 Haiyan Tang
Haiyan Tang Yingying Luo
Yingying Luo Zhenchu Tang
Zhenchu Tang Jianguang Tang2
Jianguang Tang2 Jia Fang
Jia Fang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 12 October 2023
Sec. Neurogenetics
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1270793
This article is part of the Research Topic Diagnosis, Animal Models and Therapeutic Interventions for Neuromuscular Diseases View all 5 articles
SCPx deficiency is a rare disorder of peroxisomal beta-oxidation dysfunction, and it has only been documented in two patients thus far. In the previously reported patients, both patients were primarily presented with slowly progressive dystonia or ataxia, and they both displayed symmetrical lesions in the thalamus and brainstem on magnetic resonance imaging. This study presents the third patient exhibiting a similar neuroimaging abnormality but a notably different clinical phenotype characterized by episodic psychosis. Through whole-exome sequencing, we identified a homozygous splicing mutation in SCP2 (c.674 + 1G > C), and further RNA sequencing revealed exon 8 skipping in the mature transcripts of SCP2. This study significantly expands our understanding of the genotypic and phenotypic spectrum associated with SCP2-related metabolic encephalopathy.
SCP2 gene is located on chromosome 1p32 and encodes two distinct proteins, sterol carrier protein X (SCPx) and sterol carrier protein 2 (SCP2), via separate promoters (1). SCPx functions as a peroxisomal enzyme with thiolase activity involved in peroxisomal beta-oxidation. Its primary role is in the degradation of very long-chain fatty acids (VLCFA) and branched-chain fatty acids such as phytanic and pristanic acid. In the current study, we reported the case of a patient who presented with episodic psychosis, which was determined to be caused by a novel SCP2 splicing mutation.
Ethics approval was obtained from the Second Xiangya Hospital, Central South University (Changsha, China). Written informed consent was obtained from the patient and his family members for their enrollment. A 48-year-old male was admitted to our hospital thrice between 2012 and 2017 due to episodic psychosis. During each attack, he experienced delusions, irritability, aggressive behavior, bursts of uncontrollable laughter, crying, and talking to himself. Each episode was triggered by an acute upper respiratory tract infection. He was the third child of healthy, non-consanguineous parents with no family history of relevant conditions. The patient had experienced stuttering and tremors in his lips and hands for as long as his son could remember. Physical signs were the same during each episode. Examination revealed confusion, restlessness, and disorientation to time, person, and place, with normal limb muscle strength, brisk deep tendon reflexes, positive bilateral palmomental reflex, sucking reflex, and Babinski's reflex. On each occasion, cranial magnetic resonance imaging (MRI) revealed symmetrical T2 hyperintense signals in the thalamus, mesocephalon, and pons without gadolinium enhancement (see Figure 1). The cerebrospinal fluid (CSF) showed no abnormalities. No antibodies were detected for autoimmune encephalitis, paraneoplastic neurological syndromes, and rheumatologic disorders. Ceruloplasmin, lactic acid, free carnitine, acylcarnitines, amino acids in the blood, and organic acids in the urine were all within normal ranges. Nerve conduction studies and needle electromyography showed no abnormalities. The patient was previously diagnosed with viral encephalitis, cerebral infarction, or mitochondrial encephalopathy. He received appropriate treatments, such as antiviral and antiplatelet therapy, during each hospitalization. Psychiatric symptoms gradually improved after 3–6 months of treatment with antipsychotic drugs such as olanzapine or quetiapine. Between episodes, the patient was able to care for himself and engage in communication with others independently.
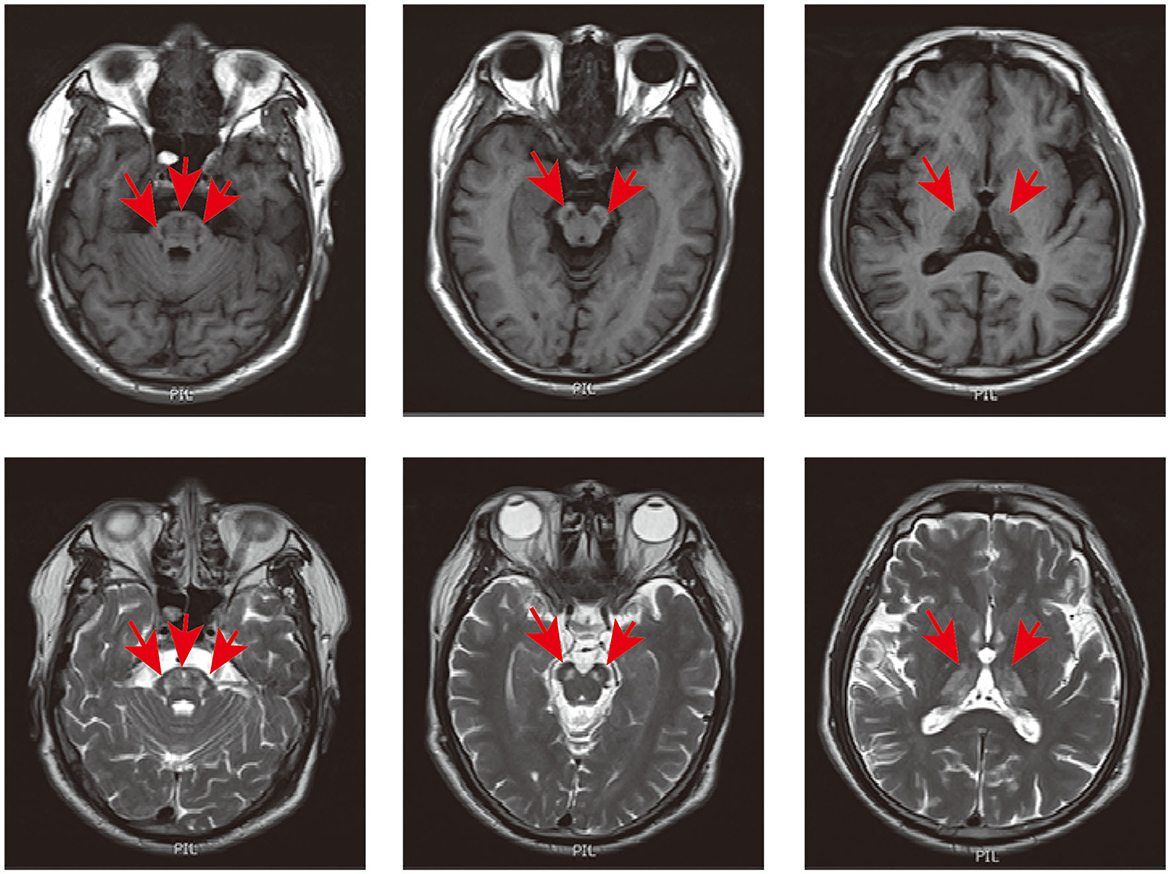
Figure 1. Cranial MRI of the patient. Axial T1 and T2-weighted imaging showed hypointense and hyperintense signals in the thalamus, mesocephalon, and pons (indicated in red arrow).
In January 2018, the patient was readmitted to the hospital for continuous generalized tonic-clonic seizures (GTCS) that lasted 18 h without recovery between seizures. The GTCS occurred every 10 min, with each seizure lasting 1–2 min. Upon admission, he presented with confusion. The physical examination was limited due to poor cooperation. Pristanic acid concentration was slightly elevated at 0.62 μmol/L, just above the control range (<0.60 μmol/L), while VLCFA and phytanic acid were within the standard limit. Cranial MRI revealed abnormalities consistent with prior findings.
Whole-exome sequencing of the proband revealed a homozygous splicing mutation, c.674 + 1G > C, in SCP2 (NM_002979.5) in chr1:53442442 (hg19). Sanger sequencing confirmed the candidate mutation in the proband and his family members. The proband's father and sisters were heterozygotes at this position (see Figures 2A, B). RNA sequencing identified two outlier junctions in the SCP2 cluster, providing support for the skipping of the eighth exon (p = 4.81E-12, FDR = 0, and p = 1.31E-4, FDR = 0.008) (see Figures 2C, D). At the mRNA level, cDNA analysis validated the aberrant splicing pattern between the sixth and ninth exons in the patient's sample through the agarose gel electrophoresis of reverse transcription polymerase chain reaction amplicons (RT-PCR) product (see Figure 2E). Sanger sequencing of the RT-PCR product confirmed the exon 8 skipping event in the patient (see Figure 2F). The eighth exon has a length of 87 base pairs. This mutation results in the loss of 29 amino acid residues in the patients' SCPx protein. The allele frequency is approximately 0.00001992 in the gnomAD database. In the case of SCP2, loss of function is a recognized disease mechanism. The variant c.674 + 1G > C in SCP2 was classified as “pathogenic” based on the ACMG guidelines.
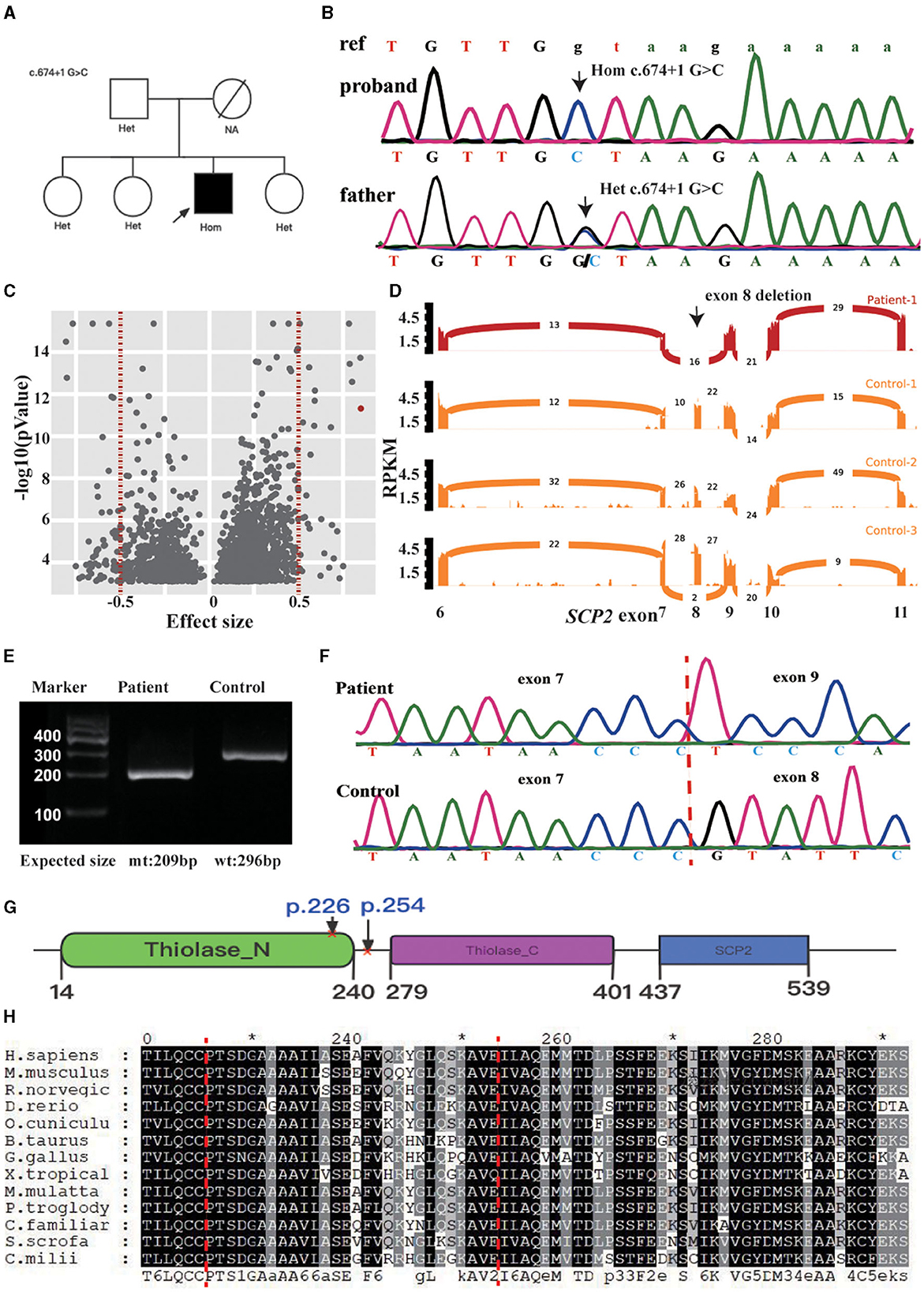
Figure 2. (A) Pedigree chart of this family. (B) Validation of SCP2 c.674 + 1 G>C by Sanger sequence in the patient and his father. The ref indicates the reference sequence, with capital letters and lowercase, representing the nucleotide in the exon and intron regions. (C) Outlier-level significance [-log10(p), y-axis] vs. effect size (junction count/total junction count-mean (total junctions count/total count of junctions in the cluster), x-axis) for the patient. There are 2030s outliers with an adjusted p < 0.05 in 1442 genes (red dots indicating two clusters in SCP2, vertical dotted lines indicating the effect size cutoff). (D) Sashimi plot of the eighth exon-skipping event (indicated in black arrow) in RNA-seq samples of the SCP2-affected (red) and three representative SCP2-unaffected (orange) individuals. The RNA-seq read coverage is given as the log10 RPKM-value (Reads Per Kilobase of transcript per Million mapped reads, y-axis), and the number of splits reads spanning an intron is indicated on the exon-connecting line. (E) The skipping event is observable in the RT-PCR product from patient blood. Mt: mtant type, wt: wide type. (F) Exon 8 skipping is validated by the Sanger sequencing of RT-PCR product. (G) schematic representation of the SCPx and the affected domain of the index patient, surrounding the amino acids region coding by the eighth exon (p. 225–254, indicated between the red cross). (H) Protein sequence alignment of SCP2 orthologs, showing the region surrounding the p. 225–254 is relatively conserved (indicated between the red dot lines). *Indicates the termination codon.
The patient's seizures were effectively halted with diazepam, and the psychiatric symptoms gradually improved. Subsequently, the patient was discharged while being placed on a phytanic acid-restricted diet.
SCPx deficiency, a rare autosomal recessive monogenic metabolic encephalopathy, has been reported in only two patients. One patient presented with leukoencephalopathy with clinical features such as slowly progressive dystonia, stutter, and motor neuropathy (2). The other patient developed hand clumsiness in his 30s, followed by gait disturbance and deafness (3). The index patient displayed episodic psychobehavioral disturbances, stutter, tremors, and epilepsy. MRI findings were consistent among all three patients, revealing symmetrical thalamus and brainstem lesions without gadolinium enhancement. As summarized in Table 1, these shared clinical features in SCPx deficiency include stutter, tremors, and symmetrical lesions in the thalamus and brainstem without gadolinium enhancement (see Table 1). Importantly, our patient first presented with episodic psychosis and epilepsy in SCPx deficiency patients highlighting the necessity for clinicians to consider the SCP2 variant during the etiological examination of individuals presenting with episodic psychosis and epilepsy.
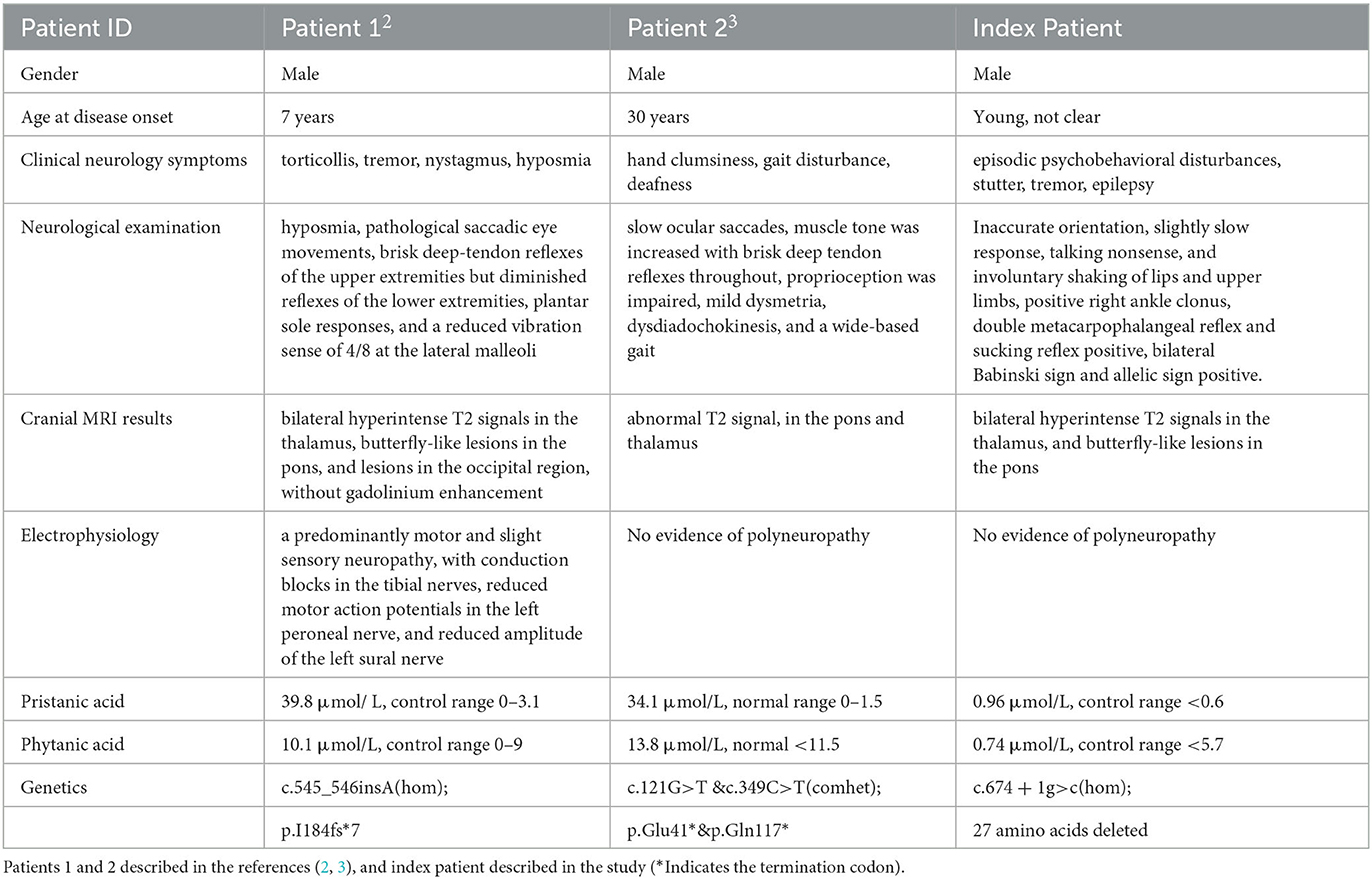
Table 1. Clinical phenotypes and laboratory investigation results of patients with SCP2 mutations.
Patients in the literature exhibited elevated pristanic acid levels, whereas the index patient showed a mild increase. Mutations in HSD17B4, which resulted in less structural damage to the D-bifunctional protein, were associated with a milder clinical and biochemical presentation (4). The D-bifunctional protein is situated upstream of SCPx in peroxisomal beta-oxidation. In our patient, the deletion of amino acid residues due to exon skipping occurs within a relatively conserved region of the protein, and this deletion region intersects with the thiolase_N domain (see Figures 2G, H). The function of the thiolase may be impaired but remains partially functional.
Intriguingly, episodic psychosis in the index patient typically follows infections, indicating that “crises” in SCPx deficiency may be provoked by factors such as infections or extended fasting, similar to certain metabolic encephalopathies, such as isolated methylmalonic acidemia (5).
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the Second Xiangya Hospital, Central South University (Changsha, China). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
HT: Formal analysis, Writing—original draft. YL: Software, Writing—review and editing. ZT: Data curation, Writing—review and editing. JT: Investigation, Writing—review and editing. JF: Investigation, Supervision, Writing—original draft.
This research was supported by the Natural Science Foundation of Hunan (Grant/Award Number: 2023JJ30750) and the National Natural Science Foundation Youth Fund Project (Grant/Award Number: 82201354).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Yamamoto R, Kallen CB, Babalola GO, Rennert H, Billheimer JT, Strauss JF. 3rd. Cloning and expression of a cDNA encoding human sterol carrier protein 2. Proc Natl Acad Sci U S A. (1991) 88:463–7. doi: 10.1073/pnas.88.2.463
2. Ferdinandusse S, Kostopoulos P, Denis S, Rusch H, Overmars H, Dillmann U, et al. Mutations in the gene encoding peroxisomal sterol carrier protein X (SCPx) cause leukencephalopathy with dystonia and motor neuropathy. Am J Hum Genet. (2006) 78:1046–52. doi: 10.1086/503921
3. Horvath R, Lewis-Smith D, Douroudis K, Duff J, Keogh M, Pyle A, et al. SCP2 mutations and neurodegeneration with brain iron accumulation. Neurology. (2015) 85:1909–11. doi: 10.1212/WNL.0000000000002157
4. Ferdinandusse S, Ylianttila MS, Gloerich J, Koski MK, Oostheim W, Waterham HR, et al. Mutational spectrum of D-bifunctional protein deficiency and structure-based genotype-phenotype analysis. Am J Hum Genet. (2006) 78:112–24. doi: 10.1086/498880
Keywords: episodic psychosis, peroxisomal beta-oxidation, SCPx deficiency, SCP2 mutation, splicing mutation
Citation: Tang H, Luo Y, Tang Z, Tang J and Fang J (2023) Case report: Episodic psychosis caused by a novel SCP2 splicing mutation. Front. Neurol. 14:1270793. doi: 10.3389/fneur.2023.1270793
Received: 03 August 2023; Accepted: 18 September 2023;
Published: 12 October 2023.
Edited by:
Muthukumar Karuppasamy, University of Alabama at Birmingham, United StatesReviewed by:
Jose Laffita Mesa, Karolinska Institutet (KI), SwedenCopyright © 2023 Tang, Luo, Tang, Tang and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia Fang, NTA3MDkyQGNzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.