 Song Xue
Song Xue Xin Sun
Xin Sun- Stroke Center, Department of Neurology, The First Hospital of Jilin University, Changchun, China
The blood–brain barrier (BBB) is a functional phenotype exhibited by the neurovascular unit (NVU). It is maintained and regulated by the interaction between cellular and non-cellular matrix components of the NVU. The BBB plays a vital role in maintaining the dynamic stability of the intracerebral microenvironment as a barrier layer at the critical interface between the blood and neural tissues. The large contact area (approximately 20 m2/1.3 kg brain) and short diffusion distance between neurons and capillaries allow endothelial cells to dominate the regulatory role. The NVU is a structural component of the BBB. Individual cells and components of the NVU work together to maintain BBB stability. One of the hallmarks of acute ischemic stroke is the disruption of the BBB, including impaired function of the tight junction and other molecules, as well as increased BBB permeability, leading to brain edema and a range of clinical symptoms. This review summarizes the cellular composition of the BBB and describes the protein composition of the barrier functional junction complex and the mechanisms regulating acute ischemic stroke-induced BBB disruption.
1. Introduction
Proper central nervous system (CNS) functioning requires a highly specific and dynamically stable intracerebral microenvironment with extremely high metabolic demands and dependence on electrical and chemical signals for transmitting and processing neural information. Therefore, the cerebral vasculature has a unique structural function as the blood–brain barrier (BBB). The structural basis of the BBB is microvascular endothelial cells, which, with astrocytes, basement membranes, pericytes, and neurons that are physically close to the endothelium, form the neurovascular unit (NVU). The corresponding cells, the accompanying junction complexes, and transport proteins constitute three main functions of the BBB: physical, transport, and metabolic barriers. These barriers strictly control the entry of water molecules, ions, proteins, lipids, and cells from the blood into brain tissues and promptly discharge and degrade metabolites or harmful substances in brain tissues to maintain brain microenvironment homeostasis and normal neurological functions. Ischemic stroke structurally and functionally disrupts the barrier function of the NVU, leading to BBB leakage and triggering a range of clinical symptoms. This review first deals with the interactions of BBB-related cell types/structures (endothelial cells, glial cells, pericytes, neurons, and extracellular matrix) in the NVU. This summary outlines the changes that occur during BBB disruption in ischemic stroke and the main regulatory mechanisms. Understanding normal BBB function and post-infarction changes in the BBB will help evaluate and treat ischemic stroke in the future.
2. Structural components of the BBB: neurovascular unit (NVU)
NVU is a functional unit consisting of neurons and microvascular endothelial cells responsible for their blood supply, interacting with surrounding astrocytes and pericytes and regulating local blood flow (1). The cerebral vasculature has a very complex and delicate microscopic structure. First, brain tissues are highly vascularized, with at least one capillary supplying blood within 15 μm of each nerve cell (2), which can provide the nutrients required for neural activity and promptly eliminate metabolic waste. Second, cerebral small vessels and microvessels, surrounding vascular wall cells (smooth muscle cells and pericytes), astrocytes, basement membrane, and nerve cells, form the NVU microstructure (Figure 1). Some experts considered oligodendrocyte precursor cells and microglia as part of the NVU. The concept of the NVU was formally introduced at the 2001 Stroke Progress Review Group meeting of the National Institute of Neurological Disorders and Stroke (3), where neuroscientists emphasize the close relationship between nerve cells and blood vessels. The microstructure of the NVU is the cellular and molecular basis for many cerebrovascular-specific functions, such as neurovascular coupling and the BBB.
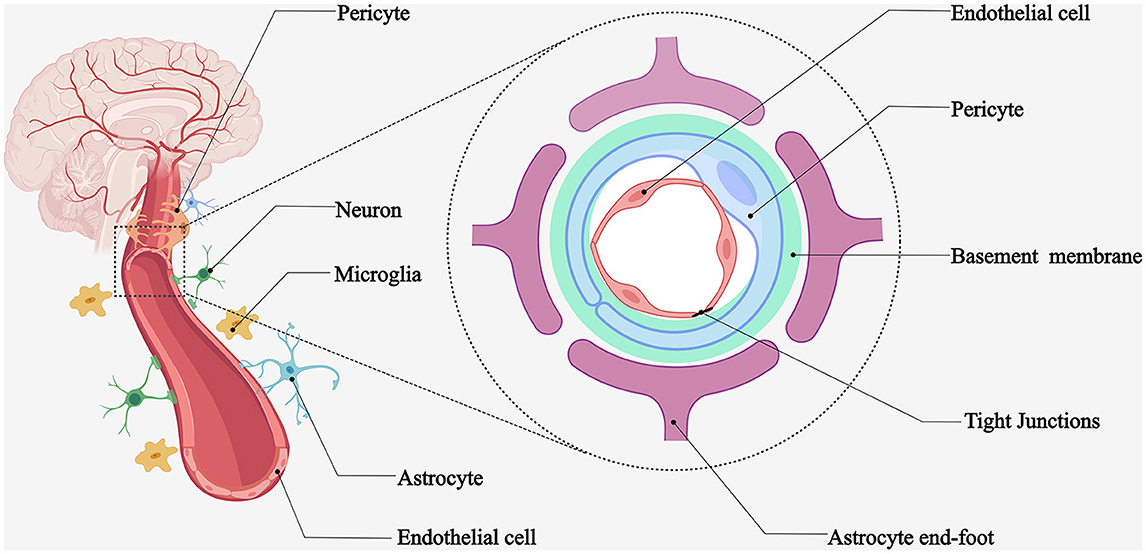
Figure 1. Schematic diagram of NVU. Microvessels adjacent to highly vascularized brain tissues provide blood flow to the surrounding nerve cells. Brain microvascular endothelial cells, pericytes, astrocytes, basement membrane, neurons, and microglia form the neurovascular unit.
The NVU components work in concert to regulate microvascular permeability, ion homeostasis, nutrient transport, metabolic toxin efflux, and cerebral hemodynamics. Disruption of these components can lead to BBB dysfunction (4). Experimental evidence has demonstrated that the interaction between developing vascular and neural tissues is crucial for BBB development, interoperability, and symbiosis (5). Astrocyte end-feet, pericytes, microglia, and neuronal protrusions surround the brain microvasculature. Such close intercellular connections mediate the BBB-specific phenotype (1). Next, we provide an overview of these NVU components and highlight the features of ischemic stroke disruption.
2.1. Brain microvascular endothelial cells
The brain microvascular endothelial cells located at the junction of brain tissues and blood have essential physiological functions, including barrier function, nutrient transport, receptor-mediated signal transduction, and leukocyte infiltration (6). Endothelial cells are characterized by a lack of fenestrae and the expression of tight junction (TJ)–protein complexes between adjacent endothelial cells (7). In addition, Peterson et al. found that endothelial cells have mechanoreceptor properties that respond to absolute fluid and transmural pressure, contributing to cerebral blood flow autoregulation (8). As the first barrier for blood components entering the nervous system, endothelial cells are the core components of the BBB and have unique structural and molecular properties, including the following five main aspects (9): high expression of TJ proteins that control the paracellular pathway between endothelial cells: TJ proteins: occludin, claudin, zonula occludens (ZO)-1, ZO-2, ZO-3, cingulin, afadin-6 (AF-6), and 7H6; adherens junctions (AJs): cadherins and associated proteins directly linked to actin filaments (6) and junctional adhesion molecules (JAMs). Lower rates of transcytosis prevent blood components from crossing the BBB via non-specific vesicular transport. In stroke, endothelial vesicles increase, representing an increased transcellular response (10, 11). The high endothelial glycocalyx layer of the cell membrane reduces the passive diffusion of blood macromolecules across the BBB. Low expression of leukocyte adhesion molecules inhibits the adhesion and transit of blood immune cells into the brain. A comprehensive molecular transport system includes the high expression of several transporter proteins (transporting glucose, amino acids, ions, and lipid molecules), membrane receptors, and efflux proteins (multidrug resistance proteins, breast cancer resistance proteins, and multidrug resistance-associated proteins) for transporting material molecules between the blood and brain tissues (12, 13).
Owing to these properties, the BBB strictly limits the non-specific entry of blood components into the brain and efficiently provides the required nutrients to the neural tissue. The TJs between adjacent endothelial cells, specific transport systems on the luminal and abluminal endothelial cell membranes, and the distribution of metabolic enzymes constitute three barrier functions of the BBB: the physical, transport, and metabolic barriers, respectively. Physical barrier: the presence of TJs allows molecules to cross the BBB via a transcellular rather than a paracellular pathway (1, 7). AJ proteins, inhibition of non-selective window pores, cellular drinking, high flow across cells, and inhibition of leukocyte adhesion molecules are also involved in the physical barrier composition (14). Most molecules cross the BBB via the transcytosis pathway (15). In contrast, the paracellular pathway, which depends on the permeability gradient (16–21), accounts for only a small percentage of TJ limitations (18, 20, 22). Transport barrier: much lower rates of endocytosis/transcytosis, mainly through the transcellular pathway. Transcellular diffusion: diffusion locations are on the luminal and abluminal membranes of endothelial cells. They are constrained to gases such as O2 and CO2 and small lipophilic molecules <400 Da (17, 19, 20, 23, 24). Carrier-mediated transport: active or passive substrate-specific transport (21, 24). Glucose transporter 1 (GLUT1), monocarboxylate transporter 1 (MCT1), L-type amino acid transporter 1 (LAT1), and major facilitator superfamily domain-containing protein 2a (Mfsd2a) carry glucose, lactic, amino, and fatty acids, respectively (25–29). GLUT1, a large neutral amino acid and nucleoside transporter protein, is essential for maintaining BBB homeostasis (30). Transcytosis remains the preferred pathway for the selective transport of plasma macromolecules such as albumin and low-density lipoproteins, despite limited transcytosis in CNS endothelial cells (31). Receptor-mediated transcytosis: major transport pathways for molecules without a specific carrier (30). These molecules bind to cell-surface receptors and form endocytic vesicles that cross the BBB (22). Adsorptive transcytosis: charge interaction between the compound and the luminal membrane of endothelial cells, inducing endocytosis (32). Efflux transport is concentrated on the luminal side of the membrane (29). To prevent the accumulation of toxic compounds across the BBB, substances are extracted from the nervous system into the bloodstream by moving a series of small molecules up to a concentration gradient (20, 21). For example, sodium-dependent amino acid transporters present in the endothelial cell membrane can remove amino acids from the brain, and these accumulated amino acids may evoke neurotoxicity during stroke (33–35). Brain microvascular endothelial cells are more abundant in the mitochondria than in the endothelial cells of other organs, which laterally reflect the energy demand that depends on adenosine triphosphate (ATP) transport (6, 36). Metabolic barrier: enzymes that constitute this barrier include ectoenzymes (peptidase and nucleotidases) and intracellular enzymes (monoamine oxidase and cytochrome p450) (37).
In contrast to the importance of TJ proteins, the role of cytosolic transport in regulating BBB permeability has been traditionally ignored or underestimated. Researchers have recognized the importance of cytosolic transport in the last few years, which has become a research hotspot in the BBB field. Low levels of cytosolic transport inhibit BBB permeability during embryonic development and adult physiological states, whereas elevated levels contribute significantly to BBB disruption under various pathological conditions, including chronic hypoperfusion, stroke, and cortical depolarization (38, 39).
Excellent and pioneering work in this area was carried out by Prof. Chenghua Gu's team at Harvard Medical School, who identified that Mfsd2a on the cell membrane is highly expressed in cerebrovascular endothelial cells, which promotes BBB formation and function mainly by maintaining a low rate of transcytosis and effectively inhibiting fossa-mediated cytosolic transport (40). Therefore, it reduces the non-specific transport of blood components into the brain and maintains the functional integrity of the BBB (41). Further studies have shown that the phospholipid transport function of Mfsd2a alters the lipid composition of endothelial cell membranes, thereby inhibiting cell membrane fossa formation, which inhibits fossa-mediated cytosolic transport (42). In addition, related studies have reported that Mfsd2a transports omega-3 fatty acids across CNS endothelial cells (43, 44). Future studies need to determine the relationship between these two functions (45, 46).
2.2. Astrocytes
Astrocytes coat more than 99% of the BBB endothelium (1, 5, 47, 48). Astrocyte end-feet form the BBB and maintain its integrity (1, 49, 50). They have multiple functions, including nutrition, structural support, BBB formation, neuronal metabolism, extracellular environment maintenance, cerebral blood flow regulation, intercellular communication stabilization, immune response control, neurotransmitter synthesis, and resistance to oxidative stress (51–54).
Astrocyte–endothelium interactions are essential for electrolyte homeostasis in the brain under both normal and pathological conditions (4). Perivascular astrocyte end-feet are highly structured and contain orthogonal arrays of particles, composed of the abundant water channel aquaporin-4 (AQP4) and the Kir4.1K+ channel, which are involved in ion volume regulation (1, 48, 55). AQP4 is associated with cytotoxic edema during ischemic stroke (4).
Astrocyte–endothelial interactions strengthen the TJ, minimize the gap junctional area of endothelial cells, and contribute to regulating cerebral blood flow (52). Both are involved in the induction and modulation of unique endothelial cell phenotypes (6, 56, 57). Astrocytes do not appear in the NVU until birth (14, 58). They are not directly involved in the physical properties of the BBB and may modulate its phenotype (6, 59). An experiment was conducted in 1969 to demonstrate this phenomenon. Horseradish peroxidase injected into the brain passes directly through astrocyte end-feet, and diffusion is blocked on the abluminal membrane of the endothelium. Studies over the past two decades have shown that astrocytes mediate barrier function, mainly by modifying the morphological composition and chemical expression of TJ (60, 61). The main molecules involved were TJ-related proteins, platelet endothelial cell adhesion molecule-1, ZO-1, and claudin-5 (60, 62). Astrocytes release several pro-barrier phenotype regulators (63), including nitric oxide (NO), which regulates vasodilation (64); sonic hedgehog, which governs TJ formation; and vascular endothelial growth factor (VEGF), which is associated with vasogenic edema in ischemic stroke (65). Astrocytes have different morphologies and phenotypes, depending on the region where they are located (66); 8 of the 11 common phenotypes were associated with vascular cross-talk (1). Multiple astrocyte morphologies can upregulate and strengthen these three barriers, making the TJ more robust, increasing the expression and polarization of transporter proteins, including Pgp24, and refining the enzyme system (67–69).
Similar to amino acid efflux transporters in endothelial cells, the excitatory amino acid transport proteins are expressed in astrocytes: excitatory amino acid transporters 1 and 2. These proteins remove extra glutamate from synapses and maintain the relative stability of excitatory neurotransmitters in the brain (70). Astrocytes play an important role in ischemic stroke-induced neuroinflammation.
2.3. Pericytes
Pericytes are kidney-shaped cells with nuclei that protrude from the lumen. They contain a small cytoplasm and are distributed irregularly along the brain microvessel wall (71–74). Pericytes cover at least 80% of the brain microvascular wall, and cell protrusions emanating from pericytes penetrate the cellular matrix to cover 20–30% of the microvascular perimeter (6). Such dimensional contact with the vasculature serves as a support for the endothelium of the NVU. It plays important physiological roles in endothelial cell development (6), BBB formation and integrity maintenance, vascular maturation and stabilization, and immune cell trafficking.
Experimental evidence suggests that pericytes have contractile properties and that contractile pericytes–coat cerebral microvessels (71, 72, 75) regulate capillary blood flow and influence cerebral blood flow autoregulation. A recent scRNA-seq study identified the molecular mechanisms by which pericytes regulate vessel diameter. Strong evidence for the role of pericytes in controlling vessel diameter is provided by the presence of receptors for vasoactive factors, such as L-type voltage-gated calcium channels, and those involved in the contraction of smooth muscle cell actin on pericytes (76). To a certain extent, pericytes act as the “vascular smooth muscle cells” of the brain's microvasculature.
Evidence for pericyte–endothelial cell interactions has also been found. The absence of pericytes in mice by experimental means revealed the downregulation of Mfsd2a expression (43). Pericyte deletion leads to increased endothelial transcytosis without affecting the integrity of TJs (59, 77). Recent studies have shown that pericytes regulate endothelial transcytosis by binding to integrins in endothelial cells through vitronectin expression (78).
2.4. Neurons
As mentioned previously, neurons and their nearby cerebral microvasculature are in close contact, and there is an intimate interaction between neurons and their matching vessels. Although direct neuron–endothelial contact has been demonstrated (66, 79), neuronal communication in the NVU is primarily mediated by astrocytes (80). Little is known about the other structural and functional contributions of neurons to the BBB (80). The suggested pathological factors, such as ischemic stroke-induced BBB disruption, are anatomical and usually accompanied by selective compensatory changes in cerebral blood flow (81–83). This indicates that interactions between neurons and microvessels can modulate BBB permeability.
2.5. Extracellular matrix (ECM)
The ECM layer generated by the basal cell membrane serves as a signaling site for cell-cell interactions (1). The ECM is divided into two layers: the vascular basement membrane produced by pericytes and endothelial cells and the glial basement membrane secreted by astrocytes (84). The structural proteins that comprise the ECM include collagen type IV, laminin, fibronectin, elastin, thrombospondin, and various proteoglycans that are readily degraded by proteases. Their degradation is associated with increased BBB permeability during ischemic stroke (85, 86). The ECM wraps around endothelial cells and pericytes. It separates and anchors cells through adhesion receptors. Astrocytes and pericytes secrete integrin and dystroglycan families of matrix adhesion receptors that are distributed in the ECM and mediate NVU function (87).
2.6. Tight junction (TJ)
TJ is an essential junction complex between adjacent epithelial cells. TJs comprise intact membrane and cytoplasmic accessory proteins, including occludin, claudins, JAMs, ZO proteins, and cingulin. Actin is a cytoskeletal protein that maintains endothelial structure and function. TJ proteins are linked to cytoskeletal actin by accessory proteins. The tightness of the TJ connections determines the paracellular permeability of water-soluble molecules (6, 49). The BBB transendothelial electrical resistance (TEER) is a measure of the degree of permeability and efficiency of TJs in the BBB (88). The TEER of peripheral microvessels is typically 2–20 ohm.cm2, while the TEER of the brain endothelium can reach 1,000 ohm.cm2 (1). The polarity of the luminal and abluminal membranes of endothelial cells contributes to barrier function. The concept of polarity is derived from quantitative biochemical studies (6). TJs perform a structural gate function (1), limiting paracellular permeability. Its fence function separates the apical and basal regions of the cell membrane (89), allowing endothelial cells to exhibit apical–basal polarization properties. The luminal and abluminal polarization of receptors, ion channels, and enzymes in endothelial cells makes the functional partitioning of endothelial cells more refined and rational (6). TJ proteins exist in different isoforms because of their tissue origins (4). Phosphorylation regulates the activities of TJ and accessory proteins (5).
2.7. Occludin
Occludin is a 65-KDa protein with 504 amino acids (6, 90). Occludin has two extracellular loops and one intracellular loop, and its carboxyl and amino terminals are anchored to the cytoplasm. The carboxy-terminal serine residues of occludin are linked to the cytoskeleton via ZO-1 and ZO-2, allowing occludin to bind to the basal cellular membrane. Occludin phosphorylation regulates its functional state (91). The volume of occludin in endothelial cells is much higher in the CNS than in the periphery (90, 92). Recent studies have shown that occludin is a key factor in regulating BBB permeability (93–96). A study in 2017 using a rat model of ischemic stroke found that the occludin protein in TJs of brain microvessels caused by cerebral ischemia rapidly degraded. Elevated blood occludin levels show a time-dependent correlation with the degree of BBB injury. Therefore, occludin proteins in the blood may serve as a clinically relevant marker of ischemic stroke (97).
2.8. Claudins
Claudins comprise numerous proteins. They have two extracellular loops through which adjacent endothelial cells are connected, forming the major sealing component of TJs (98). The two intracellular loops of claudins can bind to ZO-1, ZO-2, and ZO-3 through their carboxyl termini (99, 100). Claudins are morphologically similar to occludins but do not share sequence homology (90). Claudin-5 plays a crucial role in the early development of the CNS as a symbol of the BBB (101). The phosphorylation pathway can regulate the functional activity of claudin-5 (102, 103). The phosphorylation of claudin-5 at Thr207 increases the permeability of TJ. In experimental models of pathological conditions, such as ischemic stroke, claudin-1 is lost in the cerebral vasculature (49), claudin-5 expression is reduced (104, 105), and the interaction between claudin-5 and occludins is disrupted (106).
2.9. Junctional adhesion molecule (JAM)
JAMs are a family of immunoglobulin superfamily proteins that localize to intercellular slits. JAM-A, JAM-B, and JAM-C are expressed in endothelial cells and are involved in the construction and maintenance of the TJ (4, 107). The extracellular portion of the transmembrane structural domain of JAMs consists of two immunoglobulin-like loops formed by disulfide bonds (107). JAMs mainly bind to the intracellular components ZO-1, partitioning defective protein-3(PAR-3), AF-6, and multi-PDZ-protein-1(MUPP-1) (108). Like claudins, JAMs participate in homophilic and heterophilic interactions between cells (107). Deficiency in JAM protein expression and BBB disruption are directly related (109–112).
2.10. Membrane-associated guanylate kinase (MAGUK) proteins
MAGUK proteins are also called cytoplasmic accessory proteins. MAGUK proteins are accessory components of TJ structures. Their multiple structural domains are involved in protein–protein interactions (113), forming protein complexes attached to the cell membrane. The MAGUK proteins associated with TJ are ZO-1, ZO-2, ZO-3, and the newly identified cingulin, 7H6, and AF-6 (5, 113). ZO-1, a 220-KDa phosphoprotein, is expressed in epithelial and endothelial cells (114, 115). ZO-1 bridges the TJ proteins with the actin cytoskeleton (91, 116–118). ZO-1 transmits the TJ state to the intracellular zone and vice versa. Decreased ZO-1 expression is associated with increased BBB permeability (118). ZO-2, a 160-KDa phosphoprotein, shares homology with ZO-1 (119, 120). Immunofluorescence microscopy studies conducted in 2003 showed that ZO-3 was enriched in the TJ of epithelial cells (121). Although it is expressed in the epithelium, its role in TJ has not been fully elucidated (122). Cingulin is a 140–160-KDa phosphoprotein. It is localized to the cytoplasmic surface of TJs and is associated with the ZO proteins, myosin, JAM-A, and AF-6 (123, 124). As a vital scaffolding protein of TJs, it can form various junctions and transmit mechanical forces generated by cytoskeletal contraction, thus regulating BBB permeability (5, 125). 7H6, a 155-KDa phosphoprotein, reversibly detaches from TJ when ATP is depleted, increasing paracellular permeability (126, 127). AF-6, a 180-KDa protein, is involved in TJ regulation by linking it to ZO-1 (103).
2.11. Adherens junction (AJ)
AJ: An intercellular junction, also called zonula adherens (1), is a specific type of intercellular interaction consisting of cadherins, catenins, vinculin, and actinin (128). However, the effect of AJ on BBB permeability in pathological states has not been fully elucidated. However, its interaction with vascular endothelial growth factor (VEGF) receptor-2 and vascular endothelial (VE)-cadherin-mediated upregulation of claudin-5 suggests that AJ plays a central role in angiogenesis and the regulation of TJ integrity (129, 130).
3. Ischemic stroke
Ischemic stroke is a leading cause of death and disability worldwide. It imposes substantial economic and emotional burdens on patients, families, and society. Ischemic stroke is characterized by a sudden reduction or cessation of oxygen and blood supply due to local arterial occlusion of the brain tissues, resulting in irreversible cell death and tissue damage in the infarct core. The surrounding penumbra may regain function by restoring cerebral blood flow owing to mild ischemia. Various functions of the cerebral vasculature, such as BBB and blood flow regulation, are also affected by acute ischemic–hypoxic injury. Disruption of the BBB is a hallmark of ischemic stroke (131). The mechanisms of BBB disruption induced by stroke include phosphorylation of TJ proteins, regulation of transporter protein expression, neuroinflammation, and abnormal enzyme function (132).
3.1. Ischemic phase and biphasic nature of barrier permeability
Ischemic stroke occurs chronologically during the ischemic and reperfusion phases, during which a series of cellular changes occur (4). The BBB is incomplete long before neuronal damage occurs (133, 134), and the concept of NVU emphasizes that BBB disruption in ischemic stroke damages the cell structure and disrupts intercellular interactions (135, 136). Although called a barrier, the BBB is not a physical wall (137). In fact, the breakdown of the BBB caused by brain infarction is a dysregulation of molecular proteins associated with its three functional barrier properties.
Blockage of cerebral blood flow and rapid hypoxia in the ischemic core triggers a chain of events in the infarct area, including ATP depletion; neurotransmitter leakage: dopamine, excitotoxic glutamate, arachidonic acid, and ceramide efflux that produce extremely acute neurotoxic effects; ion imbalance: increased intracellular calcium; metabolic disorders and increased acidosis; endothelial cells, astrocytes, neurons swelling due to lactic acid accumulation; cerebral microvascular diameter reduction; oxidative stress; neuroinflammation activation; protease induction and promotion of extracellular matrix degradation at the BBB (138, 139). Hypoxia disrupts the localization or expression of TJ proteins in several in vitro models of cerebral infarction (118, 140). Fischer et al. demonstrated that ZO-1 and ZO-2 translocate to the nucleus in a hypoxic environment in vitro (141). In addition, the in vitro hypoxic environment altered the cell membrane localization and expression of claudin-5, whereas TEER decreased (142). To date, there have been some new developments regarding BBB disruption after ischemic stroke. In 2014, Knowland et al. observed that barrier function was impaired as early as 6 h after stroke, with increased endothelial vesicles and transcytosis in a transgenic mouse stroke model. In contrast, the TJ showed severe structural defects after only 2 days. This suggests that early BBB defects are caused by defective transport barriers (10). Data from a study by Haley and Lawrence subsequently demonstrated that endothelial vesicle upregulation might exacerbate BBB permeability in the early stages of cerebral ischemia and that the number of endothelial vesicles correlates with the degree of BBB disruption (143). Krueger et al. established various experimental models of focal cerebral ischemia, and their experimental data strongly suggest that ischemia-associated BBB deficits are primarily caused by endothelial cell degeneration (144).
Opening of the BBB is a biphasic phenomenon in preclinical models of ischemic stroke (145). First, after the onset of acute ischemic stroke, initial reperfusion permeability can occur at the time of acute elevation of the regional cerebral blood flow (rCBF), followed by biphasic permeability of the BBB (4). Animal models and human studies have documented that the first phase occurs in the hyperacute phase of acute ischemic stroke, usually within 6 h of onset (146–149). The second phase occurs in the acute phase of acute ischemic stroke, usually within 72/96 h after onset, when the initial cytotoxicity-induced neuroinflammation further disrupts the BBB. A second peak infiltration rate is observed during this phase (146–148, 150). It is worth noting that other studies, including those in humans, have shown that elevated BBB permeability can persist for several weeks after stroke (149, 151) (Figure 2).
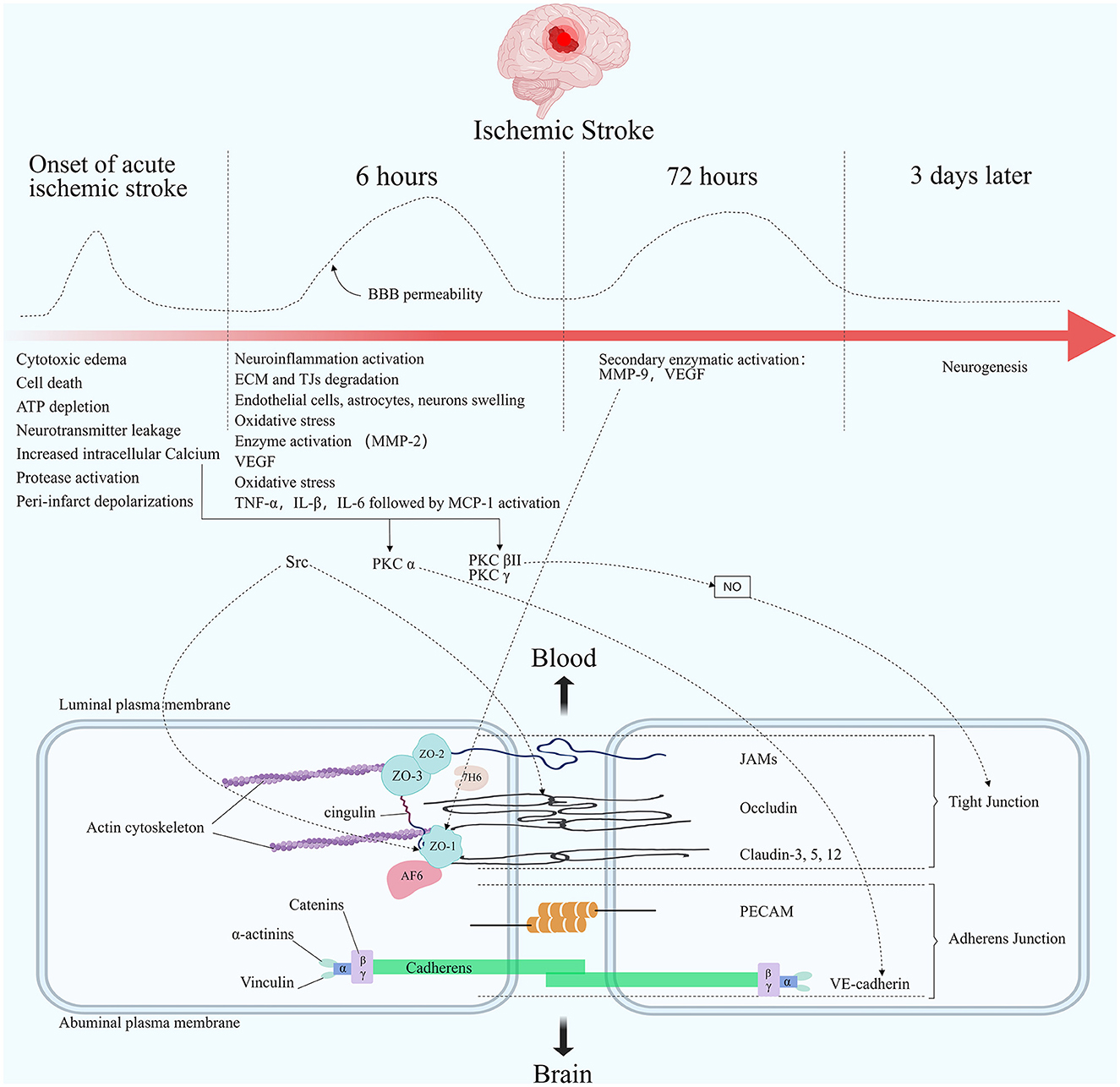
Figure 2. Molecular composition of BBB tight junctions and major changes in acute ischemic stroke. The TJ consists of three integral membrane proteins, claudins, occludin, and junction adhesion molecules (JAMs), and a number of cytoplasmic accessory proteins including zonula occludens 1, 2, and 3 (ZO-1, 2, 3), cingulin, afadin-6 (AF-6), and 7H6. Membrane proteins are connected to actin by cytoplasmic proteins. The carboxy-terminal serine residues of occludin are linked to the cytoskeleton via ZO-1 and ZO-2. Adherens junctions are formed by cadherins, catenins, vinculin, and actinin. Among the important molecules are vascular endothelial cadherin (VE-cadherin) and platelet endothelial cell adhesion molecules (PECAM). The figure shows the changes in blood-brain barrier permeability at different times after the onset of ischemic stroke, as well as the main pathophysiological processes in each stage. Acutely elevated local cerebral blood flow can lead to initial reperfusion permeability. The opening of the BBB presents a biphasic phenomenon. The first phase usually occurs within 6 h after an acute cerebral infarction. The second phase usually occurs within 72 h after the onset of an acute cerebral infarction. Elevated Ca2+ concentration in the endothelial under ischemic hypoxic conditions regulates TJ proteins' stability through multiple pathways. PKC α can act on VE-cadherin to participate in AJ degradation. PKC βII and PKC γ cause TJ changes by regulating NO activity. Src phosphorylates occludin and ZO-1. Elevated MMP-9 reduces ZO-1 expression and its cytoplasmic translocation, leading to barrier disruption.
3.2. Brain edema
Ischemic brain injury can rapidly lead to cerebral edema, with excessive fluid retention in the intracellular or extracellular spaces of the brain (36). Cerebral edema is a leading cause of clinical deterioration and death in ischemic stroke and is divided into cytotoxic and vasogenic edema (152, 153). Cytotoxic edema appears within minutes of ischemia, after which the BBB breaks down and vasogenic edema emerges (154, 155). The dysfunction of ion transporters in the BBB is an important mechanism that leads to brain edema. Cytotoxic edema: after ischemia, the activity of Na+/H+ exchangers, Na+-K+-Cl cotransporters, and calcium-activated potassium channel KCa3.1 at the BBB increases, facilitating the transport of Na+ and Cl− across the cells into the relatively intact BBB (156–158). Cerebral edema gradually develops as Na+ intake increases (156, 157). The endothelial cells showed initial nuclear swelling and moderate cytoplasmic swelling within 2 h. Swelling of astrocytic end-feet begins within 5 min of energy depletion after cerebral infarction, leading to the detachment of the end-feet from the endothelium (159–161). Tagaya observed that 2 h after middle cerebral artery occlusion, there was no expression of α1β1, α3β1, and α6β1 on endothelial cells and α6β4 on astrocytes (162). Vasogenic edema arises from changes in the TJ, which increase the permeability of macromolecules and cause fluid to flow outside the blood vessels (163).
AQP, a highly permeable water channel, is highly enriched in the astrocyte end-feet. It is involved in maintaining fluid homeostasis in the brain. In several ischemic stroke models, AQP4 contributes to brain edema formation, and its absence disables cellular water uptake (164, 165). The structure of the NVU determines the complexity of BBB disruption induced by ischemic stroke, and there is no general way to regulate it. The main modulators of this process are described below. Studies on TJ protein modifications after ischemic stroke have focused on phosphorylation (36). Phosphorylation is the primary regulatory mechanism of protein effects on the BBB (5). The phosphorylation and dephosphorylation states of TJs depend on the amino acid residues that undergo phosphorylation and the type of stimulus (6). Some functionally relevant protein kinases are summarized below.
3.3. Related protein kinases
Calcium-dependent protein kinase C (PKC) subtypes may be involved in TJ regulation, including α, βI, βII, and γ. Myosin light-chain (MLC) phosphorylation regulates endothelial cell paracellular permeability. MLC phosphorylation leads to actin–myosin contraction via calcium/calmodulin-independent MLC-kinase (MLCK) phosphorylation (166, 167). This process increases the permeability of the TJ, allowing F-actin, ZO-1, and occludins to redistribute (168). PKC inhibitors can reduce endothelial cell permeability by introducing several factors (thrombin, bradykinin, VEGF, hydrogen peroxide, platelet-activating factor, and neutrophils) (169, 170). The expression of PKC α, βI, and ε shows an increase in hypoxic or hyperglycemic environments (171). Following ischemia, hyperglycemia, and inflammatory insults, endothelial cells exhibit increased permeability via a PKC α-dependent pathway (172–175). PKC α is also involved in the catabolism of AJs via VE-cadherin, which leads to increased paracellular permeability (175). Increased expression of PKC βII and PKC γ can regulate NO synthase, indicating that PKC may affect TJ changes by regulating NO activity (176). Mitogen-activated protein kinases (MAPKs) belong to the serine/threonine kinases family. The most studied MAPKs are extracellular signal-related kinases (ERK1/2), c-Jun N-terminal kinases (JNK), and p38-protein (170). Studies in consecutive years have demonstrated that the integrity of TJ is associated with increased levels of cyclic adenosine monophosphate (cAMP) (177–179). The effect of cAMP is most likely mediated by the protein kinase A (PKA) pathway. PKA stabilizes cytoskeletal proteins, mediates the dephosphorylation of MLC, enhances cell-matrix adhesion, and detaches F-actin from myosin (180–182). Protein kinase G (PKG) is a serine/threonine-specific protein kinase activated by cyclic guanosine monophosphate (cGMP). Both in vivo and in vitro studies have demonstrated that factors relevant to ischemic stroke, including bradykinin, histamine, NO, tumor necrosis factor (TNF)-α, platelet-activating factor, and VEGF, can lead to increased NO-cGMP dependent paracellular permeability (141, 170, 183). Protein tyrosine kinases (PTKs) act as intracellular signal transduction molecules that regulate endothelial paracellular permeability. PTK can be classified as receptor and non-receptor-mediated types. Occludin phosphorylation diminishes its ability to bind to ZO-1 and ZO-2, leading to increased paracellular permeability (140, 184, 185). Phosphorylation of AJ proteins unanchors them to the cytoskeleton and affects TJ permeability (186). Among the non-receptor-mediated PTKs, the Src family plays a vital role in TJ regulation (187, 188). Interleukin (IL)-1β, reactive oxygen species (ROS), and tumor necrosis factor (TNF) modulate Src and increase endothelial permeability of the BBB (189–191). Phosphorylation of myosin MLCK by Src enhances actin-myosin interactions and increases paracellular permeability (192, 193). Studies have demonstrated that cerebral infarction-related oxidative stress-induced endothelial cell permeability is associated with Src, which phosphorylates occludin and ZO-1 (184, 194, 195). Oxidative stress can also lead to F-actin redistribution and stress fiber formation (196, 197).
3.4. Ca2+
Under physiological and pathological conditions, TJ responds rapidly to intracellular signals that regulate junctional complexes (6). Molecules that regulate BBB permeability are induced by altering intracellular Ca2+ (49). Early studies have demonstrated that Ca2+, a second messenger, is a key component in the regulation of TJ through multiple pathways (such as PKC, MAPK, and phospholipase-A2) (198). Elevated intracellular Ca2+ triggers a signaling cascade response that regulates TJ transcriptional expression and alters TJ post-translational distribution (199). The endothelial Ca2+ concentration increases in an ischemic–hypoxic environment, the ATP-dependent efflux mechanism is disrupted, and the control of intracellular Ca2+ by the cytoplasmic endoplasmic reticulum Ca2+-ATPase fails (199, 200). Increased Ca2+ in endothelial cells enables the activation of MLCK (166, 167), induces actin reorganization, changes cell shape, and increases cell permeability (201, 202). Ca2+ directly affects the stability of BBB endothelial cell membranes and TJ proteins by promoting phospholipase-A2 activation, which allows membrane phospholipids to release free fatty acids (4).
3.5. Neuroinflammation
Neuroinflammation is an integral part of the pathophysiology of cerebrovascular diseases, particularly ischemic stroke. Many studies have shown that poststroke neuroinflammation is an important factor in the long-term prognosis of ischemic stroke. After a stroke, various factors, such as ROS formation, necrotic cells, and damaged tissues, can activate inflammatory cells, resulting in an inflammatory response. Several substances have been found in the cerebrospinal fluid (CSF) of stroke patients, and studies suggest that TNF-α and IL-1β initiate neuroinflammation in ischemic stroke (4). Cyclooxygenase inhibition can eliminate TNF-α-, IL-1β-, and IL-6-induced increase in endothelial paracellular permeability, suggesting a role for arachidonic acid in increasing BBB permeability during ischemic stroke (203, 204). The production and release of chemokines chemoattractant protein monocyte chemoattractant protein-1 (MCP-1) and cytokine-induced neutrophil chemoattractant (CINC) are regulated by regulatory factors. In the pathological context of stroke, MCP-1 is a major factor associated with leukocyte entry into the brain (205). The biphasic BBB permeability that occurs during hypoxic reperfusion is consistent with an increase in the secretion of MCP-1 by astrocytes and endothelial cells in the brain (206). Biphasic BBB permeability is associated with the redistribution of occludin, ZO-1, ZO-2, and claudin-5 (206). IL-1 is associated with the induction of endothelial cell adhesion molecules during ischemic stroke. IL-β-induced increase in paracellular permeability of the BBB is related to the loss of occludin and ZO-1 in the junctional complex (207).
3.6. Enzyme activities
Matrix metalloproteinases (MMPs) are a family of zinc-dependent endopeptidases that can degrade fibronectin and laminin (50) and have been identified as key factors in BBB disruption in ischemic stroke (208). The basement membrane is a scaffold for brain endothelial cells and is composed of type IV collagen, laminin, fibronectin, elastin, thrombospondins, various proteoglycans, and heparin sulfate. MMPs exhibit substrate specificity for type IV collagen, laminin, and fibronectin, which degrade basement membranes. MMPs have been identified as the apparent initiators of BBB disruption. After the onset of ischemic stroke, MMP expression is upregulated and activated through pro-inflammatory cytokine pathways (via NF-kB) or activation of HIF-1a and furin (50, 209). A clinical study showed that MMP-9 levels were significantly elevated in patients with acute ischemic stroke (210). MMP-mediated BBB opening in ischemic stroke may be regulated by NO signaling. The activation of MMP-2 is thought to initiate TJ protein degradation (211). In the late stages of stroke-induced BBB disruption, MMP-9 expression triggers devastating injury (211). The reason for the disruption of barrier function caused by elevated MMP-2/9 levels is the reduced expression of ZO-1 and its cytoplasmic translocation (212). Another study confirmed that the degree of BBB damage is consistent with reduced MMP-2/9 expression (213). Therefore, inhibition of MMPs may benefit patients with stroke (214, 215). Notably, despite their destructive effects in the acute phase of stroke, MMPs may play a beneficial role in the recovery phase of stroke (216–218). MMP-9 is upregulated in the peri-infarct region 7–14 days after stroke and may be involved in vascular remodeling. Interestingly another study demonstrated that MMP-13 promotes stroke injury in the acute phase but improves angiogenesis during the repair phase (217).
3.7. Actin polymerization
Actin is not a tight junction protein by traditional definition, but it has an important role in BBB stabilization (219). The actin cytoskeleton normally provides anchoring sites for tight junction proteins (220, 221). Dynamic stabilization between the cytoskeleton and tight junction proteins is important for BBB maintenance (222, 223). Actin is usually distributed uniformly throughout the endothelial cell in the form of short filaments and diffuse monomers (220). Unpolymerized globular actin polymerizes into the filamentous-actin (F-actin) form via an ATP-promoted process (224). Phosphorylation of myosin light chain (MLC) promotes the formation of dense stress fibers from short F-actins and induces actin contraction (225, 226). After ischemia and hypoxia, the normal actin cytoskeleton polymerizes into linear stress fibers spanning the endothelial cell interior (220). Increased contraction and tension of the actin cytoskeleton after polymerization occurs, leading to cell shrinkage, impaired tight junctions, and ultimately disruption of the BBB (225–227). Experimental evidence shows that actin polymerization after acute ischemic stroke induces the formation of stress fibers in the cytoskeleton, exposing the BBB to greater vulnerability to damage by MMPs (221). MMP-9 is usually considered to be the initiating factor of BBB disruption after acute ischemic stroke (228). Contrary to previous perceptions, the tension generated by stress fibers promotes the breakdown of tight junction proteins (229). Preliminary studies have shown that long before MMP-9 begins to degrade tight junctions, the redistribution of junctional complex proteins induced by actin polymerization causes structural changes in endothelial cells that disrupt the BBB (221). The structural disruption makes BBB more susceptible to degradation by MMP-9. Therefore, slowing down the cytoskeletal structural changes of endothelial cells in the early phase of ischemic stroke may bring a new therapeutic target for the protection of the BBB (230, 231).
3.8. Zinc
Zinc is the second most abundant essential trace element in the human body (232). It plays a crucial role in brain growth and development as a neurotransmitter or neuromodulator. Imbalances in zinc are closely associated with brain disorders (233). Most of the zinc ions in the human body are bound to zinc-binding proteins, while another portion exists as free zinc ions in organs, tissues, body fluids, and secretions (234). Zinc transport proteins regulate and maintain zinc ion homeostasis. Zinc-regulated transporter (ZRT)/iron-regulated transporter (IRT)-like protein (ZIP) family increases zinc intake, while the zinc transporter family (ZnTs) mediates the efflux of zinc from the cytoplasm, increasing the plasma zinc concentration.
BBB regulates zinc homeostasis in the brain. Studies have shown that under normal physiological conditions, the BBB isolates zinc between the plasma and the brain (235). Studies in animal models have shown that zinc homeostasis is critical for BBB integrity. Both zinc deficiency and zinc overload lead to BBB disruption under pathological conditions. In the experiment, dynamic magnetic resonance imaging measurements revealed that zinc deficiency leads to a significant increase in BBB permeability in rats exposed to high oxygen levels (236). In a rat model of cerebral ischemia, accumulation of zinc in ischemic microvessels was observed, leading to the loss of tight junction proteins (occludin and claudin-5). Accumulated zinc mediates cerebral ischemia-induced BBB injury by upregulating superoxide and MMPs. Chelated zinc reduces BBB permeability in ischemic rats (237). Interestingly, during the recovery phase of cerebral ischemia, zinc has been shown to alleviate brain ischemic atrophy, promote neural function restoration, and facilitate angiogenesis in the process of cerebral ischemic repair through the astrocyte-mediated HIF-1α/VEGF signaling pathway (238). A study compared the serum zinc concentration levels between ischemic stroke patients and healthy controls, and the results showed a significant decrease in serum zinc concentration in stroke patients. Zinc may represent an independent risk factor for stroke (239). Another study compared the serum zinc concentrations in patients with acute ischemic stroke to those with transient ischemic attacks. The results showed that stroke patients had significantly lower serum zinc concentrations (240). The fate of reduced serum zinc needs further study. MicroRNA-30a (miR-30a) is a member of the miR-30 family and is abundant in human endothelial cells. Experiments in cellular and animal models of ischemic stroke demonstrated that miR-30a can mediate BBB damage. miR-30a can directly negatively regulate ZnT4. Inhibition of miR-30a decreases BBB permeability, prevents degradation of tight junction proteins, and reduces intracellular free zinc in endothelial cells (241). This suggests that miR-30a may be an effective therapeutic target for ischemic stroke.
4. Discussion
Over the past 40 years, studies from animal models to clinical studies have elucidated the occurrence, development, composition, and maintenance of the BBB. The emergence of the NVU concept has dramatically changed the field of cerebrovascular research, prompting researchers to study and understand BBB physiology and pathology from a systematic perspective. We now recognize that the BBB is more than a simple physical anatomical barrier; endothelial cells, astrocytes, and other cells work together as functional complexes to regulate the barrier function. The basic structure of the BBB is now well understood; however, new insights into its fine structure are emerging. New technologies, such as two-photon microscopy, may be used in future studies for further in-depth investigation (41, 59, 77). Interactions between the components of the BBB are intricate. As mentioned above, many common points of phosphorylation are concentrated in the BBB cytoskeleton. We need to identify the processes that regulate the barrier and the core of the pathway. The recently discovered Mfsd2a protein is a current research hotspot, and further clarification of its role in physiology and pathology will help us better understand the BBB (41, 42). Future preclinical studies should consider multiple cell types in the NVU and the effects of peripheral systems on the NVU.
To date, the mechanism of BBB disruption in ischemic stroke remains unclear, and significant discoveries have been made regarding barrier permeability after disruption, such as the discovery of biphasic barrier permeability. However, the timing of the occurrence and maintenance of biphasic permeability must be clarified to provide a basis for the timing of stroke treatment. We also need an in vitro system to reliably reproduce the in vivo barrier, in which changes in the BBB can be dynamically observed using a stroke model. This ideal in vitro barrier requires the expression of essential components; however, the endothelial cells of the CNS can quickly lose their barrier properties in culture (242, 243). Recently, different research teams have made progress in perfecting the in vitro BBB model. In 2011, Hatherell et al. co-cultured endothelial cells with astrocytes and pericytes to recapitulate NVUs in vitro, increasing TEER (244). In 2013, Paolinelli et al. demonstrated that activation of the Wnt/β-catenin pathway in vitro increased the restrictiveness of endothelial cell monolayers without the need for co-culture with other cell types (245). Lippmann's study showed that endothelial cells with barrier properties can be generated from human pluripotent stem cells (246, 247). These new developments provide insights for the establishment of in vitro models. In vitro BBB models derived from stem cell sources may provide an ideal experimental setting for future studies.
In ischemic stroke-induced BBB disruption, the destructive factors may exhibit protective effects at different times, for example, MMPs. We need to clarify the role of these “dual nature” factors at different times to inform the timing of treatment for cerebral infarction. Studies on BBB disruption have revealed that endogenous barrier transport proteins may be potential targets for CNS drug transport (248). Neuronal protection alone may not improve the neurological prognosis for treating cerebral infarction. Therefore, therapeutic approaches that favor multiple cell types must be considered. Astrocytes are one of the most abundant and widely exposed cellular components. Thus, these compounds can potentially serve as therapeutic targets (52).
These detectable components are clinically valuable when structural proteins leak into the blood or CSF following BBB disruption after a stroke. For example, CSF albumin, IgG per serum albumin, or IgG ratio measurement can be used as clinical biomarkers to assess the integrity of the BBB (249–253). Serum S100B is a protein expressed by astrocytes in the brain and is considered a possible candidate for detecting BBB damage caused by astrocytes (254). Future research should focus on finding easily detectable BBB-related markers of cerebral infarction injury to provide a new basis for diagnosis and treatment.
Author contributions
SX drafted the manuscript. SX, XZ, Z-HY, and X-KS illustrated the manuscript. XS revised the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by the Science and Technology Department of Jilin Province (YDZJ202301ZYTS520) to XS.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. (2006) 7:41–53. doi: 10.1038/nrn1824
2. Tsai PS, Kaufhold JP, Blinder P, Friedman B, Drew PJ, Karten HJ, et al. Correlations of neuronal and microvascular densities in murine cortex revealed by direct counting and colocalization of nuclei and vessels. J Neurosci. (2009) 29:14553–70. doi: 10.1523/JNEUROSCI.3287-09.2009
3. Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron. (2017) 96:17–42. doi: 10.1016/j.neuron.2017.07.030
4. Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. (2008) 32:200–19. doi: 10.1016/j.nbd.2008.08.005
5. Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. (2005) 57:173–85. doi: 10.1124/pr.57.2.4
6. Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood–brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. (2006) 1:223–36. doi: 10.1007/s11481-006-9025-3
7. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. (2010) 37:13–25. doi: 10.1016/j.nbd.2009.07.030
8. Peterson EC, Wang Z, Britz G. Regulation of cerebral blood flow. Int J Vasc Med. (2011) 2011:823525. doi: 10.1155/2011/823525
9. Xiong B, Li A, Lou Y, Chen S, Long B, Peng J, et al. Precise cerebral vascular atlas in stereotaxic coordinates of whole mouse brain. Front Neuroanat. (2017) 11:128. doi: 10.3389/fnana.2017.00128
10. Knowland D, Arac A, Sekiguchi KJ, Hsu M, Lutz SE, Perrino J, et al. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron. (2014) 82:603–17. doi: 10.1016/j.neuron.2014.03.003
11. Nahirney PC, Reeson P, Brown CE. Ultrastructural analysis of blood-brain barrier breakdown in the peri-infarct zone in young adult and aged mice. J Cereb Blood Flow Metab. (2016) 36:413–25. doi: 10.1177/0271678X15608396
12. Langen UH, Ayloo S, Gu C. Development and cell biology of the blood-brain barrier. Annu Rev Cell Dev Biol. (2019) 35:591–613. doi: 10.1146/annurev-cellbio-100617-062608
13. Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell. (2015) 163:1064–78. doi: 10.1016/j.cell.2015.10.067
14. Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. (2013) 19:1584–96. doi: 10.1038/nm.3407
15. Bernardo-Castro S, Sousa JA, Bras A, Cecilia C, Rodrigues B, Almendra L, et al. Pathophysiology of blood-brain barrier permeability throughout the different stages of ischemic stroke and its implication on hemorrhagic transformation and recovery. Front Neurol. (2020) 11:594672. doi: 10.3389/fneur.2020.594672
16. Laksitorini M, Prasasty VD, Kiptoo PK, Siahaan TJ. Pathways and progress in improving drug delivery through the intestinal mucosa and blood-brain barriers. Ther Deliv. (2014) 5:1143–63. doi: 10.4155/tde.14.67
17. Barar J, Rafi MA, Pourseif MM, Omidi Y. Blood-brain barrier transport machineries and targeted therapy of brain diseases. Bioimpacts. (2016) 6:225–48. doi: 10.15171/bi.2016.30
18. Georgieva JV, Hoekstra D, Zuhorn IS. Smuggling drugs into the brain: an overview of ligands targeting transcytosis for drug delivery across the blood-brain barrier. Pharmaceutics. (2014) 6:557–83. doi: 10.3390/pharmaceutics6040557
19. Moura RP, Martins C, Pinto S, Sousa F, Sarmento B. Blood-brain barrier receptors and transporters: an insight on their function and how to exploit them through nanotechnology. Expert Opin Drug Deliv. (2019) 16:271–85. doi: 10.1080/17425247.2019.1583205
20. Orthmann A, Fichtner I, Zeisig R. Improving the transport of chemotherapeutic drugs across the blood-brain barrier. Expert Rev Clin Pharmacol. (2011) 4:477–90. doi: 10.1586/ecp.11.26
21. Tajes M, Ramos-Fernandez E, Weng-Jiang X, Bosch-Morato M, Guivernau B, Eraso-Pichot A, et al. The blood-brain barrier: structure, function and therapeutic approaches to cross it. Mol Membr Biol. (2014) 31:152–67. doi: 10.3109/09687688.2014.937468
22. Lalatsa A, Butt AM. Physiology of the blood–brain barrier and mechanisms of transport across the BBB. In: Nanotechnology-Based Targeted Drug Delivery Systems for Brain Tumors. Amsterdam: Elsevier (2018) p. 49–74.
23. Xie J, Shen Z, Anraku Y, Kataoka K, Chen X. Nanomaterial-based blood-brain-barrier (BBB) crossing strategies. Biomaterials. (2019) 224:119491. doi: 10.1016/j.biomaterials.2019.119491
24. Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: from physiology to disease and back. Physiol Rev. (2019) 99:21–78. doi: 10.1152/physrev.00050.2017
25. Nguyen LN, Ma D, Shui G, Wong P, Cazenave-Gassiot A, Zhang X, et al. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature. (2014) 509:503–6. doi: 10.1038/nature13241
26. Boado RJ Li JY, Nagaya M, Zhang C, Pardridge WM. Selective expression of the large neutral amino acid transporter at the blood-brain barrier. Proc Natl Acad Sci U S A. (1999) 96:12079–84. doi: 10.1073/pnas.96.21.12079
27. Cornford EM, Hyman S, Swartz BE. The human brain Glut1 glucose transporter: ultrastructural localization to the blood-brain barrier endothelia. J Cereb Blood Flow Metab. (1994) 14:106–12. doi: 10.1038/jcbfm.1994.15
28. Kido Y, Tamai I, Okamoto M, Suzuki F, Tsuji A. Functional clarification of MCT1-mediated transport of monocarboxylic acids at the blood-brain barrier using in vitro cultured cells and in vivo BUI studies. Pharm Res. (2000) 17:55–62. doi: 10.1023/A:1007518525161
29. Shen S, Zhang W. ABC transporters and drug efflux at the blood-brain barrier. Rev Neurosci. (2010) 21:29–53. doi: 10.1515/REVNEURO.2010.21.1.29
30. Lombardo SM, Schneider M, Tureli AE, Gunday Tureli N. Key for crossing the BBB with nanoparticles: the rational design. Beilstein J Nanotechnol. (2020) 11:866–83. doi: 10.3762/bjnano.11.72
31. Xiao G, Gan LS. Receptor-mediated endocytosis and brain delivery of therapeutic biologics. Int J Cell Biol. (2013) 2013:703545. doi: 10.1155/2013/703545
32. Salameh TS, Banks WA. Delivery of therapeutic peptides and proteins to the CNS. Adv Pharmacol. (2014) 71:277–99. doi: 10.1016/bs.apha.2014.06.004
33. Cederberg HH, Uhd NC, Brodin B. Glutamate efflux at the blood-brain barrier: cellular mechanisms and potential clinical relevance. Arch Med Res. (2014) 45:639–45. doi: 10.1016/j.arcmed.2014.11.004
34. Heyes S, Pratt WS, Rees E, Dahimene S, Ferron L, Owen MJ, et al. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog Neurobiol. (2015) 134:36–54. doi: 10.1016/j.pneurobio.2015.09.002
35. Campos-Bedolla P, Walter FR, Veszelka S, Deli MA. Role of the blood-brain barrier in the nutrition of the central nervous system. Arch Med Res. (2014) 45:610–38. doi: 10.1016/j.arcmed.2014.11.018
36. Jiang X, Andjelkovic AV, Zhu L, Yang T, Bennett MVL, Chen J, et al. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol. (2018) 163–164:144–71. doi: 10.1016/j.pneurobio.2017.10.001
37. Agundez JA, Jimenez-Jimenez FJ, Alonso-Navarro H, Garcia-Martin E. Drug and xenobiotic biotransformation in the blood-brain barrier: a neglected issue. Front Cell Neurosci. (2014) 8:335. doi: 10.3389/fncel.2014.00335
38. Sadeghian H, Lacoste B, Qin T, Toussay X, Rosa R, Oka F, et al. Spreading depolarizations trigger caveolin-1-dependent endothelial transcytosis. Ann Neurol. (2018) 84:409–23. doi: 10.1002/ana.25298
39. Sun Z, Gao C, Gao D, Sun R, Li W, Wang F, et al. Reduction in pericyte coverage leads to blood-brain barrier dysfunction via endothelial transcytosis following chronic cerebral hypoperfusion. Fluids Barriers CNS. (2021) 18:21. doi: 10.1186/s12987-021-00255-2
40. Lacoste B, Comin Cesar H, Ben-Zvi A, Kaeser Pascal S, Xu X, Costa Luciano da F, et al. Sensory-related neural activity regulates the structure of vascular networks in the cerebral cortex. Neuron. (2014) 83:1117–30. doi: 10.1016/j.neuron.2014.07.034
41. Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, et al. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature. (2014) 509:507–11. doi: 10.1038/nature13324
42. Andreone BJ, Chow BW, Tata A, Lacoste B, Ben-Zvi A, Bullock K, et al. Blood-brain barrier permeability is regulated by lipid transport-dependent suppression of caveolae-mediated transcytosis. Neuron. (2017) 94:581–94 e5. doi: 10.1016/j.neuron.2017.03.043
43. Betsholtz C. Physiology: double function at the blood-brain barrier. Nature. (2014) 509:432–3. doi: 10.1038/nature13339
44. Zhao Z, Zlokovic BV. Blood-brain barrier: a dual life of Mfsd2a? Neuron. (2014) 82:728–30. doi: 10.1016/j.neuron.2014.05.012
45. Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, et al. Glut1 reductions exacerbate Alzheimer's disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci. (2015) 18:521–30. doi: 10.1038/nn.3966
46. Zheng PP, Romme E, van der Spek PJ, Dirven CM, Willemsen R, Kros JM. Glut1/SLC2A1 is crucial for the development of the blood-brain barrier in vivo. Ann Neurol. (2010) 68:835–44. doi: 10.1002/ana.22318
47. Filous AR, Silver J. “Targeting astrocytes in CNS injury and disease: a translational research approach”. Prog Neurobiol. (2016) 144:173–87. doi: 10.1016/j.pneurobio.2016.03.009
48. Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia. (2010) 58:1094–103. doi: 10.1002/glia.20990
49. Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. (2004) 16:1–13. doi: 10.1016/j.nbd.2003.12.016
50. Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. (2015) 7:a020412. doi: 10.1101/cshperspect.a020412
51. Gee JR, Keller JN. Astrocytes: regulation of brain homeostasis via apolipoprotein E. Int J Biochem Cell Biol. (2005) 37:1145–50. doi: 10.1016/j.biocel.2004.10.004
52. Liu Z, Chopp M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog Neurobiol. (2016) 144:103–20. doi: 10.1016/j.pneurobio.2015.09.008
53. Osborn LM, Kamphuis W, Wadman WJ, Hol EM. Astrogliosis: an integral player in the pathogenesis of Alzheimer's disease. Prog Neurobiol. (2016) 144:121–41. doi: 10.1016/j.pneurobio.2016.01.001
54. Rossi D. Astrocyte physiopathology: at the crossroads of intercellular networking, inflammation and cell death. Prog Neurobiol. (2015) 130:86–120. doi: 10.1016/j.pneurobio.2015.04.003
55. Neuhaus J. Orthogonal arrays of particles in astroglial cells: quantitative analysis of their density, size, and correlation with intramembranous particles. Glia. (1990) 3:241–51. doi: 10.1002/glia.440030403
56. Janzer RC, Raff MC. Astrocytes induce blood–brain barrier properties in endothelial cells. Nature. (1987) 325:253–7. doi: 10.1038/325253a0
57. Willis CL, Nolan CC, Reith SN, Lister T, Prior MJ, Guerin CJ, et al. Focal astrocyte loss is followed by microvascular damage, with subsequent repair of the blood-brain barrier in the apparent absence of direct astrocytic contact. Glia. (2004) 45:325–37. doi: 10.1002/glia.10333
58. Siegenthaler JA, Sohet F, Daneman R. “Sealing off the CNS”: cellular and molecular regulation of blood–brain barriergenesis. Curr Opin Neurobiol. (2013) 23:1057–64. doi: 10.1016/j.conb.2013.06.006
59. Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. (2010) 468:562–6. doi: 10.1038/nature09513
60. Colgan OC, Collins NT, Ferguson G, Murphy RP, Birney YA, Cahill PA, et al. Influence of basolateral condition on the regulation of brain microvascular endothelial tight junction properties and barrier function. Brain Res. (2008) 1193:84–92. doi: 10.1016/j.brainres.2007.11.072
61. Siddharthan V, Kim YV, Liu S, Kim KS. Human astrocytes/astrocyte-conditioned medium and shear stress enhance the barrier properties of human brain microvascular endothelial cells. Brain Res. (2007) 1147:39–50. doi: 10.1016/j.brainres.2007.02.029
62. Ogunshola OO, Al-Ahmad A. HIF-1 at the blood-brain barrier: a mediator of permeability? High Alt Med Biol. (2012) 13:153–61. doi: 10.1089/ham.2012.1052
63. Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia. (2013) 61:1939–58. doi: 10.1002/glia.22575
64. Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. (2007) 10:1369–76. doi: 10.1038/nn2003
65. Davis B, Tang J, Zhang L, Mu D, Jiang X, Biran V, et al. Role of vasodilator stimulated phosphoprotein in VEGF induced blood-brain barrier permeability in endothelial cell monolayers. Int J Dev Neurosci. (2010) 28:423–8. doi: 10.1016/j.ijdevneu.2010.06.010
66. Nian K, Harding IC, Herman IM, Ebong EE. Blood-brain barrier damage in ischemic stroke and its regulation by endothelial mechanotransduction. Front Physiol. (2020) 11:605398. doi: 10.3389/fphys.2020.605398
67. Hawkins RA, Peterson DR, Vina JR. The complementary membranes forming the blood-brain barrier. IUBMB Life. (2002) 54:101–7. doi: 10.1080/15216540214541
68. Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. (2002) 200:629–38. doi: 10.1046/j.1469-7580.2002.00064.x
69. Haseloff RF, Blasig IE, Bauer HC, Bauer H. In search of the astrocytic factor(s) modulating blood-brain barrier functions in brain capillary endothelial cells in vitro. Cell Mol Neurobiol. (2005) 25:25–39. doi: 10.1007/s10571-004-1375-x
70. Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem. (2006) 98:641–53. doi: 10.1111/j.1471-4159.2006.03913.x
71. Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. (2005) 97:512–23. doi: 10.1161/01.RES.0000182903.16652.d7
72. Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature. (2006) 443:700–4. doi: 10.1038/nature05193
73. Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. (2011) 14:1398–405. doi: 10.1038/nn.2946
74. Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. (2011) 21:193–215. doi: 10.1016/j.devcel.2011.07.001
75. Cipolla MJ. The cerebral circulation. In: Colloquium Series on Integrated Systems Physiology: From Molecule to Function. San Rafael, CA: Morgan & Claypool Publishers (2009) p. 1–59.
76. Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. (2018) 554:475–80. doi: 10.1038/nature25739
77. Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature. (2010) 468:557–61. doi: 10.1038/nature09522
78. Ayloo S, Lazo CG, Sun S, Zhang W, Cui B, Gu C. Pericyte-to-endothelial cell signaling via vitronectin-integrin regulates blood-CNS barrier. Neuron. (2022) 110:1641–55 e6. doi: 10.1016/j.neuron.2022.02.017
79. Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol. (2006) 100:1059–64. doi: 10.1152/japplphysiol.00954.2005
80. Koehler RC, Gebremedhin D, Harder DR. Role of astrocytes in cerebrovascular regulation. J Appl Physiol. (2006) 100:307–17. doi: 10.1152/japplphysiol.00938.2005
81. Shen Q, Du F, Huang S, Duong TQ. Spatiotemporal characteristics of postischemic hyperperfusion with respect to changes in T1, T2, diffusion, angiography, and blood-brain barrier permeability. J Cereb Blood Flow Metab. (2011) 31:2076–85. doi: 10.1038/jcbfm.2011.64
82. Vital SA, Terao S, Nagai M, Granger DN. Mechanisms underlying the cerebral microvascular responses to angiotensin II-induced hypertension. Microcirculation. (2010) 17:641–9. doi: 10.1111/j.1549-8719.2010.00060.x
83. Cucullo L, Couraud P-O, Weksler B, Romero I-A, Hossain M, Rapp E, et al. Immortalized human brain endothelial cells and flow-based vascular modeling: a marriage of convenience for rational neurovascular studies. J Cereb Blood Flow Metab. (2007) 28:312–28. doi: 10.1038/sj.jcbfm.9600525
84. Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. (2010) 10:712–23. doi: 10.1038/nri2852
85. del Zoppo GJ, Milner R. Integrin-matrix interactions in the cerebral microvasculature. Arterioscler Thromb Vasc Biol. (2006) 26:1966–75. doi: 10.1161/01.ATV.0000232525.65682.a2
86. Wang CX, Shuaib A. Critical role of microvasculature basal lamina in ischemic brain injury. Prog Neurobiol. (2007) 83:140–8. doi: 10.1016/j.pneurobio.2007.07.006
87. Baeten KM, Akassoglou K. Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev Neurobiol. (2011) 71:1018–39. doi: 10.1002/dneu.20954
88. Ronaldson PT, Davis TP. Targeting transporters: promoting blood-brain barrier repair in response to oxidative stress injury. Brain Res. (2015) 1623:39–52. doi: 10.1016/j.brainres.2015.03.018
89. Engelhardt S, Patkar S, Ogunshola OO. Cell-specific blood-brain barrier regulation in health and disease: a focus on hypoxia. Br J Pharmacol. (2014) 171:1210–30. doi: 10.1111/bph.12489
90. Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. (1993) 123:1777–88. doi: 10.1083/jcb.123.6.1777
91. Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem. (1998) 273:29745–53. doi: 10.1074/jbc.273.45.29745
92. Vorbrodt AW, Dobrogowska DH. Molecular anatomy of intercellular junctions in brain endothelial and epithelial barriers: electron microscopist's view. Brain Res Rev. (2003) 42:221–42. doi: 10.1016/S0165-0173(03)00177-2
93. Ronaldson PT, Demarco KM, Sanchez-Covarrubias L, Solinsky CM, Davis TP. Transforming growth factor-beta signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain. J Cereb Blood Flow Metab. (2009) 29:1084–98. doi: 10.1038/jcbfm.2009.32
94. McCaffrey G, Staatz WD, Quigley CA, Nametz N, Seelbach MJ, Campos CR, et al. Tight junctions contain oligomeric protein assembly critical for maintaining blood–brain barrier integrity in vivo. J Neurochemist. (2007) 103:2540–55. doi: 10.1111/j.1471-4159.2007.04943.x
95. McCaffrey G, Seelbach MJ, Staatz WD, Nametz N, Quigley C, Campos CR, et al. Occludin oligomeric assembly at tight junctions of the blood-brain barrier is disrupted by peripheral inflammatory hyperalgesia. J Neurochem. (2008) 106:2395–409. doi: 10.1111/j.1471-4159.2008.05582.x
96. Lochhead JJ, McCaffrey G, Quigley CE, Finch J, DeMarco KM, Nametz N, et al. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab. (2010) 30:1625–36. doi: 10.1038/jcbfm.2010.29
97. Pan R, Yu K, Weatherwax T, Zheng H, Liu W, Liu KJ. Blood occludin level as a potential biomarker for early blood brain barrier damage following ischemic stroke. Sci Rep. (2017) 7:40331. doi: 10.1038/srep40331
98. Piontek J, Winkler L, Wolburg H, Muller SL, Zuleger N, Piehl C, et al. Formation of tight junction: determinants of homophilic interaction between classic claudins. FASEB J. (2008) 22:146–58. doi: 10.1096/fj.07-8319com
99. Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. Direct binding of three tight junction-associated maguks, ZO-1, ZO-2, and ZO-3, with the cooh termini of claudins. J Cell Biol. (1999) 147:1351–63. doi: 10.1083/jcb.147.6.1351
100. Ruffer C, Gerke V. The C-terminal cytoplasmic tail of claudins 1 and 5 but not its PDZ-binding motif is required for apical localization at epithelial and endothelial tight junctions. Eur J Cell Biol. (2004) 83:135–44. doi: 10.1078/0171-9335-00366
101. Tam SJ, Watts RJ. Connecting vascular and nervous system development: angiogenesis and the blood-brain barrier. Annu Rev Neurosci. (2010) 33:379–408. doi: 10.1146/annurev-neuro-060909-152829
102. Soma T, Chiba H, Kato-Mori Y, Wada T, Yamashita T, Kojima T, et al. Thr207 of claudin-5 is involved in size-selective loosening of the endothelial barrier by cyclic AMP. Exp Cell Res. (2004) 300:202–12. doi: 10.1016/j.yexcr.2004.07.012
103. Yamamoto T, Harada N, Kawano Y, Taya S, Kaibuchi K. In vivo interaction of AF-6 with activated Ras and ZO-1. Biochem Biophys Res Commun. (1999) 259:103–7. doi: 10.1006/bbrc.1999.0731
104. McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. (2008) 28:9451–62. doi: 10.1523/JNEUROSCI.2674-08.2008
105. Yang Y, Rosenberg GA. Mmp-mediated disruption of claudin-5 in the blood-brain barrier of rat brain after cerebral ischemia. Methods Mol Biol. (2011) 762:333–45. doi: 10.1007/978-1-61779-185-7_24
106. Kaur J, Tuor UI, Zhao Z, Barber PA. Quantitative MRI reveals the elderly ischemic brain is susceptible to increased early blood-brain barrier permeability following tissue plasminogen activator related to claudin 5 and occludin disassembly. J Cereb Blood Flow Metab. (2011) 31:1874–85. doi: 10.1038/jcbfm.2011.79
107. Weber C, Fraemohs L, Dejana E. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol. (2007) 7:467–77. doi: 10.1038/nri2096
108. Ebnet K, Aurrand-Lions M, Kuhn A, Kiefer F, Butz S, Zander K, et al. The junctional adhesion molecule (JAM) family members JAM-2 and JAM-3 associate with the cell polarity protein PAR-3: a possible role for jams in endothelial cell polarity. J Cell Sci. (2003) 116:3879–91. doi: 10.1242/jcs.00704
109. Hoffman WH, Stamatovic SM, Andjelkovic AV. Inflammatory mediators and blood brain barrier disruption in fatal brain edema of diabetic ketoacidosis. Brain Res. (2009) 1254:138–48. doi: 10.1016/j.brainres.2008.11.100
110. Yeung D, Manias JL, Stewart DJ, Nag S. Decreased junctional adhesion molecule-A expression during blood-brain barrier breakdown. Acta Neuropathol. (2008) 115:635–42. doi: 10.1007/s00401-008-0364-4
111. Wang X, Campos CR, Peart JC, Smith LK, Boni JL, Cannon RE, et al. NRF2 upregulates ATP binding cassette transporter expression and activity at the blood-brain and blood-spinal cord barriers. J Neurosci. (2014) 34:8585–93. doi: 10.1523/JNEUROSCI.2935-13.2014
112. Wang XS, Fang HL, Chen Y, Liang SS, Zhu ZG, Zeng QY, et al. Idazoxan reduces blood-brain barrier damage during experimental autoimmune encephalomyelitis in mouse. Eur J Pharmacol. (2014) 736:70–6. doi: 10.1016/j.ejphar.2014.04.034
113. Balda MS, Gonzalez-Mariscal L, Contreras RG, Macias-Silva M, Torres-Marquez ME, Garcia-Sainz JA, et al. Assembly and sealing of tight junctions: possible participation of G-proteins, phospholipase C, protein kinase C and calmodulin. J Membr Biol. (1991) 122:193–202. doi: 10.1007/BF01871420
114. Mitic LL, Anderson JM. Molecular architecture of tight junctions. Annu Rev Physiol. (1998) 60:121–42. doi: 10.1146/annurev.physiol.60.1.121
115. Howarth AG, Hughes MR, Stevenson BR. Detection of the tight junction-associated protein ZO-1 in astrocytes and other nonepithelial cell types. Am J Physiol. (1992) 262:C461–9. doi: 10.1152/ajpcell.1992.262.2.C461
116. Abbruscato TJ, Lopez SP, Mark KS, Hawkins BT, Davis TP. Nicotine and cotinine modulate cerebral microvascular permeability and protein expression of ZO-1 through nicotinic acetylcholine receptors expressed on brain endothelial cells. J Pharm Sci. (2002) 91:2525–38. doi: 10.1002/jps.10256
117. Fischer S, Wobben M, Marti HH, Renz D, Schaper W. Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res. (2002) 63:70–80. doi: 10.1006/mvre.2001.2367
118. Mark KS, Davis TP. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. (2002) 282:H1485–94. doi: 10.1152/ajpheart.00645.2001
119. Gumbiner B, Lowenkopf T, Apatira D. Identification of a 160-kDa polypeptide that binds to the tight junction protein ZO-1. Proc Natl Acad Sci U S A. (1991) 88:3460–4. doi: 10.1073/pnas.88.8.3460
120. Itoh M, Morita K, Tsukita S. Characterization of ZO-2 as a maguk family member associated with tight as well as adherens junctions with a binding affinity to occludin and alpha catenin. J Biol Chem. (1999) 274:5981–6. doi: 10.1074/jbc.274.9.5981
121. Inoko A, Itoh M, Tamura A, Matsuda M, Furuse M, Tsukita S. Expression and distribution of ZO-3, a tight junction maguk protein, in mouse tissues. Genes Cells. (2003) 8:837–45. doi: 10.1046/j.1365-2443.2003.00681.x
122. Takenaga Y, Takagi N, Murotomi K, Tanonaka K, Takeo S. Inhibition of Src activity decreases tyrosine phosphorylation of occludin in brain capillaries and attenuates increase in permeability of the blood-brain barrier after transient focal cerebral ischemia. J Cereb Blood Flow Metab. (2009) 29:1099–108. doi: 10.1038/jcbfm.2009.30
123. Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, Dejana E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem. (2000) 275:20520–6. doi: 10.1074/jbc.M905251199
124. Cordenonsi M, D'Atri F, Hammar E, Parry DA, Kendrick-Jones J, Shore D, et al. Cingulin contains globular and coiled-coil domains and interacts with ZO-1, ZO-2, ZO-3, and myosin. J Cell Biol. (1999) 147:1569–82. doi: 10.1083/jcb.147.7.1569
125. Bangsow T, Baumann E, Bangsow C, Jaeger MH, Pelzer B, Gruhn P, et al. The epithelial membrane protein 1 is a novel tight junction protein of the blood-brain barrier. J Cereb Blood Flow Metab. (2008) 28:1249–60. doi: 10.1038/jcbfm.2008.19
126. Satoh H, Zhong Y, Isomura H, Saitoh M, Enomoto K, Sawada N, et al. Localization of 7H6 tight junction-associated antigen along the cell border of vascular endothelial cells correlates with paracellular barrier function against ions, large molecules, and cancer cells. Exp Cell Res. (1996) 222:269–74. doi: 10.1006/excr.1996.0034
127. Zhong Y, Enomoto K, Isomura H, Sawada N, Minase T, Oyamada M, et al. Localization of the 7H6 antigen at tight junctions correlates with the paracellular barrier function of Mdck cells. Exp Cell Res. (1994) 214:614–20. doi: 10.1006/excr.1994.1299
128. Redzic Z. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: similarities and differences. Fluids Barriers CNS. (2011) 8:3. doi: 10.1186/2045-8118-8-3
129. Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol. (2006) 174:593–604. doi: 10.1083/jcb.200602080
130. Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. (2008) 10:923–34. doi: 10.1038/ncb1752
131. Sifat AE, Vaidya B, Abbruscato TJ. Blood-brain barrier protection as a therapeutic strategy for acute ischemic stroke. AAPS J. (2017) 19:957–72. doi: 10.1208/s12248-017-0091-7
132. Abdullahi W, Tripathi D, Ronaldson PT. Blood-brain barrier dysfunction in ischemic stroke: targeting tight junctions and transporters for vascular protection. Am J Physiol Cell Physiol. (2018) 315:C343–C56. doi: 10.1152/ajpcell.00095.2018
133. DiNapoli VA, Huber JD, Houser K, Li X, Rosen CL. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging. (2008) 29:753–64. doi: 10.1016/j.neurobiolaging.2006.12.007
134. Sakadzic S, Lee J, Boas DA, Ayata C. High-resolution in vivo optical imaging of stroke injury and repair. Brain Res. (2015) 1623:174–92. doi: 10.1016/j.brainres.2015.04.044
135. Arai K, Lok J, Guo S, Hayakawa K, Xing C, Lo EH. Cellular mechanisms of neurovascular damage and repair after stroke. J Child Neurol. (2011) 26:1193–8. doi: 10.1177/0883073811408610
136. Posada-Duque RA, Barreto GE, Cardona-Gomez GP. Protection after stroke: cellular effectors of neurovascular unit integrity. Front Cell Neurosci. (2014) 8:231. doi: 10.3389/fncel.2014.00231
137. Profaci CP, Munji RN, Pulido RS, Daneman R. The blood-brain barrier in health and disease: important unanswered questions. J Exp Med. (2020) 217:4. doi: 10.1084/jem.20190062
138. Grossmann J. Molecular mechanisms of “detachment-induced apoptosis–anoikis”. Apoptosis. (2002) 7:247–60. doi: 10.1023/A:1015312119693
139. Mannello F, Luchetti F, Falcieri E, Papa S. Multiple roles of matrix metalloproteinases during apoptosis. Apoptosis. (2005) 10:19–24. doi: 10.1007/s10495-005-6058-7
140. Kago T, Takagi N, Date I, Takenaga Y, Takagi K, Takeo S. Cerebral ischemia enhances tyrosine phosphorylation of occludin in brain capillaries. Biochem Biophys Res Commun. (2006) 339:1197–203. doi: 10.1016/j.bbrc.2005.11.133
141. Fischer S, Wiesnet M, Marti HH, Renz D, Schaper W. Simultaneous activation of several second messengers in hypoxia-induced hyperpermeability of brain derived endothelial cells. J Cell Physiol. (2004) 198:359–69. doi: 10.1002/jcp.10417
142. Koto T, Takubo K, Ishida S, Shinoda H, Inoue M, Tsubota K, et al. Hypoxia disrupts the barrier function of neural blood vessels through changes in the expression of claudin-5 in endothelial cells. Am J Pathol. (2007) 170:1389–97. doi: 10.2353/ajpath.2007.060693
143. Haley MJ, Lawrence CB. The blood-brain barrier after stroke: structural studies and the role of transcytotic vesicles. J Cereb Blood Flow Metab. (2017) 37:456–70. doi: 10.1177/0271678X16629976
144. Krueger M, Bechmann I, Immig K, Reichenbach A, Hartig W, Michalski D. Blood-brain barrier breakdown involves four distinct stages of vascular damage in various models of experimental focal cerebral ischemia. J Cereb Blood Flow Metab. (2015) 35:292–303. doi: 10.1038/jcbfm.2014.199
145. Prakash R, Carmichael ST. Blood-brain barrier breakdown and neovascularization processes after stroke and traumatic brain injury. Curr Opin Neurol. (2015) 28:556–64. doi: 10.1097/WCO.0000000000000248
146. Durukan A, Marinkovic I, Strbian D, Pitkonen M, Pedrono E, Soinne L, et al. Post-ischemic blood-brain barrier leakage in rats: one-week follow-up by MRI. Brain Res. (2009) 1280:158–65. doi: 10.1016/j.brainres.2009.05.025
147. Lin CY, Chang C, Cheung WM, Lin MH, Chen JJ, Hsu CY, et al. Dynamic changes in vascular permeability, cerebral blood volume, vascular density, and size after transient focal cerebral ischemia in rats: evaluation with contrast-enhanced magnetic resonance imaging. J Cereb Blood Flow Metab. (2008) 28:1491–501. doi: 10.1038/jcbfm.2008.42
148. Pillai DR, Dittmar MS, Baldaranov D, Heidemann RM, Henning EC, Schuierer G, et al. Cerebral ischemia-reperfusion injury in rats–a 3 T MRI study on biphasic blood-brain barrier opening and the dynamics of edema formation. J Cereb Blood Flow Metab. (2009) 29:1846–55. doi: 10.1038/jcbfm.2009.106
149. Strbian D, Durukan A, Pitkonen M, Marinkovic I, Tatlisumak E, Pedrono E, et al. The blood-brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience. (2008) 153:175–81. doi: 10.1016/j.neuroscience.2008.02.012
150. Carmichael ST. The 3 Rs of stroke biology: radial, relayed, and regenerative. Neurotherapeutics. (2016) 13:348–59. doi: 10.1007/s13311-015-0408-0
151. Merali Z, Huang K, Mikulis D, Silver F, Kassner A. Evolution of blood-brain-barrier permeability after acute ischemic stroke. PLoS ONE. (2017) 12:e0171558. doi: 10.1371/journal.pone.0171558
152. Bounds JV, Wiebers DO, Whisnant JP, Okazaki H. Mechanisms and timing of deaths from cerebral infarction. Stroke. (1981) 12:474–7. doi: 10.1161/01.STR.12.4.474
153. Davalos A, Toni D, Iweins F, Lesaffre E, Bastianello S, Castillo J. Neurological deterioration in acute ischemic stroke: potential predictors and associated factors in the european cooperative acute stroke study (ECASS) I. Stroke. (1999) 30:2631–6. doi: 10.1161/01.STR.30.12.2631
154. Dharmasaroja PA. Fluid intake related to brain edema in acute middle cerebral artery infarction. Transl Stroke Res. (2016) 7:49–53. doi: 10.1007/s12975-015-0439-1
155. Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab. (2016) 36:513–38. doi: 10.1177/0271678X15617172
156. Chen Y-J, Wallace BK, Yuen N, Jenkins DP, Wulff H, O'Donnell ME. Blood–brain barrier Kca3.1 channels. Stroke. (2015) 46:237–44. doi: 10.1161/STROKEAHA.114.007445
157. O'Donnell ME. Blood-brain barrier Na transporters in ischemic stroke. Adv Pharmacol. (2014) 71:113–46. doi: 10.1016/bs.apha.2014.06.011
158. O'Donnell ME, Chen YJ, Lam TI, Taylor KC, Walton JH, Anderson SE. Intravenous HOE-642 reduces brain edema and Na uptake in the rat permanent middle cerebral artery occlusion model of stroke: evidence for participation of the blood-brain barrier NA/H exchanger. J Cereb Blood Flow Metab. (2013) 33:225–34. doi: 10.1038/jcbfm.2012.160
159. del Zoppo GJ, Hallenbeck JM. Advances in the vascular pathophysiology of ischemic stroke. Thromb Res. (2000) 98:73–81. doi: 10.1016/S0049-3848(00)00218-8
160. Dodson RF, Chu LW, Welch KM, Achar VS. Acute tissue response to cerebral ischemia in the Gerbil. An ultrastructural study. J Neurol Sci. (1977) 33:161–70. doi: 10.1016/0022-510X(77)90190-3
161. Kimelberg HK. Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia. (2005) 50:389–97. doi: 10.1002/glia.20174
162. Tagaya M, Haring HP, Stuiver I, Wagner S, Abumiya T, Lucero J, et al. Rapid loss of microvascular integrin expression during focal brain ischemia reflects neuron injury. J Cereb Blood Flow Metab. (2001) 21:835–46. doi: 10.1097/00004647-200107000-00009
163. Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. (1997) 17:171–80. doi: 10.1523/JNEUROSCI.17-01-00171.1997
164. Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. (2000) 6:159–63. doi: 10.1038/72256
165. Zeng XN, Xie LL, Liang R, Sun XL, Fan Y, Hu G. AQP4 knockout aggravates ischemia/reperfusion injury in mice. CNS Neurosci Ther. (2012) 18:388–94. doi: 10.1111/j.1755-5949.2012.00308.x
166. Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol. (1995) 163:510–22. doi: 10.1002/jcp.1041630311
167. Goeckeler ZM, Wysolmerski RB. Myosin light chain kinase-regulated endothelial cell contraction: the relationship between isometric tension, actin polymerization, and myosin phosphorylation. J Cell Biol. (1995) 130:613–27. doi: 10.1083/jcb.130.3.613
168. Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, et al. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J Cell Sci. (2006) 119:2095–106. doi: 10.1242/jcs.02915
169. Harhaj NS, Antonetti DA. Regulation of tight junctions and loss of barrier function in pathophysiology. Int J Biochem Cell Biol. (2004) 36:1206–37. doi: 10.1016/j.biocel.2003.08.007
170. Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol. (2002) 39:213–23. doi: 10.1016/S1537-1891(03)00010-7
171. Yang T, Roder KE, Bhat GJ, Thekkumkara TJ, Abbruscato TJ. Protein kinase C family members as a target for regulation of blood-brain barrier Na,K,2cl-cotransporter during in vitro stroke conditions and nicotine exposure. Pharm Res. (2006) 23:291–302. doi: 10.1007/s11095-005-9143-2
172. Aiello LP, Bursell SE, Clermont A, Duh E, Ishii H, Takagi C, et al. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes. (1997) 46:1473–80. doi: 10.2337/diab.46.9.1473
173. Mehta D. Serine/threonine phosphatase 2B regulates protein kinase C-alpha activity and endothelial barrier function. Am J Physiol Lung Cell Mol Physiol. (2001) 281:L544–5. doi: 10.1152/ajplung.2001.281.3.L544
174. Hempel A, Lindschau C, Maasch C, Mahn M, Bychkov R, Noll T, et al. Calcium antagonists ameliorate ischemia-induced endothelial cell permeability by inhibiting protein kinase C. Circulation. (1999) 99:2523–9. doi: 10.1161/01.CIR.99.19.2523
175. Sandoval R, Malik AB, Minshall RD, Kouklis P, Ellis CA, Tiruppathi C. Ca(2+) signalling and pkcalpha activate increased endothelial permeability by disassembly of VE-cadherin junctions. J Physiol. (2001) 533:433–45. doi: 10.1111/j.1469-7793.2001.0433a.x
176. Banan A, Farhadi A, Fields JZ, Zhang LJ, Shaikh M, Keshavarzian A. The delta-isoform of protein kinase C causes inducible nitric-oxide synthase and nitric oxide up-regulation: key mechanism for oxidant-induced carbonylation, nitration, and disassembly of the microtubule cytoskeleton and hyperpermeability of barrier of intestinal epithelia. J Pharmacol Exp Ther. (2003) 305:482–94. doi: 10.1124/jpet.102.047308
177. Deli MA, Dehouck MP, Abraham CS, Cecchelli R, Joo F. Penetration of small molecular weight substances through cultured bovine brain capillary endothelial cell monolayers: the early effects of cyclic adenosine 3′,5′-monophosphate. Exp Physiol. (1995) 80:675–8. doi: 10.1113/expphysiol.1995.sp003877
178. Ishizaki T, Chiba H, Kojima T, Fujibe M, Soma T, Miyajima H, et al. Cyclic amp induces phosphorylation of claudin-5 immunoprecipitates and expression of claudin-5 gene in blood-brain-barrier endothelial cells via protein kinase A-dependent and -independent pathways. Exp Cell Res. (2003) 290:275–88. doi: 10.1016/S0014-4827(03)00354-9
179. Hurst RD, Clark JB. Alterations in transendothelial electrical resistance by vasoactive agonists and cyclic AMP in a blood-brain barrier model system. Neurochem Res. (1998) 23:149–54.
180. Langeler EG, van Hinsbergh VW. Norepinephrine and iloprost improve barrier function of human endothelial cell monolayers: role of cAMP. Am J Physiol. (1991) 260:C1052–9. doi: 10.1152/ajpcell.1991.260.5.C1052
181. Hastie LE, Patton WF, Hechtman HB, Shepro D. H2o2-induced filamin redistribution in endothelial cells is modulated by the cyclic AMP-dependent protein kinase pathway. J Cellular Physiol. (1997) 172:373–81.
182. Moy AB, Shasby SS, Scott BD, Shasby DM. The effect of histamine and cyclic adenosine monophosphate on myosin light chain phosphorylation in human umbilical vein endothelial cells. J Clin Invest. (1993) 92:1198–206. doi: 10.1172/JCI116690
183. Wong D, Dorovini-Zis K, Vincent SR. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp Neurol. (2004) 190:446–55. doi: 10.1016/j.expneurol.2004.08.008
184. Rao RK, Basuroy S, Rao VU, Karnaky KJ, Gupta A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J. (2002) 368:471–81. doi: 10.1042/bj20011804
185. Kale G, Naren AP, Sheth P, Rao RK. Tyrosine phosphorylation of occludin attenuates its interactions with ZO-1, ZO-2, and ZO-3. Biochem Biophys Res Commun. (2003) 302:324–9. doi: 10.1016/S0006-291X(03)00167-0
186. Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. (2008) 121:2115–22. doi: 10.1242/jcs.017897
187. Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell. (1999) 4:915–24. doi: 10.1016/S1097-2765(00)80221-X
188. Paul R, Zhang ZG, Eliceiri BP, Jiang Q, Boccia AD, Zhang RL, et al. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat Med. (2001) 7:222–7. doi: 10.1038/84675
189. Akiyama C, Yuguchi T, Nishio M, Tomishima T, Fujinaka T, Taniguchi M, et al. Src family kinase inhibitor PP1 reduces secondary damage after spinal cord compression in rats. J Neurotrauma. (2004) 21:923–31. doi: 10.1089/0897715041526230
190. Kevil CG, Okayama N, Alexander JS. H(2)O(2)-mediated permeability II: importance of tyrosine phosphatase and kinase activity. Am J Physiol Cell Physiol. (2001) 281:C1940–7. doi: 10.1152/ajpcell.2001.281.6.C1940
191. Nwariaku FE, Liu Z, Zhu X, Turnage RH, Sarosi GA, Terada LS. Tyrosine phosphorylation of vascular endothelial cadherin and the regulation of microvascular permeability. Surgery. (2002) 132:180–5. doi: 10.1067/msy.2002.125305
192. Tinsley JH, Teasdale NR, Yuan SY. Myosin light chain phosphorylation and pulmonary endothelial cell hyperpermeability in burns. Am J Physiol Lung Cell Mol Physiol. (2004) 286:L841–7. doi: 10.1152/ajplung.00341.2003
193. Birukov KG, Csortos C, Marzilli L, Dudek S, Ma SF, Bresnick AR, et al. Differential regulation of alternatively spliced endothelial cell myosin light chain kinase isoforms by P60(Src). J Biol Chem. (2001) 276:8567–73. doi: 10.1074/jbc.M005270200
194. Schreibelt G, Kooij G, Reijerkerk A, Doorn R, Gringhuis SI, Pol S, et al. Reactive oxygen species alter brain endothelial tight junction dynamics via Rhoa, PI3 kinase, and PKB signaling. FASEB J. (2007) 21:3666–76. doi: 10.1096/fj.07-8329com
195. Sheth P, Basuroy S, Li C, Naren AP, Rao RK. Role of phosphatidylinositol 3-kinase in oxidative stress-induced disruption of tight junctions. J Biol Chem. (2003) 278:49239–45. doi: 10.1074/jbc.M305654200
196. Liu SM, Sundqvist T. Effects of hydrogen peroxide and phorbol myristate acetate on endothelial transport and f-actin distribution. Exp Cell Res. (1995) 217:1–7. doi: 10.1006/excr.1995.1056
197. Lee HS, Namkoong K, Kim DH, Kim KJ, Cheong YH, Kim SS, et al. Hydrogen peroxide-induced alterations of tight junction proteins in bovine brain microvascular endothelial cells. Microvasc Res. (2004) 68:231–8. doi: 10.1016/j.mvr.2004.07.005
198. Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol Neurobiol. (2000) 20:131–47. doi: 10.1023/a:1007074420772
199. Brown RC, Davis TP. Calcium modulation of adherens and tight junction function: a potential mechanism for blood-brain barrier disruption after stroke. Stroke. (2002) 33:1706–11. doi: 10.1161/01.STR.0000016405.06729.83
200. Tiruppathi C, Minshall RD, Paria BC, Vogel SM, Malik AB. Role of Ca2+ signaling in the regulation of endothelial permeability. Vascul Pharmacol. (2002) 39:173–85. doi: 10.1016/S1537-1891(03)00007-7
201. Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. (2001) 91:1487–500. doi: 10.1152/jappl.2001.91.4.1487
202. Lum H, Malik AB. Mechanisms of increased endothelial permeability. Can J Physiol Pharmacol. (1996) 74:787–800. doi: 10.1139/y96-081
203. Candelario-Jalil E, Taheri S, Yang Y, Sood R, Grossetete M, Estrada EY, et al. Cyclooxygenase inhibition limits blood-brain barrier disruption following intracerebral injection of tumor necrosis factor-alpha in the rat. J Pharmacol Exp Ther. (2007) 323:488–98. doi: 10.1124/jpet.107.127035
204. de Vries HE, Blom-Roosemalen MC, van Oosten M, de Boer AG, van Berkel TJ, Breimer DD, et al. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J Neuroimmunol. (1996) 64:37–43. doi: 10.1016/0165-5728(95)00148-4
205. Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol. (2006) 66:232–45. doi: 10.1016/j.surneu.2005.12.028
206. Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. J Cereb Blood Flow Metab. (2006) 26:797–810. doi: 10.1038/sj.jcbfm.9600229
207. Bolton SJ, Anthony DC, Perry VH. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience. (1998) 86:1245–57. doi: 10.1016/S0306-4522(98)00058-X
208. Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and timps are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. (1998) 29:2189–95. doi: 10.1161/01.STR.29.10.2189
209. Yang C, Hawkins KE, Dore S, Candelario-Jalil E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol. (2019) 316:C135–C53. doi: 10.1152/ajpcell.00136.2018
210. Brouns R, Wauters A, De Surgeloose D, Marien P, De Deyn PP. Biochemical markers for blood-brain barrier dysfunction in acute ischemic stroke correlate with evolution and outcome. Eur Neurol. (2011) 65:23–31. doi: 10.1159/000321965
211. Yang Y, Rosenberg GA. Matrix metalloproteinases as therapeutic targets for stroke. Brain Res. (2015) 1623:30–8. doi: 10.1016/j.brainres.2015.04.024
212. Zhang S, An Q, Wang T, Gao S, Zhou G. Autophagy- and MMP-2/9-mediated reduction and redistribution of ZO-1 contribute to hyperglycemia-increased blood-brain barrier permeability during early reperfusion in stroke. Neuroscience. (2018) 377:126–37. doi: 10.1016/j.neuroscience.2018.02.035
213. Zeng C, Wang D, Chen C, Chen L, Chen B, Li L, et al. Zafirlukast protects blood-brain barrier integrity from ischemic brain injury. Chemico-Biological Interact. (2020) 316:108915. doi: 10.1016/j.cbi.2019.108915
214. Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. (2005) 50:329–39. doi: 10.1002/glia.20169
215. Morancho A, Rosell A, García-Bonilla L, Montaner J. Metalloproteinase and stroke infarct size: role for anti-inflammatory treatment? Ann N Y Acad Sci. (2010) 1207:123–33. doi: 10.1111/j.1749-6632.2010.05734.x
216. Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. (2006) 311:1770–3. doi: 10.1126/science.1123933
217. Ma F, Martinez-San Segundo P, Barcelo V, Morancho A, Gabriel-Salazar M, Giralt D, et al. Matrix metalloproteinase-13 participates in neuroprotection and neurorepair after cerebral ischemia in mice. Neurobiol Dis. (2016) 91:236–46. doi: 10.1016/j.nbd.2016.03.016
218. Sood RR, Taheri S, Candelario-Jalil E, Estrada EY, Rosenberg GA. Early beneficial effect of matrix metalloproteinase inhibition on blood-brain barrier permeability as measured by magnetic resonance imaging countered by impaired long-term recovery after stroke in rat brain. J Cereb Blood Flow Metab. (2008) 28:431–8. doi: 10.1038/sj.jcbfm.9600534
219. Lai CH, Kuo KH, Leo JM. Critical role of actin in modulating BBB permeability. Brain Res Brain Res Rev. (2005) 50:7–13. doi: 10.1016/j.brainresrev.2005.03.007
220. Burridge K, Wittchen ES. The tension mounts: stress fibers as force-generating mechanotransducers. J Cell Biol. (2013) 200:9–19. doi: 10.1083/jcb.201210090
221. Shi Y, Zhang L, Pu H, Mao L, Hu X, Jiang X, et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat Commun. (2016) 7:10523. doi: 10.1038/ncomms10523
222. Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta. (2008) 1778:794–809. doi: 10.1016/j.bbamem.2007.09.003
223. Endres M, Fink K, Zhu J, Stagliano NE, Bondada V, Geddes JW, et al. Neuroprotective effects of gelsolin during murine stroke. J Clin Invest. (1999) 103:347–54. doi: 10.1172/JCI4953
224. Atkinson SJ, Hosford MA, Molitoris BA. Mechanism of actin polymerization in cellular ATP depletion. J Biol Chem. (2004) 279:5194–9. doi: 10.1074/jbc.M306973200
225. Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci. (2008) 1123:134–45. doi: 10.1196/annals.1420.016
226. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. (2006) 86:279–367. doi: 10.1152/physrev.00012.2005
227. Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, et al. Monocyte chemoattractant protein-1 regulation of blood–brain barrier permeability. J Cereb Blood Flow Metabol. (2005) 25:593–606. doi: 10.1038/sj.jcbfm.9600055
228. Khatri R, McKinney AM, Swenson B, Janardhan V. Blood-brain barrier, reperfusion injury, and hemorrhagic transformation in acute ischemic stroke. Neurology. (2012) 79:S52–7. doi: 10.1212/WNL.0b013e3182697e70
229. Stamatovic SM, Keep RF, Wang MM, Jankovic I, Andjelkovic AV. Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J Biol Chem. (2009) 284:19053–66. doi: 10.1074/jbc.M109.000521
230. Shi Y, Jiang X, Zhang L, Pu H, Hu X, Zhang W, et al. Endothelium-targeted overexpression of heat shock protein 27 ameliorates blood-brain barrier disruption after ischemic brain injury. Proc Natl Acad Sci U S A. (2017) 114:E1243–E52. doi: 10.1073/pnas.1621174114
231. Eira J, Silva CS, Sousa MM, Liz MA. The cytoskeleton as a novel therapeutic target for old neurodegenerative disorders. Prog Neurobiol. (2016) 141:61–82. doi: 10.1016/j.pneurobio.2016.04.007
232. Andreini C, Banci L, Bertini I, Rosato A. Counting the zinc-proteins encoded in the human genome. J Proteome Res. (2006) 5:196–201. doi: 10.1021/pr050361j
233. Shuttleworth CW, Weiss JH. Zinc: New clues to diverse roles in brain ischemia. Trends Pharmacol Sci. (2011) 32:480–6. doi: 10.1016/j.tips.2011.04.001
234. Hess SY, Peerson JM, King JC, Brown KH. Use of serum zinc concentration as an indicator of population zinc status. Food Nutr Bull. (2007) 28:S403–29. doi: 10.1177/15648265070283S303
235. Furuta S, Suzuki M, Toyama S, Miwa M, Sano H. Tissue distribution of polaprezinc in rats determined by the double tracer method. J Pharm Biomed Anal. (1999) 19:453–61. doi: 10.1016/S0731-7085(98)00236-2
236. Noseworthy MD, Bray TM. Zinc deficiency exacerbates loss in blood-brain barrier integrity induced by hyperoxia measured by dynamic MRI. Proc Soc Exp Biol Med. (2000) 223:175–82. doi: 10.1046/j.1525-1373.2000.22324.x
237. Qi Z, Liang J, Pan R, Dong W, Shen J, Yang Y, et al. Zinc contributes to acute cerebral ischemia-induced blood-brain barrier disruption. Neurobiol Dis. (2016) 95:12–21. doi: 10.1016/j.nbd.2016.07.003
238. Li Y, Ma T, Zhu X, Zhang M, Zhao L, Wang P, et al. Zinc improves neurological recovery by promoting angiogenesis via the astrocyte-mediated HIF-1α/VEGF signaling pathway in experimental stroke. CNS Neurosci Ther. (2022) 28:1790–9. doi: 10.1111/cns.13918
239. Munshi A, Babu S, Kaul S, Shafi G, Rajeshwar K, Alladi S, et al. Depletion of serum zinc in ischemic stroke patients. Methods Find Exp Clin Pharmacol. (2010) 32:433–6. doi: 10.1358/mf.2010.32.6.1487084
240. Bhatt A, Farooq MU, Enduri S, Pillainayagam C, Naravetla B, Razak A, et al. Clinical significance of serum zinc levels in cerebral ischemia. Stroke Res Treat. (2011) 2010:245715. doi: 10.4061/2010/245715
241. Wang P, Pan R, Weaver J, Jia M, Yang X, Yang T, et al. MicroRNA-30A regulates acute cerebral ischemia-induced blood–brain barrier damage through ZnT4/Zinc pathway. J Cereb Blood Flow Metab. (2020) 41:641–55. doi: 10.1177/0271678X20926787
242. Grant GA, Abbott NJ, Janigro D. Understanding the physiology of the blood-brain barrier: in vitro models. News Physiol Sci. (1998) 13:287–93. doi: 10.1152/physiologyonline.1998.13.6.287
243. Wilhelm I, Fazakas C, Krizbai IA. In vitro models of the blood-brain barrier. Acta Neurobiol Exp (Wars). (2011) 71:113–28.
244. Hatherell K, Couraud P-O, Romero IA, Weksler B, Pilkington GJ. Development of a three-dimensional, all-human in vitro model of the blood–brain barrier using mono-, co-, and tri-cultivation transwell models. J Neurosci Methods. (2011) 199:223–9. doi: 10.1016/j.jneumeth.2011.05.012
245. Paolinelli R, Corada M, Ferrarini L, Devraj K, Artus C, Czupalla CJ, et al. Wnt activation of immortalized brain endothelial cells as a tool for generating a standardized model of the blood brain barrier in vitro. PLoS ONE. (2013) 8:8. doi: 10.1371/journal.pone.0070233
246. Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, Shusta EV. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci Rep. (2014) 4:4160. doi: 10.1038/srep04160
247. Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, et al. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol. (2012) 30:783–91. doi: 10.1038/nbt.2247
248. Ronaldson PT, Davis TP. Blood-brain barrier integrity and glial support: mechanisms that can be targeted for novel therapeutic approaches in stroke. Curr Pharm Des. (2012) 18:3624–44. doi: 10.2174/138161212802002625
249. Reiber H. Dynamics of brain-derived proteins in cerebrospinal fluid. Clin Chim Acta. (2001) 310:173–86. doi: 10.1016/S0009-8981(01)00573-3
250. Reiber H. The Discrimination between different blood-csf barrier dysfunctions and inflammatory reactions of the CNS by a recent evaluation graph for the protein profile of cerebrospinal fluid. J Neurol. (1980) 224:89–99. doi: 10.1007/BF00313347
251. Blyth BJ, Farahvar A, He H, Nayak A, Yang C, Shaw G, et al. elevated serum ubiquitin carboxy-terminal hydrolase L1 is associated with abnormal blood-brain barrier function after traumatic brain injury. J Neurotrauma. (2011) 28:2453–62. doi: 10.1089/neu.2010.1653
252. Link H, Tibbling G. Principles of albumin and igg analyses in neurological disorders II relation of the concentration of the proteins in serum and cerebrospinal fluid. Scand J Clin Lab Invest. (1977) 37:391–6. doi: 10.3109/00365517709091497
253. Tibbling G, Link H, Ohman S. Principles of albumin and igg analyses in neurological disorders I establishment of reference values. Scand J Clin Lab Invest. (1977) 37:385–90. doi: 10.3109/00365517709091496
Keywords: blood–brain barrier, neurovascular unit, microvascular endothelial cells, astrocytes, ischemic stroke
Citation: Xue S, Zhou X, Yang Z-H, Si X-K and Sun X (2023) Stroke-induced damage on the blood–brain barrier. Front. Neurol. 14:1248970. doi: 10.3389/fneur.2023.1248970
Received: 27 June 2023; Accepted: 08 August 2023;
Published: 28 September 2023.
Edited by:
Gabriel Broocks, University of Hamburg, GermanyCopyright © 2023 Xue, Zhou, Yang, Si and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Sun, c3VuX3hpbkBqbHUuZWR1LmNu