
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 13 October 2023
Sec. Pediatric Neurology
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1254140
This article is part of the Research TopicCase Reports in Pediatric Neurology: Volume III - 2023View all 6 articles
 Julia Macintosh1,2†
Julia Macintosh1,2† Stefanie Perrier1,2†
Stefanie Perrier1,2† Maxime Pinard3Luan T. Tran1,2†Kether Guerrero1,2†
Maxime Pinard3Luan T. Tran1,2†Kether Guerrero1,2† Chitra Prasad4,5,6†
Chitra Prasad4,5,6† Asuri N. Prasad4,6†
Asuri N. Prasad4,6† Tomi Pastinen7,8,9†
Tomi Pastinen7,8,9† Isabelle Thiffault7,8,10†
Isabelle Thiffault7,8,10† Benoit Coulombe3,11†
Benoit Coulombe3,11† Geneviève Bernard1,2,12,13,14*†
Geneviève Bernard1,2,12,13,14*†RNA polymerase III-related leukodystrophy (POLR3-related leukodystrophy) is a rare, genetically determined hypomyelinating disease arising from biallelic pathogenic variants in genes encoding subunits of RNA polymerase III (Pol III). Here, we describe the first reported case of POLR3-related leukodystrophy caused by biallelic pathogenic variants in POLR3D, encoding the RPC4 subunit of Pol III. The individual, a female, demonstrated delays in walking and expressive and receptive language as a child and later cognitively plateaued. Additional neurological features included cerebellar signs (e.g., dysarthria, ataxia, and intention tremor) and dysphagia, while non-neurological features included hypodontia, hypogonadotropic hypogonadism, and dysmorphic facial features. Her MRI was notable for diffuse hypomyelination with myelin preservation of early myelinating structures, characteristic of POLR3-related leukodystrophy. Exome sequencing revealed the biallelic variants in POLR3D, a missense variant (c.541C > T, p.P181S) and an intronic splice site variant (c.656-6G > A, p.?). Functional studies of the patient’s fibroblasts demonstrated significantly decreased RNA-level expression of POLR3D, along with reduced expression of other Pol III subunit genes. Notably, Pol III transcription was also shown to be aberrant, with a significant decrease in 7SK RNA and several distinct tRNA genes analyzed. Affinity purification coupled to mass spectrometry of the POLR3D p.P181S variant showed normal assembly of Pol III subunits yet altered interaction of Pol III with the PAQosome chaperone complex, indicating the missense variant is likely to alter complex maturation. This work identifies biallelic pathogenic variants in POLR3D as a novel genetic cause of POLR3-related leukodystrophy, expanding the molecular spectrum associated with this disease, and proposes altered tRNA homeostasis as a factor in the underlying biology of this hypomyelinating disorder.
Leukodystrophies are a heterogeneous group of rare, inherited white matter disorders (1). In particular, as one of the most common hypomyelinating leukodystrophies, POLR3-related leukodystrophy presents in previously healthy children with a variety of neurological and non-neurological clinical features, together with a characteristic hypomyelination pattern on brain MRI (2, 3). The MRI pattern includes hypomyelination, appearing as hyperintensity of white matter relative to gray matter on T2-weighted images and variable signal (hyperintense, hypointense, or isointense) of white matter relative to gray matter on T1-weighted images, along with relative preservation of myelination in early myelinating structures (i.e., dentate nucleus, optic radiations, globus pallidus, anterolateral nucleus of the thalamus, and in some cases, corticospinal tracts at the level of the posterior limb of the internal capsule), with or without thinning of the corpus callosum and cerebellar atrophy (4). Neurological features are predominantly motor and can include cerebellar (e.g., abnormal extraocular movements, dysarthria, ataxia, and dysmetria), pyramidal (e.g., spasticity), and extrapyramidal (e.g., dystonia) signs, along with variable cognitive involvement (3). POLR3-related leukodystrophy is further clinically distinguished from other leukodystrophies by characteristic non-neurological features including dental abnormalities (e.g., hypodontia) and endocrine abnormalities (e.g., hypogonadotropic hypogonadism and short stature), from which the alternate disease name, 4H leukodystrophy (i.e., hypomyelination, hypodontia, and hypogonadotropic hypogonadism) was derived (5). Ocular abnormalities, typically myopia, are also seen, as is craniofacial involvement, the latter most commonly seen in individuals harboring biallelic pathogenic variants in the POLR1C gene (5–7).
RNA polymerase III (Pol III) is a 17-subunit enzymatic complex tasked, in eukaryotes, with the transcription of critical and ubiquitous non-coding RNAs, including 5S ribosomal RNA, all tRNAs, and a variety of other non-coding RNAs involved in transcription, RNA processing and localization, and/or translation (8). While autosomal recessive variants in POLR3A (OMIM *614258; OMIM #607694) and POLR3B (OMIM *614366; OMIM #614381), which encode the catalytic subunits of the Pol III enzymatic complex, were originally identified as the etiology of this disease, biallelic pathogenic variants in POLR1C (OMIM *610060; OMIM #616494) and POLR3K (OMIM *606007; OMIM #619310) have since been recognized as causative (9–12). Nonetheless, a subset of individuals with clinical and neuroradiological characteristics aligning with this disease still remain without a genetic diagnosis.
In this study, we report the first case of POLR3-related leukodystrophy arising from biallelic pathogenic variants in the POLR3D gene, in an individual with a clinical and neuroradiological phenotype consistent with POLR3-related leukodystrophy. In addition, we demonstrate that these novel variants, including a missense variant [NM_001722.3 (POLR3D): c.541C > T, p.P181S] and an intronic splice site variant [NM_001722.3 (POLR3D): c.656-6G > A, p.?], cause altered RNA-level expression of Pol III subunits and Pol III transcripts. We also demonstrate that the missense variant impacts the interaction between Pol III and its chaperone complex.
Written informed consent from the legal representative of the patient was obtained. This study was approved by the Montreal Children’s Hospital and the McGill University Health Center Research Ethic Boards (11-105-PED, 2019-4972) and the Children’s Mercy Institutional Review Board (study #11120514). A retrospective review of available medical records and brain MRI was performed.
Using genomic DNA extracted from peripheral blood leukocytes, exome sequencing was initially performed by PerkinElmer, as previously described (12), and later repeated using the Illumina TruSeq library preparation and IDT XGen Exome Enrichment protocol (13). Sequences were aligned to reference gene sequences based on the human genome build (GRCh38/hg38) and analyzed using the custom-developed software RUNES and Viking (14). Variant interpretation was performed using the American College of Medical Genetics and Genomics (ACMG) standards and guidelines for the interpretation of sequence variants (14).
To analyze transcription levels of Pol III subunit genes and Pol III-transcribed genes, RNA was collected from fibroblasts and cDNA was synthesized for RT-qPCR analysis. Primer designs are provided in the supplemental materials (Supplementary Table S1).
To assess how the missense variant in POLR3D impacts the assembly of the Pol III complex, the POLR3D WT sequence (NCBI sequence: BC004484) was amplified by PCR using the following primers (POLR3D HindIII F: 5′-CCC AAG CTT ATG TCG GAA GGA AAC GCC GCC G-3′ and POLR3D XhoI R: 5′-CCG CTC GAG TTA CCG GTG TTT GTG ATC CAA GAG G-3′) and cloned in the pCMV-3tag-1a parental plasmids (between HindIII and XhoI cloning site). The p.P181S mutation was introduced by site-directed mutagenesis by using the overlapping primers F: 5′-GAA ATA TGC CTG TGC AGC TGT CGC TGG CTC ACT CAG GAT G-3′ and R: 5′-CAT CCT GAG TGA GCC AGC GAC AGC TGC ACA GGC ATA TTT C-3′ (15). Both sequences were confirmed by Sanger sequencing. Stable clones of POLR3D WT and p.P181S were produced in HeLa cells by transfecting each digested plasmids with Jet Prime, following manufacturer’s instruction. 3x-FLAG-POLR3 WT and p.P181S expression level of the different clones was assessed via western blotting by using an anti-FLAG antibody to selected clones with equivalent expression.
Affinity purification followed by liquid chromatography–tandem mass spectrometry (LC–MS/MS) acquisition and raw data analysis were performed, as previously described (16).
Detailed methods pertaining to genetic analysis, fibroblast culture, RT-qPCR work, and proteomic studies are described in the Supplementary material.
The patient, a female, was born to non-consanguineous parents. The pregnancy was complicated by concerns over fetal size throughout the gestational period and a large blood clot was passed in the first few weeks of pregnancy. Delivery was unremarkable. In the neonatal period, the patient was noted to have feeding difficulties and required IV fluids initially, which later improved. Her development was noted to be delayed and she did not walk until 18 months. In her early years of school, teachers noticed delays in both expressive and receptive language, and she was formally diagnosed with learning disabilities. Vision and hearing were normal. She experienced a cognitive plateau and at age 26 years, was functioning at a grade 6 reading level and grade 3 comprehension level. Neuropsychological assessment revealed that most areas of cognition, including working memory and attention, were impaired. She was also noted to have cerebellar involvement, including an ataxic gait, intention tremor, and dysarthria. Silent laryngeal aspirations of thin fluids were remarked on a modified barium swallow study, and she was diagnosed with dysphagia at age 28 years. In her late 20s, she started to have trouble with ambulation and became wheelchair dependent by her mid-30s, at which time she was also considered to be anarthric. She died at the age of 37.
Of interest, she also had various non-neurological features characteristic of POLR3-related leukodystrophy. These included hypodontia, as she was missing two maxillary lateral incisors and two mandibular second premolars. Regarding endocrine abnormalities, she had hypogonadotropic hypogonadism. On MRI, her pituitary gland was noted to be small. A bone mineral density exam revealed osteopenia. She did not have short stature nor ocular involvement, including no myopia. Interestingly, she was noted to have non-specific dysmorphic facial features, with deep-set eyes and frontal prominence.
Various investigations were pursued to identify the etiology of her neurodegenerative condition. Karyotyping and multi-telomere fluorescence in situ hybridization (FISH) revealed no chromosomal or telomere abnormalities, respectively. A negative transferrin isoelectric focusing ruled out disorders of glycosylation, and mitochondrial DNA analysis was unremarkable. Metabolic diseases, including mucopolysaccharidosis and mucolipidosis type IV, were considered but ruled out upon further testing. An expansion was found in the SCA8 gene (95/25), inherited from the father (124/23), and associated with spinocerebellar ataxia type 8. However, given that expansions of SCA8 are not all associated with cerebellar ataxia, and considering her age at disease onset and the presence of hypomyelination and non-neurological features, this finding was considered unlikely to explain her disease (17, 18). At age 24 years, a clinical diagnosis of 4H leukodystrophy was suggested, prior to the identification of the genetic etiology of this disorder (19).
Initial magnetic resonance imaging (MRI) performed at age 17 years revealed diffuse hypomyelination. Repeat MRIs at 23 and 29 years likewise demonstrated these white matter abnormalities, along with a thin corpus callosum and cerebellar atrophy (Figures 1A,E). A pattern of myelin preservation was seen in certain structures, including the optic radiations, dentate nucleus, anterolateral nucleus of the thalamus, and the globus pallidus, which are known to be early-myelinating structures characteristically spared in POLR3-related leukodystrophy (Figure 1) (5, 20). Cerebellar atrophy and a thin corpus callosum were likewise seen on a computed tomography (CT) scan performed at age 24 years, which also revealed mild–moderate ventricular enlargement of both the infra- and supratentorial ventricles and cerebral atrophy.
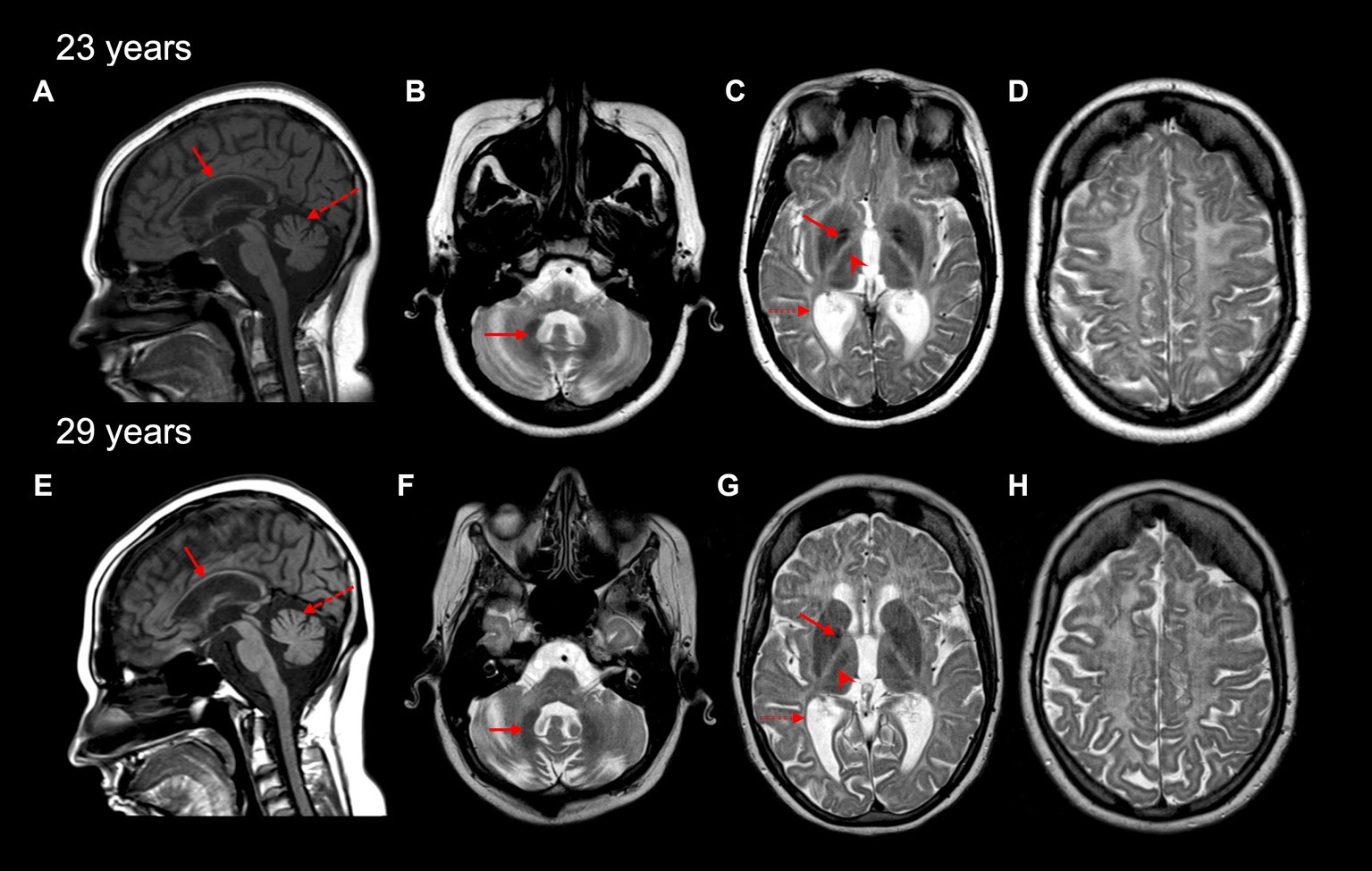
Figure 1. Brain MRI characteristics of the affected individual at 23 and 29 years old. A thin corpus callosum (red arrow) and cerebellar vermis atrophy (red dashed arrow) are seen on sagittal T1-weighted MRI images (A,E). Cerebellar hemisphere atrophy was also seen at 23 and 29 years (not shown). Diffuse hypomyelination is evident (C,D,G,H), with relative preservation of myelin seen in the dentate nucleus (red arrow in B,F), globus pallidus (red arrow in C,G), the anterolateral nucleus of the thalamus (red arrowhead in C,G), and optic radiations (dashed red arrow in C,G) on axial T2-weighted MRI images.
Initial research exome sequencing (ES) was completed by PerkinElmer in 2013, which first revealed the novel compound heterozygous variants in POLR3D (NM_001722.3): a missense variant (c.541C > T, p.P181S) and an intronic splice site variant (c.656-6G > A, p.?). To further investigate the cause of disease, proband ES was repeated on a research basis in 2022 using an updated sequencing platform, which iteratively confirmed the presence of biallelic variants in POLR3D, without detection of other candidate variants in genes encoding subunits of Pol III, including no deletions or duplications, and no detection of candidates in other leukodystrophy-associated genes. Symptom-driven analysis including genes of uncertain significance was performed, with no diagnostic or partial genotype identified for the patient. Targeted Sanger sequencing and co-segregation analysis confirmed both variants were present in the affected individual, with each parent carrying one variant (Figures 2A–C).
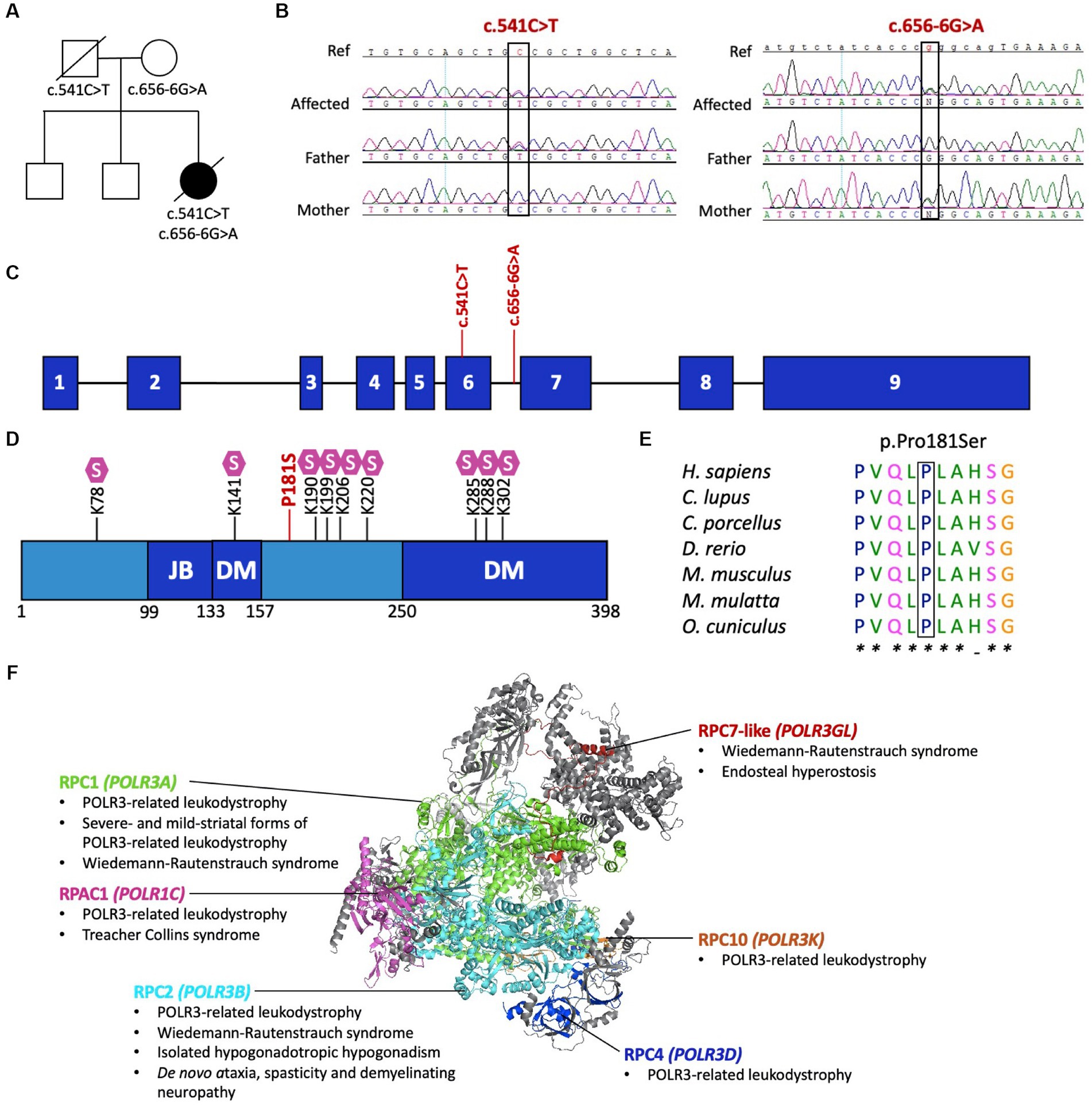
Figure 2. Pathogenic variants in POLR3D. (A) Familial segregation of the c.541C > T (p.P181S) and c.656-6G>A (p.?). (B) POLR3D sequence chromatograms of the affected individual and her parents show segregation of the variants. (C) Schematic of POLR3D genomic DNA with pathogenic variants mapped. (D) Schematic of the domain organization of the RPC4 protein; JB, Jaw binding; DM, Dimerization domain. Lysine residues subjected to sumoylation (S) are marked on the sequence. The missense mutation (red) found in the patient described here is proximal to a sumoylation hotspot. (E) The missense variant in POLR3D is at a conserved amino acid residue. (F) Modeled human structure of RNA polymerase III. Colors identify Pol III subunits encoded by genes that harbor pathogenic variants, including POLR3A (green), POLR3B (cyan), POLR1C (magenta), POLR3D (blue), POLR3K (orange), and POLR3GL (red). Phenotypes associated with the genes are listed under the protein name. In all cases inheritance is autosomal recessive, except for de novo ataxia, spasticity, and demyelinating neuropathy. Modeled on PyMOL, PDB: 7A6H.
The POLR3D missense variant (c.541C > T, p.P181S) is predicted by ACMG guidelines to be pathogenic, based on its absence from population databases (PM2) and in silico evidence of a deleterious effect (PP3). Of interest, the missense variant is proximal to a sumoylation hotspot (Figure 2D) and resides at a conserved residue (Figure 2E).
The POLR3D intronic variant (c.656-6G > A, p.?) is predicted to impact splicing through disruption of the wild-type donor site and activation of a cryptic acceptor site [change at acceptor site (6 bps downstream of exon 6) = −52.9% (range − 93.4% to −12.5%); Human Splice Finder; https://hsf.genomnis.com/] (21). Moreover, the intronic variant is predicted to result in abnormal exon 7 skipping, which would lead to a premature termination codon (p.V219Gfs*13), predicted to cause loss of function due to a truncated or absent product via nonsense mediated decay. Of the 17 genes encoding Pol III subunits, POLR3D is the fifth gene to be associated with a leukodystrophy phenotype and the sixth gene to be associated with a genetic disease (Figure 2F) (22).
To determine the molecular impact of these POLR3D variants on Pol III transcription, RT-qPCR was performed on RNA extracted from primary fibroblasts of the patient and age/sex-matched control cells. RT-qPCR assessment revealed a significant decrease in POLR3D RNA-level expression, as well as a significant decrease in expression of several other Pol III subunits (i.e., POLR3A, POLR3B, POLR1C, POLR3E, and POLR3F; Figure 3A). Assessment of Pol III transcript expression revealed a significant decrease in 7SK RNA. In contrast, U6 spliceosomal RNA was significantly increased relative to the control. The Pol III transcripts 5S, 7SL, RPPH1, and RMRP were not significantly altered. Upon analysis of distinct tRNAs, expression levels of most analyzed tRNAs were reduced. Relative to the control, tRNA-Ala-AGC-10-1, tRNA-Gly-TCC-4-1, and tRNA-Ile-TAT-3-1 showed a significant decrease in expression, with tRNA-Tyr-GTA-8-1 trending toward a decrease but not reaching statistical significance (Figure 3B).
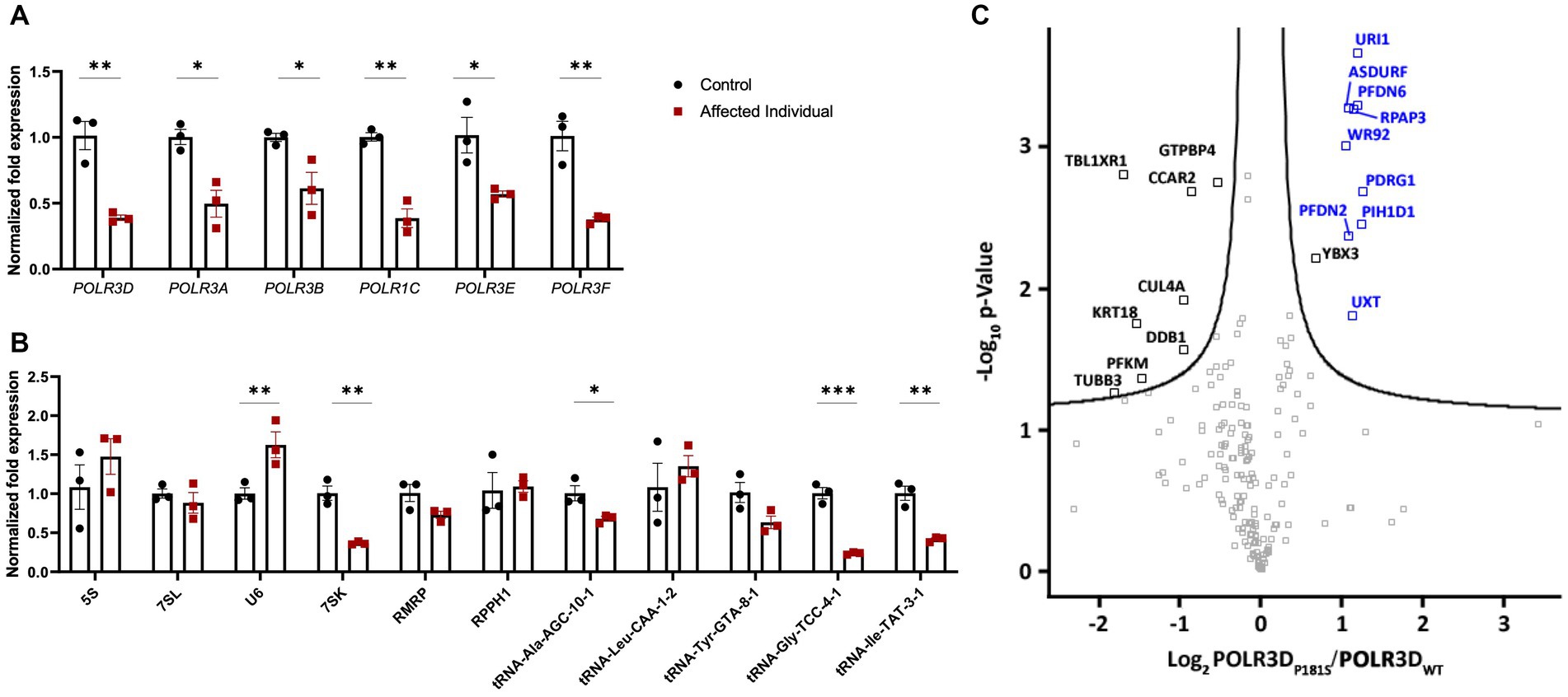
Figure 3. Molecular and complex assembly level implications of pathogenic variants in POLR3D. (A) RT-qPCR analysis of RNA derived from patient-derived fibroblasts and an age- and sex-matched control (n = 3) demonstrating significantly decreased mRNA expression of all Pol III subunits analyzed (e.g., POLR3A, POLR3B, POLR1C, POLR3D, POLR3E, and POLR3F) in the affected individual. (B) RT-qPCR analysis of Pol III transcripts reveals a differential impact of biallelic pathogenic variants on Pol III transcription, including significantly increased expression of U6 RNA and significantly decreased expression of 7SK RNA, tRNA-Ala-GTA-8-1, tRNA-Gly-TCC-4-1, and tRNA-Ile-TAT-3-1 and a trend toward a decrease in tRNA-Tyr-GTA-8-1. RT-qPCR data represent mean ± SEM normalized fold expression after normalizing to reference genes TFRC and PGK1. *p < 0.05, **p < 0.01, ***p < 0.001, two-sided Student’s t-test. (C) Volcano plot illustrating the log2 ratio between POLR3D p.P181S and POLR3D WT proteins (n = 3). PAQosome subunits are marked in blue and all other proteins that cleared the False Discovery Rate (FDR) threshold of 0.05 are marked in black.
The missense mutation seen here resides in an unstructured region of the RPC4 protein (23). In line with this, affinity purification coupled to mass spectrometry (AP-MS) analysis revealed that all Pol III subunits were detected (Supplementary Table S2) and that the missense variant did not impact interactions between RPC4 and its partnering Pol III complex subunits. Nonetheless, nearly all subunits (9/12) of the PAQosome (i.e., the chaperone complex that regulates Pol III) were significantly increased, suggestive of prolonged interaction of the complex with the p.P181S mutant Pol III complex (Figure 3C).
In this study, we report the first individual with a clinical and radiological diagnosis of POLR3-related leukodystrophy caused by biallelic pathogenic variants in POLR3D. The individual had neurological and non-neurological features characteristic of POLR3-related leukodystrophy including motor signs (e.g., cerebellar), cognitive involvement and dysphagia, for the former, and hypodontia, hypogonadotropic hypogonadism, and dysmorphic facial features, for the latter. Further, her MRI pattern was consistent with POLR3-related leukodystrophy, as described above. Based on this clinical and neuroimaging overlap, she had initially been clinically diagnosed with 4H leukodystrophy despite the absence of variants in Pol III genes known to cause this disorder (e.g., POLR3A, POLR3B, POLR1C, and POLR3K). Overall, the individual’s disease course was consistent with the typical presentation of later-onset POLR3-related leukodystrophy, involving disease onset in school years with cognitive impairment, later followed by motor features, with or without non-neurological features (3, 5).
POLR3D encodes the RPC4 subunit of Pol III, which has previously been implicated in transcription initiation and termination (23). Interestingly, RPC4 is known to be affected by some POLR3-related leukodystrophy variants in the POLR3B gene, which cause destabilization of RPC4/RPC5 dimer assembly (24). Further, RPC4 is distinguished as a source of Pol III complex regulation due to its numerous sumoylation sites (25). Sumoylation is a type of post-translational modification conserved from yeast to human, and within RPC4 specifically, sumoylation is thought to be a mechanism of negative regulation for the Pol III complex (25). Indeed, sumoylation of Rpc53, the yeast homolog of RPC4, has been shown to induce ubiquitylation of the complex, followed by Cdc48 segregase-mediated disassembly and ultimately, proteasomal degradation (25). The proximity of the p.P181S missense variant identified here to a sumoylation hotspot in PRC4 is perhaps relevant to the pathogenicity of this variant (23).
Upon RNA-level expression analysis of the affected gene, we found POLR3D to be significantly decreased. To our surprise, when we analyzed additional genes encoding Pol III subunits, these were likewise significantly decreased. Pathogenic variants in genes encoding Pol III subunits have previously been shown to be hypomorphic, and studies have demonstrated reduced protein expression of the affected subunit (9, 26). We propose that the decrease in expression of these genes could result from a compensatory mechanism. As Pol III works as an enzymatic complex, the stoichiometric balance of each subunit is of interest. In the case of a decrease in one Pol III subunit, we suggest that the transcription of non-affected Pol III genes may be dampened, as these subunits would otherwise be found in excess. However, as RNA-level expression is not a faithful representation of protein-level expression, additional work is required to support this hypothesis (27).
Functionally, Pol III plays a crucial cellular role in the expression of various non-coding RNAs. As such, we sought to assess how the variants in POLR3D may impact Pol III transcription. Expression of Pol III-transcribed non-coding RNAs in a diseased context is variable in the literature, which may be in part due to a range of cell types being analyzed in different studies (11, 28–30). Certain Pol III transcripts, classified as Type 3, have upstream promoters reminiscent of those used by Pol II genes, with previous work demonstrating that at least some Type 3 genes have promoters occupied by Pol II, raising the possibility of compensatory transcription (8, 31, 32). As U6 fits into this Type 3 category, this could explain why an increase in U6 is seen. Albeit, RMRP and RPPH1 likewise fit into this Type 3 category, and were not shown to be significantly altered. The 5S and 7SL have also shown variable changes in expression when analyzed in different cell types from individuals with POLR3-related leukodystrophy that have distinct genotypes and disease severities, while we did not see significant changes in either (11, 28, 30). The challenges associated with assessing tRNAs using techniques such as RT-qPCR, in part due to their short sizes and depth of post-transcriptional modifications, have often precluded their analysis (33). Here, we were able to assess five distinct tRNAs and found three to be significantly decreased. We likewise identified a significant decrease in the Pol III transcript 7SK, an indirect inhibitor of mRNA production.
To explore how the missense variant in POLR3D might impact the assembly and/or maturation of the Pol III complex, we performed an AP-MS analysis of the p.P181S mutant compared to WT, which revealed an enrichment of the PAQosome complex with the p.P181S mutant. As the PAQosome normally interacts rapidly with newly synthesized POLR3A and is not enriched in fully assembly complexes, these results suggest that the p.P181S POLR3D variant might perturb Pol III complex maturation, presumably contributing to the pathogenicity of this variant (16).
In summary, we describe the first case of POLR3-related leukodystrophy caused by biallelic pathogenic variants in POLR3D, expanding the genetic spectrum of this hypomyelinating leukodystrophy and further supporting proper Pol III function as critical for neurodevelopment and myelination. Finally, we identify how these pathogenic variants impact enzymatic complex maturation and transcriptional activity of Pol III to provide further insight into disease mechanisms.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
The studies involving humans were approved by Montreal Children’s Hospital and the McGill University Health Center Research Ethic Boards (11-105-PED, 2019-4972) and the Children’s Mercy Institutional Review Board (study #11120514). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
JM: Conceptualization, Data curation, Formal analysis, Writing – original draft. SP: Data curation, Formal analysis, Writing – review & editing. MP: Data curation, Formal analysis, Writing – review & editing. LT: Data curation, Writing – review & editing. KG: Data curation, Writing – review & editing. CP: Data curation, Writing – review & editing. AP: Data curation, Writing – review & editing. TP: Data curation, Writing – review & editing. IT: Data curation, Formal analysis, Writing – review & editing. BC: Data curation, Formal analysis, Writing – review & editing. GB: Conceptualization, Data curation, Formal analysis, Supervision, Writing – original draft.
This work was supported by project grants from the Canadian Institutes of Health Research (377869, 426534) as well as research grants from the Montreal Children’s Hospital Foundation and Leuco-Action. GB has received the Clinical Research Scholar Junior 1 Award from the Fonds de Recherche du Quebec-Santé (FRQS; 2012–2016), the New Investigator Salary Award from the Canadian Institutes of Health Research (2017–2022), and the Clinical Research Scholar Senior Award from the FRQS (2022–2025). This research was undertaken thanks in part to funding from the Canada First Research Excellence Fund and Fonds de recherche du Québec, awarded to the Healthy Brain Healthy Lives (HBHL) initiative at McGill University. JM is supported by HBHL. SP has been supported by the FRQS Doctoral Scholarship, the Fondation du Grand Défi Pierre Lavoie Doctoral Scholarship, the McGill Faculty of Medicine F.S.B. Miller Fellowship, and the Research Institute of the McGill University Health Center Desjardins Studentship in Child Health Research. The work by TP and IT was made possible by the generous gifts to Children’s Mercy Research Institute and Genomic Answers for Kids program at Children’s Mercy Kansas City. TP holds the Dee Lyons/Missouri Endowed Chair in Pediatric Genomic Medicine.
The authors wish to thank the patient and her family for their participation in this research. TP and IT would like to thank the GA4K team and Laura Puckett, Adam Walters, and Margaret Gibson for their work in library preparation and sequencing. MP and BC are grateful to Denis Faubert, Josée Champagne, Sylvain Tessier, and Marguerite Boulos for their assistance in sample processing and mass spectrometry data acquisition.
GB was a consultant for Passage Bio Inc (2020–2022) and Ionis (2019). She is/was a site investigator for the Alexander’s disease trial of Ionis (2021–present), Metachromatic leukodystrophy of Shire/Takeda (2020–2021), Krabbe and GM1 gene therapy trials of Passage Bio (2021–present), GM1 natural history study from the University of Pennsylvania sponsored by Passage Bio (2021-present), and Adrenoleukodystrophy/Hematopoietic stem cell transplantation natural history study of Bluebird Bio (2019), a site sub-investigator for the MPS II gene therapy trial of Regenxbio (2021–present) and the MPS II clinical trial of Denali (2022–present). She has received unrestricted educational grants from Takeda (2021–2022). LT currently manages sponsored clinical trials at the site level for Ionis Pharmaceuticals (Alexander disease clinical trial, 2021–present), Passage Bio (Krabbe disease and GM1 gangliosidosis clinical trials, 2021–present), and Teva Pharmaceuticals (chronic and episodic migraine clinical trials, 2022–present), and also manages a GM1 gangliosidosis natural history study sponsored by the University of Pennsylvania with funding from Passage Bio.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1254140/full#supplementary-material
1. Vanderver, A , Prust, M , Tonduti, D , Mochel, F , Hussey, HM , Helman, G, et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol Genet Metab. (2015) 114:494–500. doi: 10.1016/j.ymgme.2015.01.006
2. Soderholm, HE , Chapin, AB , Bayrak-Toydemir, P , and Bonkowsky, JL . Elevated Leukodystrophy incidence predicted from genomics databases. Pediatr Neurol. (2020) 111:66–9. doi: 10.1016/j.pediatrneurol.2020.06.005
4. Steenweg, ME , Vanderver, A , Blaser, S , Bizzi, A , de Koning, TJ , Mancini, GM, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain. (2010) 133:2971–82. doi: 10.1093/brain/awq257
5. Wolf, NI , Vanderver, A , van Spaendonk, RM , Schiffmann, R , Brais, B , Bugiani, M, et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology. (2014) 83:1898–905. doi: 10.1212/WNL.0000000000001002
6. Gauquelin, L , Cayami, FK , Sztriha, L , Yoon, G , Tran, LT , Guerrero, K, et al. Clinical spectrum of POLR3-related leukodystrophy caused by biallelic POLR1C pathogenic variants. Neurol Genet. (2019) 5:e369. doi: 10.1212/NXG.0000000000000369
7. Mirchi, A , Guay, SP , Tran, LT , Wolf, NI , Vanderver, A , Brais, B, et al. Craniofacial features of POLR3-related leukodystrophy caused by biallelic variants in POLR3A, POLR3B and POLR1C. J Med Genet. (2023) 60:1026–34. doi: 10.1136/jmg-2023-109223
8. Dieci, G , Fiorino, G , Castelnuovo, M , Teichmann, M , and Pagano, A . The expanding RNA polymerase III transcriptome. Trends Genet. (2007) 23:614–22. doi: 10.1016/j.tig.2007.09.001
9. Bernard, G , Chouery, E , Putorti, ML , Tetreault, M , Takanohashi, A , Carosso, G, et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet. (2011) 89:415–23. doi: 10.1016/j.ajhg.2011.07.014
10. Tetreault, M , Choquet, K , Orcesi, S , Tonduti, D , Balottin, U , Teichmann, M, et al. Recessive mutations in POLR3B, encoding the second largest subunit of pol III, cause a rare hypomyelinating leukodystrophy. Am J Hum Genet. (2011) 89:652–5. doi: 10.1016/j.ajhg.2011.10.006
11. Dorboz, I , Dumay-Odelot, H , Boussaid, K , Bouyacoub, Y , Barreau, P , Samaan, S, et al. Mutation in POLR3K causes hypomyelinating leukodystrophy and abnormal ribosomal RNA regulation. Neurol Genet. (2018) 4:e289. doi: 10.1212/NXG.0000000000000289
12. Thiffault, I , Wolf, NI , Forget, D , Guerrero, K , Tran, LT , Choquet, K, et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun. (2015) 6:7623. doi: 10.1038/ncomms8623
13. Soden, SE , Saunders, CJ , Willig, LK , Farrow, EG , Smith, LD , Petrikin, JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. (2014) 6:265ra168. doi: 10.1126/scitranslmed.3010076
14. Richards, S , Aziz, N , Bale, S , Bick, D , Das, S , Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
15. Choquet, K , Pinard, M , Yang, S , Moir, RD , Poitras, C , Dicaire, MJ, et al. The leukodystrophy mutation Polr3b R103H causes homozygote mouse embryonic lethality and impairs RNA polymerase III biogenesis. Mol Brain. (2019) 12:59. doi: 10.1186/s13041-019-0479-7
16. Pinard, M , Dastpeyman, S , Poitras, C , Bernard, G , Gauthier, MS , and Coulombe, B . Riluzole partially restores RNA polymerase III complex assembly in cells expressing the leukodystrophy-causative variant POLR3B R103H. Mol Brain. (2022) 15:98. doi: 10.1186/s13041-022-00974-z
17. Cleary, JD , Subramony, SH , and Ranum, LPW . Spinocerebellar ataxia type 8 In: MP Adam, GM Mirzaa, RA Pagon, SE Wallace, B LJH, and KW Gripp, et al., editors. GeneReviews. Seattle (WA): University of Washington (1993)
18. Worth, PF , Houlden, H , Giunti, P , Davis, MB , and Wood, NW . Large, expanded repeats in SCA8 are not confined to patients with cerebellar ataxia. Nat Genet. (2000) 24:214–5. doi: 10.1038/73411
19. Timmons, M , Tsokos, M , Asab, MA , Seminara, SB , Zirzow, GC , Kaneski, CR, et al. Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology. (2006) 67:2066–9. doi: 10.1212/01.wnl.0000247666.28904.35
20. La Piana, R , Tonduti, D , Gordish Dressman, H , Schmidt, JL , Murnick, J , Brais, B, et al. Brain magnetic resonance imaging (MRI) pattern recognition in pol III-related leukodystrophies. J Child Neurol. (2014) 29:214–20. doi: 10.1177/0883073813503902
21. Desmet, FO , Hamroun, D , Lalande, M , Collod-Beroud, G , Claustres, M , and Beroud, C . Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. (2009) 37:e67. doi: 10.1093/nar/gkp215
22. Watt, KE , Macintosh, J , Bernard, G , and Trainor, PA . RNA polymerases I and III in development and disease. Semin Cell Dev Biol. (2022) 136:49–63. doi: 10.1016/j.semcdb.2022.03.027
23. Girbig, M , Misiaszek, AD , Vorlander, MK , Lafita, A , Grotsch, H , Baudin, F, et al. Cryo-EM structures of human RNA polymerase III in its unbound and transcribing states. Nat Struct Mol Biol. (2021) 28:210–9. doi: 10.1038/s41594-020-00555-5
24. Ramsay, EP , Abascal-Palacios, G , Daiss, JL , King, H , Gouge, J , Pilsl, M, et al. Structure of human RNA polymerase III. Nat Commun. (2020) 11:6409. doi: 10.1038/s41467-020-20262-5
25. Wang, Z , Wu, C , Aslanian, A , Yates, JR 3rd, and Hunter, T . Defective RNA polymerase III is negatively regulated by the SUMO-ubiquitin-Cdc48 pathway. elife. (2018) 7:e35447. doi: 10.7554/eLife.35447
26. Perrier, S , Gauquelin, L , Fallet-Bianco, C , Dishop, MK , Michell-Robinson, MA , Tran, LT, et al. Expanding the phenotypic and molecular spectrum of RNA polymerase III-related leukodystrophy. Neurol Genet. (2020) 6:e425. doi: 10.1212/NXG.0000000000000425
27. Liu, Y , Beyer, A , and Aebersold, R . On the dependency of cellular protein levels on mRNA abundance. Cells. (2016) 165:535–50. doi: 10.1016/j.cell.2016.03.014
28. Shimojima, K , Shimada, S , Tamasaki, A , Akaboshi, S , Komoike, Y , Saito, A, et al. Novel compound heterozygous mutations of POLR3A revealed by whole-exome sequencing in a patient with hypomyelination. Brain Dev. (2014) 36:315–21. doi: 10.1016/j.braindev.2013.04.011
29. Choquet, K , Forget, D , Meloche, E , Dicaire, MJ , Bernard, G , Vanderver, A, et al. Leukodystrophy-associated POLR3A mutations down-regulate the RNA polymerase III transcript and important regulatory RNA BC200. J Biol Chem. (2019) 294:7445–59. doi: 10.1074/jbc.RA118.006271
30. Azmanov, DN , Siira, SJ , Chamova, T , Kaprelyan, A , Guergueltcheva, V , Shearwood, AJ, et al. Transcriptome-wide effects of a POLR3A gene mutation in patients with an unusual phenotype of striatal involvement. Hum Mol Genet. (2016) 25:4302–14. doi: 10.1093/hmg/ddw263
31. James Faresse, N , Canella, D , Praz, V , Michaud, J , Romascano, D , and Hernandez, N . Genomic study of RNA polymerase II and III SNAPc-bound promoters reveals a gene transcribed by both enzymes and a broad use of common activators. PLoS Genet. (2012) 8:e1003028. doi: 10.1371/journal.pgen.1003028
32. Park, JS , and Kunkel, GR . Both RNA polymerase III and RNA polymerase II accurately initiate transcription from a human U6 promoter in vitro. Biochem Biophys Res Commun. (1995) 214:934–40. doi: 10.1006/bbrc.1995.2376
Keywords: POLR3-related leukodystrophy, 4H leukodystrophy, hypomyelination, RNA polymerase III, POLR3D , RPC4
Citation: Macintosh J, Perrier S, Pinard M, Tran LT, Guerrero K, Prasad C, Prasad AN, Pastinen T, Thiffault I, Coulombe B and Bernard G (2023) Biallelic pathogenic variants in POLR3D alter tRNA transcription and cause a hypomyelinating leukodystrophy: A case report. Front. Neurol. 14:1254140. doi: 10.3389/fneur.2023.1254140
Edited by:
Hanna Mierzewska, Institute of Mother and Child, PolandReviewed by:
Luca Marsili, University of Cincinnati, United StatesCopyright © 2023 Macintosh, Perrier, Pinard, Tran, Guerrero, Prasad, Prasad, Pastinen, Thiffault, Coulombe and Bernard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geneviève Bernard, Z2VuZXZpZXZlLmJlcm5hcmRAbWNnaWxsLmNh
†ORCID: Julia Macintosh https://orcid.org/0000-0002-9444-2365
Stefanie Perrier https://orcid.org/0000-0002-6881-7573
Luan T. Tran https://orcid.org/0000-0003-4666-1755
Kether Guerrero https://orcid.org/0000-0001-8412-9925
Chitra Prasad https://orcid.org/0000-0001-6007-7251
Asuri N. Prasad https://orcid.org/0000-0002-6057-1038
Tomi Pastinen https://orcid.org/0000-0003-4016-5021
Isabelle Thiffault https://orcid.org/0000-0001-7987-6731
Benoit Coulombe https://orcid.org/0000-0003-1702-0692
Geneviève Bernard https://orcid.org/0000-0002-9634-2966
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.