALG, Lima KDF, Teixeira HS, Schmid MF, et al. Isolated area postrema syndrome preceding the diagnosis of giant cell arteritis: a case report. Neurol Neuroimmunol Neuroinflamm. (2023) 10:113. doi: 10.1212/NXI.0000000000200113
193. Nakano F, Ueno Y, Suda A, Takanashi M, Yamashita A, Abe Y, et al. Fatal ischemic stroke caused by cerebral small arteritis in a patient with giant cell arteritis. J Neurol Sci. (2018) 391:22–4. doi: 10.1016/j.jns.2018.05.010
194. Topakian R, Stieglbauer K, Nussbaumer K, Dreer B, Sonnberger M, Aichner FT. Reversibility of basilar artery stenosis following timely treatment of temporal arteritis. Cerebrovasc Dis. (2009) 27:204–6. doi: 10.1159/000193465
195. Kawamura R, Mizuma A, Kouchi M, Nagata E, Takahashi W, Takizawa S. A recurrent case of ischemic stroke caused by vasospasm due to giant cell arteritis. J Stroke Cerebrovasc Dis. (2017) 26:e216–7. doi: 10.1016/j.jstrokecerebrovasdis.2017.07.023
196. Dziadkowiak E, Chojdak-Lukasiewicz J, Paradowski B, Bladowska J. Isolated arteritis misdiagnosed as bilateral orbital tumors in a patient with acute ischemic stroke. Radiol Case Rep. (2022) 17:3927–32. doi: 10.1016/j.radcr.2022.07.023
197. Kinoshita A, Otsuka Y, Deguchi K, Wakabayashi H. Giant cell arteritis of the middle cerebral artery. BMJ Case Rep. (2022) 15. doi: 10.1136/bcr-2022-250318
198. Kjeldsen MH, Reske-Nielsen E. Pathological changes of the central nervous system in giant-cell arteritis. Acta Ophthalmol (Copenh). (1968) 46:49–56. doi: 10.1111/j.1755-3768.1968.tb02494.x
199. Russi E, Aebi M, Kraus-Ruppert R, Mumenthaler M. Intracranial giant cell arteritis. J Neurol. (1979) 221:219–24. doi: 10.1007/BF00314638
200. Shigeyasu M, Sasaki N, Nishino S, Sakai N. Giant cell arteritis with simultaneous onset of multiple intracranial vascular occlusions: a case report. Surg Neurol Int. (2022) 13:21. doi: 10.25259/SNI_1001_2021
201. Brass SD, Durand ML, Stone JH, Chen JW, Stone JR. Case records of the Massachusetts General Hospital. Case 36-2008 A 59-year-old man with chronic daily headache. N Engl J Med. (2008) 356:2267–78. doi: 10.1056/NEJMcpc0805313
202. Kobayashi S, Yano T, Matsumoto Y, Numano F, Nakajima N, Yasuda K, et al. Clinical and epidemiologic analysis of giant cell (temporal) arteritis from a nationwide survey in 1998 in Japan: the first government-supported nationwide survey. Arthritis Rheum. (2003) 49:594–8. doi: 10.1002/art.11195
203. Enzmann D, Scott WR. Intracranial involvement of giant-cell arteritis. Neurology. (1977) 27:794–7. doi: 10.1212/WNL.27.8.794
204. Pfefferkorn T, Schuller U, Cyran C, Hufner K, Fesl G, Seelos K, et al. Giant cell arteritis of the Basal cerebral arteries: correlation of MRI, dsa, and histopathology. Neurology. (2010) 74:1651–3. doi: 10.1212/WNL.0b013e3181df0a09
205. Hauenstein C, Reinhard M, Geiger J, Markl M, Hetzel A, Treszl A, et al. Effects of early corticosteroid treatment on magnetic resonance imaging and ultrasonography findings in giant cell arteritis. Rheumatology (Oxford). (2012) 51:1999–2003. doi: 10.1093/rheumatology/kes153
206. Poillon G, Collin A, Benhamou Y, Clavel G, Savatovsky J, Pinson C, et al. Increased diagnostic accuracy of giant cell arteritis using three-dimensional fat-saturated contrast-enhanced vessel-wall magnetic resonance imaging at 3 T. Eur Radiol. (2020) 30:1866–75. doi: 10.1007/s00330-019-06536-7
207. Bley TA, Uhl M, Carew J, Markl M, Schmidt D, Peter HH, et al. Diagnostic value of high-resolution MR imaging in giant cell arteritis. AJNR Am J Neuroradiol. (2007) 28:1722–7. doi: 10.3174/ajnr.A0638
208. Bley TA, Markl M, Schelp M, Uhl M, Frydrychowicz A, Vaith P, et al. Mural inflammatory hyperenhancement in MRI of giant cell (temporal) arteritis resolves under corticosteroid treatment. Rheumatology (Oxford). (2008) 47:65–7. doi: 10.1093/rheumatology/kem283
209. Reichenbach S, Adler S, Bonel H, Cullmann JL, Kuchen S, Butikofer L, et al. Magnetic resonance angiography in giant cell arteritis: results of a randomized controlled trial of tocilizumab in giant cell arteritis. Rheumatology (Oxford). (2018) 57:982–6. doi: 10.1093/rheumatology/key015
210. Unizony S, Arias-Urdaneta L, Miloslavsky E, Arvikar S, Khosroshahi A, Keroack B, et al. Tocilizumab for the treatment of large-vessel vasculitis (giant cell arteritis, Takayasu arteritis) and polymyalgia rheumatica. Arthritis Care Res (Hoboken). (2012) 64:1720–9. doi: 10.1002/acr.21750
211. Sanchez-Alvarez C, Hawkins AS, Koster MJ, Lehman VT, Crowson CS, Warrington KJ. Clinical and radiographic features of giant cell arteritis with intracranial involvement. ACR Open Rheumatol. (2020) 2:471–7. doi: 10.1002/acr2.11161
212. Schmidt WA, Kraft HE, Vorpahl K, Volker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med. (1997) 337:1336–42. doi: 10.1056/NEJM199711063371902
213. Karahaliou M, Vaiopoulos G, Papaspyrou S, Kanakis MA, Revenas K, Sfikakis PP. Colour duplex sonography of temporal arteries before decision for biopsy: a prospective study in 55 patients with suspected giant cell arteritis. Arthritis Res Ther. (2006) 8:R116. doi: 10.1186/ar2003
214. Seitz L, Christ L, Lotscher F, Scholz G, Sarbu AC, Butikofer L, et al. Quantitative ultrasound to monitor the vascular response to tocilizumab in giant cell arteritis. Rheumatology (Oxford). (2021) 60:5052–9. doi: 10.1093/rheumatology/keab484
215. Boettinger M, Sebastian S, Gamulescu MA, Grauer O, Ritzka M, Schuierer G, et al. Bilateral vertebral artery occlusion with retrograde basilary flow in three cases of giant cell arteritis. BMJ Case Rep. (2009) 2009:bcr07.2008.0488. doi: 10.1136/bcr.07.2008.0488
216. Zarar A, Zafar TT, Khan AA, Suri MF, Qureshi AI. Internal carotid artery stenosis associated with giant cell arteritis: case report and discussion. J Vasc Interv Neurol. (2014) 7:24–7.
217. Togo M, Kono T, Hoshi T, Imamura H, Todo K, Adachi H, et al. Successful endovascular therapy for multiple intracranial arterial stenosis associated with medically intractable giant cell arteritis. J Neurol Sci. (2018) 384:104–6. doi: 10.1016/j.jns.2017.11.027
218. Steiger R, Walchhofer LM, Rietzler A, Mair KJ, Knoflach M, Glodny B, et al. Cerebral phosphorus magnetic resonance spectroscopy in a patient with giant cell arteritis and endovascular therapy. Case Rep Radiol. (2018) 2018:7806395. doi: 10.1155/2018/7806395
219. Ahdab R, Thabuy F, Menager De Froberville E, Brugieres P, Hosseini H. Reversible vertebral artery stenosis following corticotherapy in giant cell arteritis. Eur Neurol. (2008) 59:331. doi: 10.1159/000121427
220. Takekawa H, Daimon Y, Takashima R, Aiba S, Tanaka H, Hirata K. Giant cell arteritis associated with lesion of the internal carotid artery: assessment of response to steroid therapy by magnetic resonance angiography. Intern Med. (2008) 47:1285–6. doi: 10.2169/internalmedicine.47.1137
221. Polomat K, Chausson N, Olindo S, Smadja D. Reversibility of vertebrobasilar stenoses following treatment with corticosteroid therapy in patients with giant cell arteritis. Rev Neurol (Paris). (2010) 166:940–3. doi: 10.1016/j.neurol.2010.03.007
222. Mahawish K, Cariga P. Multi-territory infarcts caused by intracranial giant cell arteritis. New Zealand Medical Journal. (2020) 133:129–31.
223. Koster MJ, Warrington KJ, Lehman VT. Comment on: Development of intracranial vasculitis in giant cell arteritis during tocilizumab treatment by Naderi. Clin Exp Rheumatol. (2021) 39 Suppl 129:198. doi: 10.55563/clinexprheumatol/3k20me
224. Michotte A, De Keyser J, Dierckx R, Impens N, Solheid C, Ebinger G. Brain stem infarction as a complication of giant-cell arteritis. Clin Neurol Neurosurg. (1986) 88:127–9. doi: 10.1016/S0303-8467(86)80008-7
225. Simonsen CZ, Speiser L, Hansen IT, Jayne D, Von Weitzel-Mudersbach P. Endovascular treatment of intracerebral giant cell arteritis. Front Neurol. (2020) 11:287. doi: 10.3389/fneur.2020.00287
226. Armellin L, Sammel AM, Ng B, Sarathy K, Lambros J, Amir-Nezami T, et al. Coronary artery stenting in acute coronary syndrome associated with giant cell arteritis. J Cardiol Cases. (2017) 16:77–81. doi: 10.1016/j.jccase.2017.05.008
227. Dellaripa PF, Eisenhauer AC. Bilateral percutaneous balloon angioplasty of the axillary arteries in a patient with giant cell arteritis and upper extremity ischemic symptoms not responsive to corticosteroids. J Rheumatol. (1998) 25:1429–33.
228. Espigol-Frigolé G, Gomez-Choco M, Obachm V, Sanroman L, Prieto S, Argelich R, et al. Percutaneous transluminal angioplasty of internal carotid and vertebral arteries in giant cell arteritis (GCA): report of two cases. APMIS. (2009) 117:104−105. doi: 10.1111/j.1600-0463.2009.02504.x
229. Chausson N, Olindo S, Signate A, Cohen-Tenoudji P, Aveillan M, Saint-Vil M, et al. Bilateral intracerebral angioplasty in a patient with stroke caused by giant cell arteritis. Rev Neurol (Paris). (2010) 166:328–32. doi: 10.1016/j.neurol.2009.05.011
230. Biondi A, Katz JM, Vallabh J, Segal AZ, Gobin YP. Progressive symptomatic carotid dissection treated with multiple stents. Stroke. (2005) 36:e80–82. doi: 10.1161/01.STR.0000177883.50262.cf
231. Dawson ET, Brown DA, Rabinstein AA. Headache, TIA and subarachnoid haemorrhage: dissecting an unusual cause for stroke-like symptoms. BMJ Case Rep. (2017) 2017:bcr2017219927. doi: 10.1136/bcr-2017-219927
232. Czihal M, Lottspeich C, Kohler A, Prearo I, Hoffmann U, Priglinger SG, et al. Transocular sonography in acute arterial occlusions of the eye in elderly patients: diagnostic value of the spot sign. PLoS One. (2021) 16:e0247072. doi: 10.1371/journal.pone.0247072
233. Daudin J, Bluwol E, Chaine G, Rohart C. Artérite à cellules géantes révélée par des nodules cotonneux. J Français d'Ophtalmologie. (2006) 29:1157.e1153. doi: 10.1016/S0181-5512(06)73912-2
234. Schauble B, Wijman CA, Koleini B, Babikian VL. Ophthalmic artery microembolism in giant cell arteritis. J Neuroophthalmol. (2000) 20:273–5. doi: 10.1097/00041327-200020040-00015
235. Arias-Rivas S, Santamaria Cadavid M, Rodriguez-Yanez M, Blanco M. Microembolism detection in giant cell arteritis. Neurol Clin Pract. (2016) 6:e35–6. doi: 10.1212/CPJ.0000000000000204
236. Alba MA, Espigol-Frigole G, Prieto-Gonzalez S, Tavera-Bahillo I, Garcia-Martinez A, Butjosa M, et al. Central nervous system vasculitis: still more questions than answers. Curr Neuropharmacol. (2011) 9:437–48. doi: 10.2174/157015911796557920
237. Tang V, Fantaneanu T, Chakraborty S, Patel V, Dowlatshahi D. Intracranial non-occlusive thrombus and multiple strokes in giant cell arteritis. Can J Neurol Sci. (2012) 39:116–7. doi: 10.1017/S0317167100022149
238. Valente F, Carro A, Moral S, Evangelista A. Multiple thrombi in the ascending aorta: usefulness of contrast transesophageal echocardiography in a case of Horton's aortitis. Circulation. (2013) 128:e44–45. doi: 10.1161/CIRCULATIONAHA.112.001156
239. Godoy P, Araujo Sde A, Paulino EJr, Lana-Peixoto MA. Coronary giant cell arteritis and acute myocardial infarction. Arq Bras Cardiol. (2007) 88:e84–87. doi: 10.1590/S0066-782X2007000400027
240. Andrade J, Al Ali A, Saw J, Wong GC. Acute stent thrombosis in a patient with giant cell arteritis. Can J Cardiol. (2008) 24:e25–26. doi: 10.1016/S0828-282X(08)70186-7
241. Lammie GA, Sandercock PA, Dennis MS. Recently occluded intracranial and extracranial carotid arteries. Relevance of the unstable atherosclerotic plaque. Stroke. (1999) 30:1319–25. doi: 10.1161/01.STR.30.7.1319
242. Collado A, Santamaria J, Ribalta T, Cinta Cid M, Canete JD, Tolosa E. Giant-cell arteritis presenting with ipsilateral hemiplegia and lateral medullary syndrome. Eur Neurol. (1989) 29:266–8. doi: 10.1159/000116424
243. Bonnin JM, Lander H. Giant-cell arteritis; report of a case with autopsy. J Pathol Bacteriol. (1956) 71:369–73. doi: 10.1002/path.1700710211
244. Howard GF, Ho SU, Kim KS, Wallach J. Bilateral carotid artery occlusion resulting from giant cell arteritis. Ann Neurol. (1984) 15:204–207. doi: 10.1002/ana.410150216
245. Butt Z, Cullen JF, Mutlukan E. Pattern of arterial involvement of the head, neck, and eyes in giant cell arteritis: three case reports. Br J Ophthalmol. (1991) 75:368–71. doi: 10.1136/bjo.75.6.368
246. Paccalin M, Amoura Z, Roblot P, Godeau P, Becq-Giraudon B, Piette JC. Premature cerebral vascular accidents after initiation of steroid therapy in Horton disease. Rev Med Interne. (2000) 21:550–4. doi: 10.1016/S0248-8663(00)89232-2
247. Bogousslavsky J, Deruaz JP, Regli F. Bilateral obstruction of internal carotid artery from giant-cell arteritis and massive infarction limited to the vertebrobasilar area. Eur Neurol. (1985) 24:57–61. doi: 10.1159/000115762
248. Avina-Zubieta JA, Bhole VM, Amiri N, Sayre EC, Choi HK. The risk of deep venous thrombosis and pulmonary embolism in giant cell arteritis: a general population-based study. Ann Rheum Dis. (2016) 75:148–54. doi: 10.1136/annrheumdis-2014-205665
249. Flammer J, Pache M, Resink T. Vasospasm, its role in the pathogenesis of diseases with particular reference to the eye. Prog Retin Eye Res. (2001) 20:319–49. doi: 10.1016/S1350-9462(00)00028-8
250. Hayreh, S. S. (1992). Vasospasm and transient monocular blindness. N Engl J Med 326, 837-838; author reply 838–839. doi: 10.1056/NEJM199203193261215
251. Rozen TD. Brief sharp stabs of head pain and giant cell arteritis. Headache. (2010) 50:1516–9. doi: 10.1111/j.1526-4610.2010.01718.x
252. Knash ME, Monteith TS, Raskin NH. A comment on brief sharp stabs of head pain and giant cell arteritis. Headache. (2011) 51:1010. doi: 10.1111/j.1526-4610.2011.01920.x
253. Lo Gullo A, Koster MJ, Crowson CS, Makol A, Ytterberg SR, Saitta A, et al. Venous thromboembolism and cerebrovascular events in patients with giant cell arteritis: a population-based retrospective cohort study. PLoS ONE. (2016) 11:e0149579. doi: 10.1371/journal.pone.0149579
254. Dasgupta B, Smith K, Khan AS, Coath F, Wakefield RJ. 'Slope sign': a feature of large vessel vasculitis? Ann Rheum Dis. (2019) 78:1738. doi: 10.1136/annrheumdis-2019-216213
255. Reinhard M, Schmidt D, Schumacher M, Hetzel A. Involvement of the vertebral arteries in giant cell arteritis mimicking vertebral dissection. J Neurol. (2003) 250:1006–9. doi: 10.1007/s00415-003-1155-4
256. Parra J, Domingues J, Sargento-Freitas J, Santana I. Extensive intracranial involvement with multiple dissections in a case of giant cell arteritis. BMJ Case Rep. (2014) 2014:bcr2014204130. doi: 10.1136/bcr-2014-204130
257. Proft F, Czihal M, Rémi J, Fesl G, Woischke C, Schulze-Koops H. Fatal Spontaneous Bilateral Vertebral Artery Dissection in Giant Cell Arteritis (GCA). J Vasculitis. (2017) 3. doi: 10.4172/2471-9544.1000123
258. Yashima A, Yamashita H, Yamada S, Noguchi T, Takahashi Y, Kaneko H. A case of giant cell arteritis mimicking vertebral dissection on contrast-enhanced magnetic resonance angiography. Clin Exp Rheumatol. (2018) 36 Suppl 111:178–9.
259. Marte Furment M, Antigua Jimenez S, Pabolu S. Acute stroke due to vertebral artery dissection in giant cell arteritis. Case Rep Rheumatol. (2021) 2021:5518541. doi: 10.1155/2021/5518541
260. Hashami L, Haj Mohamad Ebrahim Ketabforoush A, Nirouei M. Giant cell arteritis with rare manifestations of stroke and internal carotid artery dissection: a case study. Clin Case Rep. (2022) 10:e05597. doi: 10.1002/ccr3.5597
261. Amiri N, De Vera M, Choi HK, Sayre EC, Avina-Zubieta JA. Increased risk of cardiovascular disease in giant cell arteritis: a general population-based study. Rheumatology (Oxford). (2016) 55:33–40. doi: 10.1093/rheumatology/kev262
262. Coronel L, Rodriguez-Pardo J, Monjo I, De Miguel E. Prevalence and significance of ischemic cerebrovascular events in giant cell arteritis. Med Clin (Barc). (2021) 157:53–7. doi: 10.1016/j.medcli.2020.05.068
Keywords: giant cell (temporal) arteritis, stroke, stroke/physiopathology, embolic stroke, thrombotic stroke
Citation: Bonnan M and Debeugny S (2023) Giant-cell arteritis related strokes: scoping review of mechanisms and rethinking treatment strategy? Front. Neurol. 14:1305093. doi: 10.3389/fneur.2023.1305093
Received: 30 September 2023; Accepted: 17 November 2023;
Published: 07 December 2023.
Edited by:
Eiichiro Nagata, Tokai University Isehara Hospital, JapanReviewed by:
Christopher Carr, Augusta University, United StatesKlearchos Psychogios, Metropolitan Hospital, Greece
Copyright © 2023 Bonnan and Debeugny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mickael Bonnan, bWlja2FlbF9ib25uYW5AeWFob28uZnI=