
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 24 November 2023
Sec. Neurogenetics
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1251467
This article is part of the Research TopicConsanguinity and Rare Genetic Neurological DiseasesView all 9 articles
 Hui Zhu1
Hui Zhu1 Shuyao Zhu1Qiong Jiang1Ying Pang1Yu Huang1Yan Chen2Ting Hou2Wenxin Deng2Xingyu Liu1Lan Zeng3Ai Chen1
Shuyao Zhu1Qiong Jiang1Ying Pang1Yu Huang1Yan Chen2Ting Hou2Wenxin Deng2Xingyu Liu1Lan Zeng3Ai Chen1 Jin Wang3*†Zemin Luo1*†
Jin Wang3*†Zemin Luo1*†Vulto-van Silfhout-de Vries syndrome (VSVS; MIM 615828) is an extremely rare autosomal dominant disorder with unknown incidence. It is always caused by de novo heterozygous pathogenic variants in the DEAF1 gene, which encodes deformed epidermal autoregulatory factor-1 homology. VSVS is characterized by mild to severe intellectual disability (ID) and/or global developmental delay (GDD), seriously limited language expression, behavioral abnormalities, somnipathy, and reduced pain sensitivity. In this study, we present a Chinese boy with moderate GDD and ID, severe expressive language impairment, behavioral issues, autism spectrum disorder (ASD), sleeping dysfunction, high pain threshold, generalized seizures, imbalanced gait, and recurrent respiratory infections as clinical features. A de novo heterozygous pathogenic missense variant was found in the 5th exon of DEAF1 gene, NM_021008.4 c.782G>C (p. Arg261Pro) variant by whole exome sequencing (WES). c.782G>C had not been previously reported in genomic databases and literature. According to the ACMG criteria, this missense variant was considered to be “Likely Pathogenic”. We diagnosed the boy with VSVS both genetically and clinically. At a follow-up of 2.1 years, his seizures were well controlled after valproic acid therapy. In addition, the child’s recurrent respiratory infections improved at 3.5 years of age, which has not been reported in previous individuals. Maybe the recurrent respiratory infections like sleep problems reported in the literature are not permanent but may improve naturally over time. The literature review showed that there were 35 individuals with 28 different de novo pathogenic variants of DEAF1-related VSVS. These variants were mostly missense and the clinical manifestations were similar to our patient. Our study expands the genotypic and phenotypic profiles of de novo DEAF1.
Vulto-van Silfhout-de Vries syndrome (VSVS; MIM 615828), also known as autosomal dominant mental retardation-24 (MRD24), is an autosomal dominant disorder caused by de novo heterozygous pathogenic variant in DEAF1 gene (1). The major clinical phenotypes of VSVS include different levels (mild to severe) of intellectual disability (ID) and/or global developmental delay (GDD), serious language limitations, motor retardation, autism spectrum disorders (ASD), problems with behaving, somnipathy, and increased pain threshold. It is fairly rare with only 11 articles having described the disease and has an unknown incidence (1–11).
Herein, we report a Chinese male child with a novel de novo pathogenic missense variant in the DEAF1 gene, namely, c.782G>C (p. Arg261Pro), that was confirmed by using whole-exome sequencing (WES) and Sanger sequencing verification. The phenotype of our patient was basically consistent with those reported in the literature. This report expands the known range of VSVS due to de novo pathogenic variants of the DEAF1 gene. Our patient recurrent respiratory infections are not permanent. The phenomenon has not been described in previously reported cases. In addition, to summarize the clinical manifestations and genotypes, we also reviewed the VSVS cases reported in literature worldwide. We aim to raise clinicians’ knowledge and understanding of VSVS so that the disease gradually becomes no longer rare in the future.
A 3-year-old boy was admitted to the neurological in-patient department with ID, speech delay, and seizures in April 2021. He was born at term by cesarean section. His birth weight and length were 3,200 g and 48 cm, respectively. His non-consanguineous parents were healthy and were pregnant for the first time. There was no history of prenatal exposure to alcohol, drugs, or medications.
He presented GDD noticed since babyhood, but his development revealed no regression. He crawled at 8 months, began to grow baby teeth and was sitting at 1 year old, and could stand on his own by 1.6 years old. He could walk at the age of 2, and his gait was ataxic. He started speaking in bi-syllables (baba and mama) at 2.3 years of age, but his language ability was not improved at 3 years. He could not feed himself at 3 years old. The Gesell Developmental Scale indicated a developmental quotient score of 46, and the Standford-Binet Intelligence Scale indicated a score of 40 defined as moderate ID. He manifested insomnia after birth, and sleep disturbance disappeared when he was 4 years old. The child had a high pain threshold and the following abnormal behavioral manifestations: poor eye contact, restricted and repetitive behavior, fascination, attention deficit hyperactivity disorder (ADHD), irritability, aggressive behavior, and context-inappropriate laughter. From birth to date, he had recurrent respiratory tract infections characterized by an average of approximately once a month of upper respiratory tract infection or bronchopneumonia. Gastrointestinal infections occurred two to three rounds per year. There were no deformities of the face, limbs, and spine. His vision and hearing were normal. Muscle tone and strength were normal for four limbs. His first seizure occurred at 2.9 years of age, and by 3 years of age, he had had five rounds of seizures, and his seizure was generalized tonic–clonic.
Given the presence of poor eye contact, stereotyped behavior, fascination, ID, poor expressive speech, behavioral abnormalities, epilepsy, sleep dysfunction, and motor delay, a diagnosis of autism spectrum disorder and comorbid neurodevelopmental disorders (ASD-NDDs) was considered. Thus, blood samples of the child and his parents were collected for genetic analysis by WES.
Ethical approval was provided by the institutional ethics committee. After informed consent was given, EDTA blood samples were obtained from the boy and his parents. WES was conducted and the methods have been described in detail in previous studies (12). PolyPhen-2, SIFT, Provean, Mutation Taster, and Mutation Assessor were used to predict whether the detected variant was damaging or causing disease. Based on the homology study of the solution structure of the human Sp100b and the SAND domain by heteronuclear NMR (PDBe 1h5p), the PDB structure of the SAND domain was modeled by phyre2 server software and modified by UCSF chimera software (Figure 1). The result of the variant was interpreted by the American College of Medical Genetics and Genomics (ACMG) guidelines (Genet Med. 2015) (13).
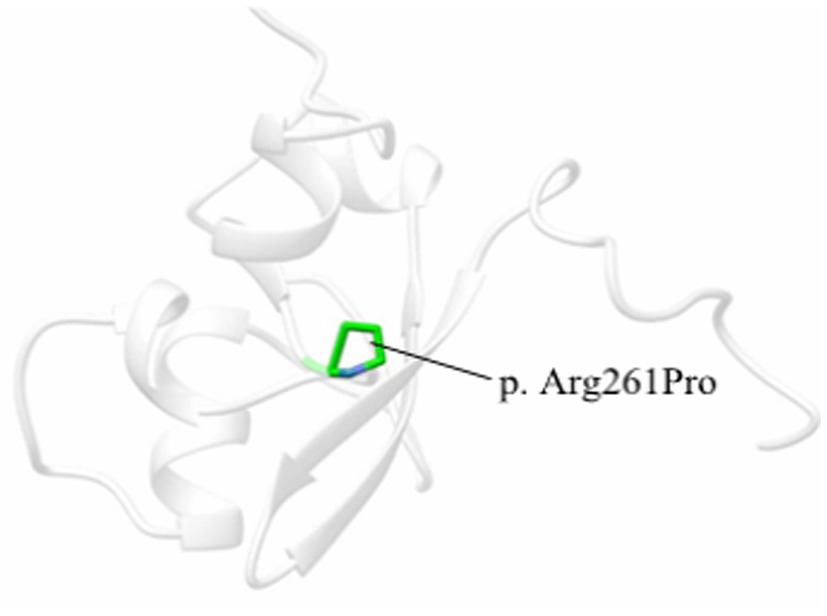
Figure 1. One 3D model of the SAND domain in this study based on the homology study of the solution structure of the human Sp100b SAND domain by heteronuclear NMR (PDBe 1h5p). The amino acid substitution is in green.
The results of Trio-WES revealed a de novo heterozygous missense variant (NM_021008.4: c.782G>C, p. Arg261Pro) in the fifth exon of DEAF1 gene, which was confirmed by Sanger sequencing (Figure 2). The variant was predicted to be damaging/disease-causing by PolyPhen-2 (score = 1), SIFT (score = 0.009), Provean (score = −5.29), Mutation Taster, and Mutation Assessor. The variant also affects evolutionary conserved amino acids within the DEAF1 SAND domain. According to ACMG (13), the missense variant was classified as “Likely Pathogenic” with PS2 + PM2_Supporting + PP3. This pathogenic variant was not reported in 1000 Genomes, ExAC, gnomAD, ClinVar Databases, and previous literature. Certainly, at present, the pathogenic variant data of our patient has been deposited into the ClinVar bank and the accession number is VCV002444491.1.
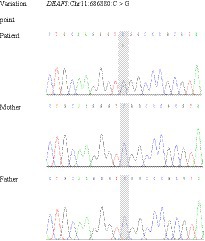
Figure 2. Sanger sequencing showing a heterozygous variant in DEAF1: the patient has a heterozygous pathogenic variant c.782G>C in exon 5, which is not present in either of the parents, suggesting the de novo occurrence of the variation.
Based on the clinical manifestations and genetic sequencing results, we diagnosed the boy with VSVS. His seizures were under treatment with valproic acid monotherapy that was started at 3 years of age and were well-controlled during the 2.1 years of follow-up. We followed up every 1–3 months by phone, WeChat chat, and clinic attendance. Every 3 months, there had to be an outpatient follow-up, detailed medical history, and physical examination to better record the child’s growth, development, and disease evolution. During the follow-up, we also found that the child’s recurrent respiratory infections disappeared at 3.5 years of age. Now, he is 5.1 years of age and is improving in his socio-adaptive skills, such as feeding himself.
A systematic literature search in PubMed and OMIM was performed. MeSH and title or abstract were used for all eligible studies that mainly focused on the DEAF1 variants in VSVS. The research strategy was as follows: “DEAF1” AND (“Vulto-van Silfhout-de Vries syndrome” or “autosomal dominant mental retardation-24” or “intellectual disability with speech impairment and behavioral problems”). Data from all eligible studies were analyzed and discussed by two reviewers. Detailed results are available in the “Discussion” section.
VSVS is a relatively new and extremely rare autosomal dominant intellectual disability disease that has been discovered in the past 13 years, with few reports since the first case description by Vissers and his colleagues in 2010 (10). In 2014, Vulto-van Silfhout et al. (8) certified de novo heterozygous variations in DEAF1 leading to VSVS in humans. The DEAF1 gene (MIM 602635) encodes deformed epidermal autoregulatory factor-1 homology, which contains 12 exons and encodes 565 amino acids. This gene is highly expressed in brain cells, especially during the extremely young fetal development stage (8). As a transcriptional activator and repressor, DEAF1 makes a vast difference in affecting the expression of diverse genes (2, 14–16). The main clinical manifestations of VSVS include different levels of GDD / ID, serious expressive language disorder, behavioral problems, autistic behaviors, somnipathy, and increased pain threshold (2).
We searched the world documentation previously reported from the first description in 2010 to March 2023. By March 2023, only 35 individuals, including adults and children, had been reported that carried de novo pathogenic variants of the DEAF1 gene (1–11). Among the 35 cases, the median age was 7.1 years (range 1.3–38 years), with 14 women and 21 men. The major clinical phenotypes are summarized in Table 1. Different levels of ID / GDD and behavioral issues were described in all cases (35/35; 100%). Of those persons with behavioral abnormalities, the commonest was ASD (30/34; 88.2%). Severe expressive language barriers were presented in most persons (33/34; 97.1%), including 14 persons with speech absence and 19 cases with speech limitation. Lag of motor development and sleeping trouble were also usual at 84.8% (28/33) and 83.3% (25/30), respectively. In particular, it needs to be mentioned that sleep–wake disorders usually begin after birth and improve as the child grows. Reduced pain sensitivity was reported in 21 persons (21/26; 80.8%). Seizures were found in 21 cases (21/27; 77.8%), and the most common was generalized (18/27; 66.7%). Twenty-two individuals had abnormal gait (22/30; 73.3%) that incorporated gait ataxia (7/30; 23.3%) and other walking patterns (18/30, 60%) of broad-based and imbalanced gait or a tip-toe gait. Relatively less common clinical manifestations and those that are neglected and not described in detail include recurrent infections, mild facial deformities, and feeding difficulties (2). The major clinical features of our patient were similar phenotypes with previously described VSVS cases. Nevertheless, what calls for special attention is that the boy’s recurrent respiratory infections improved naturally at 3.5 years old, which has not been reported in the previous literature. We speculate that the recurrent respiratory infections in VSVS which are not permanent but are similar to previously reported sleep problems may improve naturally over time. Anyway, recurrent infections may be worth further studying in the future.
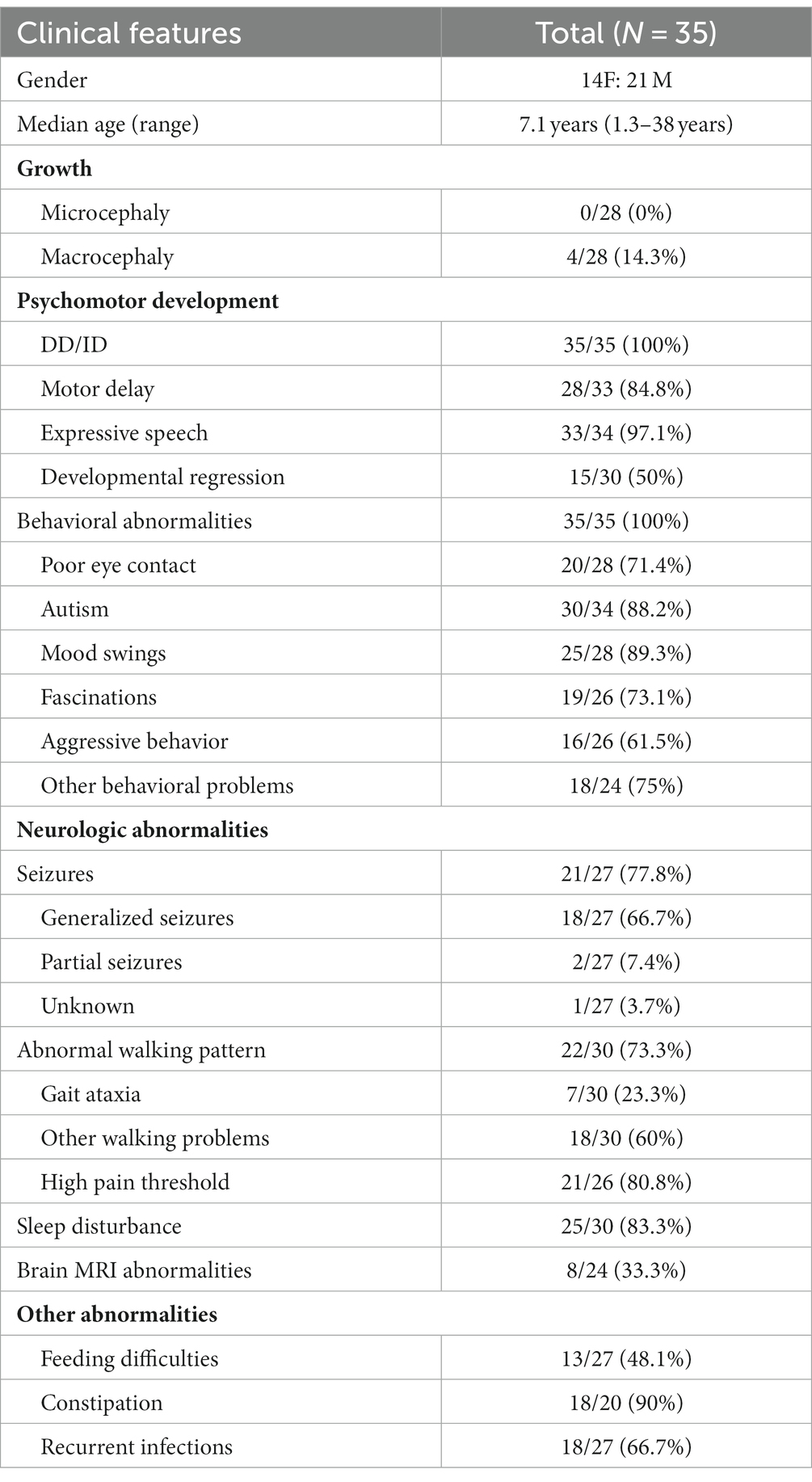
Table 1. Phenotype of patients with pathogenic de novo variants of DEAF1 that have been previously reported.
Seizures were observed in 77.8 percent of previously presented VSVS individuals. The epilepsies caused by de novo in DEAF1 genes were predominantly generalized. Regarding the curative effect observation of antiepileptic drugs (AEDs) treatment, hitherto, there have been only two studies with small samples or case series, but their conclusions were opposite. One study included six individuals with VSVS, and the result showed almost all generalized persons (5/6; 83.3%) had no epileptic seizures by the use of valproic acid therapy (1). In contrast, the other study included 10 individuals, which revealed that epilepsies were not well controlled in the overwhelming majority of individuals (9/10;90%) (2). However, we found that only one individual was given valproic acid and other individuals were given other AEDs in the study of Nabais Sa et al. (2). Owing to the seizures of VSVS being ordinarily generalized, valproic acid could be the first choice in AEDs (1). Chen et al. (1) speculated that the interaction between valproic acid and the DEAF1 gene may inhibit neuronal excitation. The difference in the results of the two studies on antiepileptic efficacy may be attributed to the function of different pathogenic variants in the DEAF1 gene leading to different responses to AEDs (1, 2). Additionally, because the individuals in the two studies were treated with different AEDs, an accurate assessment was not available. Furthermore, the number of cases in the current relevant studies is small, making it difficult to draw more precise conclusions. Therefore, every case of VSVS is important and should be described in detail. Our patient had a generalized seizure. His seizures were well controlled by taking valproic acid monotherapy, and there were no seizures during his 2.1 years of follow-up. Of course, we will continue to follow up on this child for a long time.
DEAF1 includes the following five functional domains: SAND, ZnF, NLS, NES, and MYND (2). Twenty-eight de novo pathogenic variants of the DEAF1 gene were reported in the literature including 23 missense variants, 3 splice-site variants, and 2 in-frame deletion variants (Table 2). Most variants (27/28;96.4%) are located in the SAND or adjacent ZnF domain. Only the p. Lys305del variant is in the NLS domain. The de novo pathogenic mutations in DEAF1 caused VSVS to be located in or around the SAND domain. It has been demonstrated that de novo pathogenic variants damaged the DEAF1 transcriptional inhibitory activity of DEAF1 and reverse DEAF1-mediated transcriptional activation at the Eif4g3 promoter (2, 8). A de novo heterozygous pathogenic missense variant of the DEAF1 gene that was located within the SAND domain was detected in our patient. In addition, no candidate genes were detected other than DEAF1 in this child by WES. It has been reported that a dominant negative effect caused by missense mutations of DEAF1 genes has been regarded as a possible pathogenic mechanism of VSVS (2, 8). Vulto-van Silfhout and his colleagues put forward that the dominant negative effect of missense mutations in individuals of VSVS could cause severe clinical presentations (8). The in-frame deletion mutations could result in amino acid change (2). The splicing site variations play an extremely important role during the alternative splicing of DEAF1 mRNA transcriptions (2). Sharma et al. (3) proposed that the splice-site variants disrupt gene function by forming a truncated protein impeding further downstream reactions rather than having a dominant negative effect that leads to relatively less serious clinical manifestations.
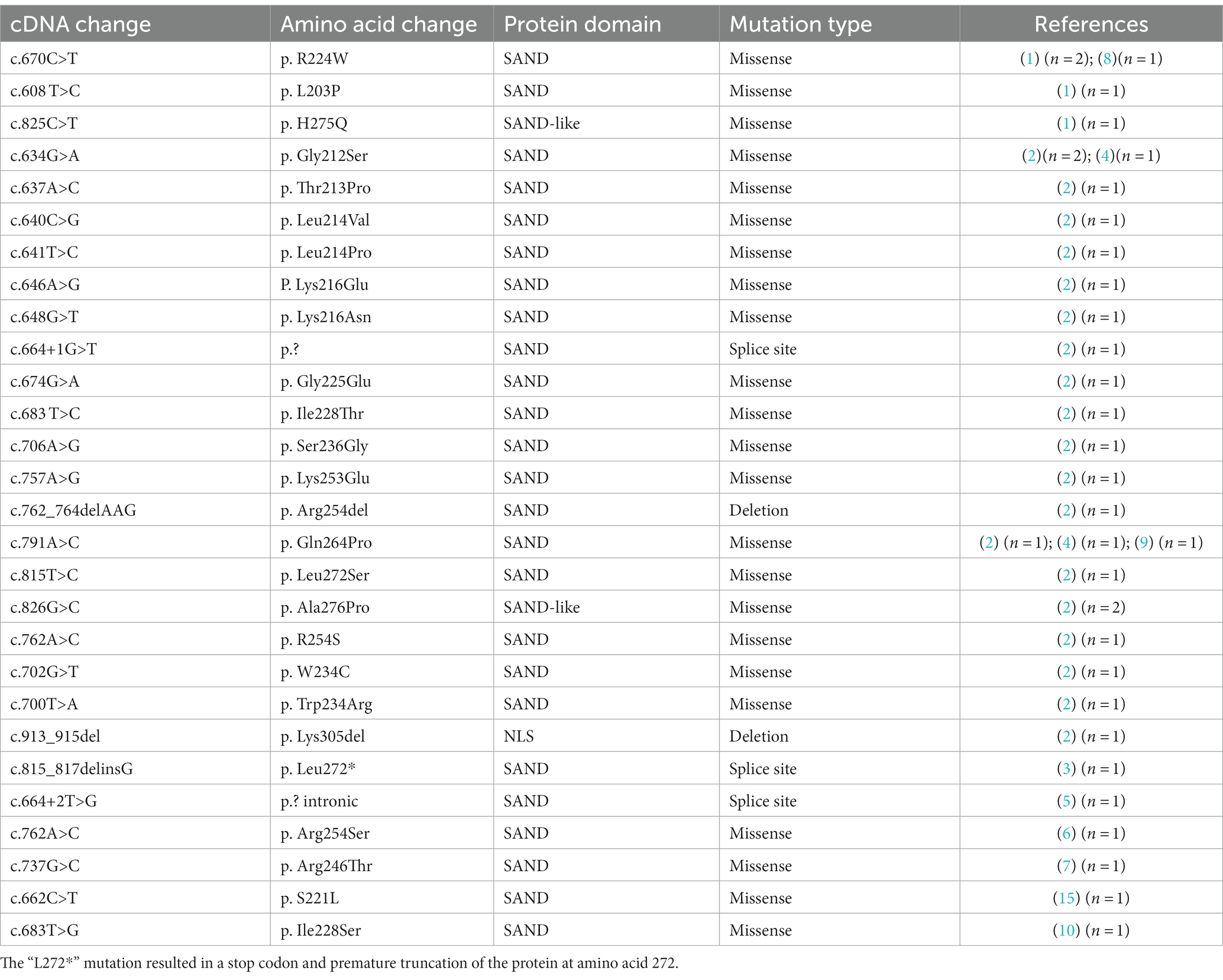
Table 2. Pathogenic de novo variants of DEAF1 that have been previously reported.
What needs to be specifically identified is the recessively inherited dyskinesia, seizures, and intellectual developmental disorder syndrome (DYSEIDD). In terms of clinical findings, with the exception of microcephaly, the clinical features of DYSEIDD and VSVS were basically the same (2). DYSEIDD is caused by the biallelic pathogenic variants in the DEAF1 gene (MIM 617171) (2). In the DEAF1 gene mutations, compared to the de novo variants, only pathogenic biallelic variants can lead to microcephaly. However, it is regrettable that not all individuals with DYSEIDD have microcephaly (2). Therefore, genetic sequencing, such as WES or whole genome sequencing, is required to distinguish the two diseases.
In summary, our patient and prior reports showed that different AEDs revealed different results in managing seizures in those with VSVS. This issue needs further research. Our patient provides additional information on the phenotype, genotype, and effective therapy in VSVS.
We described a Chinese VSVS boy with a de novo novel pathogenic missense variant of DEAF1 located within the SAND domain. It enriches the known range of DEAF1 de novo pathogenic variants and adds to the few reports of VSVS. In addition, we speculate that recurrent respiratory infections like previously reported sleep problems are not permanent but may improve naturally over time.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of Sichuan Provincial Maternity and Child Health Care Hospital (Approval Code:20230331-026). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
HZ designed this study, collected and integrated data, and wrote the article. SZ integrated the data. QJ, YP, YH, YC, TH, WD, XL, LZ, and AC participated in the evaluation and treatment of the patient. JW and ZL participated in the study’s design and coordination, revised article. All authors contributed to the article and approved the submitted version.
This research was funded by Natural Science Foundation of Sichuan Province (no. 2022NSFSC0784) and Science and Technology Project of Chengdu Bureau (no. 2021-YF05-01658-SN).
The authors are grateful to the child and his family for their contributions to this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chen, S, Deng, X, Xiong, J, He, F, Yang, L, Chen, B, et al. De novo variants of DEAF1 cause intellectual disability in six Chinese patients. Clin Chim Acta. (2021) 518:17–21. doi: 10.1016/j.cca.2021.02.026
2. Nabais Sa, MJ, Jensik, PJ, McGee, SR, Parker, MJ, Lahiri, N, McNeil, EP, et al. De novo and biallelic DEAF1 variants cause a phenotypic spectrum. Genet Med. (2019) 21:2059–69. doi: 10.1038/s41436-019-0473-6
3. Sharma, P, Gambhir, PS, Phadke, SR, and Mandal, K. Expanding the phenotype in autosomal dominant mental retardation-24: a novel variation in DEAF1 gene. Clin Dysmorphol. (2019) 28:94–7. doi: 10.1097/MCD.0000000000000252
4. Chen, L, Jensik, PJ, Alaimo, JT, Walkiewicz, M, Berger, S, Roeder, E, et al. Functional analysis of novel DEAF1 variants identified through clinical exome sequencing expands DEAF1-associated neurodevelopmental disorder (DAND) phenotype. Hum Mutat. (2017) 38:1774–85. doi: 10.1002/humu.23339
5. Li, SJ, Yu, SS, Luo, HY, Li, X, Rao, B, Wang, Y, et al. Two de novo variations identified by massively parallel sequencing in 13 Chinese families with children diagnosed with autism spectrum disorder. Clin Chim Acta. (2018) 479:144–7. doi: 10.1016/j.cca.2018.01.025
6. Berger, SI, Ciccone, C, Simon, KL, Malicdan, MC, Vilboux, T, Billington, C, et al. Exome analysis of Smith-Magenis-like syndrome cohort identifies de novo likely pathogenic variants. Hum Genet. (2017) 136:409–20. doi: 10.1007/s00439-017-1767-x
7. Wenger, AM, Guturu, H, Bernstein, JA, and Bejerano, G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. (2017) 19:209–14. doi: 10.1038/gim.2016.88
8. Vulto-van Silfhout, AT, Rajamanickam, S, Jensik, PJ, Vergult, S, de Rocker, N, Newhall, KJ, et al. Mutations affecting the SAND domain of DEAF1 cause intellectual disability with severe speech impairment and behavioral problems. Am J Hum Genet. (2014) 94:649–61. doi: 10.1016/j.ajhg.2014.03.013
9. Rauch, A, Wieczorek, D, Graf, E, Wieland, T, Endele, S, Schwarzmayr, T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. (2012) 380:1674–82. doi: 10.1016/S0140-6736(12)61480-9
10. Vissers, LE, de Ligt, J, Gilissen, C, Janssen, I, Steehouwer, M, de Vries, P, et al. A de novo paradigm for mental retardation. Nat Genet. (2010) 42:1109–12. doi: 10.1038/ng.712
11. Bodunova, N, Vorontsova, M, Khatkov, I, Baranova, E, Bykova, S, Degterev, D, et al. A unique observation of a patient with Vulto-van Silfhout-de Vries syndrome. Diagnostics. (2022) 12:1887. doi: 10.3390/diagnostics12081887
12. Zhu, H, Zhao, ZH, Zhu, SY, Xiong, F, He, LH, Zhang, Y, et al. Renal-hepatic-pancreatic dysplasia-1 with a novel NPHP3 genotype: a case report and review of the literature. BMC Pediatr. (2022) 22:603. doi: 10.1186/s12887-022-03659-7
13. Richards, S, Aziz, N, Bale, S, Bick, D, Das, S, Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
14. Yip, L, Creusot, RJ, Pager, CT, Sarnow, P, and Fathman, CG. Reduced DEAF1 function during type 1 diabetes inhibits translation in lymph node stromal cells by suppressing Eif4g3. J Mol Cell Biol. (2013) 5:99–110. doi: 10.1093/jmcb/mjs052
15. Barker, HE, Smyth, GK, Wettenhall, J, Ward, TA, Bath, ML, Lindeman, GJ, et al. Deaf-1 regulates epithelial cell proliferation and side-branching in the mammary gland. BMC Dev Biol. (2008) 8:94. doi: 10.1186/1471-213X-8-94
Keywords: Vulto-van Silfhout-de Vries syndrome, dominant mental retardation-24, DEAF1, de novo, intellectual disability, behavioral abnormalities
Citation: Zhu H, Zhu S, Jiang Q, Pang Y, Huang Y, Chen Y, Hou T, Deng W, Liu X, Zeng L, Chen A, Wang J and Luo Z (2023) Vulto-van Silfhout-de Vries syndrome caused by de novo variants of DEAF1 gene: a case report and literature review. Front. Neurol. 14:1251467. doi: 10.3389/fneur.2023.1251467
Edited by:
Suzanne Lesage, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Alan Kenneth Percy, University of Alabama at Birmingham, United StatesCopyright © 2023 Zhu, Zhu, Jiang, Pang, Huang, Chen, Hou, Deng, Liu, Zeng, Chen, Wang and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Wang, amlud2o1MjFAMTI2LmNvbQ==; Zemin Luo, NDA4NDcxOTIxQHFxLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.