 Samantha P. Martin1,2,3,4*
Samantha P. Martin1,2,3,4* Beth A. Leeman-Markowski1,2,4
Beth A. Leeman-Markowski1,2,4- 1Comprehensive Epilepsy Center, New York University Langone Health, New York, NY, United States
- 2Department of Neurology, New York University Langone Health, New York, NY, United States
- 3New York University Grossman School of Medicine, New York, NY, United States
- 4VA New York Harbor Healthcare System, New York, NY, United States
Traumatic brain injury (TBI), Alzheimer’s disease (AD), and epilepsy share proposed mechanisms of injury, including neuronal excitotoxicity, cascade signaling, and activation of protein biomarkers such as tau. Although tau is typically present intracellularly, in tauopathies, phosphorylated (p-) and hyper-phosphorylated (hp-) tau are released extracellularly, the latter leading to decreased neuronal stability and neurofibrillary tangles (NFTs). Tau cleavage at particular sites increases susceptibility to hyper-phosphorylation, NFT formation, and eventual cell death. The relationship between tau and inflammation, however, is unknown. In this review, we present evidence for an imbalanced endoplasmic reticulum (ER) stress response and inflammatory signaling pathways resulting in atypical p-tau, hp-tau and NFT formation. Further, we propose tau as a biomarker for neuronal injury severity in TBI, AD, and epilepsy. We present a hypothesis of tau phosphorylation as an initial acute neuroprotective response to seizures/TBI. However, if the underlying seizure pathology or TBI recurrence is not effectively treated, and the pathway becomes chronically activated, we propose a “tipping point” hypothesis that identifies a transition of tau phosphorylation from neuroprotective to injurious. We outline the role of amyloid beta (Aβ) as a “last ditch effort” to revert the cell to programmed death signaling, that, when fails, transitions the mechanism from injurious to neurodegenerative. Lastly, we discuss targets along these pathways for therapeutic intervention in AD, TBI, and epilepsy.
1 Introduction
TBI and CTE are characterized by abnormal tau deposition in brain tissue. Epilepsy can also represent a form of tauopathy, as a result of cellular injury due to repetitive seizures. Seizure-induced injury responses include neuronal excitotoxicity and inflammatory cascades, which can lead to tau deposition and cell death (1–3). Tau is crucial for neuronal structural integrity and intracellular axonal transport (4, 5). Although tau is most commonly present intracellularly, p-tau is also found in the synaptic cleft (6, 7). Hp-tau leads to decreased neuronal stability and extracellular NFT formation, seen in neurodegenerative disorders including AD, CTE, TBI, and epilepsy. Tau cleaved by caspases, a family of enzymes involved in programmed cell death, is also present in NFTs (8, 9). Tau cleavage at specific sites by caspases increases susceptibility to hyper-phosphorylation and NFT formation, suggesting that cell death pathways contribute to the pathology of tauopathies (9).
The role of inflammation in this cascade, however, is unknown. We briefly outline key inflammatory proteins involved in molecular signaling in TBI, AD, and epilepsy; discuss ER stress and its differing roles in TBI, AD, and epilepsy; and summarize how inflammatory signaling imbalances the ER stress response post-injury. We propose that, in response to acute moderate–severe TBI or single seizures, both inflammatory signaling and an overwhelmed ER stress response activate tau-induced signaling pathways to prevent further cellular dysfunction and restore intracellular homeostasis. Furthermore, we propose that in response to repeated injury, there is chronic activation of pro-inflammatory pathways and continual imbalance of the ER stress response, along with chronic activation of tau-induced signaling pathways.
We discuss three distinct processes, neuroprotection, injury, and degeneration, where injury is potentially reversible, and degeneration represents the spread of toxic effects to neighboring neurons and a lower likelihood of reversibility. We propose pathways by which the neuroinflammatory response to injury (seizures or TBI) contributes to tau hyper-phosphorylation and NFT formation, ultimately presenting our final hypothesis: tau phosphorylation plays a key role in neuroprotection, responding to recurrent seizures/injury, but there is a “tipping point” from neuroprotective to injurious effects – the repeated or sustained induction of an imbalanced ER stress response (specifically, the unfolded protein response [UPR]) and tau phosphorylation/hyper-phosphorylation. The ER stress response stimulates tau phosphorylation and continued tau cleavage; further phosphorylation/hyper-phosphorylation of tau promotes a continued UPR response and promotes neurodegeneration. This chronic dysregulation results in a shift from a tau-induced signaling pathway as a compensatory, neuroprotective response – which once reduced cellular dysfunction and attempted to restore apoptotic-necrotic dynamics and cellular homeostasis – to an injurious mechanism that is unable to maintain intracellular homeostasis, nor dynamically revert to mechanisms of programmed cell death (apoptosis).
Lastly, we propose a role for Aβ and outline its “last ditch effort” to mediate the injurious effects of excitotoxicity and chronic tau pathway activation, reverting the cell to pro-death signaling. However, due to (1) sustained UPR signaling interacting with tau and Aβ (2) the inability of reactive astrocytes and microglia to successfully break down toxic tau and Aβ aggregates, this leads to further tau hyper-phosphorylation resulting in NFT formation, as well as Aβ plaque accumulation – the hallmarks of neurodegeneration seen in AD pathology.
2 Injury response: molecular signaling
Inflammatory signaling, excitotoxic propagation, and ER stress play key roles in the atypical activation of cell death cascades and excessive phosphorylation of tau, resulting in downstream toxic tau aggregates and eventual neurodegeneration.
2.1 Inflammatory proteins and neurotransmission
Inflammatory proteins, including receptor-interacting kinases (e.g., RIP1/RIP3) and cytokines (e.g., interleukin-1 [IL-1], caspases), modulate inflammatory function and regulate forms of cell death such as necroptosis and apoptosis (10–12). Effects of inflammatory mediators are complex, in that they differ based on injury type, location, and chronicity. Even a single, acute TBI can cause sustained inflammatory signaling, measured by interleukin (IL) protein levels (13). A continued inflammatory response may lead to secondary neuronal injury and a decreased likelihood of spontaneous recovery over time, with persistent neuropsychological deficits. Additional injuries may contribute to chronic functional deficits, due to shortened recovery time between injuries and long-term neurodegeneration.
Neurotransmitters can modulate inflammatory responses in brain injury by disrupting pro-inflammatory cytokines, microglial production, and calcium signaling (14). Glutamate and γ-aminobutyric acid (GABA) are the major excitatory and inhibitory neurotransmitters, respectively. Glutamate release into the synaptic cleft occurs via calcium influx and intracellular calcium-dependent signaling (15). Once glutamate acts upon post-synaptic neurons, astrocytes collect and convert it to glutamine which is transported back to pre-synaptic neurons (16). Neuronal excitotoxicity due to altered glutamate and GABA receptor expression and function is evident in models of TBI (17, 18).
N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) are glutamate receptors responsible for neuronal influx of calcium in post-synaptic neurons. Table 1 summarizes NMDA and AMPA functions during typical neuronal depolarization and action potential propagation. The net effect of selectively activating these receptors and regulating their post-synaptic densities is to potentiate a non-toxic glutamate response, which promotes synaptic plasticity, long-term potentiation, and learning and memory (17, 19, 20). However, if these receptors are unselectively trafficked to/from key synaptic regions in brain injury, the result is an acute disruption of these signaling processes. In mechanical models of injury, down-regulation of the AMPA GluR2 and NMDA N2A receptors, along with up-regulation of the NMDA N2B receptor, lead to atypical calcium influx resulting in acute excitotoxic cell death (21–23).
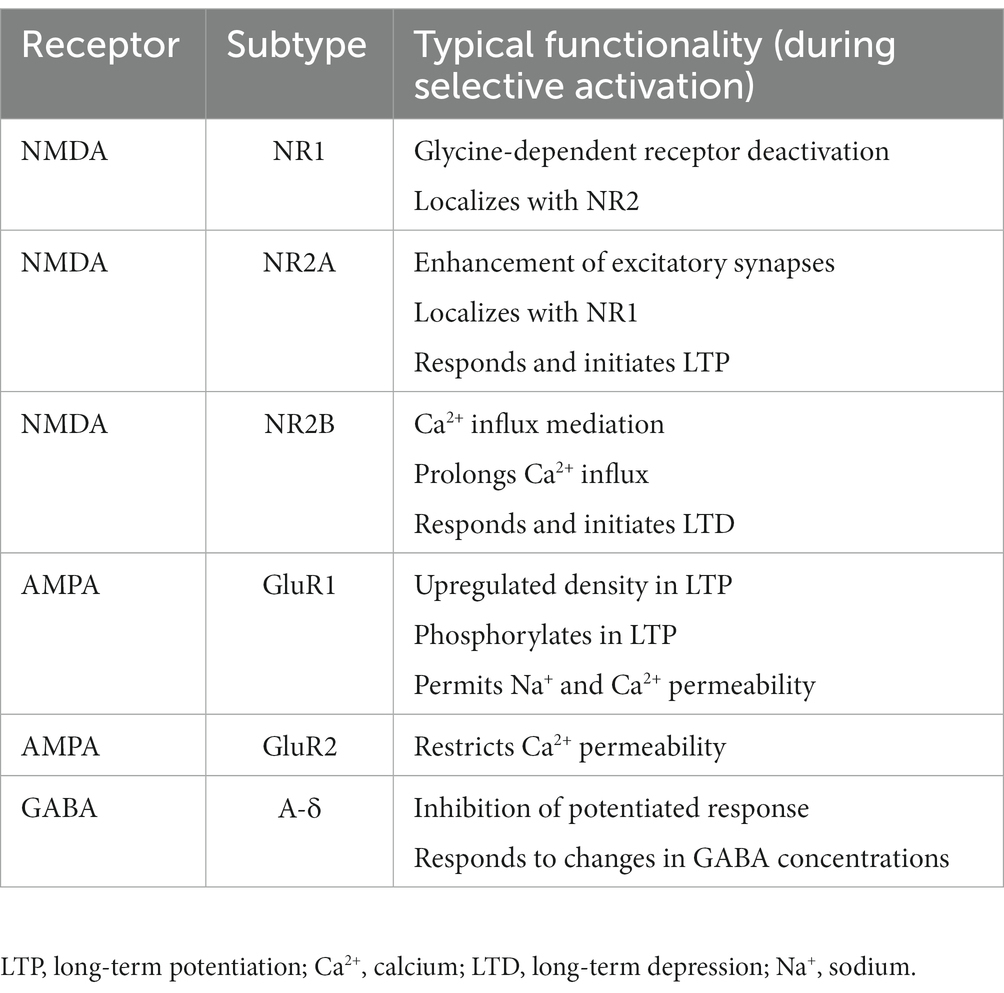
Table 1. Typical functionality (during selective activation) of glutamate and GABA receptors.
2.2 Molecular signaling in TBI
Although TBI primarily leads to neocortical cell death, hippocampal vulnerability is also apparent. In a controlled cortical impact (CCI) mouse model of moderate TBI, apoptosis of immature hippocampal neurons was observed 24–72 h after injury (24). Limited inflammatory markers may be observed up to 7 days post-CCI, and necrosis of immature hippocampal neurons was evident for at least 14 days post-injury (25, 26). These results demonstrate hippocampal vulnerability in response to TBI that may clinically present as memory complaints.
Both altered excitatory glutamate signaling and reduced GABA-mediated inhibition contribute to excitotoxicity in brain injury (27). In a mouse CCI model, glutamate expression correlated with epileptiform activity within injured and adjacent cortex in the setting of decreased GABAergic interneurons. Further, there was significant reduction of the GABAA γ2-subunit in CCI-injured rats with post-traumatic epilepsy (18). In a mouse CCI model of severe TBI, GABAA δ and GABAB B2 receptor subunit expression in dentate gyrus granule cells was reduced by 40–43% (24). In contrast, human studies of chronic, repetitive injuries in athletes (closed head injury [CHI] model) found a compensatory increase in GABAB receptor expression (28). Decreased GABAA receptor expression disrupts the inhibitory response (29), while increased GABAB receptor expression, responsible for membrane hyperpolarization, may serve to avoid further depolarization and excitotoxic effects.
2.3 Molecular signaling in AD
AD pathology includes Aβ plaque accumulation and NFT formation, with tau aggregation and hyper-phosphorylation contributing to dysregulated microtubule dynamics and neuronal functioning (30). Necroptosis activation by RIP1/RIP3 kinases was found in postmortem AD brains (31). Elevated levels of inflammatory markers IL-1β, IL-6, and tumor necrosis factor-alpha (TNF-α) were found in postmortem AD and transgenic animal brains, and microglial and astrocytic activation was observed in response to neurotoxic cytokine expression (32–36).
Excitotoxicity due to dysregulated Ca2+-mediated NMDA receptor functioning decreases cell survival (37, 38). Aβ regulates synaptic vesicle release and affects NMDA receptor structure, density, and electrophysiology – ultimately affecting glutamate transmission and resulting in cognitive changes (39–43). In AD patients with severe cognitive deterioration, decreased glutamate and GABA levels were noted in temporal cortex and CSF compared to AD patients with mild cognitive deterioration and age-matched controls (44, 45), and decreased concentrations of GABAergic terminals in cortical neurons adjacent to Aβ plaques were found in AD patients and transgenic AD mouse models (46, 47). These findings suggest impaired receptor function and neurotransmission and an imbalance between excitatory and inhibitory activity in AD.
2.4 Molecular signaling in epilepsy
Inflammatory responses in epilepsy can contribute to recurrent seizures, secondary neuronal injury, and chronic neurodegeneration (2). During focal to bilateral tonic–clonic seizures, cytokines exert effects through increased AMPA receptor density, NMDA-dependent calcium influx, and reduction of GABAA receptor density, resulting in greater synaptic glutamate and decreased synaptic GABA concentrations (48–51). Excess glutamate increases the likelihood of neuronal depolarization, excitotoxicity, and eventual cell death (52–54), particularly in models of temporal lobe epilepsy (55). Glia rapidly produce interleukins, particularly interleukin-1 beta (IL-1β), postictally. IL-1β enhances neuronal excitability and sustains inflammatory responses (56, 57). Increased IL-1β activity leads to neuronal degeneration in epileptogenic regions, while astrocytes that express its receptor have neuroprotective functions (1, 58). Astrocytes can mediate the effect of IL-1β on hippocampal neurons, contributing to their likelihood of survival. The presence of astrocytes in epileptogenic regions is a compensatory response to excess synaptic glutamate (59, 60). Increased astrocytes in regions of post-ictal neuronal injury suggest IL-1β involvement in the initiation and continuation of local seizure activity (59).
We propose that during a single seizure and mild TBI (Figure 1), excitotoxic depolarization enhances IL-1β signaling and increases NMDA receptor activity, leading to local propagation of excitotoxic depolarization and extracellular glutamate accumulation. This process, along with increased Aβ and cytokine secretion, recruits astrocytes into the synapse (61) to collect glutamate post-seizure. Excess glutamate also recruits microglia to clear cellular debris, remove excess Aβ, and return to neuronal homeostasis (60, 62, 63). If neuronal homeostasis is not achieved, further excitotoxic injury and cell death signaling can occur.
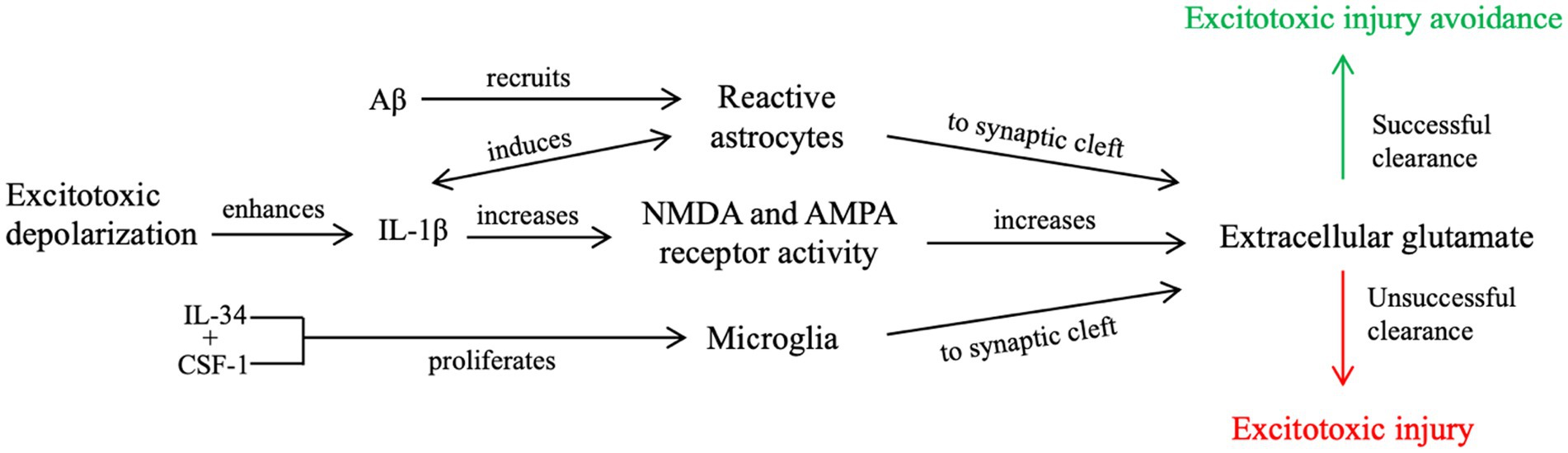
Figure 1. Our proposed contributory mechanism of IL-1β signaling during a single, brief seizure or mild TBI. Enhanced IL-1β signaling from excitotoxic depolarization results in increased glutamate receptor activity and further propagation of excitotoxic signaling, resulting in an accumulation of post-synaptic glutamate. Increased neuroinflammatory signaling, including upregulated cytokine and Aβ secretion and increased concentrations of extracellular glutamate, recruit microglia and reactive astrocytes to the post-synaptic cleft. Unsuccessful clearance of extracellular glutamate, cellular debris, and Aβ from the synaptic cleft by reactive astrocytes and microglia leads to further excitotoxic propagation and places the cell at risk for excitotoxic injury. Successful clearance, however, reduces the risk of excitotoxic injury, as it attempts to revert the cell to neuronal homeostasis.
Neuronal damage in TBI, AD, and epilepsy can result from secondary inflammatory responses and neuronal excitotoxicity. Interleukins, particularly IL-1β, are key modulators of pro-inflammatory responses and apoptosis. Additionally, dysregulation of the glutamate-GABA/excitation-inhibition balance leads to excitotoxic injury and neuronal death.
2.5 ER stress and its role in TBI, AD, and epilepsy
ER stress occurs when there is an imbalance between the ability of the ER to fold proteins and the cellular demand for protein folding (64). In response to ER stress, the UPR signals to either (1) protect the cell by correcting the imbalance between folding ability and demand (65) via the protein kinase R-like ER kinase (PERK) pathway or (2) promote programmed cell death. Cell death occurs via C/EBP homologous protein (CHOP) and Apaf-1-dependent apoptosis or via necroptosis involving RIP1/RIP3-activation and rapid ATP depletion (66–68). Acute UPRs are protective to the cell. Sustained UPRs, however, induce caspase-dependent apoptosis (69), deplete intracellular ATP (70), and induce necrosis (70).
ER stress contributes to neuronal loss in TBI (26, 71, 72), AD (73), and epilepsy (74) and correlates with tau phosphorylation in TBI and AD (75, 76). In a CCI rat model, markers of reactive ER stress were associated with increased tau oligomers and tau kinase (GSK-3β) activation (77). To study the relationship between tau phosphorylation and ER-stress in promoting AD-like pathogenesis, tau phosphorylation was induced in rat cortical neurons, resulting in a UPR response with elevation of p-PERK and other modulator proteins. In the same study, an ER stress inducer enhanced tau phosphorylation at specific sites (75).
In human AD autopsy material, PERK correlated with atypical tau phosphorylation (78), and tau interacted with ER proteins leading to neuronal dysfunction and neurotoxicity (79). In epilepsy, the relationship between ER stress and tau phosphorylation is unknown, although relationships between epilepsy and unfolded proteins have been established. A mouse model of epilepsy suggested that acute, reactive ER stress responses may reduce seizure recurrence or severity (80). In resected tissue from patients with epilepsy due to focal cortical dysplasia, however, there were greater accumulations of unfolded proteins and increased levels of CHOP in patients who were not rendered seizure-free (81). Hence, acute, reactive stress responses may be protective, while chronically increased ER stress may contribute to seizure recurrence.
Aβ can trigger ER stress, just as ER stress can promote Aβ formation, leading to excitotoxicity and apoptosis (82–84). While amyloid precursor protein (APP) increases resistance to ER stress-induced apoptosis in specific cell cultures (85), intracellular Aβ counteracts APP by activating ER stress and pre-disposing cells to other pathways of programmed cell death (86). In brain endothelial cells, Aβ increased concentrations of UPR signaling mediators, increased intracellular Ca2+, and upregulated pro-apoptotic transcription factors (87). The relationship between Aβ and excitotoxicity is complex, however, in that Aβ also acts directly on the ER stress response protein XBP1 to reduce intracellular Ca2+ concentrations and limit excitotoxic injury (88).
Data suggest initial neuroprotective effects of reactive ER stress, activation of the PERK pathway, and APP (89). However, we postulate that sustained, repeated, or anticipatory (i.e., in the face of chronic injury) induction of the ER stress response may increase atypical tau phosphorylation and Aβ concentrations, with deleterious effects. Aβ has both pro-apoptotic and excitotoxic effects, but to limit neural injury, it acts feeds back on the ER stress response to interrupt it. If Aβ fails to halt its excitotoxic effects, and microglia and reactive astrocytes cannot successfully clear toxic tau and Aβ aggregates, neurodegeneration follows.
3 Injury response: the role of tau
Tau plays a key role in ER stress and Aβ pathways. Tau is a neuronal protein that supports axonal transport and microtubule dynamics (4). In neurodegenerative diseases, tau is abnormally present within subcortical neurons, including the hippocampus. Tau hyper-phosphorylation results in deposits of neurofibrillary tangles (NFTs), corresponding with diminished neuronal stability and subsequent aberrant neuronal communication (4, 5). These structural abnormalities lead to cognitive deficits, including memory loss (90–93). Elevated levels of total- (t-), phosphorylated- (p-), and hyperphosphorylated- (hp-) tau are detected in CSF at various time points post-TBI/seizure (91, 94–98). Accumulation and spread of tau aggregates occurs in various cortical and subcortical areas post-injury/seizure and in AD (93, 94, 99–101).
To explain the role of tau in brain injury and its relationship to the above inflammatory and excitotoxic processes, we posit two distinct signaling mechanisms, combining components of various pathways described in the literature: (1) an acute injury response (AIR; Figure 2), and (2) a recurrent injury response (RIR; Figure 3). AIR and RIR propose varying degrees of interleukin, NMDA/AMPA receptor, and Ca2+/calmodulin-dependent protein kinase (CaMK) involvement. We also propose a slower neuroprotective tau (NPT) response mechanism shared by acute seizures and TBI. However, with repeated seizures/TBI leading to chronically activated/sustained ER stress responses, the NPT pathway will become dysregulated, resulting in neural injury (Figure 4).
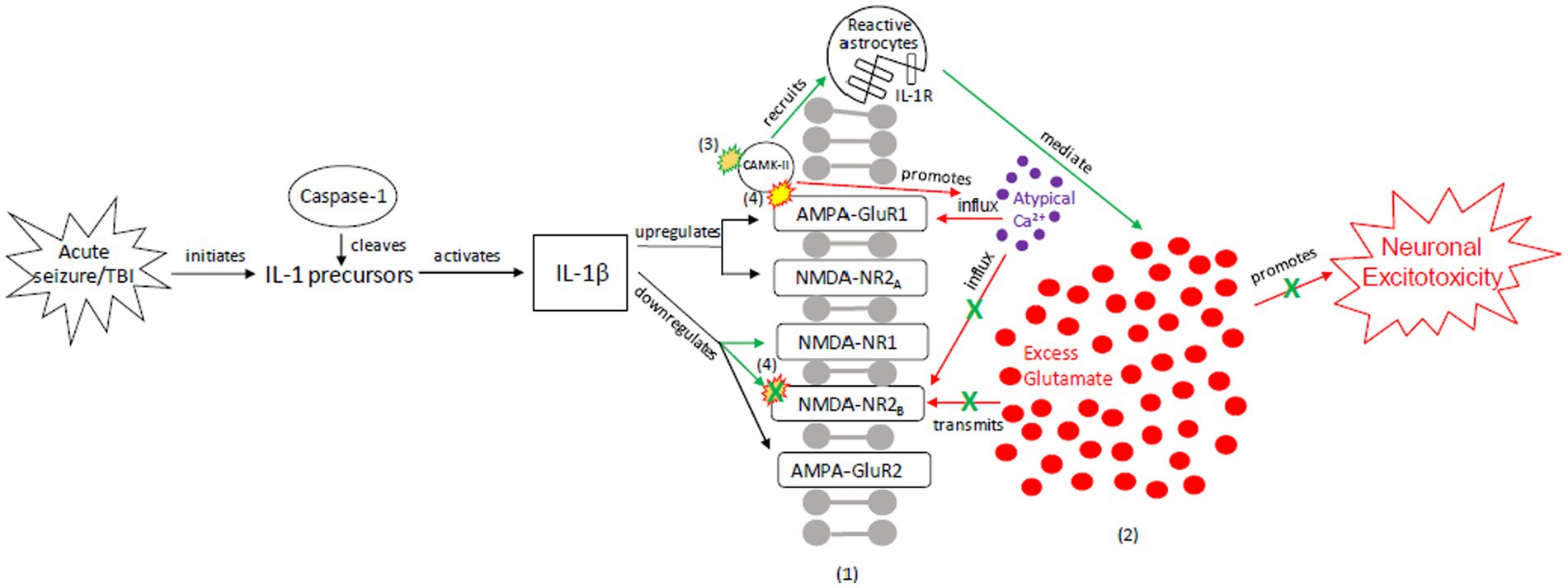
Figure 2. Our proposed acute injury response (AIR) mechanism outlining reactive signaling to an acute, brief seizure or acute, mild TBI. This mechanism shunts cellular signaling away from pro-death response pathways and toward cellular protection, with the goals of restoring the balance between glutamate release and reuptake, intracellular Ca2+-driven ER stress responses, and apoptotic-necrotic dynamics. In response to acute injury, caspase-1 cleaves IL-1 precursors, resulting in IL-1β formation. IL-1β unselectively up-or down-regulates glutamate receptor subunit densities, resulting in an acute disruption of balanced glutamate release and reuptake. There is increased CaMK-II activation that promotes increased glutamate release (102, 103). Unselective CaMK-II phosphorylation and autophosphorylation occurs at upregulated AMPA-GluR1 (104) but not at down-regulated NMDA-R2B (105), resulting in increased AMPA-GluR1 Ca2+ influx/channel conductance and decreased NMDA-NR2B Ca2+ influx/channel conductance, respectively. However, CaMK-II also recruits astrocytes into the affected region (59, 60, 106, 107). Increased astrocytes/IL-1 receptor density aid in clearing excess glutamate and ILs, inhibiting further glutamate release, thereby limiting excitotoxic propagation. (1) = Neuronal membrane, (2) = Synaptic cleft, (3) = CaMK-II autophosphorylation, (4) = CaMK-II-Glutamate receptor phosphorylation. Red = Excitotoxic signaling, Green = Neuroprotective signaling. X = response reduction/down-regulation.
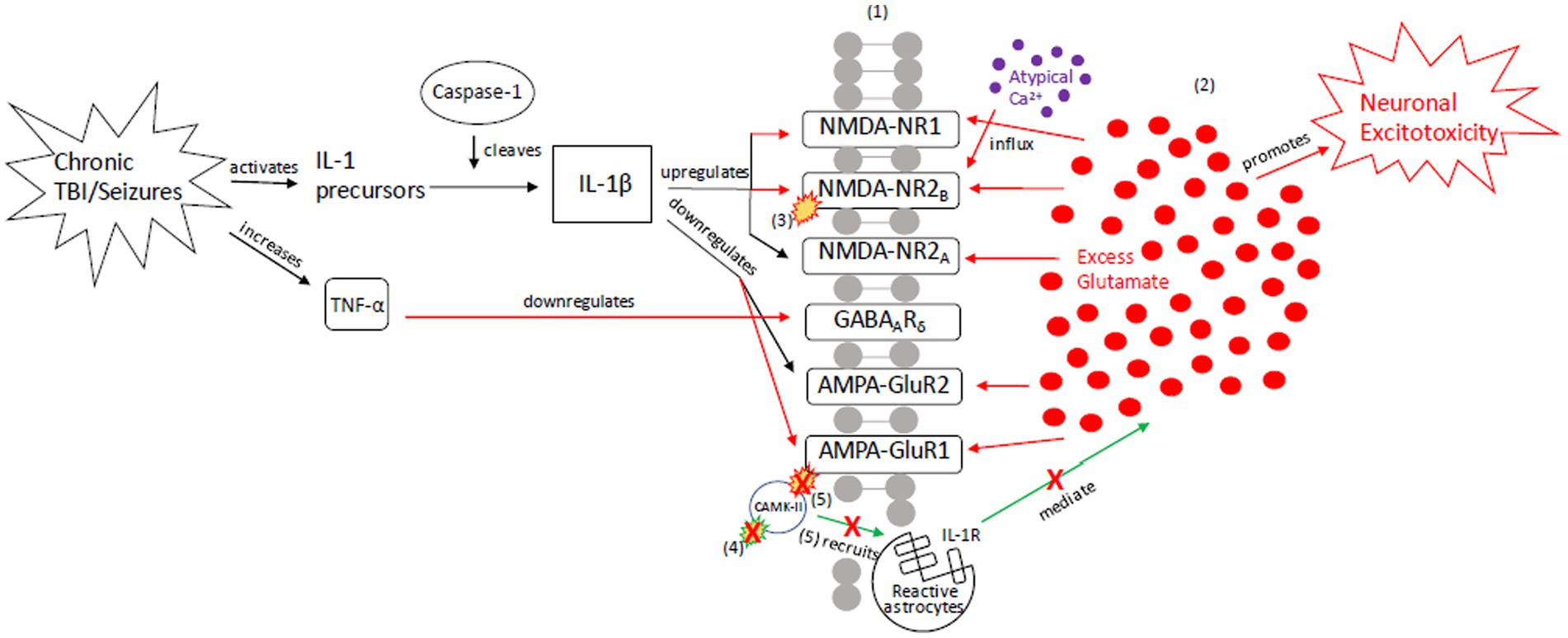
Figure 3. Our proposed recurrent injury response (RIR) mechanism outlining reactive signaling to chronic and/or moderate–severe TBI and chronic and/or prolonged seizures. This mechanism shunts cellular signaling toward pro-death response pathways of apoptosis and necrosis due to imbalanced glutamate release and reuptake, Ca2+-driven ER stress responses, and apoptotic-necrotic dynamics. In response to chronic injury, caspase-1 cleaves IL-1 precursors, resulting in IL-1β formation, and TNF-α downregulates GABAA receptors (18, 108–113). However, unlike the AIR mechanism, IL-1β increases NMDA receptor activity via GluNR2B phosphorylation (112). Increased NMDA receptor densities contribute to atypical Ca2+ influx and prolonged excitotoxic signaling. Concurrently, AMPA-GluR1 and-GluR2 receptors are down-regulated in response to chronic injury, resulting in dysregulated CaMK-II autophosphorylation and AMPA-GluR1 site phosphorylation (21–23, 49, 114–116). Due to disrupted CaMK-II phosphorylation and autophosphorylation, reactive astrocytes cannot be successfully recruited to the synapse to clear excess glutamate and proteasome recruitment into dendritic spines is impaired, respectively (117). The result is neuronal excitotoxic depolarization and propagation, neurotoxic release of ATP, and preferential apoptotic signaling (118). (1) = Neuronal membrane, (2) = Synaptic cleft, (3) = IL-1β-activated NMDA-NR2B phosphorylation, (4) CaMK-II autophosphorylation, (5) CaMK-II-AMPA-GluR1 phosphorylation. Red = Excitotoxic signaling, Green = Neuroprotective signaling. X = response reduction/down-regulation.
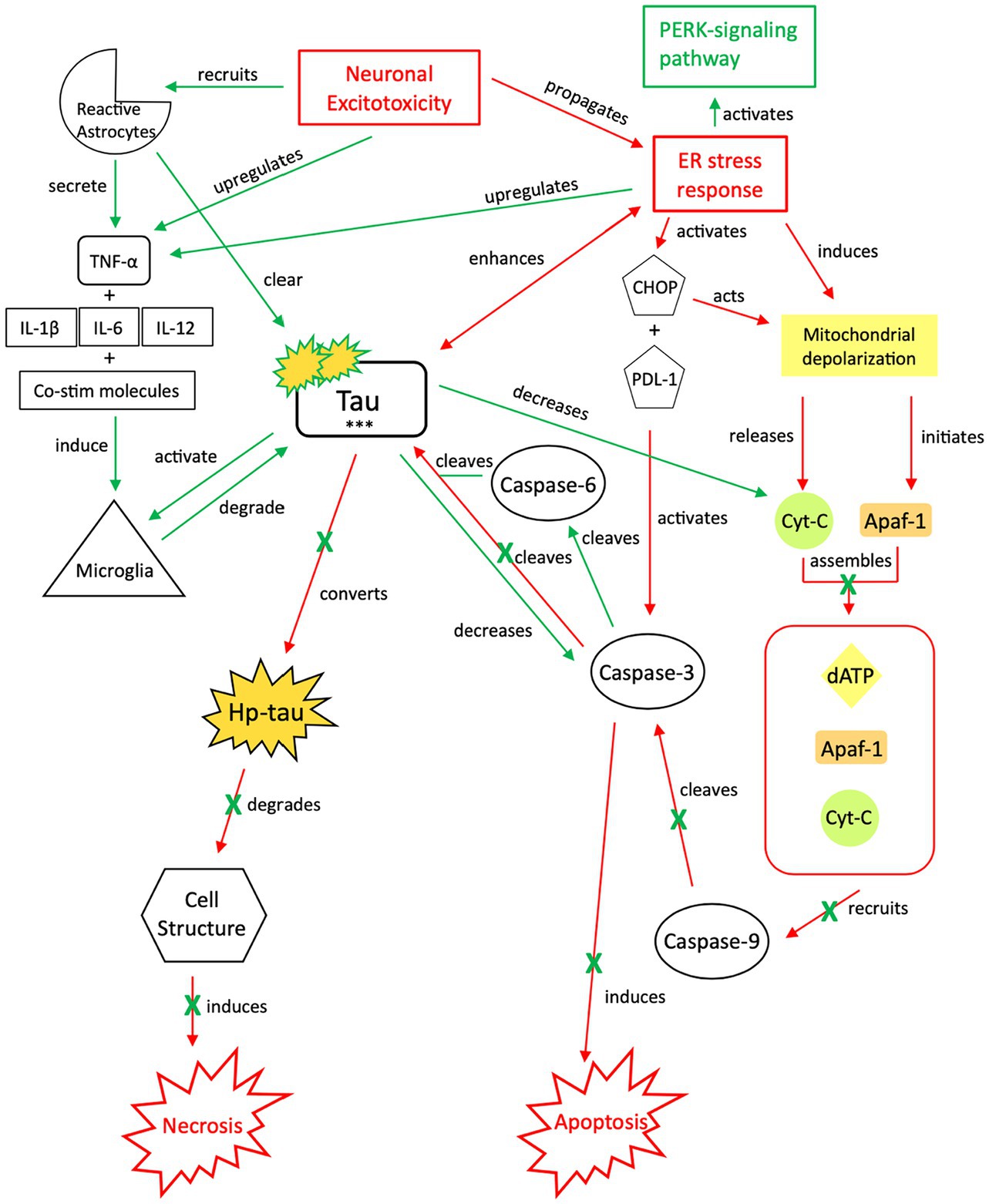
Figure 4. Our proposed neuroprotective response mechanism involving tau (NPT). Neuronal excitotoxicity imbalances the ER stress response, which activates two pathways: the PERK pathway, responsible for reverting the cell to homeostasis and preserving its integrity, and pro-cell death signaling cascades via CHOP and rapid mitochondrial depolarization, such as apoptosis. In typical apoptotic signaling, mitochondrial depolarization initiates Apaf-1 and releases cyt-c. Cyt-c, with Apaf-1 and dATP, assembles into an apoptosome complex (119–121). The apoptosome complex recruits caspase-9, caspase-9 cleaves caspase-3, and caspase-3 activates apoptosis (122, 123). Tau preserves cellular integrity and reverts cellular signaling away from pro-cell death signaling cascades. Although reduction of caspase-3 cleavage of tau reverts the cell away from apoptotic signaling, tau is cleaved by additional caspases such as caspase-6, resulting in tau phosphorylation (124–128). The increased presence of p-tau decreases the concentration of cyt-c and caspase-3, thereby further inhibiting apoptotic signaling (122, 129, 130). To avoid additional cell death pathways (i.e., necrosis), increases in cytokine expression, TNF-α, and tau concentrations recruit reactive astrocytes and microglia to break down excess tau into non-toxic components (131–136). Successful breakdown of accumulated tau by microglia and reactive astrocytes downregulates pro-death signaling pathways and restores cellular homeostasis. PERK, protein kinase R-like ER kinase; TNF, tumor necrosis factor; IL, interleukin; Co-stim, co-stimulatory (molecules); cyt-c, cytochrome-c; Apaf-1, apoptotic peptidase activating factor-1; dATP, deoxyadenosine triphosphate; NFTs, neurofibrillary tangles; Red, Pro-death signaling; Green, Neuroprotective signaling; o-tau, tau oligomers; t-tau, total tau; p-tau, phosphorylated tau; ***, O-tau, t-tau, p-tau; X, response reduction/down-regulation.
3.1 Acute injury response (AIR)
The AIR pathway is a pro-inflammatory mechanism that minimizes the likelihood of acute excitotoxic effects and cell death. In the AIR pathway, an acute TBI or brief seizure leads to IL-1β formation (108–110, 122), which has multiple effects on NMDA and AMPA receptors (Figure 2), including downregulation of NMDA receptors NR1 and NR2B. Unselective CAMK-II activation, coupled with the IL-1β signaling, promotes atypical calcium influx and excitotoxic glutamate release. As a result, there is an increased probability of cell death unless excess glutamate can be cleared from the synapse. CAMK, however, also recruits astrocytes into the affected region, evidenced by reactive astrocytes and phosphorylated CAMK-II in the hippocampal CA3 region of a kainic acid mouse model (106). The inflow of reactive astrocytes, coupled with increased IL-1 receptor density, clears excess synaptic glutamate (59, 60, 107).
3.2 Recurrent injury response (RIR)
The RIR pathway results in excitotoxity and apoptosis (Figure 3). Recurrent TBI activates -IL-1 precursors, which are cleaved into IL-1β by proteases such as caspase-1 (108–110). Similarly, recurrent seizures, through excitotoxic neuronal depolarization, activate caspase-1 and lead to IL-1β signaling (108–111). IL-1β, however, does not down-regulate NMDA receptors as in AIR. Instead, IL-1β hyper-activates NMDA receptors via GluNR2B subunit phosphorylation in response to chronic injury (112). The resultant increase in NMDA receptor density contributes to atypical calcium influx, prolongs excitatory synaptic enhancement, and propagates pathologic signaling from excess glutamate.
Further, there is decreased GABAA receptor density (50) and downregulation of the GABAA receptor δ-subunit (18, 113), contributing to extracellular glutamate accumulation and excitotoxicity (18, 113). AMPA-GluR1 and GluR2 receptors are also down-regulated in response to injury (21–23, 49, 114–116). As a result of AMPA dysregulation, CAMK-II autophosphorylation is impaired and recruitment of proteasomes – highly active enzyme complexes that play a role in cell-cycle progression – into dendritic spines is blocked, resulting in apoptosis (117). Additionally, subsequent phosphorylation at AMPA receptors also indirectly decreases astrocytic recruitment and clearance of excess glutamate (118).
If the AIR pathway (Figure 2) is unsuccessful in mediating excitotoxicity or if the RIR pathway is activated in chronic injury/seizures (Figure 3), apoptosis (acute programmed cell death) and necrosis (passive cellular degradation and death) result (142). Oxygen free radical production, caspase activation (e.g., caspase-3 and caspase-6), mitochondrial membrane depolarization, and further neurotoxicity occur (143–145). To minimize the possibility of cell death and preserve structural and functional integrity of surrounding neurons, an additional neuro-protective response is needed. We posit that tau signaling pathways first respond to recurrent seizures/injury in attempt to preserve cellular integrity; however, there is a “tipping point” that transitions the mechanism from neuroprotective to injurious – the repeated or sustained induction of an imbalanced ER stress response (specifically, the unfolded protein response [UPR]) and resultant aberrant tau phosphorylation. The ER stress response stimulates tau phosphorylation and continued tau cleavage; further phosphorylation/hyper-phosphorylation of tau promotes a continued UPR response and promotes neurodegeneration. This chronic dysregulation results in a shift from a tau-induced signaling pathway as a compensatory, neuroprotective response – which once reduced cellular dysfunction and restored apoptotic-necrotic dynamics and cellular homeostasis – to an injurious mechanism that is unable to maintain intracellular homeostasis, nor revert to mechanisms of programmed cell death.
3.3 Neuroprotective response (NPT): the expression and consumption of tau
In apoptosis, caspase-3 is activated by multiple mechanisms, including inflammatory responses, mitochondrial-based pathways, and an imbalanced ER stress response (119–123) (Figure 4). To divert the cell away from this apoptotic pathway and attempt to restore cellular homeostasis while maintaining structural integrity, caspases and ATP processes that induce apoptosis must be downregulated, TNF-α expression must be promoted, and tau phosphorylation must be induced, in conjunction with ER stress-induced PERK-pathway activation. Decreasing available caspases and apoptotic signaling reduces the likelihood of further neurotoxic depolarization and cell death, while increasing the likelihood that cellular homeostasis is restored (146). Induction of tau phosphorylation via caspase-6 cleavage indirectly reduces apoptotic signaling while preserving cellular integrity; tau also indirectly activates microglia, which are responsible for tau degradation to its non-toxic components.
Both caspase-3 and caspase-6 cleave tau (124–126) at multiple sites, which increases the susceptibility of tau to phosphorylation (9, 126–128). However, increased tau phosphorylation will also decrease caspase-3 activation in a negative feedback loop (122, 129, 130, 147). We posit that although the imbalanced ER stress response induces atypical tau phosphorylation (75), its acute effect is minimal due to this reduction in caspase-3 activation. As caspase-3 activation is required by apoptosis (119–121, 148), we posit that there is a transition from apoptosis to cellular preservation. However, with a halt of apoptotic signaling in the setting of increased tau concentrations, microglial and reactive astrocyte activation via upregulation of TNF-α, pro-inflammatory cytokines (IL-1β, IL-6, IL-12) and enzymes, and co-stimulatory molecules (131, 132) is required to break down tau. Additionally, tau oligomers (o-tau) and aggregates activate microglia to phagocytize tau and process its isoforms into non-toxic components (133–136). The ER stress response also upregulates Ca2+-ATPases in microglia, enhancing their capacity for phagocytosis and tau breakdown (149). Tau clearance is crucial to reestablishing cellular homeostasis and re-balancing the ER stress response post-seizure/injury.
3.4 Neuro-injurious tau response (NIT): transitioning from neuroprotection to injury
We posit that in an acute, mild TBI or brief seizure, tau will assist the cell in reverting to balanced ER stress response signaling and intracellular homeostasis. However, chronic or sustained activation of tau signaling cascades due to severe and/or recurrent injury will eventually transition this mechanism from neuroprotective to injurious (Figure 5). While tau expression benefits microtubule dynamics, overexpression of phosphorylated, cleaved isoforms disrupts microtubule transport and increases the risk of toxic tau aggregates (150). The overexpression of tau, atypical accumulation of p-tau and hp-tau from caspase-3 cleavage and apoptosis inhibition, and tau deposition due to the inability of microglia to successfully break down toxic tau aggregates, could be a result of the cell’s failed attempt to maintain homeostatic microtubule dynamics.
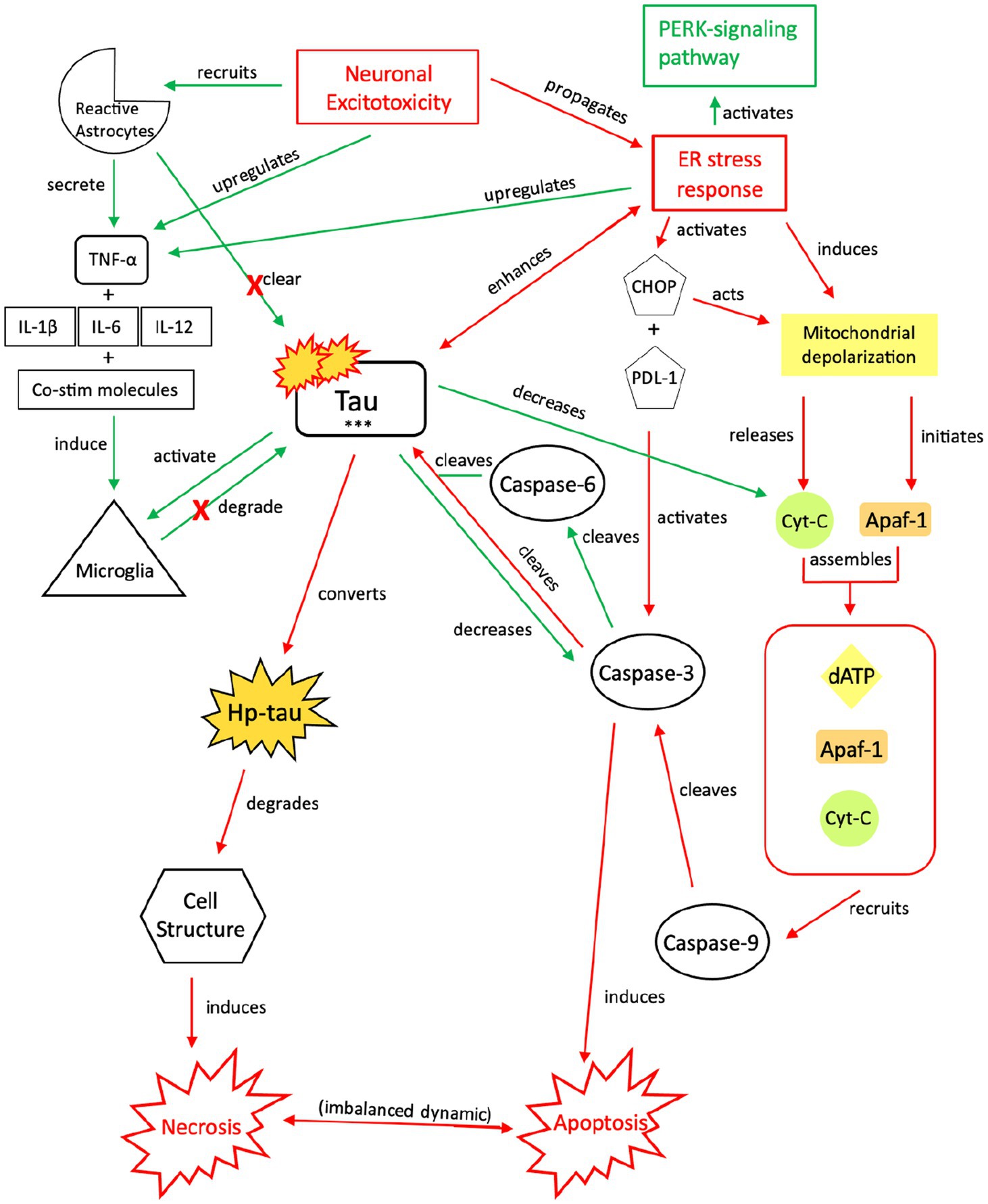
Figure 5. Our proposed injurious response mechanism involving tau (NIT) outlining injurious tau signaling and the resulting imbalance in apoptotic-necrotic signaling due to a chronic or sustained injury response from TBI or seizures. Similar to the NPT response, neuronal excitotoxicity imbalances the ER stress response, which activates two pathways: the PERK pathway and pro-cell death signaling pathway. Increased presence of p-tau decreases the concentration of cyt-c and caspase-3, inhibiting apoptotic signaling; although downregulated, caspase-3 still cleaves tau and contributes to tau’s toxic effects, which further reverts the cell away from apoptotic signaling toward necrosis (119–121). To compensate for this shift, increased cytokine expression, increased TNF-α, and increased tau concentrations recruit reactive astrocytes and microglia to break down excess tau into non-toxic components (107, 131, 132). However, unsuccessful breakdown of tau by microglia and reactive astrocytes results in a build-up of toxic tau aggregates that are secreted extracellularly (137, 138). Adjacent cells attempt to break down the toxic tau into non-toxic components (99), but chronic activation of the NIT pathway due to recurrent or sustained injury dysregulates this response, resulting in an injurious build-up of toxic levels of tau, hp-tau, and NFTs, which reinforce necrotic signaling (139–141). PERK, protein kinase R-like ER kinase; TNF, tumor necrosis factor; IL, interleukin; Co-stim, co-stimulatory (molecules); cyt-c, cytochrome-c; Apaf-1, apoptotic peptidase activating factor-1; dATP, deoxyadenosine triphosphate; NFTs, neurofibrillary tangles; Red, Pro-death signaling; Green, Neuroprotective signaling; o-tau, tau oligomers; t-tau, total tau; p-tau, phosphorylated tau; ***, O-tau, t-tau, p-tau; X, response reduction/down-regulation.
The NPT process depends upon the ability of the cell to revert to balanced ER stress responses, balanced apoptotic-necrotic dynamics, and intracellular homeostasis. Successful reactive astrocytic phagocytosis of tau and microglial clearance of tau play key roles in restoring intracellular dynamics. We posit that in the setting of sustained or recurrent injury, however, the ability of reactive astrocytes and microglia to break down tau becomes dysregulated. A resultant buildup of intra-microglial toxic tau occurs (99), which inhibits microglial and reactive astrocytic phagocytosis, threatens neuronal integrity, and drives expulsion of toxic tau aggregates from the cell via exosomal packaging and secretion. However, these secreted toxic tau aggregates are misfolded (151) and therefore more resistant to microglial break down. These exosomal tau aggregates have injurious effects (99) due to increased likelihood of exosomal leakage and surrounding neuronal uptake (152, 153). Further, the recurrent or sustained activation of the ER stress response reinforces microglial migration and dysregulation and limits the ability of microglia to actively break down tau. The inter-neuronal spread of toxic tau may mark the initial transition from a neuroprotective to a more widespread injurious process.
The NPT response may be an attempt to preserve cellular integrity, by avoiding further injury from apoptosis through tau phosphorylation and limiting effects of necrosis through astrocytic and microglial involvement. Over time, however, the NPT mechanism will still result in cell death if the underlying chronic pathology remains untreated. Further, with recurrent injury (e.g., repetitive seizures, repeated head trauma), the NPT response will become overwhelmed, and an aberrant, injurious process will ensue. Over time, repeated activation of injurious pathways will require a “last ditch effort” to revert the cell to pro-apoptotic signaling cascades and avoid further transition to a widespread neurodegenerative process, which leaves the question – what is the role of Aβ?
4 The role of amyloid-β in the transition from neuroprotection to tauopathy
With chronic pathology, Aβ concentrations are also increased by caspase-3-mediated APP cleavage and an imbalanced ER stress response (88, 154, 155). We posit that, in response to recurrent or severe injury, sustained Aβ signaling is a “last ditch effort” by the cell to restore cell death signaling and reduce the injurious effects of an imbalanced ER stress response and atypical tau (Figures 6, 7). Although Aβ induction increases plaque formation, it also has neuroprotective effects, recruiting additional reactive astrocytes and microglia for toxic aggregate breakdown (157, 167, 168, 170). However, if the cell cannot degrade toxic tau and Aβ aggregates and restore cell death signaling, Aβ’s relationship with tau further transitions the NPT response to a neurodegenerative process because it prevents tau from appropriately binding to microtubules and induces atypical tau phosphorylation (154, 156, 171, 172) (Figures 8, 9).
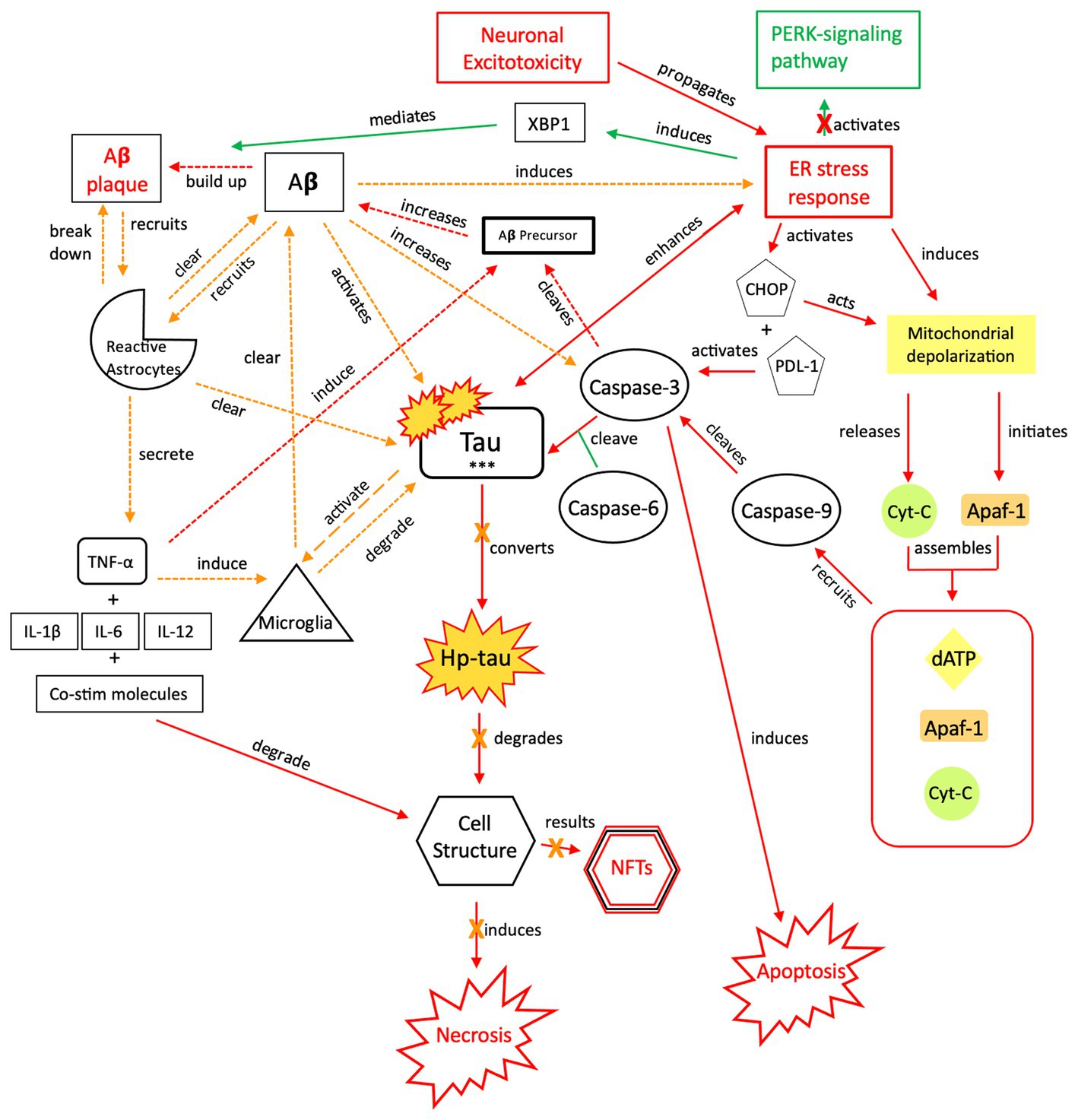
Figure 6. The neuroprotective response of Aβ, aka the “last ditch effort” to revert the cell to programmed death signaling and rebalance the apoptotic-necrotic signaling dynamic. In response to a recurrent or sustained ER stress response, imbalanced apoptotic-necrotic signaling dynamic, and atypical tau phosphorylation, Aβ activation both induces the ER stress response and increases caspase-3 cleavage of Aβ precursor protein (155). However, Aβ also recruits microglia and reactive astrocytes in response to excitotoxic signaling and increased tau concentrations (156). Breakdown of toxic tau aggregates and Aβ by microglia and reactive astrocytes mitigates the effect of Aβ-associated tau seeding and propagation (133, 157). As increased microglial trafficking is indirectly induced by the presence of Aβ, this mechanism also has detrimental effects due to shared apolipoprotein E (APOE), amyloidosis, and microglial transcript pathways and sustained neuroinflammation (158). Due to microglial inflammation and activation, reactive astrocytes are upregulated and recruited in attempts to clear toxic tau and Aβ and further orient the cell toward apoptotic signaling (159–162). Ultimately, a reduction in both inflammatory signaling and tau phosphorylation are needed once apoptotic-necrotic signaling dynamics have been reestablished, to prevent transition to an irreversible, degenerative pathway. PERK, protein kinase R-like ER kinase; Aβ, amyloid beta; XBP1, X-box binding protein 1; TNF, tumor necrosis factor; IL, interleukin; Co-stim, co-stimulatory (molecules); Cyt-c, cytochrome-c; Apaf-1, apoptotic peptidase activating factor-1; dATP, deoxyadenosine triphosphate; NFTs, neurofibrillary tangles; Red = Pro-death signaling, Green = Neuroprotective signaling, Orange = Aβ-involved signaling; o-tau, tau oligomers; t-tau, total tau; p-tau, phosphorylated tau; *** = O-tau, t-tau, p-tau. X = reduction/down-regulation. Solid line = signaling cascade induced/propagated by the ER stress response and tau; dashed line = signaling cascades resulting from Aβ involvement.
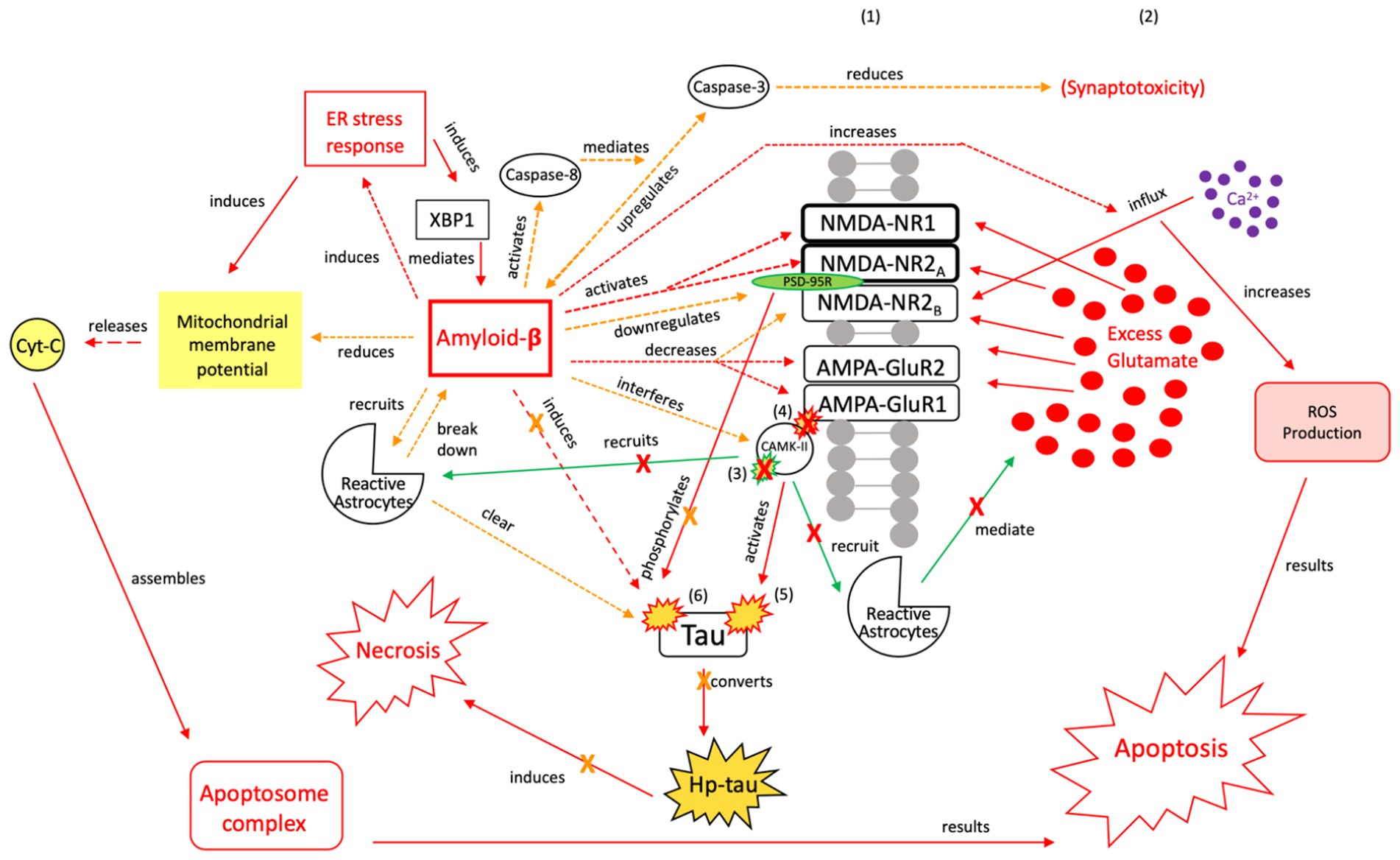
Figure 7. The neuroprotective response of Aβ, aka the “last ditch effort” to revert the cell to pro-apoptotic signaling and reduce tau and Aβ toxicity. Selective NMDA regulation, downregulating scaffolding protein PSD-95, and activating caspase-8 reduce the excitotoxic effects of Aβ and atypical tau phosphorylation (154). PSD-95 receptor downregulation results in reduced tau phosphorylation and protection of synapses from the effects of Aβ, while caspase-8 activation indirectly reduces synaptotoxicity by mediating the relationship between Aβ and caspase-3 (163, 164). Aβ also reduces mitochrondrial membrane potential and directly induces the ER stress response, resulting in apoptosome complex formation and eventual ROS-induced apoptosis (165). Reactive astrocytes are recruited to break down Aβ and clear tau aggregates. However, Aβ also has injurious effects, as it increases intracellular Ca2+ and ROS production, while also acting directly on tau (154). Therefore, this mechanism is considered a “last ditch effort” to acutely kill the cell via apoptotic signaling to minimize the negative effects from toxic tau, Aβ accumulation, and necrotic signaling. We posit that limiting the effects of tau and Aβ toxicity is predicated on the acute nature of this response and treatment of the underlying pathology to avoid irreversible injury and/or a transition to a more widespread neurodegenerative process. XBP1, X-box binding protein 1; Cyt-c, cytochrome-c; ROS, reactive oxygen species; Red = Pro-death signaling, Green = Neuroprotective signaling, Orange = Aβ-involved signaling; X = reduction/down-regulation. Solid line = signaling cascade induced/propagated by the ER stress response and tau; dashed line = signaling cascades resulting from Aβ involvement. (1) = Neuronal membrane, (2) = Synaptic cleft, (3) = CaMK-II-autophosphorylation, (4) = CaMK-II-Glutamate receptor phosphorylation, (5) = CaMK-II-tau-phosphorylation, (6) = PSD95-NMDA receptor complex-tau phosphorylation.
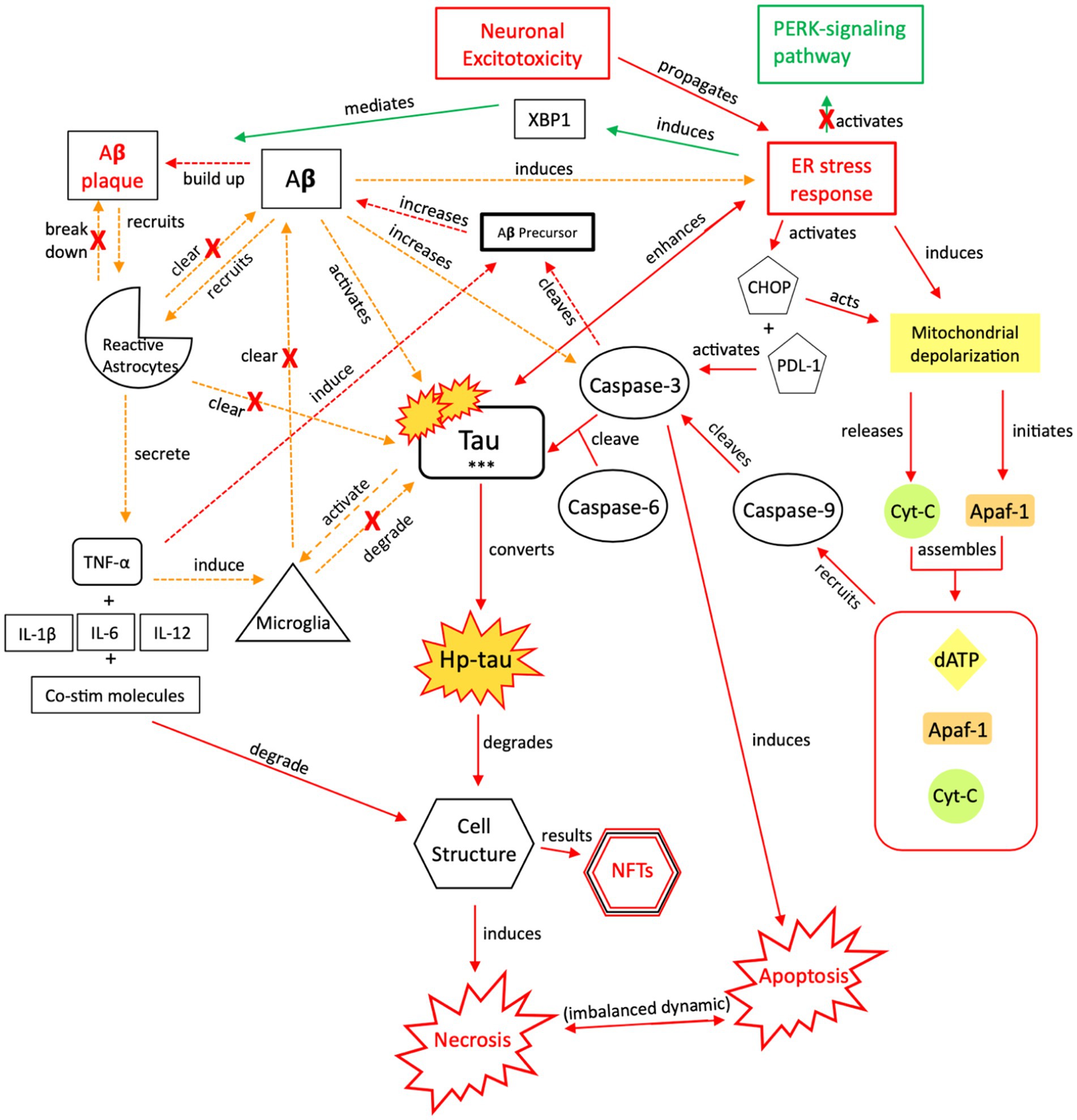
Figure 8. The injurious response of Aβ, aka its failed “last ditch effort” to downregulate propagation of toxic tau and rebalance apoptotic-necrotic signaling dynamics. Due to accumulated toxic tau aggregates and dysregulated tau clearance by reactive astrocytes and microglia, pro-death signaling mechanisms become favored over cellular preservation signaling. However, a recurrent, reactive ER stress response leads to an imbalance of apoptotic-necrotic signaling and enhances atypical tau phosphorylation. Further, Aβ precursor protein is cleaved by caspase-3, and Aβ concentrations increase, further propagating the ER stress response (88, 154, 155). Due to the Aβ precursor overexpression and increased Aβ production, defective mitochondria are produced, mitochondrial dynamics are altered, and their trafficking is reduced, leading to further intracellular Ca2+ influx and apoptotic-necrotic imbalance (166). However, the ER stress response also has neuroprotective effects, inducing selective transcription factor XBP1, which mediates Aβ plaque formation (88). Simultaneously, Aβ directly recruits reactive astrocytes and indirectly recruits microglia, through TNF-α and pro-inflammatory signaling, which cluster around Aβ plaques to clear them (157, 167, 168). Yet, the induction of pro-inflammatory signaling from astrocytic recruitment further induces Aβ precursor protein; increased Aβ concentrations result in increased atypical tau phosphorylation/hyper-phosphorylation and further ER stress response induction (169). Thus, reactive astrocytes have both neuroprotective and injurious effects (170). Continued apoptotic-necrotic signaling imbalance, degradation in cell structure, and NFT formation results from atypical activation of these pathways. PERK, protein kinase R-like ER kinase; Aβ, amyloid beta; XBP1, X-box binding protein 1; TNF, tumor necrosis factor; IL, interleukin; Co-stim, co-stimulatory (molecules); Cyt-c, cytochrome-c; Apaf-1, apoptotic peptidase activating factor-1; dATP, deoxyadenosine triphosphate; NFTs, neurofibrillary tangles; Red = Pro-death signaling, Green = Neuroprotective signaling, Orange = Aβ-involved signaling; o-tau, tau oligomers; t-tau, total tau; p-tau, phosphorylated tau; *** = O-tau, t-tau, p-tau. X = reduction/down-regulation. Solid line = signaling cascade induced/propagated by the ER stress response and tau; dashed line = signaling cascades resulting from Aβ involvement.
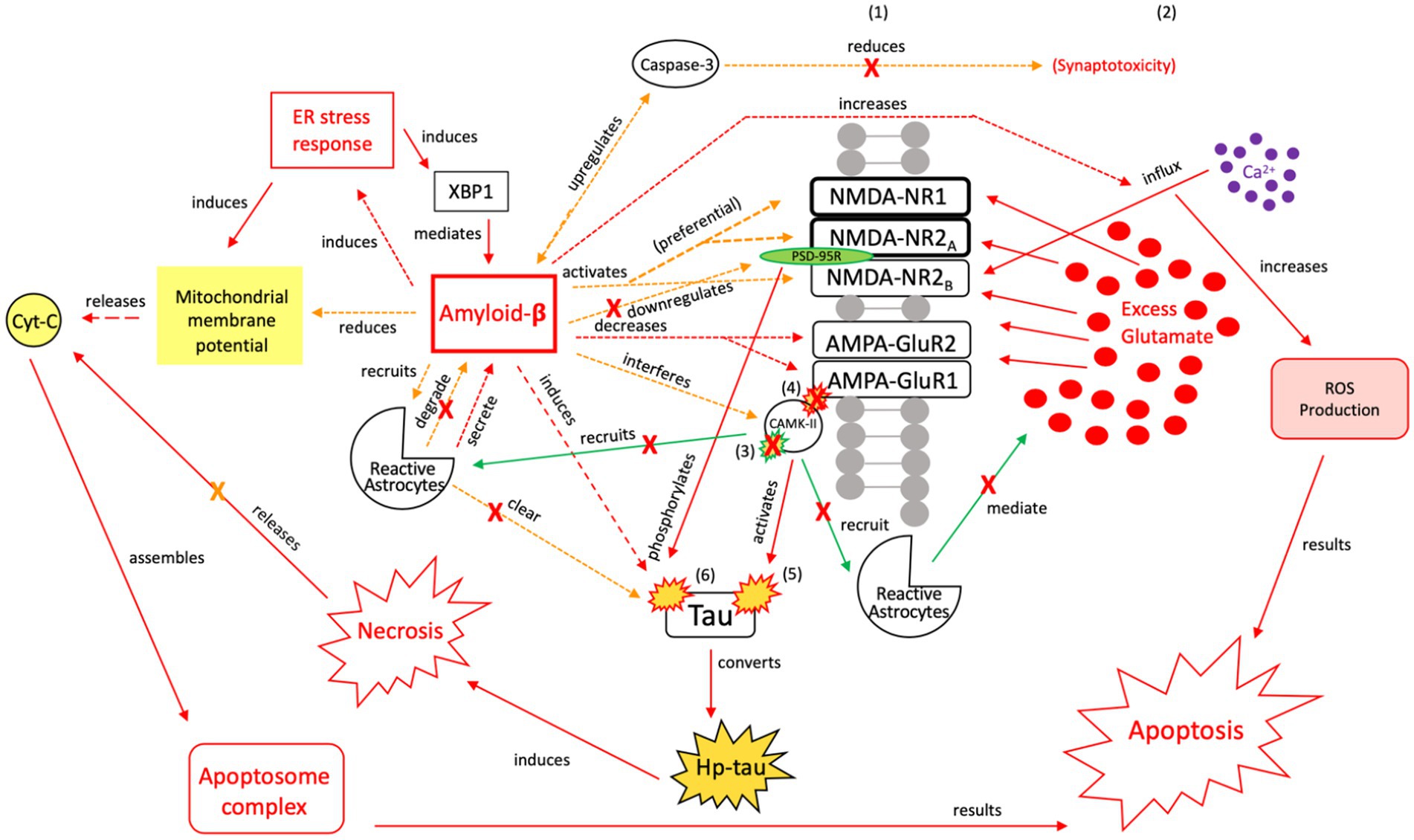
Figure 9. The injurious response of Aβ, aka the failed “last ditch effort” to revert the cell to pro-apoptotic signaling and rebalance apoptosis-necrosis, due to recurrent or sustained Aβ signaling. Unlike neuroprotective Aβ responses, preferential activation of NMDA-R1 and-2A/B receptor subunits by Aβ (171, 172), and their increased surface expression regulated by PSD-95, adversely affects channel assembly and conductance (173), promoting further neuroexcitotoxicity, atypical tau phosphorylation, and increased susceptibility to Aβ (164). Unsuccessful toxic tau aggregate and Aβ breakdown by microglia [seen in (A)] propagates the injurious effects of Aβ-associated tau seeding and propagation (133). In the presence of dysregulated tau and Aβ mechanisms, as well as dysregulated microglial and reactive astrocytic clearance, the failure to reduce neuroinflammation and excitotoxic propagation results in a transition from neuroprotection to neurodegeneration. We posit that this point marks the transition from a injurious mechanism to a more widespread neurodegenerative process. XBP1, X-box binding protein 1; Cyt-c, cytochrome-c; ROS, reactive oxygen species; Red = Pro-death signaling, Green = Neuroprotective signaling, Orange = Aβ-involved signaling; X = reduction/down-regulation. Solid line = signaling cascade induced/propagated by the ER stress response and tau; dashed line = signaling cascades resulting from Aβ involvement. (1) = Neuronal membrane, (2) = Synaptic cleft, (3) = CaMK-II-autophosphorylation, (4) = CaMK-II-Glutamate receptor phosphorylation, (5) = CaMK-II-tau-phosphorylation, (6) = PSD95-NMDA receptor complex-tau phosphorylation.
In both typical functioning and in response to acute neuronal injury, we postulate that tau and Aβ signaling processes occur in parallel. In acute neuronal injury, however, we propose greater initial reliance on tau signaling in comparison to Aβ signaling, in avoidance of necrotic processes and reorientation toward cellular preservation and stabilization. With recurrent or severe neuronal injury, we postulate that the “last ditch effort” of Aβ indicates a “cellular switch” to greater reliance on Aβ signaling, for the purpose of activating apoptotic signaling and limiting neurotoxic spread. If the underlying injurious pathology is not reduced/halted, the result is a transition of the “at risk” neuroprotective response to one of neurodegeneration.
The ER stress response can induce apoptotic signaling cascades (169) in addition to promoting Aβ formation. Aβ formation comes with several costs, in that Aβ will activate pro-inflammatory responses and caspase-3 activity, in attempts to revert the cell to pro-apoptotic signaling; however, increased caspase-3-selective tau cleavage by Aβ and dysregulated mitochondrial production and recruitment results in further tau-related toxicity and an imbalanced intracellular dynamic (156, 157). While caspase-3 typically promotes tau cleavage and phosphorylation during apoptotic signaling, Aβ increases aberrant caspase-3 activity during necrosis (Figures 6, 7) (155). Hence, Aβ initiates atypical tau cleavage. It renders tau increasingly susceptible to hyperphosphorylation and toxic aggregates, because it atypically alters tau at specific phosphorylation sites (154), ultimately leading to microglial injury and neurotoxicity (Figure 8) (156). In AD, soluble Aβ induces tau hyperphosphorylation in hippocampal neurons, disrupting microtubule stability. De-phosphorylation of Aβ-induced p-tau results in the restoration of tau microtubule binding capacity (154), suggesting that the process is at least partially reversible, and suggests some initial benefit of Aβ formation.
Extracellular insoluble Aβ aggregates, however, are associated with neurotoxicity and degeneration (155). Both soluble and insoluble Aβ1-40 and Aβ1-42 levels are elevated in patients with AD compared to typical aging brains (174). The soluble forms comprise the greatest proportion of total Aβ in typical aging brains but the lowest in AD brains (174). Acute cell death is highly dependent upon the relationship between soluble Aβ and soluble cytoplasmic tau, which can propagate extracellularly (175). The relationship between Aβ and tau suggests that each can act on the other in a negative feedback loop, triggering the transition from non-toxic to toxic aggregates (175). Therefore, it is possible that soluble Aβ reflects typical brain functioning, but with neuronal injury, neurons are “at-risk” for soluble toxic tau formation and toxic tau/Aβ aggregate propagation extracellularly, resulting in an eventual transition to an insoluble state. This, in turn, reduces the proportion of soluble to insoluble Aβ and soluble phosphorylated to abnormally phosphorylated tau, further transitioning the mechanism to one of eventual degeneration (176).
Toxic Aβ accumulation results from several mechanisms, with prominent roles of microglia and astrocytes. Similar to tau, Aβ clearance requires microglial and reactive astrocytic degradation (Figure 6) (157). Aβ plaques can result from microglial dysregulation and increased Aβ-induced caspase-3 activity, as caspase-3 cleaves APP-β (177). Aβ also activates reactive astrocytes, which cluster around Aβ plaques (Figure 6) (167, 168). The astrocytes secrete interleukins and TNF-α, promoting further inflammation to break down Aβ (167, 168), however, these pro-inflammatory proteins also induce APP-β (178, 179), resulting in increased Aβ concentrations. Further neurodegeneration can also occur due to astrocytic secretion of Aβ (Figure 9) (180). Aβ deposits were found in the hippocampus with progression to the cortex prior to the formation of NFTs in a transgenic AD model, supporting neurodegenerative signaling cascades outlined in Figure 5 (181). Aβ deposits were also found in ~30% of severe TBI cases postmortem (182, 183). This, coupled with Aβ-promoted tau cleavage (9), indicates a relationship between amyloid-β, tau, and NFTs.
In vitro and in vivo, microglia clear soluble extracellular Aβ via micropinocytosis, in which successful uptake and degradation depends on actin and tubulin dynamics (184). Inflammatory processes promote signaling cascades and the recruitment of microglia to initiate soluble Aβ uptake and degradation (157). In an acute injury model, this process is postulated to be neuroprotective. However, with recurrent or sustained injury, this process may be dysregulated due to unstable actin/tubulin dynamics and imbalanced ATP involvement, leading to further neural injury. The transition from soluble to insoluble Aβ has yet to be fully understood. However, data suggest that a progressive Aβ transition from soluble to insoluble takes place in the ER/intermediate compartment pathway, and that the degree of insolubility correlates with overexpressed APP-β concentration (185). The uptake and degradation of insoluble Aβ, comprised of neurotoxic, soluble Aβ oligomers, occur through different endocytic mechanisms that are microglia and astrocytic receptor mediated (186–188). Further, simultaneous intra-astrocytic accumulation of soluble and neurotoxic Aβ for degradation promotes vesicle-induced neuronal apoptosis (189). Resulting from cell death, cellular contents, including neurotoxic Aβ, are released into cytoplasm and quickly re-phagocytosed by surrounding neurons. In acute injury, this process would be neuroprotective for the prevention of necrosis; however, with recurrent injury, it is a mechanism for further neurotoxic propagation and eventual systemic degradation.
Aβ activity disrupts cellular integrity, but we posit that Aβ attempts to minimize neurodegenerative damage by targeting NMDA/AMPA receptors and mitochondrial membrane potential (MMP) as part of a “last ditch effort” to reactivate apoptotic signaling (Figures 6, 7). Aβ recruits reactive astrocytes to compensate for microglial dysregulation and clear toxic Aβ (Figure 6). However, because shared biochemical mechanisms associated with neuronal homeostasis and cell death are dysregulated, and neuroprotective mechanisms such as reactive astrocytic phagocytosis of Aβ are functioning abnormally (180), these pathways promote further excitotoxic signaling and neurodegeneration (Figures 8, 9). If Aβ is not properly cleared, it can cause further atypical tau hyperphosphorylation, microtubule destabilization, and assembly of tau into filament structures seen in AD (190).
Aβ oligomers preferentially activate NMDA NR1/NR2A receptor subunits, which initiate LTP and regulate NMDA NR2B-mediated calcium influx (191). Aβ oligomers can induce a rapid increase in intracellular Ca2+ via NR2B influx and cause mitochondrial damage leading to hippocampal cell death (191). Aβ peptides interfere with CaMK-II activity and decrease AMPA receptor trafficking, leading to atypical synaptic distribution and LTP/LTD disruption (171, 172). The Aβ and NMDA relationships may explain a sustained excitotoxic response seen post-TBI/post-seizure. Due to sustained NR1/NR2A responses to high frequency stimulation, disrupted NR2B-mediated calcium influx, and diminished AMPA receptor activity (171, 172), downstream effects of RIR continue, along with a failure to clear excess synaptic glutamate (Figure 3). AMPA receptors are crucial for synaptic plasticity, learning, and memory (192, 193). Loss of AMPA receptors results in diminished synaptic transmission, long-term depression, and difficulties with learning and memory (193). In both brain tissue from AD patients and Aβ-treated neurons, there are significant decreases in AMPA receptor densities, with higher receptor turnover (194). In the presence of Aβ, decreased AMPA receptor expression and greater receptor turnover may be early indicators of atypical mechanistic changes associated with AD and resultant cognitive decline. If Aβ is acutely activated, we posit that the cell reorients to apoptotic signaling, minimizing injurious effects of Aβ; however, chronic Aβ activation further imbalances apoptotic-necrotic signaling and initiates a transition of this “last ditch effort” from injurious to neurodegenerative.
Several mechanisms act in concert to increase phosphorylation of tau in the setting of repeated injuries. (1) Aβ induces caspase-3 activation (195) (Figure 5). (2) Aβ-42 reduces MMP in cortical neurons (122, 146, 196), thereby increasing ATP production and cyt-c release. Cyt-c mediates caspase-3 activation that leads to tau cleavage and phosphorylation (197). (3) Endogenous tau interacts with the PSD95-NMDA receptor complex, which selectively phosphorylates tau (198). To efficiently kill the cell via apoptosis, Aβ must activate alternative apoptotic pathways while reducing the tau response. (1) Caspase-8 recruitment by Aβ mediates the relationship between Aβ and caspase-3, resulting in decreased synaptic excitotoxicity and a reorientation toward apoptotic signaling (163). (2) NMDA NR1/NR2A receptor activity affects downstream ROS production resulting in apoptosis (163, 198). (3) Aβ downregulates the PSD95-NMDA receptor complex, decreasing tau phosphorylation. In cultured cells, Aβ-induced apoptosis increased reactive oxygen species (ROS) production but not hp-tau (165). While ROS-produced apoptosis has detrimental effects, as a “last ditch” neuroprotective effort of Aβ, it limits further hp-tau and NFT formation. Limiting the effects of tau and Aβ toxicity is predicated on the acute nature of this response and treatment of the underlying pathology to avoid long-term neurodegeneration.
4.1 Summary of NPT, NIT, and Aβ hypotheses
Tau phosphorylation antagonizes apoptotic processes in response to increased ER stress and imbalanced homeostatic dynamics (147). Tau hyperphosphorylation is a reactive response activated when faced with apoptotic cell death (120, 129). The build-up of hp-tau, therefore, could represent a failed neuroprotective mechanism. NFTs, a hallmark of tauopathies, form as a downstream result of the RIR and NPT pathways. In addition to containing hp-tau, NFTs contain active caspase-6, caspase-6-cleaved tau, and Aβ, further supporting that NFTs are the end result of a neuronal degradation pathway – one that initially includes a neuroprotective pathway preferred by the cell over acute apoptotic death (9), but over time, becomes overwhelmed by the accumulation of repeated injuries.
The NPT response suggests that excess tau is phosphorylated in attempts to preserve cellular integrity in the short-term. In response, microglia and reactive astrocytes are triggered to reinstate homeostasis and break down intracellular tau into non-toxic isoforms. However, in the setting of repeated injury, when excess tau phosphorylation exceeds microglial and astrocytic capacity for tau degradation, toxic tau accumulates. This, along with aberrant tau cleavage, aggregation, and hyper-phosphorylation, propagates a dysregulated microglial response. To combat this, the toxic tau must be expelled from the cell and is done so through exosomal packaging and secretion.
We posit that the response mechanism is neuroprotective to the point of halting apoptosis, phosphorylating tau, and clearing tau via microglia and reactive astrocytes, and that it would continue to be neuroprotective if repetitive seizures or head injuries did not (1) lead to Aβ accumulation and its production of toxic tau and (2) outpace the ability to clear tau. Because epilepsy and repeated TBIs are plagued with recurrent cellular injury and ER response activation, however, a buildup of cleaved, phosphorylated, and hyperphosphorylated tau results in toxic tau aggregates. These toxic aggregates are then propagated to surrounding neurons, adversely affecting these neighboring neurons and increasing the likelihood for localized neuronal degeneration. The extracellular leakage of toxic tau also contributes to NFT formation and induces tauopathy-related necrosis, transitioning the mechanism over time from neuroprotective to neurodegenerative.
The role of Aβ is pivotal in the development of neurodegeneration. Aβ induces tau phosphorylation, contributing to toxic tau aggregates that cannot be cleared by microglia and reactive astrocytes. Additionally, microglia cannot clear the excess Aβ, leading to inflammatory signaling, excitotoxicity, and Aβ plaque accumulation. Microglial and reactive astrocytic dysregulation results in further tau and Aβ leakage that contributes to injury. We posit that, although increased Aβ concentration has a deleterious effect on cellular integrity and microglial functioning, increased Aβ also reactivates preferential apoptotic signaling by targeting NMDA/AMPA receptor functioning, CaMK-II phosphorylation, astrocytic recruitment, and mitochondrial membrane permeability (Figure 6). Because glutamate transmission, apoptosis, and necrotic signaling share related pathways, this Aβ compensatory mechanism cannot differentiate between typical and atypical activation, such that excitotoxicity continues. Recurrent or sustained activation of these mechanisms results in the necrotic cell death seen in tauopathies.
5 Clinical correlations: tau and TBI
Early studies lacked an association between TBI and cerebrospinal fluid (CSF) p-tau levels, likely because of insufficient sensitivity of the assay, requiring development of novel techniques (92, 199). An enhanced immunoassay using multi-arrayed fiber optics (EIMAF) detected acutely increased t-tau and p-tau levels in brain and blood following CCI in rodents and in CSF following severe TBI in humans. T-tau and p-tau levels remained significantly elevated during the chronic stage of CCI in rodents. While t-tau and p-tau levels decreased during the chronic stage of severe TBI in humans, elevated levels were still detected in subsequent months post-injury. T-tau levels approached normal limits approximately one-month post-injury, while p-tau levels remained elevated six months post-injury (200). EIMAF also demonstrated increased p-tau levels, t-tau levels, and p-tau/t-tau ratios in individuals with acute or chronic TBI compared to healthy controls (201). Using a single-molecule enzyme-linked immunosorbent assay (SIMOA), blood t-tau levels were greater in professional hockey players across multiple time points post-head injury (from one to 48 h) compared to preseason (pre-injury) (91). Recent studies have also measured tau within exosomes isolated from plasma (202, 203). This technique has been applied in remote repetitive TBI, with elevated exosomal t-tau and p-tau levels negatively correlating with neuropsychological measures (202, 203).
Tau levels correlate with clinical recovery, with a negative association between CSF tau and clinical improvement (204). Ventricular CSF t-tau concentrations in the setting of severe TBI negatively correlated with clinical improvement over one year (205). Plasma p-and t-tau levels measured in patients ~24-h post-acute head injury were associated with short-and long-term outcomes; p-tau and p-tau/t-tau ratios in blood negatively correlated with recovery in participants with chronic TBI (201). Human data concur with a rat model, in which serum and CSF tau levels positively correlated with traumatic spinal cord injury severity and negatively correlated with locomotor function (206). These results support p-tau as a biomarker that reflects a broad picture of axonal injury, TBI severity, cognitive functioning, and long-term outcomes.
6 Clinical correlations: tau and AD
Pathological p-tau aggregation is a biomarker of neurodegeneration in AD. In a transgenic mouse model of AD, microglial activation occurs in a progressive fashion, correlating with increased tau hyper-phosphorylation and Aβ plaque accumulation (207). Human and animal models of AD and other dementias identify atypical tau processes that contribute to increased hyper-phosphorylation, microglial activation, NFT formation, and neurodegeneration (208), including genetic mutations and post-translational modifications (209–214). Atypical tau phosphorylation and APP mutations correlate with NFT formation in animal models and human AD (215, 216). In human AD brain tissue, tau pathology was divided into early and late stages, with tau deposition first observed in entorhinal cortex and hippocampus. Later tau aggregates correlated with cognitive decline (217). In human lateral temporal cortex obtained from late-stage AD brains, increased markers of the ER stress response correlated with decreased post-synaptic PSD-95 markers and increased tau (218).
Increased CSF t-tau levels were also found in patients with AD (219). Elevated CSF tau levels demonstrated a strong association with AD and improved discrimination of AD from other dementias, while Aβ levels failed to improve diagnostic accuracy (220). CSF p-tau181, 217, and 231 concentrations accurately predicted cognitive impairment in patients with AD, but not in patients with other dementias or controls (221). P-tau231 was the earliest detector of increased Aβ in AD pathology, preceding Aβ identification by position emission tomography (PET) (221). Further, increased levels of tau and decreased levels of Aβ1-42 in CSF were reported (222–226), highlighting their contrasting CSF profiles as biomarkers for AD. In plasma, tau levels were significantly higher in patients with AD compared to MCI patients and controls, however, use of plasma tau as a diagnostic test is not yet validated (227).
7 Clinical correlations: tau and epilepsy
A link between AD and temporal lobe epilepsy (TLE) is demonstrated by a bidirectional increase in risk, hippocampal damage (228), and cognitive deficits in both disorders, in part due to shared cortical networks, tau deposition, and amyloid pathology. Current research explores the influence of seizure activity on tau levels in brain, CSF, and blood, proposing that epilepsy is a tauopathy like AD and CTE – with proposed mechanisms of tau deposition including production during ictal and interictal activity, axonal sprouting and formation of aberrant connections in response to injury, cell death, physical injury during seizures, and decreased clearance (94). Studying the relationship between tau and epilepsy may address how seizure activity results in neuronal injury.
Limited data are available regarding tau levels in people with epilepsy. Hp-tau deposits were identified in resected temporal lobe tissue from patients with hippocampal sclerosis, evident in nearly 94% of cases and correlating with post-operative declines in verbal memory and naming, though this finding was not seen in all resection studies (94). In late-onset epilepsy of unknown origin, CSF t-tau levels were increased in comparison to controls, with t-tau and p-tau levels predicting onset of dementia (229). Elevated CSF t-tau and p-tau levels were detected in patients with status epilepticus when tested at a median of 72 h from admission (95). In the setting of status, t-tau levels positively correlated with medication resistance, status duration, disability, and development of chronic epilepsy (95). While a transient increase of CSF t-tau was reported within four days of a single, new-onset generalized convulsion, tau elevations in isolated or repeated seizures that respond promptly to medications are controversial (96, 97). Increased CSF t-tau levels were seen with symptomatic convulsions (of acute or remote etiology), but not in subjects with seizures of idiopathic or cryptogenic cause when levels were obtained within 48 h (98). CSF t-tau levels were decreased, and p-tau unchanged, when CSF was collected at least seven days after the last seizure, but seizure frequency was unknown (230). Blood–brain barrier disruption during seizures may release tau to the periphery, suggested by small, transient elevations of serum T-tau following convulsions (231). Studies of peripheral p-tau and exosomal analyses have not yet been applied to people with epilepsy, and the impact of epilepsy-related factors (e.g., seizure type, epilepsy duration) on peripheral tau levels is unknown.
The relationship between epilepsy and p-tau levels should be explored as a potential marker of neural injury severity and predictor of cognitive function and seizure control. Given the above similarities in injury pathophysiology between AD, TBI, and epilepsy, AD and TBI may serve as guides to identifying overlapping markers of neuronal damage and cognition.
8 Treating AD, TBI, and epilepsy: pharmacological interventions
8.1 Cytokine targets
A better understanding of tau deposition lends insight into AD, TBI, and epilepsy pathophysiology and presents possible targets for intervention. Potential approaches include neuroprotection, inhibition of inflammatory processes, and disruption of excitotoxic mechanisms. Trials focused on various portions of these pathways. In animal models of TBI, minocycline and statins demonstrate beneficial anti-inflammatory and neuroprotective properties, limit the expression of pro-inflammatory cytokines, and render cell death-associated astrocytes and microglia inactive (232–234). In vitro and in vivo rat brain TBI and immune system studies identified human-cultured mesenchymal stem cells coupled with purified immune cells as a promising treatment that increases production of anti-inflammatory ILs, while decreasing TNF-α (235–237). IL-34 selectively enhances microglial neuroprotective effects, homeostasis, and neuronal survival by promoting Aβ oligomeric clearance and inducing microglial enzymatic activity. These effects reduce oxidative stress without promoting neurotoxicity (61). Promotion of IL-34 receptor binding or activity may benefit those with recurrent seizures/TBI by enhancing microglial function.
8.2 NMDA receptor antagonists
Data regarding NMDA antagonists are mixed. Drugs like amantadine, a weak NMDA antagonist, are commonly used in acute brain injury rehabilitation, although supporting data are limited (238). In some TBI studies, NMDA receptor antagonists lacked efficacy and raised safety concerns (239). A trial of the competitive NMDA antagonist D-CCP-ene for the treatment of intractable focal-onset seizures led to severe adverse events in all eight subjects, including sedation, ataxia, depression, amnesia, and poor concentration (240). Seizure frequency worsened in three subjects and remained unchanged in four subjects; one participant demonstrated improved seizure frequency, yet experienced status epilepticus upon D-CCP-ene withdrawal (240). All subjects withdrew from participation, leading to premature termination of the study. However, in a large, randomized, double-blind, placebo-controlled trial of traxoprodil, an NMDA NR2B subunit antagonist, was found to be well-tolerated in adults with severe TBI; they demonstrated improved Glasgow Coma Scale outcomes 6-months post-injury compared to placebo (241).
In an animal model of hippocampal seizures, MK-801 decreased seizure severity at low doses (242). In 68 patients with super-refractory status epilepticus, ketamine infusions administered for a length of one to four days reduced seizure burden by 50% (243). Upregulated NMDA receptor trafficking in the post-synaptic membrane contributes to super-refractory status epilepticus; NMDA receptor antagonists like MK-801 and ketamine may be effective due to improved penetration of the blood brain barrier and maintain their function even in the presence of increased concentrations of intra-and extra-cellular glutamate (244–246).
Memantine, a low-affinity voltage-dependent uncompetitive NMDA antagonist, approved for use in AD, reduced tau phosphorylation and improved functional outcomes after repetitive mild TBI in adult mice (247). In patients with TLE, memantine improved cognition compared to donepezil (248). In a double-blinded, placebo-controlled trial, once-daily memantine significantly improved episodic memory and quality of life in patients with epilepsy, although confounded by reduced seizure frequency (248, 249). In contrast, in subjects with focal-onset seizures of unchanged frequency, memantine yielded no significant improvement in cognition compared to placebo (250). However, in an open-label extension phase, there were improvements in verbal memory, memory-related quality of life, and executive functioning (250). Overall, NMDA antagonists deserve further study in TBI, AD, and epilepsy (238, 241).
8.3 AMPA receptor antagonists
Alternatively, perampanel is highly selective for AMPA receptors and inhibits AMPA-induced calcium influx in rat cortical neurons (251). Pharmacological dampening of AMPA receptor function eliminated interictal-like activity in human lateral amygdala in vivo, without reducing AMPA receptor densities observed in vitro (252). It is efficacious for treatment of focal-onset seizures with a neutral cognitive profile in adult, geriatric, and pediatric patients (253–255). In a rat CCI model, perampanel preserved neurological function, inhibited apoptosis and microglial activation, reduced brain edema, and preserved blood–brain-barrier functioning post-injury, thereby protecting neuro-vasculature (256). It also reduced brain contusion volume and decreased expression of pro-inflammatory TNF-α and IL-1β (257).
The effects of perampanel on neurological functioning, inflammatory markers, and cognition in patients with AD has yet to be comprehensively studied, outside of isolated case reports. In a case study of an 89 year old woman with severe AD, intractable myoclonic epilepsy, and psychiatric symptoms of circadian rhythm disorder and irritability, perampanel improved both myoclonus and psychiatric symptoms (258). An additional case report demonstrated improved cognitive functioning in a patient with non-convulsive seizures and AD, supporting the case for early administration (259). In transgenic AD mice, inhibition of AMPA receptors by perampanel reduced hippocampal Aβ40 and Aβ42 levels and decreased levels of the soluble peptide APPβ by suppressing β-cleavage of APP (260). Further research is needed into the potential effect of perampanel in targeting Aβ pathology by reducing Aβ production in AD.
8.4 Metabotropic glutamate receptor treatments
Metabotropic glutamate receptors (mGluR) may also be a target of interest in generalized and focal seizures, as the group II and III mGluR agonists may decrease NMDA receptor function and the risk of excitotoxicity. Animal studies showed anticonvulsant effects of the group II mGluR agonist, DCG-IV, in models of limbic and generalized motor seizures (261–263). Anticonvulsant effects were also noted in DBA/2 rodent models using agonists that target group III mGluR8 and mGluR4, 4A (L-AP4, RS-4-PPG, and ACPT-1) and the antagonist, MPPG. These agents were not found to affect group I, which contribute to epileptogenesis (264, 265). However, mixed responses to mGluR-based treatments have been noted, with proconvulsant effects of group III agonists (L-AP4 and L-SOP) and the MGluR antagonist, MAP4 (266, 267). Further research is needed into safe and effective therapeutic concentrations of mGluR-targeting agents, as well as their role in seizure activity (266–270).
In our proposed RIR model, upregulation of selected NMDA receptors and downregulation of selected AMPA receptors occurs as a result of neuroinflammation in response to sustained or recurrent injury. As a result, there is an increased likelihood of seizure occurrence and atypically high concentrations of intra-and extra-cellular glutamate. At low doses, NMDA receptor antagonists can reduce seizure severity and frequency, but with mixed results. Higher doses, however, risk significant adverse effects. AMPA receptor antagonists, such as perampanel, may show greater promise due to their potential effects on hyperexcitability, underlying pathophysiology of neurodegenerative disorders, and tolerability. MGluR agonists and antagonists showed varied pro-vs. anti-convulsant effects with limited research into safe and effective therapeutic concentrations. Caution in targeting glutamate receptors is warranted.
8.5 Monoclonal antibody treatments
Anti-amyloid monoclonal antibodies, such as lecanemab and aducanumab, represent another treatment approach, possibly as maintenance drugs to slow the progression of cognitive decline over the course of the disease. Lecanemab demonstrated high affinity binding to soluble Aβ, and particularly to Aβ soluble protofibrils, which are seen in early AD (271–273). Approved for use in Alzheimer’s disease (274), lecanemab reduced Aβ markers and moderately slowed cognitive decline over 18 months compared to placebo (271, 272), although its effectiveness has been questioned. In a transgenic mouse model, aducanumab decreased both soluble and insoluble Aβ in a dose-dependent manner (275). To evaluate the safety and efficacy of aducanumab in reducing cognitive decline in patients with MCI and mild AD, two large, double-blind, placebo-controlled studies were conducted. Results indicated that aducanumab was associated with dose-dependent amyloid related imaging abnormalities (ARIA), with cerebral edema and increased risk of intracerebral hemorrhage, particularly in ApoE-ɛ4 carriers (276). Infusion-related reactions and other adverse events (including ARIA) make anti-amyloid antibodies a controversial approach in the setting of uncertain benefits. Lecanemab and aducanumab have not yet been tested in patients with TBI or epilepsy, and safety and efficacy clinical trials for both drugs are on-going.
8.6 Tau-centric treatments
Reduction of tau levels showed promise in tau-expressing transgenic mice with repetitive mild CHI. Mice were treated with kinase-targeting lithium chloride and R-roscovitine, leading to p-tau reduction that correlated with improved cognition (200, 277). Alternatively, phosphatases dephosphorylate toxic tau into non-toxic isoforms. Phosphatase 2A (PP2A) dephosphorylates hp-tau, but PP2A activity is decreased in AD brain (278, 279). In AD, GSK-3 activation inhibits PP2A (280), and PP2A inhibitory proteins (inhibitor-1 and -2) are upregulated (281). Pharmacological interventions that inhibit GSK-3, such as SAR502250 (282), or support mRNA-based downregulation of PP2A inhibitors-1/2, are promising approaches (281). Drugs for approved for other indications may also be “repurposed” given their effects on tau. Suvorexant, an FDA-approved drug for insomnia, reduces tau phosphorylation at selective sites such as −181 and decreases Aβ concentrations compared to placebo (283); its use should be investigated in other disorders. Angiotensin receptor blockers, FDA-approved for hypertension, have anticonvulsant effects in rats (284–286) and decrease incidence of epilepsy in humans (287), while decreasing CSF t-tau and p-tau in MCI patients (288) and improving cognition in hypertensive older adults with early executive impairment (289) and prodromal AD (290). These results support the need to further investigate the safety and efficacy of tau-targeting drugs in epilepsy.
8.7 ER stress response inhibition
Based on our proposed mechanism, drugs that impair the PERK pathway would have injurious effects. In a mouse TBI model, for example, inhibition of the PERK signaling pathway by GSK2606414 exacerbated immature cell loss, dendritic loss, and cell death (26).
Conversely, pharmacological upregulation of the PERK pathway may be an effective treatment target to avoid atypical ER stress response activation, reduce tau phosphorylation by ER stress response signaling (75), and mediate tau hyper-phosphorylation and Aβ neurotoxicity (291).
Drugs that target the ER stress response cell death pathways may also aid in the restoration of intracellular homeostasis, apoptotic-necrotic signaling dynamics, and ER folding capacity. In a rat lateral fluid percussion model of TBI, administration of the ER stress response inhibitor, salubrinal, 30 min prior to injury significantly reduced the ER stress response, promoted mitochondrial functioning, and inhibited downstream apoptotic signaling (292). In a mouse model of autosomal dominant lateral TLE, 4-phenylbutyric acid restored LGI1 protein function and reduced seizure susceptibility (293). In a mouse model of epilepsy, taurursodiol also reduced seizure susceptibility and mitigated repeated stress-induced neurodegeneration (294). In reducing seizure susceptibility, the likelihood of repeated or chronic activation of the ER stress response and tau-induced pathways decreases. This benefits the cell by favoring restoration of homeostasis, PERK pathway activation, and tau-involvement for maintenance of cellular dynamics; this also reduces the likelihood of repeated/chronic activation of Aβ, resulting in avoidance of irreversible or long-term neurodegeneration.
The numerous proteins and pathways involved in the brain’s inflammatory response make it challenging to identify the most appropriate target. Development of inflammatory modulators must also consider that acute inflammation can serve to protect neuronal integrity and avoid cell death, while chronic inflammation may decrease the likelihood of maximal recovery and cell survival. Further research is needed to find preventative and therapeutic agents for AD, TBI, and epilepsy.
9 Conclusion
AD, TBI, and epilepsy disrupt neuronal function and promote atypical response signaling. This review examined inflammatory and excitotoxic pathways common to AD, TBI, and epilepsy, the role of the ER stress response in the face of excitotoxicity, and tau and Aβ signaling. We proposed a mechanism by which these pathways can lead to tau deposition. We posit that tau accumulation represents an attempt to shunt the injury response from apoptosis toward neuroprotective signaling that preserves the cell, in attempts to restore homeostasis. This could be viewed as an acute “neuroprotective” response, although, if the underlying pathology is not treated, its recurrent or sustained activation will result in neurodegeneration. Our proposed mechanism supports the case for early intervention. In patients with AD, we must identify risk factors that impact tau and Aβ processes prior to the appearance of cognitive decline. In patients with TBI, this means reducing the likelihood of recurrent injury, reducing injury severity through preventative measures, and providing ample recovery time. In patients with epilepsy, we need to identify the underlying etiologies and reduce seizure frequency and severity. These pathways may present targets for intervention in AD, TBI, and epilepsy. Studies that examine mediators of these signaling cascades are needed.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
SM: Conceptualization, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. BAL-M: Conceptualization, Funding acquisition, Methodology, Supervision, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Career Development Award, IK2 CX-001255-01, from the United States Department of Veteran Affairs Clinical Sciences R&D Service and the NYU Langone Health Finding a Cure for Epilepsy and Seizures foundation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Roth, TL, Nayak, D, Atanasijevic, T, Koretsky, AP, Latour, LL, and McGavern, DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature. (2014) 505:223–8. doi: 10.1038/nature12808
2. Vezzani, A, French, J, Bartfai, T, and Baram, TZ. The role of inflammation in epilepsy. Nat Rev Neurol. (2011) 7:31–40. doi: 10.1038/nrneurol.2010.178
3. Lucke-Wold, BP, Nguyen, L, Turner, RC, Logsdon, AF, Chen, YW, Smith, KE, et al. Traumatic brain injury and epilepsy: underlying mechanisms leading to seizure. Seizure. (2015) 33:13–23. doi: 10.1016/j.seizure.2015.10.002
4. Hernandez, F, and Avila, J. Tauopathies. Cell Mol Life Sci. (2007) 64:2219–33. doi: 10.1007/s00018-007-7220-x
5. Johnson, GV, and Stoothoff, WH. Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci. (2004) 117:5721–9. doi: 10.1242/jcs.01558
6. Ittner, LM, Ke, YD, Delerue, F, Bi, M, Gladbach, A, van Eersel, J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cells. (2010) 142:387–97. doi: 10.1016/j.cell.2010.06.036
7. Kaech, S, and Banker, G. Culturing hippocampal neurons. Nat Protoc. (2006) 1:2406–15. doi: 10.1038/nprot.2006.356
8. Mandelkow, E, von Bergen, M, Biernat, J, and Mandelkow, EM. Structural principles of tau and the paired helical filaments of Alzheimer's disease. Brain Pathol. (2007) 17:83–90. doi: 10.1111/j.1750-3639.2007.00053.x
9. Gamblin, TC, Chen, F, Zambrano, A, Abraha, A, Lagalwar, S, Guillozet, AL, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci U S A. (2003) 100:10032–7. doi: 10.1073/pnas.1630428100
10. Boraschi, D, Italiani, P, Weil, S, and Martin, MU. The family of the interleukin-1 receptors. Immunol Rev. (2018) 281:197–232. doi: 10.1111/imr.12606
11. Moriwaki, K, and Chan, FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. (2013) 27:1640–9. doi: 10.1101/gad.223321.113
12. Kelliher, MA, Grimm, S, Ishida, Y, Kuo, F, Stanger, BZ, and Leder, P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. (1998) 8:297–303. doi: 10.1016/S1074-7613(00)80535-X
13. Kumar, RG, Boles, JA, and Wagner, AK. Chronic inflammation after severe traumatic brain injury: characterization and associations with outcome at 6 and 12 months Postinjury. J Head Trauma Rehabil. (2015) 30:369–81. doi: 10.1097/HTR.0000000000000067
14. Hodo, TW, de Aquino, MTP, Shimamoto, A, and Shanker, A. Critical neurotransmitters in the Neuroimmune network. Front Immunol. (2020) 11:1869. doi: 10.3389/fimmu.2020.01869
15. Neher, E, and Sakaba, T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. (2008) 59:861–72. doi: 10.1016/j.neuron.2008.08.019
16. Walls, AB, Waagepetersen, HS, Bak, LK, Schousboe, A, and Sonnewald, U. The glutamine-glutamate/GABA cycle: function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem Res. (2015) 40:402–9. doi: 10.1007/s11064-014-1473-1
17. Kumar, A, Zou, L, Yuan, X, Long, Y, and Yang, K. N-methyl-D-aspartate receptors: transient loss of NR1/NR2A/NR2B subunits after traumatic brain injury in a rodent model. J Neurosci Res. (2002) 67:781–6. doi: 10.1002/jnr.10181
18. Kharlamov, EA, Lepsveridze, E, Meparishvili, M, Solomonia, RO, Lu, B, Miller, ER, et al. Alterations of GABA(a) and glutamate receptor subunits and heat shock protein in rat hippocampus following traumatic brain injury and in posttraumatic epilepsy. Epilepsy Res. (2011) 95:20–34. doi: 10.1016/j.eplepsyres.2011.02.008
19. Chater, TE, and Goda, Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front Cell Neurosci. (2014) 8:401. doi: 10.3389/fncel.2014.00401
20. Atkins, CM, Chen, S, Alonso, OF, Dietrich, WD, and Hu, BR. Activation of calcium/calmodulin-dependent protein kinases after traumatic brain injury. J Cereb Blood Flow Metab. (2006) 26:1507–18. doi: 10.1038/sj.jcbfm.9600301
21. Cao, F, Zhou, Z, Cai, S, Xie, W, and Jia, Z. Hippocampal Long-term depression in the presence of calcium-permeable AMPA receptors. Front Synaptic Neurosci. (2018) 10:41. doi: 10.3389/fnsyn.2018.00041
22. Patel, TP, Ventre, SC, Geddes-Klein, D, Singh, PK, and Meaney, DF. Single-neuron NMDA receptor phenotype influences neuronal rewiring and reintegration following traumatic injury. J Neurosci. (2014) 34:4200–13. doi: 10.1523/JNEUROSCI.4172-13.2014
23. Isaac, JT, Ashby, MC, and McBain, CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. (2007) 54:859–71. doi: 10.1016/j.neuron.2007.06.001
24. Parga Becerra, A, Logsdon, AF, Banks, WA, and Ransom, CB. Traumatic brain injury broadly affects GABAergic signaling in dentate gyrus granule cells. eNeuro. (2021) 8:ENEURO.0055–20.2021. doi: 10.1523/ENEURO.0055-20.2021
25. Zhou, H, Chen, L, Gao, X, Luo, B, and Chen, J. Moderate traumatic brain injury triggers rapid necrotic death of immature neurons in the hippocampus. J Neuropathol Exp Neurol. (2012) 71:348–59. doi: 10.1097/NEN.0b013e31824ea078
26. Hood, KN, Zhao, J, Redell, JB, Hylin, MJ, Harris, B, Perez, A, et al. Endoplasmic reticulum stress contributes to the loss of newborn hippocampal neurons after traumatic brain injury. J Neurosci. (2018) 38:2372–84. doi: 10.1523/JNEUROSCI.1756-17.2018
27. Bao, YH, Bramlett, HM, Atkins, CM, Truettner, JS, Lotocki, G, Alonso, OF, et al. Post-traumatic seizures exacerbate histopathological damage after fluid-percussion brain injury. J Neurotrauma. (2011) 28:35–42. doi: 10.1089/neu.2010.1383
28. De Beaumont, L, Tremblay, S, Poirier, J, Lassonde, M, and Théoret, H. Altered bidirectional plasticity and reduced implicit motor learning in concussed athletes. Cereb Cortex. (2012) 22:112–21. doi: 10.1093/cercor/bhr096
29. Farrant, M, and Nusser, Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(a) receptors. Nat Rev Neurosci. (2005) 6:215–29. doi: 10.1038/nrn1625
30. Mamun, AA, Uddin, MS, Mathew, B, and Ashraf, GM. Toxic tau: structural origins of tau aggregation in Alzheimer's disease. Neural Regen Res. (2020) 15:1417–20. doi: 10.4103/1673-5374.274329
31. Caccamo, A, Branca, C, Piras, IS, Ferreira, E, Huentelman, MJ, Liang, WS, et al. Necroptosis activation in Alzheimer's disease. Nat Neurosci. (2017) 20:1236–46. doi: 10.1038/nn.4608
32. Ii, M, Sunamoto, M, Ohnishi, K, and Ichimori, Y. beta-amyloid protein-dependent nitric oxide production from microglial cells and neurotoxicity. Brain Res. (1996) 720:93–100. doi: 10.1016/0006-8993(96)00156-4
33. Griffin, WS, Sheng, JG, Roberts, GW, and Mrak, RE. Interleukin-1 expression in different plaque types in Alzheimer's disease: significance in plaque evolution. J Neuropathol Exp Neurol. (1995) 54:276–81. doi: 10.1097/00005072-199503000-00014
34. Griffin, WS, Sheng, JG, Royston, MC, Gentleman, SM, McKenzie, JE, Graham, DI, et al. Glial-neuronal interactions in Alzheimer's disease: the potential role of a 'cytokine cycle' in disease progression. Brain Pathol. (1998) 8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x
35. Prehn, JH, Bindokas, VP, Jordán, J, Galindo, MF, Ghadge, GD, Roos, RP, et al. Protective effect of transforming growth factor-beta 1 on beta-amyloid neurotoxicity in rat hippocampal neurons. Mol Pharmacol. (1996) 49:319–28.
36. Decourt, B, Lahiri, DK, and Sabbagh, MN. Targeting tumor necrosis factor alpha for Alzheimer's disease. Curr Alzheimer Res. (2017) 14:412–25. doi: 10.2174/1567205013666160930110551
37. Choi, DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. (1987) 7:369–79. doi: 10.1523/JNEUROSCI.07-02-00369.1987
38. Choi, DW, Koh, JY, and Peters, S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. (1988) 8:185–96. doi: 10.1523/JNEUROSCI.08-01-00185.1988
39. Abramov, E, Dolev, I, Fogel, H, Ciccotosto, GD, Ruff, E, and Slutsky, I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. (2009) 12:1567–76. doi: 10.1038/nn.2433
40. Domingues, A, Almeida, S, da Cruz Silva, EF, Oliveira, CR, and Rego, AC. Toxicity of beta-amyloid in HEK293 cells expressing NR1/NR2A or NR1/NR2B N-methyl-D-aspartate receptor subunits. Neurochem Int. (2007) 50:872–80. doi: 10.1016/j.neuint.2007.03.001
41. Le, WD, Colom, LV, Xie, WJ, Smith, RG, Alexianu, M, and Appel, SH. Cell death induced by beta-amyloid 1-40 in MES 23.5 hybrid clone: the role of nitric oxide and NMDA-gated channel activation leading to apoptosis. Brain Res. (1995) 686:49–60. doi: 10.1016/0006-8993(95)00450-5
42. De Felice, FG, Velasco, PT, Lambert, MP, Viola, K, Fernandez, SJ, Ferreira, ST, et al. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. (2007) 282:11590–601. doi: 10.1074/jbc.M607483200
43. Lacor, PN, Buniel, MC, Furlow, PW, Sanz Clemente, A, Velasco, PT, Wood, M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. (2007) 27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007
44. Gueli, MC, and Taibi, G. Alzheimer's disease: amino acid levels and brain metabolic status. Neurol Sci. (2013) 34:1575–9. doi: 10.1007/s10072-013-1289-9
45. Bareggi, SR, Franceschi, M, Bonini, L, Zecca, L, and Smirne, S. Decreased CSF concentrations of homovanillic acid and gamma-aminobutyric acid in Alzheimer's disease. Age-or disease-related modifications? Arch Neurol. (1982) 39:709–12. doi: 10.1001/archneur.1982.00510230035010
46. Garcia-Marin, V, Blazquez-Llorca, L, Rodriguez, J-R, Boluda, S, Muntane, G, Ferrer, I, et al. Diminished perisomatic GABAergic terminals on cortical neurons adjacent to amyloid plaques. Front Neuroanat. (2009) 3:28. doi: 10.3389/neuro.05.028.2009
47. Ramos-Miguel, A, Hercher, C, Beasley, CL, Barr, AM, Bayer, TA, Falkai, P, et al. Loss of Munc18-1 long splice variant in GABAergic terminals is associated with cognitive decline and increased risk of dementia in a community sample. Mol Neurodegener. (2015) 10:65. doi: 10.1186/s13024-015-0061-4
48. Jara, JH, Singh, BB, Floden, AM, and Combs, CK. Tumor necrosis factor alpha stimulates NMDA receptor activity in mouse cortical neurons resulting in ERK-dependent death. J Neurochem. (2007) 100:1407–20. doi: 10.1111/j.1471-4159.2006.04330.x
49. Stellwagen, D, Beattie, EC, Seo, JY, and Malenka, RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. (2005) 25:3219–28. doi: 10.1523/JNEUROSCI.4486-04.2005
50. Vezzani, A, Conti, M, de Luigi, A, Ravizza, T, Moneta, D, Marchesi, F, et al. Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J Neurosci. (1999) 19:5054–65. doi: 10.1523/JNEUROSCI.19-12-05054.1999
51. During, MJ, and Spencer, DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. (1993) 341:1607–10. doi: 10.1016/0140-6736(93)90754-5
52. Caro-Maldonado, A, Tait, SWG, Ramírez-Peinado, S, Ricci, JE, Fabregat, I, Green, DR, et al. Glucose deprivation induces an atypical form of apoptosis mediated by caspase-8 in Bax-Bak-deficient cells. Cell Death Differ. (2010) 17:1335–44. doi: 10.1038/cdd.2010.21
53. Henshall, DC, and Simon, RP. Epilepsy and apoptosis pathways. J Cereb Blood Flow Metab. (2005) 25:1557–72. doi: 10.1038/sj.jcbfm.9600149
54. Holmes, GL. Seizure-induced neuronal injury: animal data. Neurology. (2002) 59:S3–6. doi: 10.1212/WNL.59.9_suppl_5.S3
55. Pitkanen, A, and Sutula, TP. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol. (2002) 1:173–81. doi: 10.1016/S1474-4422(02)00073-X
56. Gahring, LC, White, HS, Skradski, SL, Carlson, NG, and Rogers, SW. Interleukin-1alpha in the brain is induced by audiogenic seizure. Neurobiol Dis. (1997) 3:263–9. doi: 10.1006/nbdi.1996.0123
57. Vezzani, A, and Baram, TZ. New roles for interleukin-1 Beta in the mechanisms of epilepsy. Epilepsy Curr. (2007) 7:45–50. doi: 10.1111/j.1535-7511.2007.00165.x
58. Johnson, VE, Stewart, JE, Begbie, FD, Trojanowski, JQ, Smith, DH, and Stewart, W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. (2013) 136:28–42. doi: 10.1093/brain/aws322
59. Ravizza, T, and Vezzani, A. Status epilepticus induces time-dependent neuronal and astrocytic expression of interleukin-1 receptor type I in the rat limbic system. Neuroscience. (2006) 137:301–8. doi: 10.1016/j.neuroscience.2005.07.063
60. Sul, JY, Orosz, G, Givens, RS, and Haydon, PG. Astrocytic connectivity in the hippocampus. Neuron Glia Biol. (2004) 1:3–11. doi: 10.1017/S1740925X04000031
61. Mizuno, T, Doi, Y, Mizoguchi, H, Jin, S, Noda, M, Sonobe, Y, et al. Interleukin-34 selectively enhances the neuroprotective effects of microglia to attenuate oligomeric amyloid-beta neurotoxicity. Am J Pathol. (2011) 179:2016–27. doi: 10.1016/j.ajpath.2011.06.011
62. Czapski, GA, and Strosznajder, JB. Glutamate and GABA in microglia-neuron cross-talk in Alzheimer's disease. Int J Mol Sci. (2021) 22:1677. doi: 10.3390/ijms222111677
63. Green, KN, Crapser, JD, and Hohsfield, LA. To kill a microglia: a case for CSF1R inhibitors. Trends Immunol. (2020) 41:771–84. doi: 10.1016/j.it.2020.07.001
64. Lin, JH, Walter, P, and Yen, TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. (2008) 3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434
65. Lindner, P, Christensen, SB, Nissen, P, Møller, JV, and Engedal, N. Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun Signal. (2020) 18:12. doi: 10.1186/s12964-019-0499-z
66. Smith, MI, and Deshmukh, M. Endoplasmic reticulum stress-induced apoptosis requires bax for commitment and Apaf-1 for execution in primary neurons. Cell Death Differ. (2007) 14:1011–9. doi: 10.1038/sj.cdd.4402089
67. Iurlaro, R, and Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. (2016) 283:2640–52. doi: 10.1111/febs.13598
68. Walter, F, O'Brien, A, Concannon, CG, Düssmann, H, and Prehn, JHM. ER stress signaling has an activating transcription factor 6alpha (ATF6)-dependent "off-switch". J Biol Chem. (2018) 293:18270–84. doi: 10.1074/jbc.RA118.002121
69. Walter, P, and Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. (2011) 334:1081–6. doi: 10.1126/science.1209038
70. Livezey, M, Huang, R, Hergenrother, PJ, and Shapiro, DJ. Strong and sustained activation of the anticipatory unfolded protein response induces necrotic cell death. Cell Death Differ. (2018) 25:1796–807. doi: 10.1038/s41418-018-0143-2
71. Logsdon, AF, Turner, RC, Lucke-Wold, BP, Robson, MJ, Naser, ZJ, Smith, KE, et al. Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front Cell Neurosci. (2014) 8:421. doi: 10.3389/fncel.2014.00421
72. Rubovitch, V, Shachar, A, Werner, H, and Pick, CG. Does IGF-1 administration after a mild traumatic brain injury in mice activate the adaptive arm of ER stress? Neurochem Int. (2011) 58:443–6. doi: 10.1016/j.neuint.2011.01.009
73. West, MJ, Coleman, PD, Flood, DG, and Troncoso, JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet. (1994) 344:769–72. doi: 10.1016/S0140-6736(94)92338-8
74. Dam, AM. Epilepsy and neuron loss in the Hippocampus. Epilepsia. (1980) 21:617–29. doi: 10.1111/j.1528-1157.1980.tb04315.x
75. Ho, Y-S, Yang, X, Lau, JCF, Hung, CHL, Wuwongse, S, Zhang, Q, et al. Endoplasmic reticulum stress induces tau pathology and forms a vicious cycle: implication in Alzheimer's disease pathogenesis. J Alzheimers Dis. (2012) 28:839–54. doi: 10.3233/JAD-2011-111037
76. Lucke-Wold, BP, Logsdon, AF, Turner, RC, Huber, JD, and Rosen, CL. Endoplasmic reticulum stress modulation as a target for ameliorating effects of blast induced traumatic brain injury. J Neurotrauma. (2017) 34:S-62–70. doi: 10.1089/neu.2016.4680
77. Hylin, MJ, Holden, RC, Smith, AC, Logsdon, AF, Qaiser, R, and Lucke-Wold, BP. Juvenile traumatic brain injury results in cognitive deficits associated with impaired endoplasmic reticulum stress and early Tauopathy. Dev Neurosci. (2018) 40:175–88. doi: 10.1159/000488343
78. Unterberger, U, Höftberger, R, Gelpi, E, Flicker, H, Budka, H, and Voigtländer, T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J Neuropathol Exp Neurol. (2006) 65:348–57. doi: 10.1097/01.jnen.0000218445.30535.6f
79. Meier, S, Bell, M, Lyons, DN, Ingram, A, Chen, J, Gensel, JC, et al. Identification of novel tau interactions with endoplasmic reticulum proteins in Alzheimer's disease brain. J Alzheimers Dis. (2015) 48:687–702. doi: 10.3233/JAD-150298
80. Liu, DC, Eagleman, DE, and Tsai, NP. Novel roles of ER stress in repressing neural activity and seizures through Mdm2-and p53-dependent protein translation. PLoS Genet. (2019) 15:e1008364. doi: 10.1371/journal.pgen.1008364
81. Madhamanchi, K, Madhamanchi, P, Jayalakshmi, S, Panigrahi, M, Patil, A, and Phanithi, PB. Endoplasmic reticulum stress and unfolded protein accumulation correlate to seizure recurrence in focal cortical dysplasia patients. Cell Stress Chaperones. (2022) 27:633–43. doi: 10.1007/s12192-022-01301-0
82. Hitomi, J, Katayama, T, Eguchi, Y, Kudo, T, Taniguchi, M, Koyama, Y, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol. (2004) 165:347–56. doi: 10.1083/jcb.200310015
83. Ferreiro, E, Resende, R, Costa, R, Oliveira, CR, and Pereira, CMF. An endoplasmic-reticulum-specific apoptotic pathway is involved in prion and amyloid-beta peptides neurotoxicity. Neurobiol Dis. (2006) 23:669–78. doi: 10.1016/j.nbd.2006.05.011
84. Salminen, A, Kauppinen, A, Suuronen, T, Kaarniranta, K, and Ojala, J. ER stress in Alzheimer's disease: a novel neuronal trigger for inflammation and Alzheimer's pathology. J Neuroinflammation. (2009) 6:41. doi: 10.1186/1742-2094-6-41
85. Kogel, D, Schomburg, R, Schürmann, T, Reimertz, C, König, H-G, Poppe, M, et al. The amyloid precursor protein protects PC12 cells against endoplasmic reticulum stress-induced apoptosis. J Neurochem. (2003) 87:248–56. doi: 10.1046/j.1471-4159.2003.02000.x
86. Esposito, L, Gan, L, Yu, GQ, Essrich, C, and Mucke, L. Intracellularly generated amyloid-beta peptide counteracts the antiapoptotic function of its precursor protein and primes proapoptotic pathways for activation by other insults in neuroblastoma cells. J Neurochem. (2004) 91:1260–74. doi: 10.1111/j.1471-4159.2004.02816.x
87. Fonseca, AC, Ferreiro, E, Oliveira, CR, Cardoso, SM, and Pereira, CF. Activation of the endoplasmic reticulum stress response by the amyloid-beta 1-40 peptide in brain endothelial cells. Biochim Biophys Acta. (2013) 1832:2191–203. doi: 10.1016/j.bbadis.2013.08.007
88. Casas-Tinto, S, Zhang, Y, Sanchez-Garcia, J, Gomez-Velazquez, M, Rincon-Limas, DE, and Fernandez-Funez, P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum Mol Genet. (2011) 20:2144–60. doi: 10.1093/hmg/ddr100
89. Hoozemans, JJ, Veerhuis, R, Van Haastert, ES, Rozemuller, JM, Baas, F, Eikelenboom, P, et al. The unfolded protein response is activated in Alzheimer's disease. Acta Neuropathol. (2005) 110:165–72. doi: 10.1007/s00401-005-1038-0
90. Lace, G, Savva, GM, Forster, G, de Silva, R, Brayne, C, Matthews, FE, et al. MRC-CFAS, Hippocampal tau pathology is related to neuroanatomical connections: an ageing population-based study. Brain. (2009) 132:1324–34. doi: 10.1093/brain/awp059
91. Shahim, P, Tegner, Y, Wilson, DH, Randall, J, Skillbäck, T, Pazooki, D, et al. Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol. (2014) 71:684–92. doi: 10.1001/jamaneurol.2014.367
92. Zetterberg, H, Smith, DH, and Blennow, K. Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nat Rev Neurol. (2013) 9:201–10. doi: 10.1038/nrneurol.2013.9
93. Delacourte, A. Pathological tau proteins of Alzheimer's disease as a biochemical marker of neurofibrillary degeneration. Biomed Pharmacother. (1994) 48:287–95. doi: 10.1016/0753-3322(94)90174-0
94. Tai, XY, Koepp, M, Duncan, JS, Fox, N, Thompson, P, Baxendale, S, et al. Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: a study of temporal lobe resections. Brain. (2016) 139:2441–55. doi: 10.1093/brain/aww187
95. Monti, G, Tondelli, M, Giovannini, G, Bedin, R, Nichelli, PF, Trenti, T, et al. Cerebrospinal fluid tau proteins in status epilepticus. Epilepsy Behav. (2015) 49:150–4. doi: 10.1016/j.yebeh.2015.04.030
96. Shahim, P, Rejdak, R, Ksiazek, P, Blennow, K, Zetterberg, H, Mattsson, N, et al. Cerebrospinal fluid biomarkers of beta-amyloid metabolism and neuronal damage in epileptic seizures. Eur J Neurol. (2014) 21:486–91. doi: 10.1111/ene.12336
97. Matsui, T, Maruyama, M, Matsushita, S, Arai, H, Higuchi, S, and Maruyama, K. A transient increase in cerebrospinal fluid tau level after epileptic seizure in an elderly patient. J Am Geriatr Soc. (2007) 55:2096–7. doi: 10.1111/j.1532-5415.2007.01440.x
98. Palmio, J, Suhonen, J, Keränen, T, Hulkkonen, J, Peltola, J, and Pirttilä, T. Cerebrospinal fluid tau as a marker of neuronal damage after epileptic seizure. Seizure. (2009) 18:474–7. doi: 10.1016/j.seizure.2009.04.006
99. Hopp, SC, Lin, Y, Oakley, D, Roe, AD, DeVos, SL, Hanlon, D, et al. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer's disease. J Neuroinflammation. (2018) 15:269. doi: 10.1186/s12974-018-1309-z
100. Edwards, G 3rd, Zhao, J, Dash, PK, Soto, C, Moreno-Gonzalez, I, et al. Traumatic brain injury induces tau aggregation and spreading. J Neurotrauma. (2020) 37:80–92. doi: 10.1089/neu.2018.6348
101. Thom, M, Liu, JYW, Thompson, P, Phadke, R, Narkiewicz, M, Martinian, L, et al. Neurofibrillary tangle pathology and Braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: a post-mortem study. Brain. (2011) 134:2969–81. doi: 10.1093/brain/awr209
102. Kristensen, AS, Jenkins, MA, Banke, TG, Schousboe, A, Makino, Y, Johnson, RC, et al. Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat Neurosci. (2011) 14:727–35. doi: 10.1038/nn.2804
103. Barria, A, Muller, D, Derkach, V, Griffith, LC, and Soderling, TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. (1997) 276:2042–5. doi: 10.1126/science.276.5321.2042
104. Lee, HK, Barbarosie, M, Kameyama, K, Bear, MF, and Huganir, RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. (2000) 405:955–9. doi: 10.1038/35016089
105. Mao, LM, Jin, D-Z, Xue, B, Chu, X-P, and Wang, JQ. Phosphorylation and regulation of glutamate receptors by CaMKII. Sheng Li Xue Bao. (2014) 66:365–72. doi: 10.13294/j.aps.2014.0044
106. Suh, HW, Lee, HK, Seo, YJ, Kwon, MS, Shim, EJ, Lee, JY, et al. Kainic acid (KA)-induced Ca2+/calmodulin-dependent protein kinase II (CaMK II) expression in the neurons, astrocytes and microglia of the mouse hippocampal CA3 region, and the phosphorylated CaMK II only in the hippocampal neurons. Neurosci Lett. (2005) 381:223–7. doi: 10.1016/j.neulet.2005.01.089
107. Moynagh, PN. The interleukin-1 signalling pathway in astrocytes: a key contributor to inflammation in the brain. J Anat. (2005) 207:265–9. doi: 10.1111/j.1469-7580.2005.00445.x
108. Denes, A, Lopez-Castejon, G, and Brough, D. Caspase-1: is IL-1 just the tip of the ICEberg? Cell Death Dis. (2012) 3:e338. doi: 10.1038/cddis.2012.86
109. Ghayur, T, Banerjee, S, Hugunin, M, Butler, D, Herzog, L, Carter, A, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. (1997) 386:619–23. doi: 10.1038/386619a0
110. Gu, Y, Kuida, K, Tsutsui, H, Ku, G, Hsiao, K, Fleming, MA, et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science. (1997) 275:206–9. doi: 10.1126/science.275.5297.206
111. Hewett, SJ, Jackman, NA, and Claycomb, RJ. Interleukin-1beta in central nervous system injury and repair. Eur J Neurodegener Dis. (2012) 1:195–211.
112. Viviani, B, Bartesaghi, S, Gardoni, F, Vezzani, A, Behrens, MM, Bartfai, T, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. (2003) 23:8692–700. doi: 10.1523/JNEUROSCI.23-25-08692.2003
113. Peng, Z, Huang, CS, Stell, BM, Mody, I, and Houser, CR. Altered expression of the delta subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J Neurosci. (2004) 24:8629–39. doi: 10.1523/JNEUROSCI.2877-04.2004
114. Gardoni, F, Boraso, M, Zianni, E, Corsini, E, Galli, CL, Cattabeni, F, et al. Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1beta and NMDA stimulation. J Neuroinflammation. (2011) 8:14. doi: 10.1186/1742-2094-8-14
115. Mohamed, NE, Zhao, Y, Lee, JH, Tan, MG, Esiri, MM, Wilcock, GK, et al. Upregulation of AMPA receptor GluR2 (GluA2) subunits in subcortical ischemic vascular dementia is repressed in the presence of Alzheimer's disease. Neurochem Int. (2011) 58:820–5. doi: 10.1016/j.neuint.2011.03.010
116. Bell, JD, Park, E, Ai, J, and Baker, AJ. PICK1-mediated GluR2 endocytosis contributes to cellular injury after neuronal trauma. Cell Death Differ. (2009) 16:1665–80. doi: 10.1038/cdd.2009.106
117. Bingol, B, Wang, CF, Arnott, D, Cheng, D, Peng, J, and Sheng, M. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cells. (2010) 140:567–78. doi: 10.1016/j.cell.2010.01.024
118. Ashpole, NM, Chawla, AR, Martin, MP, Brustovetsky, T, Brustovetsky, N, and Hudmon, A. Loss of calcium/calmodulin-dependent protein kinase II activity in cortical astrocytes decreases glutamate uptake and induces neurotoxic release of ATP. J Biol Chem. (2013) 288:14599–611. doi: 10.1074/jbc.M113.466235
119. Kim, HE, du, F, Fang, M, and Wang, X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc Natl Acad Sci U S A. (2005) 102:17545–50. doi: 10.1073/pnas.0507900102
120. Zeiss, CJ. The apoptosis-necrosis continuum: insights from genetically altered mice. Vet Pathol. (2003) 40:481–95. doi: 10.1354/vp.40-5-481
121. Zou, H, Henzel, WJ, Liu, X, Lutschg, A, and Wang, X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cells. (1997) 90:405–13. doi: 10.1016/S0092-8674(00)80501-2
122. Ghavami, S, Hashemi, M, Ande, SR, Yeganeh, B, Xiao, W, Eshraghi, M, et al. Apoptosis and cancer: mutations within caspase genes. J Med Genet. (2009) 46:497–510. doi: 10.1136/jmg.2009.066944
123. Salvesen, GS. Caspases: opening the boxes and interpreting the arrows. Cell Death Differ. (2002) 9:3–5. doi: 10.1038/sj.cdd.4400963
124. Wang, Y, Garg, S, Mandelkow, EM, and Mandelkow, E. Proteolytic processing of tau. Biochem Soc Trans. (2010) 38:955–61. doi: 10.1042/BST0380955
125. Guo, H, Albrecht, S, Bourdeau, M, Petzke, T, Bergeron, C, and LeBlanc, AC. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer's disease. Am J Pathol. (2004) 165:523–31. doi: 10.1016/S0002-9440(10)63317-2
126. Cotman, CW, Poon, WW, Rissman, RA, and Blurton-Jones, M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol. (2005) 64:104–12. doi: 10.1093/jnen/64.2.104
127. Horowitz, PM, Patterson, KR, Guillozet-Bongaarts, AL, Reynolds, MR, Carroll, CA, Weintraub, ST, et al. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer's disease. J Neurosci. (2004) 24:7895–902. doi: 10.1523/JNEUROSCI.1988-04.2004
128. Rissman, RA, Poon, WW, Blurton-Jones, M, Oddo, S, Torp, R, Vitek, MP, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. (2004) 114:121–30. doi: 10.1172/JCI200420640
129. Wang, HH, Li, HL, Liu, R, Zhang, Y, Liao, K, Wang, Q, et al. Tau overexpression inhibits cell apoptosis with the mechanisms involving multiple viability-related factors. J Alzheimers Dis. (2010) 21:167–79. doi: 10.3233/JAD-2010-091279
130. Qin, H, Srinivasula, SM, Wu, G, Fernandes-Alnemri, T, Alnemri, ES, and Shi, Y. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature. (1999) 399:549–57. doi: 10.1038/21124
131. Leyns, CEG, and Holtzman, DM. Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener. (2017) 12:50. doi: 10.1186/s13024-017-0192-x
132. Pais, TF, Figueiredo, C, Peixoto, R, Braz, MH, and Chatterjee, S. Necrotic neurons enhance microglial neurotoxicity through induction of glutaminase by a MyD88-dependent pathway. J Neuroinflammation. (2008) 5:43. doi: 10.1186/1742-2094-5-43
133. Gratuze, M, Chen, Y, Parhizkar, S, Jain, N, Strickland, MR, Serrano, JR, et al. Activated microglia mitigate Abeta-associated tau seeding and spreading. J Exp Med. (2021) 218:542. doi: 10.1084/jem.20210542
134. Asai, H, Ikezu, S, Tsunoda, S, Medalla, M, Luebke, J, Haydar, T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. (2015) 18:1584–93. doi: 10.1038/nn.4132
135. Bolós, M, Llorens-Martín, M, Jurado-Arjona, J, Hernández, F, Rábano, A, and Avila, J. Direct evidence of internalization of tau by microglia in vitro and in vivo. J Alzheimers Dis. (2015) 50:77–87. doi: 10.3233/JAD-150704
136. Luo, W, Liu, W, Hu, X, Hanna, M, Caravaca, A, and Paul, SM. Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep. (2015) 5:11161. doi: 10.1038/srep11161
137. Guo, JL, and Lee, VM. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. (2011) 286:15317–31. doi: 10.1074/jbc.M110.209296
138. Frost, B, Jacks, RL, and Diamond, MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. (2009) 284:12845–52. doi: 10.1074/jbc.M808759200
139. DeVos, SL, Corjuc, BT, Oakley, DH, Nobuhara, CK, Bannon, RN, Chase, A, et al. Synaptic tau seeding precedes tau pathology in human Alzheimer's disease brain. Front Neurosci. (2018) 12:267. doi: 10.3389/fnins.2018.00267
140. Furman, JL, Vaquer-Alicea, J, White, CL III, Cairns, NJ, Nelson, PT, and Diamond, MI. Widespread tau seeding activity at early Braak stages. Acta Neuropathol. (2017) 133:91–100. doi: 10.1007/s00401-016-1644-z
141. Kaufman, SK, del Tredici, K, Thomas, TL, Braak, H, and Diamond, MI. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer's disease and PART. Acta Neuropathol. (2018) 136:57–67. doi: 10.1007/s00401-018-1855-6
142. Ndountse, LT, and Chan, HM. Role of N-methyl-D-aspartate receptors in polychlorinated biphenyl mediated neurotoxicity. Toxicol Lett. (2009) 184:50–5. doi: 10.1016/j.toxlet.2008.10.013
143. Kuhlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. (2015) 13:89. doi: 10.1186/s12915-015-0201-x
144. Jung, KH, Chu, K, Lee, ST, Park, HK, Kim, JH, Kang, KM, et al. Augmentation of nitrite therapy in cerebral ischemia by NMDA receptor inhibition. Biochem Biophys Res Commun. (2009) 378:507–12. doi: 10.1016/j.bbrc.2008.11.081
145. Fan, MM, and Raymond, LA. N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington's disease. Prog Neurobiol. (2007) 81:272–93. doi: 10.1016/j.pneurobio.2006.11.003
146. Leist, M, Single, B, Castoldi, AF, Kühnle, S, and Nicotera, P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. (1997) 185:1481–6. doi: 10.1084/jem.185.8.1481
147. Li, HL, Wang, HH, Liu, SJ, Deng, YQ, Zhang, YJ, Tian, Q, et al. Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer's neurodegeneration. Proc Natl Acad Sci U S A. (2007) 104:3591–6. doi: 10.1073/pnas.0609303104
148. Fujikawa, DG, Ke, X, Trinidad, RB, Shinmei, SS, and Wu, A. Caspase-3 is not activated in seizure-induced neuronal necrosis with internucleosomal DNA cleavage. J Neurochem. (2002) 83:229–40. doi: 10.1046/j.1471-4159.2002.01152.x
149. Morales-Ropero, JM, Arroyo-Urea, S, Neubrand, VE, Martín-Oliva, D, Marín-Teva, JL, Cuadros, MA, et al. The endoplasmic reticulum ca(2+) -ATPase SERCA2b is upregulated in activated microglia and its inhibition causes opposite effects on migration and phagocytosis. Glia. (2021) 69:842–57. doi: 10.1002/glia.23931
150. Ebneth, A, Godemann, R, Stamer, K, Illenberger, S, Trinczek, B, Mandelkow, EM, et al. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol. (1998) 143:777–94. doi: 10.1083/jcb.143.3.777
151. Shi, Y, Zhang, W, Yang, Y, Murzin, AG, Falcon, B, Kotecha, A, et al. Structure-based classification of tauopathies. Nature. (2021) 598:359–63. doi: 10.1038/s41586-021-03911-7
152. Wang, Y, Balaji, V, Kaniyappan, S, Krüger, L, Irsen, S, Tepper, K, et al. The release and trans-synaptic transmission of tau via exosomes. Mol Neurodegener. (2017) 12:5. doi: 10.1186/s13024-016-0143-y
153. Saman, S, Kim, WH, Raya, M, Visnick, Y, Miro, S, Saman, S, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. (2012) 287:3842–9. doi: 10.1074/jbc.M111.277061
154. Busciglio, J, Lorenzo, A, Yeh, J, and Yankner, BA. beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. (1995) 14:879–88. doi: 10.1016/0896-6273(95)90232-5
155. Behl, C, Davis, JB, Klier, FG, and Schubert, D. Amyloid beta peptide induces necrosis rather than apoptosis. Brain Res. (1994) 645:253–64. doi: 10.1016/0006-8993(94)91659-4
156. Garwood, CJ, Pooler, AM, Atherton, J, Hanger, DP, and Noble, W. Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. (2011) 2:e167. doi: 10.1038/cddis.2011.50
157. Lee, CY, and Landreth, GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm (Vienna). (2010) 117:949–60. doi: 10.1007/s00702-010-0433-4
158. Kang, SS, Ebbert, MTW, Baker, KE, Cook, C, Wang, X, Sens, JP, et al. Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J Exp Med. (2018) 215:2235–45. doi: 10.1084/jem.20180653
159. Lian, H, Litvinchuk, A, Chiang, ACA, Aithmitti, N, Jankowsky, JL, and Zheng, H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer's disease. J Neurosci. (2016) 36:577–89. doi: 10.1523/JNEUROSCI.2117-15.2016
160. Lian, H, Yang, L, Cole, A, Sun, L, Chiang, ACA, Fowler, SW, et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer's disease. Neuron. (2015) 85:101–15. doi: 10.1016/j.neuron.2014.11.018
161. Litvinchuk, A, Wan, YW, Swartzlander, DB, Chen, F, Cole, A, Propson, NE, et al. Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in Tauopathy models and Alzheimer's disease. Neuron. (2018) 100:1337–1353.e5. doi: 10.1016/j.neuron.2018.10.031
162. Liddelow, SA, Guttenplan, KA, Clarke, LE, Bennett, FC, Bohlen, CJ, Schirmer, L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
163. Liu, J, Chang, L, Roselli, F, Almeida, OFX, Gao, X, Wang, X, et al. Amyloid-β induces caspase-dependent loss of PSD-95 and Synaptophysin through NMDA receptors. J Alzheimers Dis. (2010) 22:541–56. doi: 10.3233/JAD-2010-100948
164. Dore, K, Carrico, Z, Alfonso, S, Marino, M, Koymans, K, Kessels, HW, et al. PSD-95 protects synapses from β-amyloid. Cell Rep. (2021) 35:109194. doi: 10.1016/j.celrep.2021.109194
165. Ekinci, FJ, Linsley, MD, and Shea, TB. Beta-amyloid-induced calcium influx induces apoptosis in culture by oxidative stress rather than tau phosphorylation. Brain Res Mol Brain Res. (2000) 76:389–95. doi: 10.1016/S0169-328X(00)00025-5
166. Reddy, PH. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer's disease. Exp Neurol. (2009) 218:286–92. doi: 10.1016/j.expneurol.2009.03.042
167. White, JA, Manelli, AM, Holmberg, KH, van Eldik, LJ, and LaDu, MJ. Differential effects of oligomeric and fibrillar amyloid-beta 1-42 on astrocyte-mediated inflammation. Neurobiol Dis. (2005) 18:459–65. doi: 10.1016/j.nbd.2004.12.013
168. Wyss-Coray, T, Loike, JD, Brionne, TC, Lu, E, Anankov, R, Yan, F, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. (2003) 9:453–7. doi: 10.1038/nm838
169. Ajoolabady, A, Lindholm, D, Ren, J, and Pratico, D. ER stress and UPR in Alzheimer’s disease: mechanisms, pathogenesis, treatments. Cell Death Dis. (2022) 13:706. doi: 10.1038/s41419-022-05153-5
170. Jiwaji, Z, Tiwari, SS, Avilés-Reyes, RX, Hooley, M, Hampton, D, Torvell, M, et al. Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to tau and Aß pathology. Nat Commun. (2022) 13:135. doi: 10.1038/s41467-021-27702-w
171. Gu, Z, Liu, W, and Yan, Z. beta-amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J Biol Chem. (2009) 284:10639–49. doi: 10.1074/jbc.M806508200
172. Walsh, DM, and Selkoe, DJ. A beta oligomers – a decade of discovery. J Neurochem. (2007) 101:1172–84. doi: 10.1111/j.1471-4159.2006.04426.x
173. Lin, Y, Skeberdis, VA, Francesconi, A, Bennett, MVL, and Zukin, RS. Postsynaptic density protein-95 regulates NMDA channel gating and surface expression. J Neurosci. (2004) 24:10138–48. doi: 10.1523/JNEUROSCI.3159-04.2004
174. Wang, J, Dickson, DW, Trojanowski, JQ, and Lee, VMY. The levels of soluble versus insoluble brain Aβ distinguish Alzheimer's disease from Normal and pathologic aging. Exp Neurol. (1999) 158:328–37. doi: 10.1006/exnr.1999.7085
175. Bloom, GS. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. (2014) 71:505–8. doi: 10.1001/jamaneurol.2013.5847
176. Arnsten, AFT, Datta, D, del Tredici, K, and Braak, H. Hypothesis: tau pathology is an initiating factor in sporadic Alzheimer's disease. Alzheimers Dement. (2021) 17:115–24. doi: 10.1002/alz.12192
177. Gervais, FG, Xu, D, Robertson, GS, Vaillancourt, JP, Zhu, Y, Huang, JQ, et al. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic a beta peptide formation. Cells. (1999) 97:395–406. doi: 10.1016/S0092-8674(00)80748-5
178. Heinrich, PC, Castell, JV, and Andus, T. Interleukin-6 and the acute phase response. Biochem J. (1990) 265:621–36. doi: 10.1042/bj2650621
179. Goldgaber, D, Harris, HW, Hla, T, Maciag, T, Donnelly, RJ, Jacobsen, JS, et al. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc Natl Acad Sci U S A. (1989) 86:7606–10. doi: 10.1073/pnas.86.19.7606
180. Zhao, J, O'Connor, T, and Vassar, R. The contribution of activated astrocytes to Abeta production: implications for Alzheimer's disease pathogenesis. J Neuroinflammation. (2011) 8:150. doi: 10.1186/1742-2094-8-150
181. Oddo, S, Caccamo, A, Kitazawa, M, Tseng, BP, LaFerla, FM, et al. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. (2003) 24:1063–70. doi: 10.1016/j.neurobiolaging.2003.08.012
182. Roberts, GW, Gentleman, SM, Lynch, A, Murray, L, Landon, M, and Graham, DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry. (1994) 57:419–25. doi: 10.1136/jnnp.57.4.419
183. Roberts, GW, Gentleman, SM, Lynch, A, and Graham, DI. beta A4 amyloid protein deposition in brain after head trauma. Lancet. (1991) 338:1422–3. doi: 10.1016/0140-6736(91)92724-G
184. Mandrekar, S, Jiang, Q, Lee, CYD, Koenigsknecht-Talboo, J, Holtzman, DM, and Landreth, GE. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J Neurosci. (2009) 29:4252–62. doi: 10.1523/JNEUROSCI.5572-08.2009
185. Skovronsky, DM, Doms, RW, and Lee, VMY. Detection of a novel Intraneuronal Pool of insoluble amyloid β protein that accumulates with time in culture. J Cell Biol. (1998) 141:1031–9. doi: 10.1083/jcb.141.4.1031
186. Ries, M, and Sastre, M. Mechanisms of Aβ clearance and degradation by glial cells. Front Aging Neurosci. (2016) 8:160. doi: 10.3389/fnagi.2016.00160
187. Tolar, M, Hey, J, Power, A, and Abushakra, S. Neurotoxic soluble amyloid oligomers drive Alzheimer's pathogenesis and represent a clinically validated target for slowing disease progression. Int J Mol Sci. (2021) 22:6355. doi: 10.3390/ijms22126355
188. Haass, C, and Selkoe, DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat Rev Mol Cell Biol. (2007) 8:101–12. doi: 10.1038/nrm2101
189. Söllvander, S, Nikitidou, E, Brolin, R, Söderberg, L, Sehlin, D, Lannfelt, L, et al. Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol Neurodegener. (2016) 11:38. doi: 10.1186/s13024-016-0098-z
190. Wang, JZ, Grundke-Iqbal, I, and Iqbal, K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. (2007) 25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x
191. Texido, L, Martín-Satué, M, Alberdi, E, Solsona, C, and Matute, C. Amyloid beta peptide oligomers directly activate NMDA receptors. Cell Calcium. (2011) 49:184–90. doi: 10.1016/j.ceca.2011.02.001
192. Malinow, R, and Malenka, RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. (2002) 25:103–26. doi: 10.1146/annurev.neuro.25.112701.142758
193. Song, I, and Huganir, RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. (2002) 25:578–88. doi: 10.1016/S0166-2236(02)02270-1
194. Zhang, Y, Guo, O, Huo, Y, Wang, G, and Man, HY. Amyloid-β induces AMPA receptor ubiquitination and degradation in primary neurons and human brains of Alzheimer's disease. J Alzheimers Dis. (2018) 62:1789–801. doi: 10.3233/JAD-170879
195. Marin, N, Romero, B, Bosch-Morell, F, Llansola, M, Felipo, V, Romá, J, et al. Beta-amyloid-induced activation of caspase-3 in primary cultures of rat neurons. Mech Ageing Dev. (2000) 119:63–7. doi: 10.1016/S0047-6374(00)00172-X
196. Han, XJ, Hu, YY, Yang, ZJ, Jiang, LP, Shi, SL, Li, YR, et al. Amyloid beta-42 induces neuronal apoptosis by targeting mitochondria. Mol Med Rep. (2017) 16:4521–8. doi: 10.3892/mmr.2017.7203
197. Garrido, C, Galluzzi, L, Brunet, M, Puig, PE, Didelot, C, and Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. (2006) 13:1423–33. doi: 10.1038/sj.cdd.4401950
198. Mondragon-Rodriguez, S, Trillaud-Doppia, E, Dudilot, A, Bourgeois, C, Lauzon, M, Leclerc, N, et al. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J Biol Chem. (2012) 287:32040–53. doi: 10.1074/jbc.M112.401240
199. Neselius, S, Brisby, H, Theodorsson, A, Blennow, K, Zetterberg, H, and Marcusson, J. CSF-biomarkers in Olympic boxing: diagnosis and effects of repetitive head trauma. PLoS One. (2012) 7:e33606. doi: 10.1371/journal.pone.0033606
200. Rubenstein, R, Chang, B, Davies, P, Wagner, AK, Robertson, CS, and Wang, KKW. A novel, ultrasensitive assay for tau: potential for assessing traumatic brain injury in tissues and biofluids. J Neurotrauma. (2015) 32:342–52. doi: 10.1089/neu.2014.3548
201. Rubenstein, R, Chang, B, Yue, JK, Chiu, A, Winkler, EA, Puccio, AM, et al. Comparing plasma Phospho tau, Total tau, and Phospho tau-Total tau ratio as acute and chronic traumatic brain injury biomarkers. JAMA Neurol. (2017) 74:1063–72. doi: 10.1001/jamaneurol.2017.0655
202. Kenney, K, Qu, BX, Lai, C, Devoto, C, Motamedi, V, Walker, WC, et al. Higher exosomal phosphorylated tau and total tau among veterans with combat-related repetitive chronic mild traumatic brain injury. Brain Inj. (2018) 32:1276–84. doi: 10.1080/02699052.2018.1483530
203. Stern, RA, Tripodis, Y, Baugh, CM, Fritts, NG, Martin, BM, Chaisson, C, et al. Preliminary study of plasma Exosomal tau as a potential biomarker for chronic traumatic encephalopathy. J Alzheimers Dis. (2016) 51:1099–109. doi: 10.3233/JAD-151028
204. Zemlan, FP, Rosenberg, WS, Luebbe, PA, Campbell, TA, Dean, GE, Weiner, NE, et al. Quantification of axonal damage in traumatic brain injury: affinity purification and characterization of cerebrospinal fluid tau proteins. J Neurochem. (1999) 72:741–50. doi: 10.1046/j.1471-4159.1999.0720741.x
205. Ost, M, Nylen, K, Csajbok, L, Ohrfelt, AO, Tullberg, M, Wikkelso, C, et al. Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology. (2006) 67:1600–4. doi: 10.1212/01.wnl.0000242732.06714.0f
206. Tang, Y, Liu, HL, Min, LX, Yuan, HS, Guo, L, Han, PB, et al. Serum and cerebrospinal fluid tau protein level as biomarkers for evaluating acute spinal cord injury severity and motor function outcome. Neural Regen Res. (2019) 14:896–902. doi: 10.4103/1673-5374.249238
207. Kitazawa, M, Oddo, S, Yamasaki, TR, Green, KN, and LaFerla, FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer's disease. J Neurosci. (2005) 25:8843–53. doi: 10.1523/JNEUROSCI.2868-05.2005
208. Iqbal, K, Liu, F, Gong, CX, and Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. (2010) 7:656–64. doi: 10.2174/156720510793611592
209. Götz, J, Chen, F, Barmettler, R, and Nitsch, RM. Tau filament formation in transgenic mice expressing P301L tau *. J Biol Chem. (2001) 276:529–34. doi: 10.1074/jbc.M006531200
210. Poorkaj, P, Bird, TD, Wijsman, E, Nemens, E, Garruto, RM, Anderson, L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. (1998) 43:815–25. doi: 10.1002/ana.410430617
211. Hutton, M, Lendon, CL, Rizzu, P, Baker, M, Froelich, S, Houlden, H, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. (1998) 393:702–5. doi: 10.1038/31508
212. Wesseling, H, Mair, W, Kumar, M, Schlaffner, CN, Tang, S, Beerepoot, P, et al. Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cells. (2020) 183:1699–1713.e13. doi: 10.1016/j.cell.2020.10.029
213. Baker, M, Mackenzie, IR, Pickering-Brown, SM, Gass, J, Rademakers, R, Lindholm, C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. (2006) 442:916–9. doi: 10.1038/nature05016
214. Cruts, M, Gijselinck, I, van der Zee, J, Engelborghs, S, Wils, H, Pirici, D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. (2006) 442:920–4. doi: 10.1038/nature05017
215. Augustinack, JC, Schneider, A, Mandelkow, EM, and Hyman, BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. (2002) 103:26–35. doi: 10.1007/s004010100423
216. Lewis, J, Dickson, DW, Lin, WL, Chisholm, L, Corral, A, Jones, G, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. (2001) 293:1487–91. doi: 10.1126/science.1058189
217. Braak, H, and Braak, E. Staging of alzheimer's disease-related neurofibrillary changes. Neurobiol Aging. (1995) 16:271–8. doi: 10.1016/0197-4580(95)00021-6
218. Buchanan, H, Mackay, M, Palmer, K, Tothová, K, Katsur, M, Platt, B, et al. Synaptic loss, ER stress and neuro-inflammation emerge late in the lateral temporal cortex and associate with progressive tau pathology in Alzheimer's disease. Mol Neurobiol. (2020) 57:3258–72. doi: 10.1007/s12035-020-01950-1
219. Visser, PJ, Reus, LM, Gobom, J, Jansen, I, Dicks, E, van der Lee, SJ, et al. Cerebrospinal fluid tau levels are associated with abnormal neuronal plasticity markers in Alzheimer’s disease. Mol Neurodegener. (2022) 17:27. doi: 10.1186/s13024-022-00521-3
220. Clark, CM, Xie, S, Chittams, J, Ewbank, D, Peskind, E, Galasko, D, et al. Cerebrospinal fluid tau and β-amyloid: how well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch Neurol. (2003) 60:1696–702. doi: 10.1001/archneur.60.12.1696
221. Ashton, NJ, Benedet, AL, Pascoal, TA, Karikari, TK, Lantero-Rodriguez, J, Brum, WS, et al. Cerebrospinal fluid p-tau231 as an early indicator of emerging pathology in Alzheimer's disease. EBioMedicine. (2022) 76:103836. doi: 10.1016/j.ebiom.2022.103836
222. Andreasen, N, Vanmechelen, E, van de Voorde, A, Davidsson, P, Hesse, C, Tarvonen, S, et al. Cerebrospinal fluid tau protein as a biochemical marker for Alzheimer's disease: a community based follow up study. J Neurol Neurosurg Psychiatry. (1998) 64:298–305. doi: 10.1136/jnnp.64.3.298
223. Sunderland, T, Linker, G, Mirza, N, Putnam, KT, Friedman, DL, Kimmel, LH, et al. Decreased β-Amyloid1-42 and increased tau levels in cerebrospinal fluid of patients with Alzheimer disease. JAMA. (2003) 289:2094–103. doi: 10.1001/jama.289.16.2094
224. Galasko, D, Chang, L, Motter, R, Clark, CM, Kaye, J, Knopman, D, et al. High cerebrospinal fluid tau and low amyloid β42 levels in the clinical diagnosis of Alzheimer disease and relation to apolipoprotein E genotype. Arch Neurol. (1998) 55:937–45. doi: 10.1001/archneur.55.7.937
225. Mehta, PD, Pirttilä, T, Mehta, SP, Sersen, EA, Aisen, PS, and Wisniewski, HM. Plasma and cerebrospinal fluid levels of amyloid β proteins 1-40 and 1-42 in Alzheimer disease. Arch Neurol. (2000) 57:100–5. doi: 10.1001/archneur.57.1.100
226. Motter, R, Vigo-Pelfrey, C, Kholodenko, D, Barbour, R, Johnson-Wood, K, Galasko, D, et al. Reduction of β-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer's disease. Ann Neurol. (1995) 38:643–8. doi: 10.1002/ana.410380413
227. Zetterberg, H, Wilson, D, Andreasson, U, Minthon, L, Blennow, K, Randall, J, et al. Plasma tau levels in Alzheimer's disease. Alzheimers Res Ther. (2013) 5:9. doi: 10.1186/alzrt163
228. Sanchez, MP, García-Cabrero, AM, Sánchez-Elexpuru, G, Burgos, DF, Serratosa, JM, et al. Tau-induced pathology in epilepsy and dementia: notions from patients and animal models. Int J Mol Sci. (2018) 19:1092. doi: 10.3390/ijms19041092
229. Costa, C, Romoli, M, Liguori, C, Farotti, L, Eusebi, P, Bedetti, C, et al. Alzheimer's disease and late-onset epilepsy of unknown origin: two faces of beta amyloid pathology. Neurobiol Aging. (2019) 73:61–7. doi: 10.1016/j.neurobiolaging.2018.09.006
230. Mo, L, Ding, X, Tan, C, Liu, X, Wei, X, Wang, H, et al. Association of cerebrospinal fluid zinc-alpha2-glycoprotein and tau protein with temporal lobe epilepsy and related white matter impairment. Neuroreport. (2019) 30:586–91. doi: 10.1097/WNR.0000000000001252
231. Nass, RD, Akgün, K, Elger, C, Reichmann, H, Wagner, M, Surges, R, et al. Serum biomarkers of cerebral cellular stress after self-limiting tonic clonic seizures: an exploratory study. Seizure. (2021) 85:1–5. doi: 10.1016/j.seizure.2020.12.009
232. Lozano, D, Gonzales-Portillo, GS, Acosta, S, de la Pena, I, Tajiri, N, Kaneko, Y, et al. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat. (2015) 11:97–106. doi: 10.2147/NDT.S65815
233. Kovesdi, E, Kamnaksh, A, Wingo, D, Ahmed, F, Grunberg, NE, Long, JB, et al. Acute minocycline treatment mitigates the symptoms of mild blast-induced traumatic brain injury. Front Neurol. (2012) 3:111. doi: 10.3389/fneur.2012.00111
234. Homsi, S, Federico, F, Croci, N, Palmier, B, Plotkine, M, Marchand-Leroux, C, et al. Minocycline effects on cerebral edema: relations with inflammatory and oxidative stress markers following traumatic brain injury in mice. Brain Res. (2009) 1291:122–32. doi: 10.1016/j.brainres.2009.07.031
235. Lee, ST, Chu, K, Jung, KH, Kim, SJ, Kim, DH, Kang, KM, et al. Anti-inflammatory mechanism of intravascular neural stem cell transplantation in haemorrhagic stroke. Brain. (2008) 131:616–29. doi: 10.1093/brain/awm306
236. Aggarwal, S, and Pittenger, MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. (2005) 105:1815–22. doi: 10.1182/blood-2004-04-1559
237. Chen, X, Katakowski, M, Li, Y, Lu, D, Wang, L, Zhang, L, et al. Human bone marrow stromal cell cultures conditioned by traumatic brain tissue extracts: growth factor production. J Neurosci Res. (2002) 69:687–91. doi: 10.1002/jnr.10334
238. Ma, HM, and Zafonte, RD. Amantadine and memantine: a comprehensive review for acquired brain injury. Brain Inj. (2020) 34:299–315. doi: 10.1080/02699052.2020.1723697
239. Wang, KK, Larner, SF, Robinson, G, and Hayes, RL. Neuroprotection targets after traumatic brain injury. Curr Opin Neurol. (2006) 19:514–9. doi: 10.1097/WCO.0b013e3280102b10
240. Sveinbjornsdottir, S, Sander, JWAS, Upton, D, Thompson, PJ, Patsalos, PN, Hirt, D, et al. The excitatory amino acid antagonist D-CPP-ene (SDZ EAA-494) in patients with epilepsy. Epilepsy Res. (1993) 16:165–74. doi: 10.1016/0920-1211(93)90031-2
241. Yurkewicz, L, Weaver, J, Bullock, MR, and Marshall, LF. The effect of the selective NMDA receptor antagonist traxoprodil in the treatment of traumatic brain injury. J Neurotrauma. (2005) 22:1428–43. doi: 10.1089/neu.2005.22.1428
242. Minabe, Y, Emori, K, Shibata, R, and Kurachi, M. Antiepileptic effects of MK-801, a noncompetitive NMDA-receptor antagonist, in the low-frequency kindling model of epilepsy. Jpn J Psychiatry Neurol. (1992) 46:755–61.
243. Alkhachroum, A, der-Nigoghossian, CA, Mathews, E, Massad, N, Letchinger, R, Doyle, K, et al. Ketamine to treat super-refractory status epilepticus. Neurology. (2020) 95:e2286–94. doi: 10.1212/WNL.0000000000010611
244. Rogawski, MA. Therapeutic potential of excitatory amino acid antagonists: channel blockers and 2,3-benzodiazepines. Trends Pharmacol Sci. (1993) 14:325–31. doi: 10.1016/0165-6147(93)90005-5
245. Goodkin, HP, Yeh, JL, and Kapur, J. Status epilepticus increases the intracellular accumulation of GABAA receptors. J Neurosci. (2005) 25:5511–20. doi: 10.1523/JNEUROSCI.0900-05.2005
246. Naylor, DE, Liu, H, Niquet, J, and Wasterlain, CG. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol Dis. (2013) 54:225–38. doi: 10.1016/j.nbd.2012.12.015
247. Mei, Z, Qiu, J, Alcon, S, Hashim, J, Rotenberg, A, Sun, Y, et al. Memantine improves outcomes after repetitive traumatic brain injury. Behav Brain Res. (2018) 340:195–204. doi: 10.1016/j.bbr.2017.04.017
248. Oustad, M, Najafi, M, Mehvari, J, Rastgoo, A, Mortazavi, Z, and Rahiminejad, M. Effect of donepezil and memantine on improvement of cognitive function in patients with temporal lobe epilepsy. J Res Med Sci. (2020) 25:29. doi: 10.4103/jrms.JRMS_209_19
249. Marimuthu, P, Varadarajan, S, Krishnan, M, Shanmugam, S, Kunjuraman, G, Ravinder, JR, et al. Evaluating the efficacy of memantine on improving cognitive functions in epileptic patients receiving anti-epileptic drugs: a double-blind placebo-controlled clinical trial (phase IIIb pilot study). Ann Indian Acad Neurol. (2016) 19:344–50. doi: 10.4103/0972-2327.179971
250. Leeman-Markowski, BA, Meador, KJ, Moo, LR, Cole, AJ, Hoch, DB, Garcia, E, et al. Does memantine improve memory in subjects with focal-onset epilepsy and memory dysfunction? A randomized, double-blind, placebo-controlled trial. Epilepsy Behav. (2018) 88:315–24. doi: 10.1016/j.yebeh.2018.06.047
251. Bialer, M, Johannessen, SI, Levy, RH, Perucca, E, Tomson, T, and White, HS. Progress report on new antiepileptic drugs: a summary of the tenth EILAT conference (EILAT X). Epilepsy Res. (2010) 92:89–124. doi: 10.1016/j.eplepsyres.2010.09.001
252. Graebenitz, S, Kedo, O, Speckmann, EJ, Gorji, A, Panneck, H, Hans, V, et al. Interictal-like network activity and receptor expression in the epileptic human lateral amygdala. Brain. (2011) 134:2929–47. doi: 10.1093/brain/awr202
253. Chappell, AS, Sander, JW, Brodie, MJ, Chadwick, D, Lledo, A, Zhang, D, et al. A crossover, add-on trial of talampanel in patients with refractory partial seizures. Neurology. (2002) 58:1680–2. doi: 10.1212/WNL.58.11.1680
254. Steinhoff, BJ, Ben-Menachem, E, Ryvlin, P, Shorvon, S, Kramer, L, Satlin, A, et al. Efficacy and safety of adjunctive perampanel for the treatment of refractory partial seizures: a pooled analysis of three phase III studies. Epilepsia. (2013) 54:1481–9. doi: 10.1111/epi.12212
255. Witt, JA, and Helmstaedter, C. The impact of perampanel on cognition: a systematic review of studies employing standardized tests in patients with epilepsy. Seizure. (2022) 94:107–11. doi: 10.1016/j.seizure.2021.12.001
256. Chen, T, Liu, WB, Qian, X, Xie, KL, and Wang, YH. The AMPAR antagonist perampanel protects the neurovascular unit against traumatic injury via regulating Sirt3. CNS Neurosci Ther. (2021) 27:134–44. doi: 10.1111/cns.13580
257. Chen, T, Dai, SH, Jiang, ZQ, Luo, P, Jiang, XF, Fei, Z, et al. The AMPAR antagonist Perampanel attenuates traumatic brain injury through anti-oxidative and anti-inflammatory activity. Cell Mol Neurobiol. (2017) 37:43–52. doi: 10.1007/s10571-016-0341-8
258. Kumamoto, A, Chiba, Y, Suda, A, Hishimoto, A, and Kase, A. A severe dementia case in end of life care with psychiatric symptoms treated by Perampanel. J Epilepsy Res. (2021) 11:93–5. doi: 10.14581/jer.21012
259. Chen, YS, Chen, TS, and Huang, CW. Dementia with non-convulsive seizures: a case report. J Int Med Res. (2021) 49:3000605211062453. doi: 10.1177/03000605211062453
260. Ueda, S, Kuzuya, A, Kawata, M, Okawa, K, Honjo, C, Wada, T, et al. Acute inhibition of AMPA receptors by perampanel reduces amyloid β-protein levels by suppressing β-cleavage of APP in Alzheimer's disease models. FASEB J. (2023) 37:e23252. doi: 10.1096/fj.202300837R
261. Attwell, PJE, Singh Kent, N, Jane, DE, Croucher, MJ, and Bradford, HF. Anticonvulsant and glutamate release-inhibiting properties of the highly potent metabotropic glutamate receptor agonist (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV). Brain Res. (1998) 805:138–43. doi: 10.1016/S0006-8993(98)00698-2
262. Kłodzińska, A, Bijak, M, Chojnacka-Wójcik, E, Kroczka, B, Świąder, M, Czuczwar, SJ, et al. Roles of group II metabotropic glutamate receptors in modulation of seizure activity. Naunyn Schmiedeberg's Arch Pharmacol. (2000) 361:283–8. doi: 10.1007/s002109900197
263. Miyamoto, M, Ishida, M, and Shinozaki, H. Anticonvulsive and neuroprotective actions of a potent agonist (DCG-IV) for group II metabotropic glutamate receptors against intraventricular kainate in the rat. Neuroscience. (1997) 77:131–40. doi: 10.1016/S0306-4522(96)00442-3
264. Tang, FR, Lee, WL, Yang, J, Sim, MK, and Ling, EA. Expression of metabotropic glutamate receptor 1alpha in the hippocampus of rat pilocarpine model of status epilepticus. Epilepsy Res. (2001) 46:179–89. doi: 10.1016/S0920-1211(01)00276-5
265. Merlin, LR, Bergold, PJ, and Wong, RK. Requirement of protein synthesis for group I mGluR-mediated induction of epileptiform discharges. J Neurophysiol. (1998) 80:989–93. doi: 10.1152/jn.1998.80.2.989
266. Ghauri, M, Chapman, AG, and Meldrum, BS. Convulsant and anticonvulsant actions of agonists and antagonists of group III mGluRs. Neuroreport. (1996) 7:1469–74. doi: 10.1097/00001756-199606170-00005
267. Chapman, AG, Talebi, A, Yip, PK, and Meldrum, BS. Anticonvulsant activity of a mGlu4α receptor selective agonist, (1S,3R,4S)-1-aminocyclopentane-1,2,4-tricarboxylic acid. Eur J Pharmacol. (2001) 424:107–13. doi: 10.1016/S0014-2999(01)01013-5
268. Abdul-Ghani, A-S, Attwell, PJE, Singh Kent, N, Bradford, HF, Croucher, MJ, and Jane, DE. Anti-epileptogenic and anticonvulsant activity of l-2-amino-4-phosphonobutyrate, a presynaptic glutamate receptor agonist. Brain Res. (1997) 755:202–12. doi: 10.1016/S0006-8993(97)00098-X
269. Chapman, AG, Nanan, K, Yip, P, and Meldrum, BS. Anticonvulsant activity of a metabotropic glutamate receptor 8 preferential agonist, (R,S)-4-phosphonophenylglycine. Eur J Pharmacol. (1999) 383:23–7. doi: 10.1016/S0014-2999(99)00615-9
270. Moldrich, RX, Beart, PM, Jane, DE, Chapman, AG, and Meldrum, BS. Anticonvulsant activity of 3,4-dicarboxyphenylglycines in DBA/2 mice. Neuropharmacology. (2001) 40:732–5. doi: 10.1016/S0028-3908(01)00002-8
271. van Dyck, CH, Swanson, CJ, Aisen, P, Bateman, RJ, Chen, C, Gee, M, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. (2022) 388:9–21. doi: 10.1056/NEJMoa2212948
272. Swanson, CJ, Zhang, Y, Dhadda, S, Wang, J, Kaplow, J, Lai, RYK, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther. (2021) 13:80. doi: 10.1186/s13195-021-00813-8
273. McDade, E, Cummings, JL, Dhadda, S, Swanson, CJ, Reyderman, L, Kanekiyo, M, et al. Lecanemab in patients with early Alzheimer's disease: detailed results on biomarker, cognitive, and clinical effects from the randomized and open-label extension of the phase 2 proof-of-concept study. Alzheimers Res Ther. (2022) 14:191. doi: 10.1186/s13195-022-01124-2
275. Sevigny, J, Chiao, P, Bussière, T, Weinreb, PH, Williams, L, Maier, M, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. (2016) 537:50–6. doi: 10.1038/nature19323
276. Vaz, M, Silva, V, Monteiro, C, and Silvestre, S. Role of Aducanumab in the treatment of Alzheimer’s disease: challenges and opportunities. Clin Interv Aging. (2022) 17:797–810. doi: 10.2147/CIA.S325026
277. Rubenstein, R, Sharma, DR, Chang, B, Oumata, N, Cam, M, Vaucelle, L, et al. Novel mouse Tauopathy model for repetitive mild traumatic brain injury: evaluation of Long-term effects on cognition and biomarker levels after therapeutic inhibition of tau phosphorylation. Front Neurol. (2019) 10:124. doi: 10.3389/fneur.2019.00124
278. Wang, JZ, Grundke-Iqbal, I, and Iqbal, K. Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and-1. Brain Res Mol Brain Res. (1996) 38:200–8. doi: 10.1016/0169-328X(95)00316-K
279. Gong, CX, Shaikh, S, Wang, JZ, Zaidi, T, Grundke-Iqbal, I, and Iqbal, K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. (1995) 65:732–8. doi: 10.1046/j.1471-4159.1995.65020732.x
280. Liu, GP, Zhang, Y, Yao, XQ, Zhang, CE, Fang, J, Wang, Q, et al. Activation of glycogen synthase kinase-3 inhibits protein phosphatase-2A and the underlying mechanisms. Neurobiol Aging. (2008) 29:1348–58. doi: 10.1016/j.neurobiolaging.2007.03.012
281. Tanimukai, H, Grundke-Iqbal, I, and Iqbal, K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer's disease. Am J Pathol. (2005) 166:1761–71. doi: 10.1016/S0002-9440(10)62486-8
282. Griebel, G, Stemmelin, J, Lopez-Grancha, M, Boulay, D, Boquet, G, Slowinski, F, et al. The selective GSK3 inhibitor, SAR502250, displays neuroprotective activity and attenuates behavioral impairments in models of neuropsychiatric symptoms of Alzheimer's disease in rodents. Sci Rep. (2019) 9:18045. doi: 10.1038/s41598-019-54557-5
283. Lucey, BP, Liu, H, Toedebusch, CD, Freund, D, Redrick, T, Chahin, SL, et al. Suvorexant acutely decreases tau phosphorylation and Aβ in the human CNS. Ann Neurol. (2023) 94:27–40. doi: 10.1002/ana.26641
284. Tchekalarova, JD, Ivanova, NM, Pechlivanova, DM, Atanasova, D, Lazarov, N, Kortenska, L, et al. Antiepileptogenic and neuroprotective effects of losartan in kainate model of temporal lobe epilepsy. Pharmacol Biochem Behav. (2014) 127:27–36. doi: 10.1016/j.pbb.2014.10.005
285. Bar-Klein, G, Cacheaux, LP, Kamintsky, L, Prager, O, Weissberg, I, Schoknecht, K, et al. Losartan prevents acquired epilepsy via TGF-β signaling suppression. Ann Neurol. (2014) 75:864–75. doi: 10.1002/ana.24147
286. Hong, S, JianCheng, H, JiaWen, W, ShuQin, Z, GuiLian, Z, HaiQin, W, et al. Losartan inhibits development of spontaneous recurrent seizures by preventing astrocyte activation and attenuating blood-brain barrier permeability following pilocarpine-induced status epilepticus. Brain Res Bull. (2019) 149:251–9. doi: 10.1016/j.brainresbull.2019.05.002
287. Doege, C, Luedde, M, and Kostev, K. Association between angiotensin receptor blocker therapy and incidence of epilepsy in patients with hypertension. JAMA Neurol. (2022) 79:1296–302. doi: 10.1001/jamaneurol.2022.3413
288. Hajjar, I, and Levey, A. Association between angiotensin receptor blockers and longitudinal decline in tau in mild cognitive impairment. JAMA Neurol. (2015) 72:1069–70. doi: 10.1001/jamaneurol.2015.1001
289. Hajjar, I, Brown, L, Mack, WJ, and Chui, H. Impact of angiotensin receptor blockers on Alzheimer disease neuropathology in a large brain autopsy series. Arch Neurol. (2012) 69:1632–8. doi: 10.1001/archneurol.2012.1010
290. Hajjar, I, Okafor, M, Wan, L, Yang, Z, Nye, JA, Bohsali, A, et al. Safety and biomarker effects of candesartan in non-hypertensive adults with prodromal Alzheimer's disease. Brain Commun. (2022) 4:fcac270. doi: 10.1093/braincomms/fcac270
291. Lee, DY, Lee, KS, Lee, HJ, Kim, DH, Noh, YH, Yu, K, et al. Activation of PERK signaling attenuates Abeta-mediated ER stress. PLoS One. (2010) 5:e10489. doi: 10.1371/journal.pone.0010489
292. Tan, HP, Guo, Q, Hua, G, Chen, JX, and Liang, JC. Inhibition of endoplasmic reticulum stress alleviates secondary injury after traumatic brain injury. Neural Regen Res. (2018) 13:827–36. doi: 10.4103/1673-5374.232477
293. Yokoi, N, Fukata, Y, Kase, D, Miyazaki, T, Jaegle, M, Ohkawa, T, et al. Chemical corrector treatment ameliorates increased seizure susceptibility in a mouse model of familial epilepsy. Nat Med. (2015) 21:19–26. doi: 10.1038/nm.3759
Keywords: epilepsy, Alzheimer’s disease, tau phosphorylation, amyloid-beta, TBI, endoplasmic reticulum stress
Citation: Martin SP and Leeman-Markowski BA (2024) Proposed mechanisms of tau: relationships to traumatic brain injury, Alzheimer’s disease, and epilepsy. Front. Neurol. 14:1287545. doi: 10.3389/fneur.2023.1287545
Edited by:
Luisa Lilia Rocha, National Polytechnic Institute of Mexico (CINVESTAV), MexicoReviewed by:
Melissa Barker-Haliski, University of Washington, United StatesP. Hemachandra Reddy, Texas Tech University Health Sciences Center, United States
Copyright © 2024 Martin and Leeman-Markowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samantha P. Martin, U2FtYW50aGEuTWFydGluQG55dWxhbmdvbmUub3Jn