 Parisa Alizadeh
Parisa Alizadeh Cinthia Terroba-Chambi
Cinthia Terroba-Chambi Beatrice Achen1
Beatrice Achen1 Veronica Bruno
Veronica Bruno- 1Department of Clinical Neurosciences, University of Calgary, Calgary, AB, Canada
- 2Hotchkiss Brain Institute, Calgary, AB, Canada
Pain, a challenging symptom experienced by individuals diagnosed with Parkinson’s disease (PD), still lacks a comprehensive understanding of its underlying pathophysiological mechanisms. A systematic investigation of its prevalence and impact on the quality of life in patients affected by monogenic forms of PD has yet to be undertaken. This comprehensive review aims to provide an overview of the association between pain and monogenic forms of PD, specifically focusing on pathogenic variants in SNCA, PRKN, PINK1, PARK7, LRRK2, GBA1, VPS35, ATP13A2, DNAJC6, FBXO7, and SYNJ1. Sixty-three articles discussing pain associated with monogenic PD were identified and analyzed. The included studies exhibited significant heterogeneity in design, sample size, and pain outcome measures. Nonetheless, the findings of this review suggest that patients with monogenic PD may experience specific types of pain depending on the pathogenic variant present, distinguishing them from non-carriers. For instance, individuals with SNCA pathogenic variants have reported painful dystonia, lower extremity pain, dorsal pain, and upper back pain. However, these observations are primarily based on case reports with unclear prevalence. Painful lower limb dystonia and lower back pain are prominent symptoms in PRKN carriers. A continual correlation has been noted between LRRK2 mutations and the emergence of pain, though the conflicting research outcomes pose challenges in reaching definitive conclusions. Individuals with PINK1 mutation carriers also frequently report experiencing pain. Pain has been frequently reported as an initial symptom and the most troublesome one in GBA1-PD patients compared to those with idiopathic PD. The evidence regarding pain in ATP13A2, PARK7, VPS35, DNAJC6, FBXO7, and SYNJ1pathogenic variants is limited and insufficient. The potential linkage between genetic profiles and pain outcomes holds promising clinical implications, allowing for the potential stratification of patients in clinical trials and the development of personalized treatments for pain in monogenic PD. In conclusion, this review underscores the need for further research to unravel the intricate relationship between pain and monogenic forms of PD. Standardized methodologies, larger sample sizes, and longitudinal studies are essential to elucidate the underlying mechanisms and develop targeted therapeutic interventions for pain management in individuals with monogenic PD.
1. Introduction
Parkinson’s disease (PD) is a complex disorder with significant clinical variability, potentially influenced by genetic factors, affecting not only motor but also non-motor symptoms (NMS) including pain (1, 2). Pain in PD encompasses various categories, including musculoskeletal pain, chronic pain, fluctuation-related pain, nocturnal pain, orofacial pain, discoloration, edema/swelling, and radicular pain, as categorized by the King’s Parkinson’s Disease Pain Scale (3).
Despite of its high prevalence, with reports of up to 85% of PD patients experiencing pain (4–6), it remains underdiagnosed and undertreated, even if it significantly impacts the quality of life (7–12). Lower Back Pain (LBP) is the most common pain site in PD, surpassing its prevalence in healthy older adults (13). Shoulder pain can even precede the PD diagnosis (14), with 12% of PD patients reporting it as their initial symptom (15). Motor complications in PD patients correlate with a higher risk of pain, and pain potentially exacerbates parkinsonian symptoms (5, 16). Given the complexity of pain etiology and the limited therapeutic options for its management, a comprehensive and accurate classification of pain types are crucial for improved patient outcomes (12).
Pathogenic variants in PD-causative genes have been associated with diverse disease symptoms (17, 18). For example, cognitive decline affects 70% of PD patients with pathogenic alpha-synuclein (SNCA) gene variants, while only 23% of Leucine-Rich Repeat Kinase 2 (LRRK2) carriers exhibit cognitive impairment (18). Rigidity and bradykinesia are nearly universal in SNCA patients, with dystonia less frequently observed (18). Despite these observations, data on pain in monogenic forms of PD remain limited. Case reports and small case–control studies indicate variations in pain presentation among different genetic forms of PD. For instance, a PD patient with an SNCA pathogenic variant exhibited dorsal pain as a primary symptom (19), while another patient with the Leu347Pro PTEN-induced putative protein kinase 1 (PINK1) pathogenic variant developed long-term right-sided pain following right-hand tremor onset (20).
Reports also suggest pain as an initial symptom in PD patients with the G2019S LRRK2 variant (21). Yet, these reports rarely explore the longitudinal progression of pain or compare pain experiences among carriers of different pathogenic variants under similar conditions. Consequently, the question of whether genetic status directly leads to the emergence of pain or is associated with different types in PD remains unanswered.
This review aims to investigate the hypothesis that genotypes may influence the pain phenotype in PD patients. We assess the presence, types, severity, and onset time of pain in PD patients and their relationship with different variants in pathogenic genes. By synthetizing existing literature, this review seeks to enhance our understanding of pain in monogenic forms of PD and offer insights for future research and clinical management.
2. Methods
2.1. Search strategy
To ensure methodological rigor, this review adhered to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses extension for Scoping Reviews (PRISMA-ScR) (22). The initial literature search encompassed databases such as MEDLINE (PubMed), Embase (Elsevier), and Cochrane databases. We sought articles published from inception until March 2023. The search strategy employed a combination of subject headings and MeSH terms, including “Parkinson’s disease” OR “Parkinson disease” OR “PD,” AND “gene” OR “genetic” OR “monogenic” OR “SNCA” OR “PRKN” OR “Parkin” OR “PINK1” OR “DJ1” OR “LRRK2” OR “ATP13A2” OR “GBA1” OR “DNAJC6” OR “FBXO7” OR “SYNJ1” OR “PARK1” OR “PARK2” OR “PARK4” OR “PARK6” OR “PARK7” OR “PARK8” OR “PARK9” OR “PARK15” OR “PARK19” OR “PARK20,” AND “pain” OR “pain sensation” OR “somatosensory discomfort.” The search was conducted without restrictions on language, year of publication, study type, or publication status.
Two independent investigators (PA and CTC) conducted the search, and the search results, including abstracts and full-text articles, were organized using reference management software. In addition to the electronic search, references from included studies and review articles were screened to augment the dataset.
Monogenic forms of PD result from the inheritance of a pathogenic variant of a single gene, contributing to approximately 30% of familial cases and 3–5% of sporadic cases (23). While the PD causative gene landscape has sparked some debate, several genes, including, SNCA, Parkin RBR E3 Ubiquitin Protein Ligase (PRKN), PINK1, LRRK2, and deglycase DJ-1 (PARK7) are widely as acknowledged as monogenic PD genes by most experts (24, 25). This review incorporates vacuolar protein sorting 35 (VPS35), with only one confirmed pathogenic variant (26), ATPase Cation Transporting 13A2 (ATP13A2), F- box protein 7 (FBXO7), DnaJ Heat Shock Protein Family (Hsp40) Member C6 (DNAJC6), and Synaptojanin-1 (SYNJ1) based on a recent comprehensive review (25).
We have included findings related to the GBA1 gene (Glucosylceramidase) which elevate the risk of developing PD. Pain reports are common among GBA1 carriers, making this addition significant to our review (27).
Specific pathogenic variants linked to PD and pain syndromes are discussed within the text, while those variants reported only in individual cases are further summarized in Table 1 for comprehensive reference.
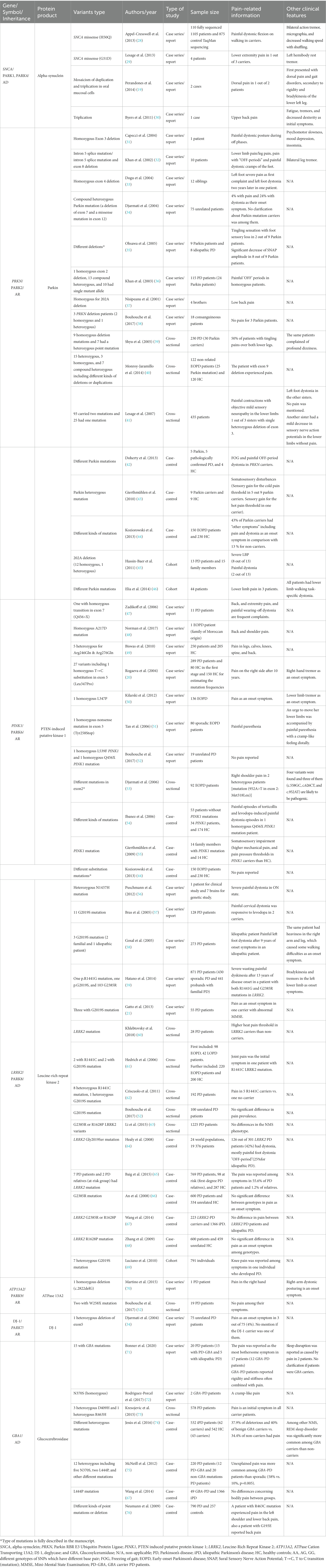
Table 1. Pain in monogenic PD: summary of the data extracted from the included studies concerning clinical studies.
2.2. Selection criteria
To ensure the relevance and quality of selected articles, the following inclusion criteria were applied: (A) Published papers that specifically focused on pain symptoms in patients with monogenic pathogenic variants associated with PD and (B) Articles that provided information on autosomal dominant (AD) or recessive (AR) forms of PD or on patients carrying at least one pathogenic variant in the SNCA, PRKN, PINK1, PARK7, LRRK2, GBA1, VPS35, ATP13A2, DNAJC6, FBXO7, or SYNJ1 genes, and reporting cases of PD-related pain. Furthermore, a meta-summary was conducted to consolidate the overall findings from the selected studies.
The exclusion criteria were as follows: (A) Articles that included PD patients carrying gene pathogenic variants other than those listed above or patients with other pain-related diseases, X-linked dystonia-parkinsonism, or rapid-onset dystonia-parkinsonism, and (B) Redundant publications.
The assessment of the retrieved occurred in two phases. Initially, titles and abstracts were screened based on the inclusion/exclusion criteria. Subsequently, the full text of the remaining articles was reviewed for final selection. Any articles that did not provide pertinent information regarding pain in monogenic forms of PD, even after a thorough full-text revision, were excluded from the analysis.
2.3. Specific aim
This review aimed to investigate the presence of pain in individuals with monogenic variants associated with PD. The primary objectives were to ascertain whether particular gene pathogenic variants within the spectrum of monogenic PD genes correlate with the presence of pain and, more specifically, to explore whether these pathogenic variants are associated with specific types of pain.
3. Results
3.1. Identification of studies
A consolidated master list comprising 541 potentially eligible articles was generated from the contributions of the two reviewers. Duplicate entries within the list were identified and removed, resulting in 534 unique articles. A preliminary screening of the titles and abstracts was conducted by the study team, which led to the exclusion of 3 due to the lack of relevance to the search terms. Furthermore, several papers were flagged for full-text review but were subsequently excluded as they did not contain any mention of pain within the reported symptoms. This process led to the exclusion of 463 articles.
In total, 63 articles met eligibility criteria for inclusion in this review. These selected articles encompassed six studies conducted on animal models, twenty-eight case series or case reports, eleven cross-sectional studies, fifteen case–control studies, and three prospective cohort studies (Figure 1). Each selected study underwent a comprehensive review, and pertinent data were extracted.
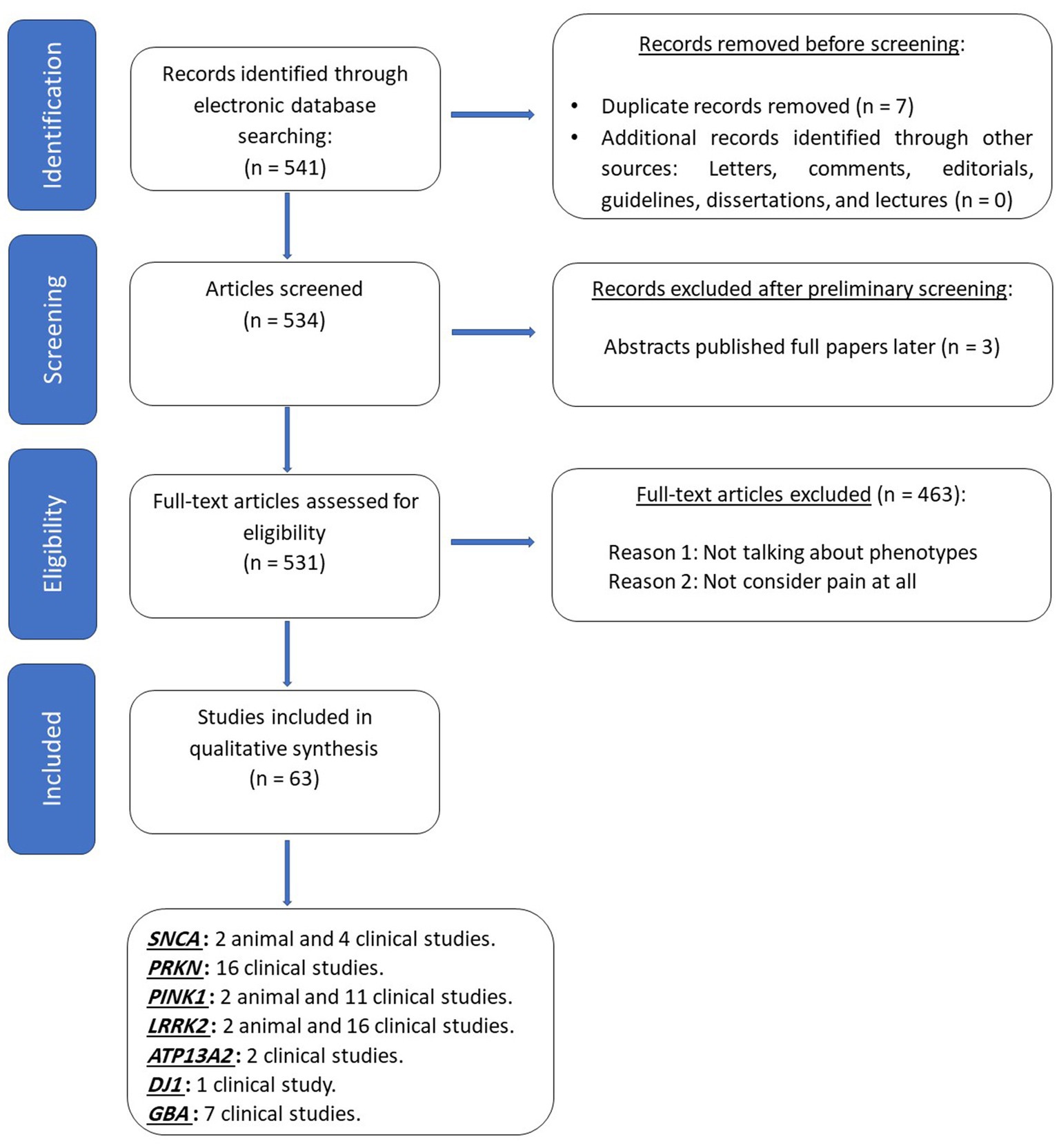
Figure 1. PRISMA’s diagram depicts the flow of information through the different phases of the comprehensive review, which included searches of databases. SNCA, alpha- synuclein; PRKN, Parkin RBR E3 Ubiquitin Protein Ligase; PINK1, PTEN induced putative protein kinase 1; LRRK2, Leucine-Rich Repeat Kinase 2; ATP13A2, ATPase Cation Transporting 13A2; PARK7, deglycase; GBA, Glucosylceramidase.
For each gene, we provide a general description and a summary of clinical studies, while details related to animal models will be presented in Table 2.
3.2. Monogenic forms of PD
More than 40 distinct chromosomal loci and 21 disease-causing genes associated with PD have been identified (77, 78). Among these, specific regions house known genes responsible for monogenic PD. Recognized monogenic PD genes include SNCA, PRKN, PINK1, PARK7, LRRK2, GBA1, VPS35, ATP13A2, DNAJC6, FBXO7, and SYNJ1 (25, 78). In monogenic PD, a pathogenic variant in a single gene is sufficient to manifest the PD phenotype (79).
All of the above mentioned genes exhibit autosomal inheritance patterns (79). In general, phenotypes resembling idiopathic PD (iPD) are more commonly observed in cases of AD inheritance, whereas young-onset parkinsonism resembling iPD or parkinsonism with atypical features is more commonly associated with AR inheritance (78).
Our review encompasses 11 out of 19 known PD-causing genes identified in the most recent comprehensive genetic database of PD (80). The exclusion of the remaining genes is due to insufficient data supporting their pathogenic role in PD and subsequent studies failed to replicate the pathogenic variant (81, 82).
Approximately 15% of individuals with PD have a family history of the disorder, with the current known monogenic forms accounting for approximately 30% of familial PD cases (16, 83).
The selected articles included in this review primarily focus on genes SNCA, PRKN, LRRK2, PINK1, PARK7, and GBA1 which are associated with either Early Onset PD (EOPD) or Late Onset PD (LOPD). We also sought information on ATP13A2, DNAJC6, FBXO7, and SYNJ1, which are rare causes of atypical PD However, our search did not yield any reports specifically addressing pain in relation to these genes.
A summary of the review results is provided in Table 1, providing an overview of key findings related to of pain in the context of monogenic PD.
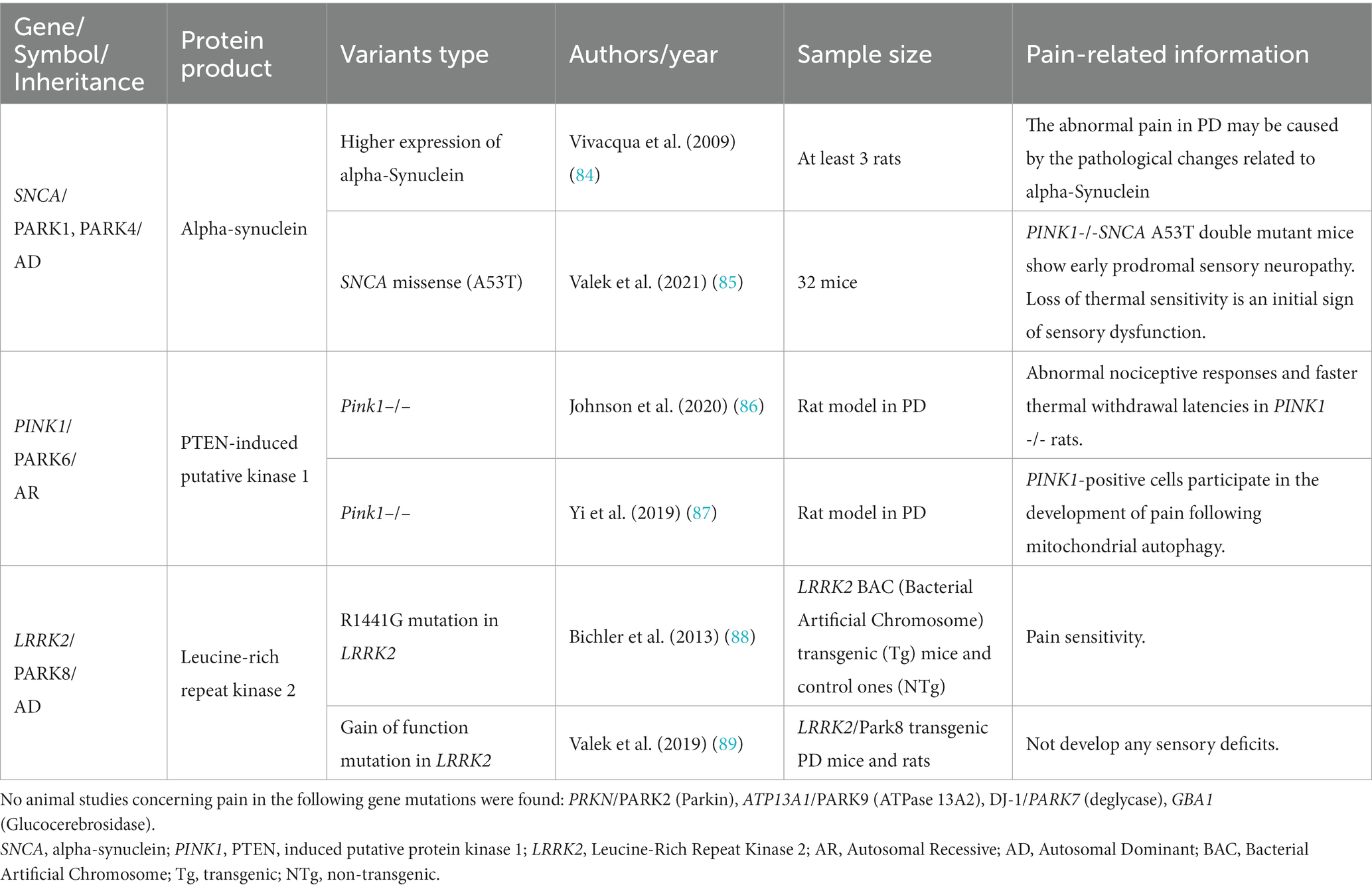
Table 2. Pain in monogenic PD: summary of the data extracted from the included studies concerning animal models.
3.2.1. SNCA (PARK1, 4)
The SNCA gene plays an important role in AD PD, with missense mutations and copy number gains (duplication or triplication) being established causes of PD. While pathogenic missense variants in SNCA are rare in the general population, duplications and triplications are also rare but more frequent, with approximately 60 reported families to date (90).
SNCA has six exons that encode alpha-synuclein, a 140-amino acid cytoplasmic protein highly abundant in neurons, particularly in the substantia nigra pars compacta (SNc), where it regulates dopamine neurotransmission (83, 91). Pathogenic SNCA variantscan lead to cytoplasmic accumulation of alpha-synuclein, promoting oxidative stress and metabolic dysfunction in the SNc (92).
Specific pathogenic variants in the SNCA that are associated with PD. Parkinson’s disease-1 (PARK1) results from a point pathogenic variant (missense) in SNCA, while SNCA gene duplication and triplication lead to Parkinson’s disease-4 (PARK-4) (93). To date, eight pathogenic missense variants have been identified in SNCA, all located within the N-terminal amphipathic region of alpha-synuclein. These variants interfere with the alpha-helix-mediated interaction with membranes, contributing to the pathogenesis of PD (94–100).
The most common of these eight missense variants is A53T (18, 80). The number of cases for other missense variants (A30G, A30P, E46K, H50Q, G51D, A53E, and A53V) are small, and some, like H50Q, are not significantly enriched in cases compared to controls (18). Therefore, sufficient evidence may not exist to classify all of them as pathogenic variants (18).
PD patients with SNCA gene pathogenic variants typically exhibite earlier age of disease onset than iPD, rapid disease progression, positive response to levodopa treatment, and often present with prominent NMS (101).
This review included six studies, two animal models and four clinical studies (all case series/reports), that investigated SNCA pathogenic variants and their potential association with pain. Out of the initially identified articles, 61 were excluded as they did not mention pain or other phenotypes related to SNCA pathogenic variants.
Regarding animal studies, Vivacqua et al. reported that higher levels of alpha-synuclein expression in spinal cord areas known to be involved in pain modulation and transmission (84). Another study showed that PINK1−/− SNCA A53T double mutant mice, which develop a PD-like disease, exhibited a loss of thermal sensitivity as an initial sign of sensory dysfunction (85).
Among clinical studies, one case report study described a novel SNCA missense pathogenic variant, G51D, in a patient with lower extremity pain among her symptoms (29). Additionally, two other case reports detailed dorsal and upper back pain in carriers with SNCA duplication and triplication (19, 30).Another case report described painful dystonic flexion of the toes while walking in a patient with H50Q variant, although this variant’s pathogenicity remains under debate (28).
3.2.2. PRKN (PARK2)
The PRKN gene is associated with an AR form of the disorder (79, 102, 103). Homozygous and compound-heterozygous pathogenic variants in PRKN are causative of PD, while heterozygous pathogenic variants may predispose to PD symptoms with low penetrance, making them potential genetic risk factors (104, 105).
PRKN comprises12 exons and encodes Parkin, a 465 amino acid protein (79, 106). Parkin is widely expressed in human tissues, with significant abundance in the brain, especially the substantia nigra pars compacta (SNc) (106).
Approximately 60 PRKN pathogenic and non-pathogenic variants have been identified, including deletions and duplications, which can complicate PRKN genotyping (107, 108). According to published data, up to 18% of EOPD patients globally and 27.6% of AR families carry PRKN pathogenic variants (23, 109). Among PD-PRKN patients, exon 3 deletion is the most frequent pathogenic variant (17). All PRKN pathogenic variants result in the loss of Parkin function, leading to a loss of Ubiquitin E3 ligase activity and subsequent neurodegeneration (110).
Monogenic PD associated with PRKN typically presents with early onset, slow symptom progression, and a positive response to dopaminergic treatment but is often accompanied by complications such as dystonia and prominent freezing of gait (111, 112).
Sixteen studies in this review provided information on the association between PRKN pathogenic variants and pain, including clinical data. These studies included eight case series/reports, three cross-sectional, three case–control studies, and two cohorts. One hundred fifty-six eight clinical studies and one animal study were excluded because they did not mention pain as a symptom or did not involve carriers pathogenic variant carriers.
A cohort study has reported painful limb dystonia as a symptom among PRKN pathogenic variant carriers (45). Besides, another cohort study described eight cases with lower limb dystonia activated by walking, Three of those cases also presented with lower limb pain unrelated to dystonic spasms (46).
A case–control study mentioned painful dystonia among three PRKN missense pathogenic carriers and general pain in another missense carrier, while one case report study described painful dystonic posture during off phases in a homozygous exon 3 deletion patient (31, 42). Three case reports described painful foot dystonia among deletion carriers, with four patients in one study also experiencing LBP (32, 33, 37).
Notably, a case series reported 24% dystonia among the patients but did without specifying whether they were PRKN pathogenic variant carriers (34). A case–control study reported “other symptoms” including pain and dystonia, as an onset symptoms in 43% of PRKN carriers compared to 13% of non-carriers (44).
Musculoskeletal pain has also been associated with PRKN pathogenic variants (R275W, exon 3 duplication and homozygous deletion) in two studies (40, 41).
In a cross-sectional study, a carrier of PRKN exon 9 deletion reported experiencing pain as a symptom, although the specific type of pain was not described (40). Another cross-sectional study noted painful contractions as an onset symptom in a patient with exon 3 deletion (41). Additionally, a homozygous PRKN pathogenic variant carrier required hospitalization due to painful OFF periods (36).
Sensory symptoms and signs, such as tingling sensation and a significant decrease in Sural Sensory Nerve Action Potential (SNAP) amplitude, were reported in two studies (35, 39). Besides, a case–control study observed a sensory gain in cold and hot pain thresholds among carriers (43).
On the other hand, there was a study screened 145 PD patients for LRRK2 pathogenic variant, 19 of whom carried a PRKN pathogenic variant. They reported their clinical data, including pain, and no specific mention of pain was reported for any of the pathogenic variant carriers (38).
3.2.3. PINK1 (PARK6)
Pathogenic variants in the PINK1 gene are the second most common cause of AR EOPD (79). PINK1 comprises 6 exons encoding PTEN-induced putative kinase 1, a 581 amino acid serine/threonine kinase (113). In normal conditions, wild-type PINK1 plays a protective role against neuronal apoptosis in neural cell lines. However, pathogenic variants associated with PARK6 disrupt this protective function, leading to the degeneration of dopaminergic neurons (114). Most PINK1 pathogenic variants are located in exon 7, with Q456X variant being the most frequent (79).
Individuals with PINK1 monogenic PD typically have an onset age of around 32 years and experience slow disease progression, often with a favorable response to levodopa treatment and sleep benefits (115).
Thirteen studies have investigated the potential relationship between pain and PINK1 pathogenic variants. These studies include two animal studies and eleven clinical research articles (seven case series/reports, one cross-sectional study, and three case–control studies). Forty-one clinical studies were excluded from the analysis because they did not mention pain as a symptom.
Regarding animal studies, a study found that thermal withdrawal latencies were significantly shorter in PINK1−/− rats than in wild-type rats over time, indicating altered pain responses (86). The second team used a rat model with neuropathic pain to investigate the role of PINK1 and observed increased expression of PINK1 in pain-related areas compared to control rats (87).
A case–control study reported painful episodes of torticollis and painful dystonia in a homozygous Q456X PINK1 pathogenic variant carrier (54). Similarly, Zadikoff et al. described a homozygous Q456X PINK1 pathogenic variant carrier who frequently experienced back and limb pain and painful wearing-off dystonia (47).
Multiple case reports, one case–control, and one cross-sectional study support the observation that pain is frequently encountered in patients with PINK1 pathogenic variants, often manifesting in various body regions, particularly the neck, back, and shoulders. However, these studies did not provide detailed descriptions of the specific type and characteristics of the reported pain (20, 48–50, 53, 55).
Additionally, a case report study highlighted a patient with a novel homozygous nonsense PINK1 pathogenic variant. This patient exhibited an urge to move her lower limbs accompanied by painful paresthesia and a sensation of distal cramping (51).
On the other hand, one case report (a homozygous Q456X carrier) and one case–control study specifically assessed pain symptoms in individuals carrying PINK1 mutations, but neither of them reported pain as a symptom among the PINK1 carriers (38, 44). In the case–control study, the prevalence of pain among PINK1 carriers was 0%, while it was 13% in the non-carrier group, it was 13, and 43% in PRKN carriers (44).
3.2.4. LRRK2 (PARK8)
Pathogenic variants in the LRRK2 gene are the most common genetic cause of AD PD (24, 116), affecting both familial and sporadic forms of the disease (117). The LRRK2 gene is a large gene consisting of 51 exons and encodes a 2,527 amino acid cytoplasmic protein called leucine-rich repeat kinase 2 (118). One of the critical functions of LRRK2 is its regulation of protein synthesis through the miRNA pathway, and impairment in this pathway has been implicated in LRRK2-related pathogenesis (119).
More than 40 pathogenic variants have been identified in the LRRK2 gene among PD patients, with eight of them known to cause PD (93). Among them, the most common and well-characterized LRRK2 pathogenic variant is G2019S, with a prevalence ranging from 0 to 42% depending on ethnicity, followed by R1441C (120, 121).
LRRK2-PD patients typically presents as a LOPD, often respond well to levodopa treatment, and have fewer NMS than iPD cases (122, 123).
Among the identified studies involving LRRK2-associated PD patients, 18 included information on pain symptoms, while 124 studies did not mention pain or other sensory symptoms and were excluded from the analysis. Of the 18 studies, two were animal studies, and the remaining 16 were clinical consisting of five case series/reports, five cross-sectional studies, five case–control studies, and one cohort study.
Evidence from mouse studies investigating the association between LRRK2 and pain observed similar pain sensitivity than controls without developing sensory deficits (88, 89).
Multiple studies have described the presence of painful dystonia in different cohorts of LRRK2 pathogenic variant carriers. Three studies reported painful dystonia among G2019S pathogenic variant carriers. A case–control study reported that 42% of carriers experienced painful foot dystonia during the “OFF period” (64). Another case report described a patient who reported painful foot dystonia (58). Furthermore, a study reported painful cervical dystonia in one individual, which showed a positive response to levodopa treatment (57).
Severe wearing-off and dyskinesia with off-time pain have been reported in a LRRK2 pathogenic variant carrier (59). Unspecified pain and joint pain have been reported as onset symptoms by three PD patients, all carriers of pathogenic variants, two G2019S and one R1441C (21, 61, 69). A case–control study found that pain is one of the most common NMS experienced by PD patients with LRRK2 pathogenic mutations, affecting over half of the subjects (65). In a cross-sectional study, pain was observed in five R1441C carriers but only in one non-carrier, although the difference was not statistically significant (p = 0.155). The specific type and characteristics of the reported pain were not described in detail (62).
Notably, a case report documented a patient with severe and painful ON-dystonia who carried a LRRK2 N1437H variant which is not recognized among the established pathogenic variants (56). Furthermore, McGill test recorded neuropathic disturbances were reported for LRRK2 pathogenic variant carriers with a mean of 8.3 8.3 ± 14 compared to 0 for non-carriers. Additionally, LRRK2 mutation carriers displayed a higher heat pain threshold compared to non-carriers (44.1 ± 4.82 vs. 40.6 ± 4.5°C, p = 0.058), suggesting a clear difference in terms of pain perception (60).
The final five included studies for this gene reported pain among individual carrying LRRK2 pathogenic variants. However, after analysis, no statistically significant difference in the prevalence of pain emerged between the carrier and non-carrier groups (52, 63, 66–68).
3.2.5. PARK7 (DJ-1)
PARK7 pathogenic variants are associated with AR PD and are relatively uncommon, constituting approximately 1 to 2% of EOPD cases (124). The PARK7 gene is comprised of 8 exons, with the initial two being noncoding, and it encodes DJ-1, a 189 amino acid protein which exhibits neuroprotective and antioxidant properties (125, 126). Pathogenic variants within PARK7 result in the production of a mutated DJ-1 protein characterized by reduced activity due to misfolding (127, 128).
Individual carrying PARK7 pathogenic variant typically experience disease onset at an average age of 27 years and often exhibit prominent NMS, including mental health disorders and cognitive decline. Dystonia is highly prevalent, affecting approximately 73% of those with DJ-1 pathogenic variant (129).
Among the 26 studies examining PARK7 pathogenic variants, only one case series/report study mentioned the presence of pain.
Djarmati et al. conducted a screening of 75 unrelated PD patients and identified one individual carrying a heterozygous deletion of exon 5 in PARK7. Among the 75 cases, three individuals presented pain as an onset symptom (4%); however, the authors did not specify whether the PARK7 pathogenic variant carrier was one of the three PD patients reporting pain (34).
3.2.6. VPS35 (PARK17)
VPS35 is responsible for encoding the vacuolar protein sorting ortholog 35, which is a critical component of a large complex involved in the transportation of proteins from endosomes to the trans-Golgi network. Pathogenic variants in VPS35 were initially identified in 2011 and represent a rare cause of AD LOPD (18, 130).
Exome analysis has revealed that the D620N is the sole confirmed pathogenic variant associated wit PD thus far (131). Monogenic PD linked to VPS35 exhibits high heritability but low penetrance. The clinical phenotype of VPS35-related PD closely resembles that of iPD, although the average age of onset is typically around 50 years old (132).
Among the twelve studies conducted on individual carrying VPS35 variants, 67 patients were included, all of whom were heterozygous carriers (18). Out of these 67 heterozygous patients, who presented a total of 10 different potentially disease-causing variants, 50 (75%) carried the pathogenic D620N variant (18). While these studies did report various symptoms in these patients, NMS were the least commonly reported (6.2%) and none of the studies specifically mentioned the presence of pain (18).
3.2.7. GBA1
Pathogenic variants in the GBA1 gene are not considered causative for PD, but they represent the most prevalent genetic susceptibility factor for the development of the disease (133, 134). While GBA1 pathogenic variants do not exhibit complete penetrance, heterozygote carriers face a fivefold increased risk of developing PD, while homozygotes have a 10- to 20-fold elevated risk (135). The penetrance of GBA pathogenic variant carriers to develop PD has been estimated as 13.7% by the age of 60 years and 29.7% by the age of 80 years (135).
Furthermore, due to their higher frequency in most PD populations compared to known monogenic PD genes such as LRRK2, SNCA, and PRKN (136), GBA1 pathogenic variants are regarded as the most significant genetic risk factor for PD (137). Recent genome-wide association studies have confirmed that approximately 8–12% of PD patients carry GBA1 pathogenic variants (138).
The GBA1 gene, located on chromosome 1q22, encodes the enzyme glucocerebrosidase, and is associated with AR Gaucher disease (GD) (136). Approximately 130 GBA1 pathogenic variants have been reported in PD patients (27, 139). Similar to GD, L444P and N370S are the two most frequent pathogenic variants. Severe pathogenic variants such as L444P are associated with a higher risk of developing PD, earlier age of onset, and more severe motor and NMS (140).
We included seven articles (comprising four case–control studies, two case series/reports, and one cross-sectional study). Eighteen articles were excluded because they did not mention pain among the reported symptoms.
While no distinctive symptoms have been reported to differentiate GBA1 pathogenic variant carriers from individuals with iPD (141), pain appears to be an exception. Some patients with GD develop progressive parkinsonian symptoms (142), and notably, pain has been more frequently reported as an initial symptom in GBA1-PD patients compared to individuals with iPD.
Shoulder pain and LBP (76), unexplained pain (58% vs. 10%, p = 0.005) (75), and cramp-like pain as the primary source of disability at a young age (72) have all been reported more frequently among GBA1 pathogenic variant carriers compared to non-carriers. In a case series pain was identified as the most bothersome symptom in 12 out of 15 GBA1-PD patients also reporting rigidity and stiffness, often accompanied by pain. One patient described painful dyskinesia as the most bothersome symptom, and two reported pain-related sleep problems (71).
In a cross-sectional study, pain was reported more frequently as an initial symptom in the GBA1-PD compared to the iPD 10.3 vs. (3.0%) (p = 0.039), with four patients reporting shoulder pain as their initial symptom. The most significant finding of this study is that the presenting symptoms of PD are similar in GBA1 carriers and non-carriers for all parameters except for pain (73).
On the other hand, two case–control studies mentioned that GBA1-PD patients experienced bodily pain among their symptoms, although no statistically significant differences were reported between GBA1-PD and the iPD group (p = 0.7) (67, 74).
Considering that symptoms tend not be more severe among patients carrying pathological variants like L444P, it becomes intriguing to explore whether pain is more closely associated with severe variants (137). Contrary to this argument, based on existing studies, while two patients with N370S variant (considered mild) reported cramp-like pain (72), 49 patients with L444P variant (classified as severe) found no significant differences in terms of bodily pain compared to individuals with iPD (67).
3.2.8. ATP13A2 (PARK9)
Pathogenic variants in the ATP13A2 gene are responsible for Kufor-Rakeb syndrome (KRS), an AR atypical form of PD (143). The ATP13A2 gene consists of 29 exons and encodes a protein of 1,180 amino acids (144). The ATP13A2 protein plays a role in reducing intracellular concentrations of manganese ions (Mn2+), thereby offering protection against apoptosis (145). Pathogenic variants in ATP13A2 lead to disruptions in the proteasomal pathway and premature degradation of ATP13A2 mRNA, contributing to the development of KRS (146).
Since the discovery of ATP13A2 pathogenic variants in 2006 (144), only a limited number of studies have been conducted and published. Among the 16 studies we assessed, one cross-sectional study and one case series/report did mention pain as a symptom, while the remaining 14 studies did not mention pain or other associated phenotypes and were consequently excluded.
In one cross-sectional study, a ATP13A2 pathogenic variant was identified in two patients who did not have pain as one of their symptoms (38). Additionally, a case report documented arm dystonic posturing as the onset symptom in a homozygous patient with 2822delG variant who was unresponsive to anticholinergics and levodopa; however this variant has not yet been definitively established as a pathogenic variant for PD (70).
3.2.9. DNAJC6 (PARK19A, b)
DNAJC6, located on 1p31.3, encodes auxilin, and its loss of function can lead to EOPD (147). In animal studies, the absence of auxilin has been linked to synaptic vesicle endocytosis disruptions, which have adverse effects on synaptic neurotransmission, homeostasis, and signaling (148). However, the precise mechanism by which auxilin deficiency induces dopaminergic neurodegeneration and unusual neurological symptoms remains incompletely understood (148).
Homozygous pathogenic variants in DNAJC6 are responsible for atypical parkinsonism, exhibiting AR inheritance pattern (149, 150). PARK19A is characterized by onset in the first or second decade of life and rapid disease progression, while PARK-19B onset occurs between the third and fifth decades, featuring a slower progressive course, and similar features to classic iPD (149–151).
Three separate case-report studies identified Juvenile-onset PD, PARK-19A, among patients with homozygous pathogenic variant in the DNAJC6 gene, including two loss-of-function and one nonsense variant; however, none of these cases reported pain as a symptom (149, 150, 152). Another study reported homozygous pathogenic variants in two unrelated families with PARK-19B and no instances of pain were among their reported symptoms (151).
Finally, in a comprehensive analysis utilizing whole exome sequencing DNAJC6 potential pathogenic variants were explored in 6 juvenile parkinsonism patients. Homozygous nonsense R256* DNAJC6 pathogenic variants were confirmed for all affected children and none of them reported pain among their symptoms (153).
3.2.10. FBXO7 (PARK15)
FBXO7 a gene comprising ten exons is located on chromosome 22q12.3, encodes a member of the F-box protein family known as F-Box Protein 7, characterized by an approximately 40 amino acid motif (154). Pathogenic variants in FBXO7 are responsible for an AR parkinsonian syndrome. The typical presenting symptoms include bradykinesia and tremor, and patients affected by this disorder frequently exhibit pyramidal signs, dysarthria, and dyskinesia (155).
To date, eight studies have identified cases carrying FBXO7 variants, predominantly associated with an early-onset parkinsonian and pyramidal syndrome (155–161). Notably, only one study reported a classical PD presentation in two siblings, caused by a new FBXO7 pathogenic variant, L34R (162). None of these studies discuss or mention pain as one of the associated symptoms.
3.2.11. SYNJ1 (PARK20)
SYNJ1, located on 21q22.11 and comprised of 33 exons, encodes Synaptojanin 1 protein. Pathogenic variant in SYNJ1 Are associated to AR EOPD (163).
Remarkably, independently and simultaneously, two studies identified the same homozygous missense pathogenic variant in the SYNJ1 gene, R258Q. In both studies affected patients were thoroughly screened for all known genes, and R258Q SYNJ1 was the sole pathogenic variant identified. Two affected siblings in each study suffered from EOPD, and none of them mention pain among their symptoms (164, 165).
4. Discussion
This review was conducted to assess the presence of pain in patients with monogenic PD-related pathogenic variants, encompassing genes such as SNCA, PRKN, PINK1, LRRK2, ATP13A2, PARK7, VPS35, GBA1, DNAJC6, FBXO7, and SYNJ1. The central findings of this review offer valuable insights into the connection between specific gene pathogenic variants and the occurrence of pain in individuals with PD.
As a summary: (1) for the SNCA gene, two point mutations were associated with lower extremity pain and painful foot dystonic flexion while walking (28, 29). Gene duplications and triplications were also linked to dorsal and upper back pain (19, 30); (2) PRKN carriers reported painful lower limb dystonia and lower back pain as prominent symptoms (31–33, 36, 37, 42, 45, 46). Additionally, musculoskeletal pain, sensory loss, tingling sensation, and reduced SNAP amplitude suggested a central origin for abnormal sensitivity in PRKN pathogenic variant carriers (35, 39–41, 43); (3) Pain was observed in PINK1 pathogenic variants. Homozygote carriers of the Q456X as the most frequent pathogenic variant experienced painful dystonia (47, 54), and pain was reported in various body parts, with a preference for the neck, back, and shoulders (20, 48–50, 53, 55). PINK1 pathogenic variants were also associated with abnormal central somatosensory processing (51); (4) The LRRK2 pathogenic variants are associated with pain, with painful dystonia reported in G2019S carriers (57, 58, 64) and G2019S and R1441C carriers reporting unspecified joint pain as their onset symptom (21, 61, 69). Multiple studies indicated that LRRK2 pathogenic variant carriers experienced different types of pain as part of their symptoms (52, 56, 59, 60, 62, 63, 66–68); (5) Limited studies have assessed pain in ATP13A2 pathogenic variant carriers. However, pain was reported in a few cases, suggesting a potential link between ATP13A2 and pain (70); (6) Among GBA1 pathogenic variant carriers, pain was reported as one of the most prevalent early symptoms, with some patients exclusively experiencing shoulder pain as an initial presentation (73). A case series study found that almost all GBA1-PD patients reported pain as their most bothersome symptom (71). Glucosylceramide accumulation, associated with GBA1 pathogenic variants, may contribute to PD-associated sensory neuropathies and pain (166). Low GBA1 activity has also been observed in PD patients without GBA1 pathogenic variants, indicating its involvement in developing or progressing PD-associated sensory neuropathy (167). It would be indeed interesting to explore whether pain is more associated with severe pathological variants in PD-related genes. However, based on the available studies, there does not appear to be a significant difference in the prevalence of pain between individuals with severe pathological variants and those with iPD (67, 137); (7) No results were available regarding pain in PARK7, VPS35, DNAJC6, FBXO7, and SYNJ1 pathogenic variants. The most common pain subtypes linked with Monogenic Parkinson’s disease are summarized in Figure 2.
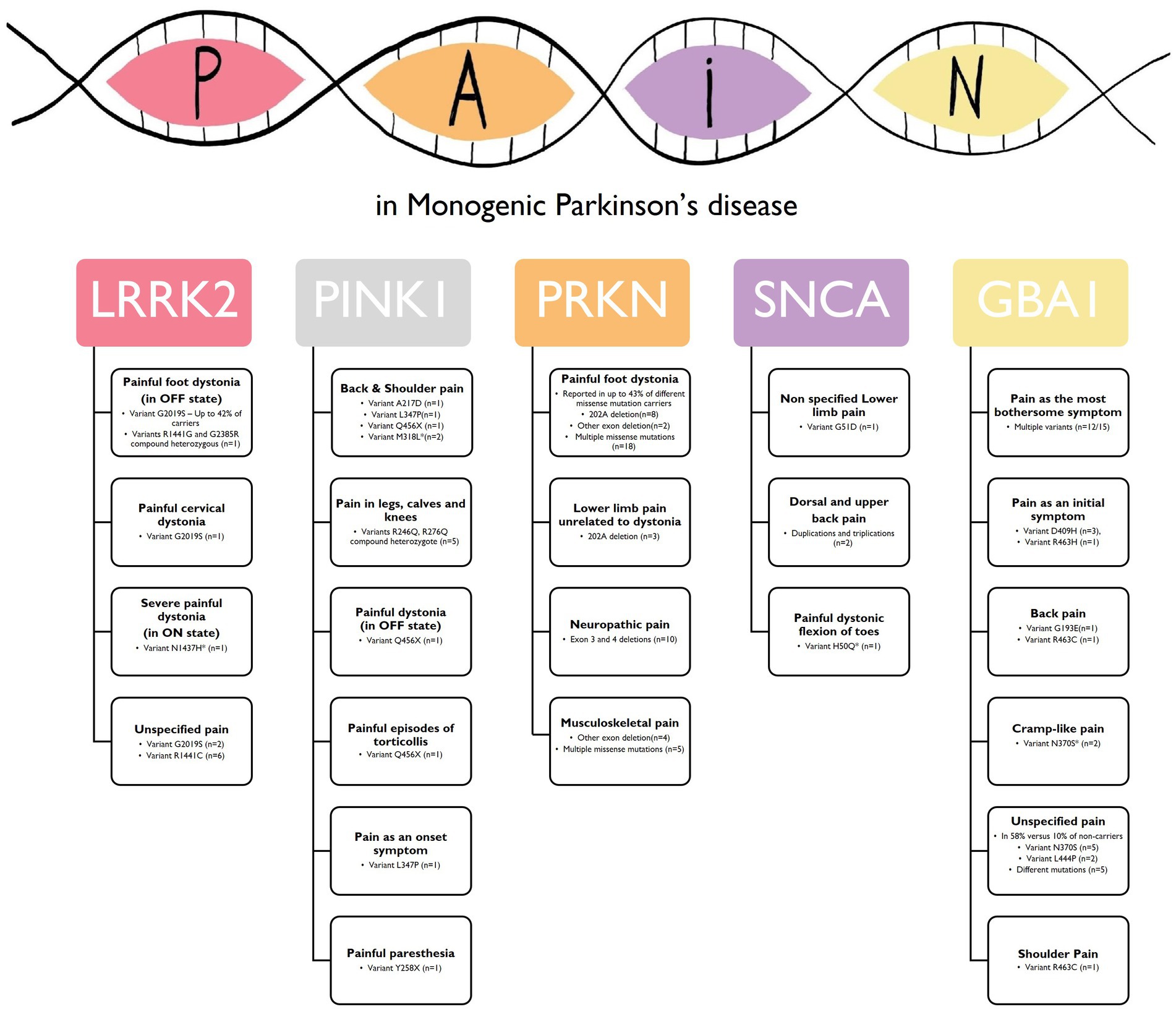
Figure 2. Common pain subtypes linked with Monogenic Parkinson’s disease. This figure illustrates the most commonly pain subtypes reported in individuals with monogenic forms of Parkinson’s disease (PD) associated with specific genes. The figure provides an overview of the pain profiles observed in relation to each gene pathogenic variant using the descriptors used in the original articles. LRRK2, Leucine-Rich Repeat Kinase 2; PINK1, PTEN induced putative protein kinase 1; PRKN, Parkin RBR E3 Ubiquitin Protein Ligase; SNCA, alpha-synuclein; GBA, Glucosylceramidase – *Variants that are not yet established as pathogenic for PD.
The findings discussed in this review provide valuable insights into the connection between specific monogenic variants in PD-related genes and pain in PD. Certain genes roles, including SNCA, PRKN, PINK1, and LRRK2, have been extensively studied providing potential perspectives into the underlying mechanisms of pain in PD.
In the case of SNCA, animal studies have indicated that abnormal pain in PD may be attributed to pathological changes related to alpha-synuclein- presence in unmyelinated areas of the spinal cord (84, 85). Clinical studies further support the presence of various pain manifestations in SNCA pathogenic variant carriers, such as painful dystonic flexion while walking and dorsal and upper back pain (19, 28, 29).
Similarly, PRKN pathogenic variant carriers have been found to experience painful lower limb dystonia and lower back pain, accompanied by reduced SNAP amplitude (31–33, 35–37, 39–43, 45, 46). These findings suggest the involvement of sensory axonal neuropathy and suggest that reduced SNAP amplitude may serve as a diagnostic indicator for PRKN-related PD.
PINK1 pathogenic variant carriers, they exhibit distinct somatosensory profiles and clinical entities compared to iPD, suggesting a primary hypofunction of nociceptive and non-nociceptive systems in PINK1-associated PD (43). Studies have proposed that specific PINK1 pathogenic variants, such as the L347P, may be associated with pain in PD patients (20). Moreover, abnormalities in nociceptive processing have been reported in PINK1 pathogenic variant carriers, indicating a potential role of abnormal central somatosensory processing in pain generation (43). Interestingly, these abnormalities seem to lead to hypoalgesia rather than hyperalgesia, contrasting with the findings in sporadic PD cases (168). The study of E3 ligase dysfunction has provided insights into the pathophysiology of PD, particularly about the PRKN gene (169). The single-base pair deletion in PRKN observed in four brothers with refractory back pain may be attributed to a lack of E3 activity, potentially contributing to lower back pain in PD patients (37). E3 ligases play a crucial role in the ubiquitin-proteasome pathway involved in protein turnover, and dysfunction in this pathway has been implicated in PD (169).
Additional research is needed to better understand the connection between LRRK2 and pain as while multiple clinical studies have suggested that individuals with LRRK2 pathogenic variants experience different pain types (21, 58, 64, 88), animal models findings also suggest that pain sensitivity remains unchanged in the presence of LRRK2 pathogenic variants (89, 123).
In the context of other monogenic variants, such as ATP13A2, PARK7, VPS35, DNAJC6, FBXO7 and SYNJ1 the current literature provides inconclusive results or insufficient data regarding their association with pain in PD. Additional studies are required to clarify the potential links between these genes and pain symptoms in PD patients.
Genetic associations with GBA1 variants have demonstrated an influence the occurrence of pain in PD. Patients carrying GBA1 variants have reported higher rates of pain compared to non-carriers (71, 73). Recent studies suggest that approaches targeting glucocerebrosidase activity or refolding may reduce PD pain and sensory loss (166). Even in PD patients without GBA1 variants, low GBA1 activity has been observed, indicating a prevalent loss of GBA1 function that may contribute to developing or progressing PD-associated sensory neuropathy (135). These findings suggest that elevated levels of glucosylceramides may underlie sensory neuropathies characterized by the loss of thermal sensation and mechanical hypersensitivity in PD patients, irrespective of the presence of chronic pain.
Overall, this comprehensive review underscores the complex relationship between monogenic pathogenic variants in PD-related genes and the presence of pain in PD. To advance our understanding of the underlying mechanisms and identify potential targets for the treatment of pain in PD further investigations are essential. While this review provides a solid foundation for future research, it also sheds light on several limitations that require attention. The absence of pain assessment in numerous studies and the lack of detailed pain characteristics impede a comprehensive understanding of pain in monogenic PD-related pathogenic variants. Furthermore, the predominance of case series/reports and the limited information available for specific gene pathogenic variants underscores the necessity for more robust studies with larger sample sizes and systematic evaluation of pain symptoms. It is because of these limitation specific frequencies or data about the prevalence of pain in monogenic forms of PD remain unclear. Addressing this knowledge gap is of paramount importance and needs the implementation of more focused and structured study designs regarding pain in PD. Finally, gaining a deeper understanding of pain as a potential prodromal symptom in monogenic PD could provide insights into early indicators and predictive markers, allowing for more timely and targeted interventions.
5. Conclusion
In conclusion, the existing evidence suggests that specific types of pain are commonly observed in individuals with monogenic forms of PD, particularly those associated with SNCA, PRKN, PINK1, LRRK2, and GBA1 genes. Pain in PD can potentially serve as a clinical marker, sometimes as a prodromal symptom as in individuals with PRKN and GBA1 pathogenic variants, but also as a potential marker of progression other genes pathogenic variants.
Given the subjective nature of pain, its effective management requires standardized and objective standards of care. Future investigations should prioritize the collection of high-quality, standardized pain data, to enable direct comparison across studies and facilitate large-scale meta-analyses. Establishing connections between genetic profiles with pain symptoms could have significant clinical implications, such as guiding the selection of diagnostic tests, facilitating patient stratification for clinical trials, and ultimately enabling personalized treatment approaches for individuals with monogenic PD.
Author contributions
VB contributed to the study’s design, planning, supervision, and manuscript review. PA and CT-C were involved in the planning, literature search, and manuscript writing. BA participated in the study’s original design and search strategy. All authors critically reviewed and approved the final version of the manuscript.
Funding
T-CC has been awarded a postdoctoral Denise Lajoie-Lake fellowship in Brain Research, facilitated by the Hotchkiss Brain Institute and the Department of Clinical Neurosciences, University of Calgary.
Acknowledgments
The artwork featured in Figure 2 was crafted by the illustrator, Rosario Oliva – @rosarioolivailustradora.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Ha, AD , and Jankovic, J . Pain in Parkinson’s disease. Mov Disord. (2012) 27:485–91. doi: 10.1002/mds.23959
3. Chaudhuri, KR , Rizos, A , Trenkwalder, C , Rascol, O , Pal, S , Martino, D, et al. King’s Parkinson’s disease pain scale, the first scale for pain in PD: an international validation: King’s PD pain scale validation. Mov Disord. (2015) 30:1623–31. doi: 10.1002/mds.26270
4. Broen, MPG , Braaksma, MM , Patijn, J , and Weber, WEJ . Prevalence of pain in Parkinson’s disease: a systematic review using the modified QUADAS tool: prevalence of pain in Parkinson’s disease. Mov. Disord. (2012) 27:480–4. doi: 10.1002/mds.24054
5. Defazio, G , Gigante, A , Mancino, P , and Tinazzi, M . The epidemiology of pain in Parkinson’s disease. J Neural Transm. (2013) 120:583–6. doi: 10.1007/s00702-012-0915-7
6. Toda, K , and Harada, T . Prevalence, classification, and etiology of pain in Parkinson’s disease: association between Parkinson’s disease and fibromyalgia or chronic widespread pain. Tohoku J Exp Med. (2010) 222:1–5. doi: 10.1620/tjem.222.1
7. Ford, B . Pain in Parkinson disease: the hidden epidemic. Nat Rev Neurol. (2009) 5:242–3. doi: 10.1038/nrneurol.2009.50
8. Mao, CJ , Chen, JP , Zhang, XY , Chen, Y , Li, SJ , Li, J, et al. Parkinson’s disease patients with pain suffer from more severe non-motor symptoms. Neurol Sci. (2015) 36:263–8. doi: 10.1007/s10072-014-1942-y
9. Quittenbaum, BH , and Grahn, B . Quality of life and pain in Parkinson’s disease: a controlled cross-sectional study. Parkinsonism Relat Disord. (2004) 10:129–36. doi: 10.1016/j.parkreldis.2003.12.001
10. Rodríguez-Violante, M , Alvarado-Bolaños, A , Cervantes-Arriaga, A , Martinez-Martin, P , Rizos, A , and Chaudhuri, KR . Clinical determinants of Parkinson’s disease-associated pain using the King’s Parkinson’s disease pain scale. Mov Disord Clin Pract. (2017) 4:545–51. doi: 10.1002/mdc3.12469
11. Tinazzi, M . Pain and motor complications in Parkinson’s disease. J Neurol Neurosurg Psychiatry. (2006) 77:822–5. doi: 10.1136/jnnp.2005.079053
12. Buhmann, C , Kassubek, J , and Jost, WH . Management of Pain in Parkinson’s disease. J Parkinsons Dis. (2020) 10:S37–48. doi: 10.3233/JPD-202069
13. Tueth, LE , and Duncan, RP . Musculoskeletal pain in Parkinson’s disease: a narrative review. Neurodegener Dis Manag. (2021) 11:373–85. doi: 10.2217/nmt-2021-0011
14. Stamey, W , Davidson, A , and Jankovic, J . Shoulder pain: a presenting symptom of Parkinson disease. JCR J Clin Rheumatol. (2008) 14:253–4. doi: 10.1097/RHU.0b013e3181826d43
15. Cleeves, L , and Findley, L . Frozen shoulder and other shoulder disturbancies in Parkinson’s disease. J Neurol Neurosurg Psychiatry. (1989) 52:813–4. doi: 10.1136/jnnp.52.6.813-b
16. Barbeau, A , and Roy, M . Familial subsets in idiopathic Parkinson’s disease. Can J Neurol Sci J Can Sci Neurol. (1984) 11:144–50. doi: 10.1017/S0317167100046308
17. Kasten, M , Hartmann, C , Hampf, J , Schaake, S , Westenberger, A , Vollstedt, EJ, et al. Genotype-phenotype relations for the Parkinson’s disease genes Parkin, PINK1, DJ1: MDSGene systematic REVIEW: MDSGene REVIEW: Parkin, PINK1, DJ1. Mov Disord. (2018) 33:730–41. doi: 10.1002/mds.27352
18. Trinh, J , Zeldenrust, FMJ , Huang, J , Kasten, M , Schaake, S , Petkovic, S, et al. Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review: MDSGene systematic review: SNCA, LRRK2, VPS35. Mov Disord. (2018) 33:1857–70. doi: 10.1002/mds.27527
19. Perandones, C , Giugni, JC , Calvo, DS , Raina, GB , De Jorge, LL , Volpini, V, et al. Mosaicism of alpha-synuclein gene rearrangements: report of two unrelated cases of early-onset parkinsonism. Parkinsonism Relat Disord. (2014) 20:558–61. doi: 10.1016/j.parkreldis.2013.11.014
20. Rogaeva, E , Johnson, J , Lang, AE , Gulick, C , Gwinn-Hardy, K , Kawarai, T, et al. Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch Neurol [Internet]. (2004) 61:1898–904. doi: 10.1001/archneur.61.12.1898
21. Gatto, EM , Parisi, V , Converso, DP , Poderoso, JJ , Carreras, MC , Martí-Massó, JF, et al. The LRRK2 G2019S mutation in a series of Argentinean patients with Parkinson’s disease: clinical and demographic characteristics. Neurosci Lett. (2013) 537:1–5. doi: 10.1016/j.neulet.2013.01.011
22. Tricco, AC , Lillie, E , Zarin, W , O’Brien, KK , Colquhoun, H , Levac, D, et al. PRISMA extension for scoping reviews (PRISMA-ScR): checklist and explanation. Ann Intern Med. (2018) 169:467–73. doi: 10.7326/M18-0850
23. Domingo, A , and Klein, C . Genetics of Parkinson disease. Handb Clin Neurol. (2018):211–27. doi: 10.1016/B978-0-444-63233-3.00014-2
24. Cook, L , Schulze, J , Verbrugge, J , Beck, JC , Marder, KS , Saunders-Pullman, R, et al. The commercial genetic testing landscape for Parkinson’s disease. Parkinsonism Relat Disord. (2021) 92:107–11. doi: 10.1016/j.parkreldis.2021.10.001
25. Jia, F , Fellner, A , and Kumar, KR . Monogenic Parkinson’s disease: genotype, phenotype, pathophysiology, and genetic testing. Genes. (2022) 13:471. doi: 10.3390/genes13030471
26. Zimprich, A , Benet-Pagès, A , Struhal, W , Graf, E , Eck, SH , Offman, MN, et al. A mutation in VPS35, encoding a subunit of the Retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. (2011) 89:168–75. doi: 10.1016/j.ajhg.2011.06.008
27. Zhang, Y , Shu, L , Sun, Q , Zhou, X , Pan, H , Guo, J, et al. Integrated genetic analysis of racial differences of common GBA variants in Parkinson’s disease: a meta-analysis. Front Mol Neurosci. (2018) 11:43. doi: 10.3389/fnmol.2018.00043
28. Appel-Cresswell, S , Vilarino-Guell, C , Encarnacion, M , Sherman, H , Yu, I , Shah, B, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease: α- Synuclein p.H50q, a novel mutation for Pd. Mov Disord. (2013) 28:811–3. doi: 10.1002/mds.25421
29. Lesage, S , Anheim, M , Letournel, F , Bousset, L , Honoré, A , Rozas, N, et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome: SNCA G51D in parkinsonism. Ann Neurol. (2013) 73:459–71. doi: 10.1002/ana.23894
30. Byers, B , Cord, B , Nguyen, HN , Schüle, B , Fenno, L , Lee, PC, et al. SNCA triplication Parkinson’s Patient’s iPSC-derived DA neurons accumulate α-Synuclein and are susceptible to oxidative stress. PLoS One. (2011) 6:e26159 doi: 10.1371/journal.pone.0026159
31. Capecci, M , Passamonti, L , Annesi, F , Annesi, G , Bellesi, M , Candiano, ICC, et al. Chronic bilateral subthalamic deep brain stimulation in a patient with homozygous deletion in the Parkin gene. Mov Disord. (2004) 19:1450–2. doi: 10.1002/mds.20250
32. Khan, NL . Progression of nigrostriatal dysfunction in a parkin kindred: an [18F]dopa PET and clinical study. Brain. (2002) 125:2248–56. doi: 10.1093/brain/awf237
33. Dogu, O , Johnson, J , Hernandez, D , Hanson, M , Hardy, J , Apaydin, H, et al. A consanguineous Turkish family with early-onset Parkinson’s disease and an exon 4 parkin deletion: a Turkish Parkin family. Mov Disord. (2004) 19:812–6. doi: 10.1002/mds.20028
34. Djarmati, A , Hedrich, K , Svetel, M , Schäfer, N , Juric, V , Vukosavic, S, et al. Detection of Parkin (PARK2) and DJ1 (PARK7) mutations in early-onset Parkinson disease: Parkin mutation frequency depends on ethnic origin of patients: MUTATION IN BRIEF. Hum Mutat. (2004) 23:525–5. doi: 10.1002/humu.9240
35. Ohsawa, Y , Kurokawa, K , Sonoo, M , Yamada, H , Hemmi, S , Iwatsuki, K, et al. Reduced amplitude of the sural nerve sensory action potential in PARK2 patients. Neurology. (2005) 65:459–62. doi: 10.1212/01.wnl.0000171859.85078.3d
36. Khan, NL , Graham, E , Critchley, P , Schrag, AE , Wood, NW , Lees, AJ, et al. Parkin disease: a phenotypic study of a large case series. Brain. (2003) 126:1279–92. doi: 10.1093/brain/awg142
37. Nisipeanu, P , Inzelberg, R , Abo Mouch, S , Carasso, RL , Blumen, SC , Zhang, J, et al. Parkin gene causing benign autosomal recessive juvenile parkinsonism. Neurology. (2001) 56:1573–5. doi: 10.1212/WNL.56.11.1573
38. Bouhouche, A , Tesson, C , Regragui, W , Rahmani, M , Drouet, V , Tibar, H, et al. Mutation analysis of consanguineous Moroccan patients with Parkinson’s disease combining microarray and gene panel. Front Neurol. (2017) 8:567. doi: 10.3389/fneur.2017.00567
39. Shyu, WC , Lin, SZ , Chiang, MF , Pang, CY , Chen, SY , Hsin, YL, et al. Early-onset Parkinson’s disease in a Chinese population: 99mTc-TRODAT-1 SPECT, Parkin gene analysis and clinical study. Parkinsonism Relat Disord. (2005) 11:173–80. doi: 10.1016/j.parkreldis.2004.12.004
40. Monroy-Jaramillo, N , Guerrero-Camacho, JL , Rodríguez-Violante, M , Boll-Woehrlen, MC , Yescas-Gómez, P , Alonso-Vilatela, ME, et al. Genetic mutations in early-onset Parkinson’s disease Mexican patients: molecular testing implications. Am J Med Genet B Neuropsychiatr Genet. (2014) 165:235–44. doi: 10.1002/ajmg.b.32228
41. Lesage, S , Magali, P , Lohmann, E , Lacomblez, L , Teive, H , Janin, S, et al. Deletion of the parkin and PACRG gene promoter in early-onset parkinsonism. Hum Mutat. (2007) 28:27–32. doi: 10.1002/humu.20436
42. Doherty, KM , Silveira-Moriyama, L , Parkkinen, L , Healy, DG , Farrell, M , Mencacci, NE, et al. Parkin disease: a Clinicopathologic entity? JAMA Neurol. (2013) 70:571–9. doi: 10.1001/jamaneurol.2013.172
43. Gierthmühlen, J , Schumacher, S , Deuschl, G , Fritzer, E , Klein, C , Baron, R, et al. Somatosensory function in asymptomatic Parkin- mutation carriers: sensory symptoms and Parkinson’s disease. Eur J Neurol. (2010) 17:513–7. doi: 10.1111/j.1468-1331.2009.02797.x
44. Koziorowski, D , Hoffman-Zacharska, D , Sławek, J , Jamrozik, Z , Janik, P , Potulska-Chromik, A, et al. Incidence of mutations in the PARK2, PINK1, PARK7 genes in polish early-onset Parkinson disease patients. Neurol Neurochir Pol. (2013) 47:319–24. doi: 10.5114/ninp.2013.36756
45. Hassin-Baer, S , Hattori, N , Cohen, OS , Massarwa, M , Israeli-Korn, SD , and Inzelberg, R . Phenotype of the 202 adenine deletion in the parkin gene: 40 years of follow-up: phenotype of the 202A deletion in the parkin gene. Mov Disord. (2011) 26:719–22. doi: 10.1002/mds.23456
46. Elia, AE , Del Sorbo, F , Romito, LM , Barzaghi, C , Garavaglia, B , and Albanese, A . Isolated limb dystonia as presenting feature of Parkin disease. J Neurol Neurosurg Psychiatry. (2014) 85:827–8. doi: 10.1136/jnnp-2013-307294
47. Zadikoff, C , Rogaeva, E , Djarmati, A , Sato, C , Salehi-Rad, S , St. George-Hyslop, P, et al. Homozygous and heterozygous PINK1 mutations: considerations for diagnosis and care of Parkinson’s disease patients. Mov Disord. (2006) 21:875–9. doi: 10.1002/mds.20854
48. Norman, BP , Lubbe, SJ , Tan, M , Warren, N , and Morris, HR . Early onset Parkinson’s disease in a family of Moroccan origin caused by a p.A217D mutation in PINK1: a case report. BMC Neurol. (2017) 17:153. doi: 10.1186/s12883-017-0933-z
49. Biswas, A , Sadhukhan, T , Majumder, S , Misra, AK , Das, SK, Variation Consortium IG, et al. Evaluation of PINK1 variants in Indian Parkinson’s disease patients. Parkinsonism Relat Disord. (2010) 16:167–71. doi: 10.1016/j.parkreldis.2009.10.005
50. Kilarski, LL , Pearson, JP , Newsway, V , Majounie, E , Knipe, MDW , Misbahuddin, A, et al. Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson’s disease: PARK2, PINK1, PARK7, LRRK2 in EOPD. Mov Disord. (2012) 27:1522–9. doi: 10.1002/mds.25132
51. Tan, EK , Yew, K , Chua, E , Puvan, K , Shen, H , Lee, E, et al. PINK1 mutations in sporadic early-onset Parkinson’s disease. Mov Disord. (2006) 21:789–93. doi: 10.1002/mds.20810
52. Bouhouche, A , Tibar, H , Ben El Haj, R , El Bayad, K , Razine, R , Tazrout, S, et al. LRRK2 G2019S mutation: prevalence and clinical features in Moroccans with Parkinson’s disease. Park Dis. (2017) 2017:1–7. doi: 10.1155/2017/2412486
53. Djarmati, A , Hedrich, K , Svetel, M , Lohnau, T , Schwinger, E , Romac, S, et al. HeterozygousPINK1 mutations: a susceptibility factor for Parkinson disease? Mov Disord. (2006) 21:1526–30. doi: 10.1002/mds.20977
54. Ibáñez, P , Lesage, S , Lohmann, E , Thobois, S , Michele, GD , Borg, M, et al. Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain. (2006) 129:686–94. doi: 10.1093/brain/awl005
55. Gierthmuhlen, J , Lienau, F , Maag, R , Hagenah, JM , Deuschl, G , Fritzer, E, et al. Somatosensory processing in a German family with PINK1 mutations: its potential role in Parkinson disease. J Neurol Neurosurg Psychiatry. (2009) 80:571–4. doi: 10.1136/jnnp.2008.158659
56. Puschmann, A , Englund, E , Ross, OA , Vilariño-Güell, C , Lincoln, SJ , Kachergus, JM, et al. First neuropathological description of a patient with Parkinson’s disease and LRRK2 p.N1437H mutation. Parkinsonism Relat Disord. (2012) 18:332–8. doi: 10.1016/j.parkreldis.2011.11.019
57. Bras, JM , Guerreiro, RJ , Ribeiro, MH , Januario, C , Morgadinho, A , Oliveira, CR, et al. G2019S dardarin substitution is a common cause of Parkinson’s disease in a Portuguese cohort. Mov Disord. (2005) 20:1653–5. doi: 10.1002/mds.20682
58. Gosal, D , Ross, OA , Wiley, J , Irvine, GB , Johnston, JA , Toft, M, et al. Clinical traits of LRRK2-associated Parkinson’s disease in Ireland: a link between familial and idiopathic PD. Parkinsonism Relat Disord. (2005) 11:349–52. doi: 10.1016/j.parkreldis.2005.05.004
59. Hatano, T , Funayama, M , Kubo, S , Ichiro, MIF , Oji, Y , Mori, A, et al. Identification of a Japanese family with LRRK2 p.R1441G-related Parkinson’s disease. Neurobiol Aging. (2014) 35:2656.e17–23. doi: 10.1016/j.neurobiolaging.2014.05.025
60. Khlebtovsky, A , Roditi, Y , and Djaldetti, R . Comparison of thermal sensation and pain thresholds in LRRK2 carriers and non carriers with Parkinson’s disease. (2023) Available at: https://www.mdsabstracts.org/abstract/comparison-of-thermal-sensation-and-pain-thresholds-in-lrrk2-carriers-and-non-carriers-with-parkinsons-disease/
61. Hedrich, K , Winkler, S , Hagenah, J , Kabakci, K , Kasten, M , Schwinger, E, et al. RecurrentLRRK2 (Park8) mutations in early-onset Parkinson’s disease. Mov Disord. (2006) 21:1506–10. doi: 10.1002/mds.20990
62. Criscuolo, C , De Rosa, A , Guacci, A , Simons, EJ , Breedveld, GJ , Peluso, S, et al. The LRRK2 R1441C mutation is more frequent than G2019S in Parkinson’s disease patients from southern Italy: LRRK2 R1441C mutation more frequent in southern Italy. Mov Disord. (2011) 26:1732–6. doi: 10.1002/mds.23735
63. Li, DW , Gu, Z , Wang, C , Ma, J , Tang, BS , Chen, SD, et al. Non-motor symptoms in Chinese Parkinson’s disease patients with and without LRRK2 G2385R and R1628P variants. J Neural Transm. (2015) 122:661–7. doi: 10.1007/s00702-014-1281-4
64. Healy, DG , Falchi, M , O’Sullivan, SS , Bonifati, V , Durr, A , Bressman, S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. (2008) 7:583–90. doi: 10.1016/S1474-4422(08)70117-0
65. Baig, F , Lawton, M , Rolinski, M , Ruffmann, C , Nithi, K , Evetts, SG, et al. Delineating nonmotor symptoms in early Parkinson’s disease and first-degree relatives. Mov Disord. (2015) 30:1759–66. doi: 10.1002/mds.26281
66. An, XK , Peng, R , Li, T , Burgunder, JM , Wu, Y , Chen, WJ, et al. LRRK2 Gly2385Arg variant is a risk factor of Parkinson’s disease among Han-Chinese from mainland China. Eur J Neurol. (2008) 15:301–5. doi: 10.1111/j.1468-1331.2007.02052.x
67. Wang, C , Cai, Y , Gu, Z , Ma, J , Zheng, Z , Tang, BS, et al. Clinical profiles of Parkinson’s disease associated with common leucine-rich repeat kinase 2 and glucocerebrosidase genetic variants in Chinese individuals. Neurobiol Aging. (2014) 35:725.e1–6. doi: 10.1016/j.neurobiolaging.2013.08.012
68. Zhang, Z , Burgunder, JM , An, X , Wu, Y , Chen, W , Zhang, J, et al. LRRK2 R1628P variant is a risk factor of Parkinson’s disease among Han-Chinese from mainland China: LRRK2 R1628P variant. Mov Disord. (2009) 24:1902–5. doi: 10.1002/mds.22371
69. Luciano, MS , Lipton, RB , Wang, C , Katz, M , Zimmerman, ME , Sanders, AE, et al. Clinical expression of LRRK2 G2019S mutations in the elderly: clinical LRRK2 G2019S expression in the elderly. Mov Disord. (2010) 25:2571–6. doi: 10.1002/mds.23330
70. Martino, D , Melzi, V , Franco, G , Kandasamy, N , Monfrini, E , and Di Fonzo, A . Juvenile dystonia-parkinsonism syndrome caused by a novel p.S941Tfs1X ATP13A2 (PARK9) mutation. Parkinsonism Relat Disord. (2015) 21:1378–80. doi: 10.1016/j.parkreldis.2015.09.036
71. Bonner, N , Bozzi, S , Morgan, L , Mason, B , Peterschmitt, MJ , Fischer, TZ, et al. Patients’ experiences of Parkinson’s disease: a qualitative study in glucocerebrosidase and idiopathic Parkinson’s disease. J Patient Rep Outcomes. (2020) 4:65. doi: 10.1186/s41687-020-00230-9
72. Rodriguez-Porcel, F , Espay, AJ , and Carecchio, M . Parkinson disease in Gaucher disease. J Clin Mov Disord. (2017) 4:7. doi: 10.1186/s40734-017-0054-2
73. Kresojević, N , Janković, M , Petrović, I , Kumar, KR , Dragašević, N , Dobričić, V, et al. Presenting symptoms of GBA-related Parkinson’s disease. Parkinsonism Relat Disord. (2015) 21:804–7. doi: 10.1016/j.parkreldis.2015.04.028
74. Jesús, S , Huertas, I , Bernal-Bernal, I , Bonilla-Toribio, M , Cáceres-Redondo, MT , Vargas-González, L, et al. GBA variants influence motor and non-motor features of Parkinson’s disease. PLoS One. (2016) 11:e0167749 doi: 10.1371/journal.pone.0167749
75. McNeill, A , Duran, R , Hughes, DA , Mehta, A , and Schapira, AHV . A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers. J Neurol Neurosurg Psychiatry. (2012) 83:853–4. doi: 10.1136/jnnp-2012-302402
76. Neumann, J , Bras, J , Deas, E , O’Sullivan, SS , Parkkinen, L , Lachmann, RH, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain. (2009) 132:1783–94. doi: 10.1093/brain/awp044
77. Deng, H , Wang, P , and Jankovic, J . The genetics of Parkinson disease. Ageing Res Rev. (2018) 42:72–85. doi: 10.1016/j.arr.2017.12.007
78. Marras, C , Lang, A , van de Warrenburg, BP , Sue, CM , Tabrizi, SJ , Bertram, L, et al. Nomenclature of genetic movement disorders: recommendations of the international Parkinson and movement disorder society task force: nomenclature of genetic movement disorders. Mov Disord. (2016) 31:436–57. doi: 10.1002/mds.26527
79. Klein, C , and Westenberger, A . Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med. (2012) 2:a008888–8. doi: 10.1101/cshperspect.a008888
80. Li, B , Zhao, G , Zhou, Q , Xie, Y , Wang, Z , Fang, Z, et al. Gene4PD: a comprehensive genetic database of Parkinson’s disease. Front Neurosci. (2021) 15:679568. doi: 10.3389/fnins.2021.679568
81. Elwan, M , Schaefer, AM , Craig, K , Hopton, S , Falkous, G , Blakely, EL, et al. Changing faces of mitochondrial disease: autosomal recessive POLG disease mimicking myasthenia gravis and progressive supranuclear palsy. BMJ Neurol Open. (2022) 4:e000352. doi: 10.1136/bmjno-2022-000352
82. Bilguvar, K , Tyagi, NK , Ozkara, C , Tuysuz, B , Bakircioglu, M , Choi, M, et al. Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci. (2013) 110:3489–94. doi: 10.1073/pnas.1222732110
83. Selvaraj, S , and Piramanayagam, S . Impact of gene mutation in the development of Parkinson’s disease. Genes Dis. (2019) 6:120–8. doi: 10.1016/j.gendis.2019.01.004
84. Vivacqua, G , Yin, JJ , Casini, A , Li, X , Li, YH , D’Este, L, et al. Immunolocalization of alpha-synuclein in the rat spinal cord by two novel monoclonal antibodies. Neuroscience. (2009) 158:1478–87. doi: 10.1016/j.neuroscience.2008.12.001
85. Valek, L , Tran, B , Wilken-Schmitz, A , Trautmann, S , Heidler, J , Schmid, T, et al. Prodromal sensory neuropathy in Pink1 −/− SNCA A53T double mutant Parkinson mice. Neuropathol Appl Neurobiol. (2021) 47:1060–79. doi: 10.1111/nan.12734
86. Johnson, RA , Kelm-Nelson, CA , and Ciucci, MR . Changes to ventilation, vocalization, and thermal nociception in the Pink1−/− rat model of Parkinson’s disease. J Parkinsons Dis. (2020) 10:489–504. doi: 10.3233/JPD-191853
87. Yi, MH , Shin, J , Shin, N , Yin, Y , Lee, SY , Kim, CS, et al. PINK1 mediates spinal cord mitophagy in neuropathic pain. J Pain Res. (2019) 12:1685–99. doi: 10.2147/JPR.S198730
88. Bichler, Z , Lim, HC , Zeng, L , and Tan, EK . Non-motor and motor features in LRRK2 transgenic mice. PLoS One. (2013) 8:e70249 doi: 10.1371/journal.pone.0070249
89. Valek, L , Auburger, G , and Tegeder, I . Sensory neuropathy and nociception in rodent models of Parkinson’s disease. Dis Model Mech. (2019) 12. doi: 10.1242/dmm.039396
90. Blauwendraat, C , Makarious, MB , Leonard, HL , Bandres-Ciga, S , Iwaki, H , Nalls, MA, et al. A population scale analysis of rare SNCA variation in the UK biobank. Neurobiol Dis. (2021) 148:105182. doi: 10.1016/j.nbd.2020.105182
91. Giasson, BI , Duda, JE , Murray, IVJ , Chen, Q , Souza, JM , Hurtig, HI, et al. Oxidative damage linked to neurodegeneration by selective α-Synuclein nitration in Synucleinopathy lesions. Science. (2000) 290:985–9. doi: 10.1126/science.290.5493.985
92. Lotharius, J . Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson’s disease. Hum Mol Genet. (2002) 11:2395–407. doi: 10.1093/hmg/11.20.2395
93. Lesage, S , and Brice, A . Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. (2009) 18:R48–59. doi: 10.1093/hmg/ddp012
94. Hoffman-Zacharska, D , Koziorowski, D , Ross, OA , Milewski, M , Poznański, J , Jurek, M, et al. Novel A18T and pA29S substitutions in α-synuclein may be associated with sporadic Parkinson’s disease. Parkinsonism Relat Disord. (2013) 19:1057–60. doi: 10.1016/j.parkreldis.2013.07.011
95. Kiely, AP , Asi, YT , Kara, E , Limousin, P , Ling, H , Lewis, P, et al. α-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol (Berl). (2013) 125:753–69. doi: 10.1007/s00401-013-1096-7
96. Krüger, R , Kuhn, W , Müller, T , Woitalla, D , Graeber, M , Kösel, S, et al. AlaSOPro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet. (1998) 18:106–8. doi: 10.1038/ng0298-106
97. Pasanen, P , Myllykangas, L , Siitonen, M , Raunio, A , Kaakkola, S , Lyytinen, J, et al. A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging. (2014) 35:2180.e1–5. doi: 10.1016/j.neurobiolaging.2014.03.024
98. Polymeropoulos, MH , Lavedan, C , Leroy, E , Ide, SE , Dehejia, A , Dutra, A, et al. Mutation in the α-Synuclein gene identified in families with Parkinson’s disease. Science. (1997) 276:2045–7. doi: 10.1126/science.276.5321.2045
99. Proukakis, C , Dudzik, CG , Brier, T , MacKay, DS , Cooper, JM , Millhauser, GL, et al. A novel -synuclein missense mutation in Parkinson disease. Neurology. (2013) 80:1062–4. doi: 10.1212/WNL.0b013e31828727ba
100. Zarranz, JJ , Alegre, J , Gómez-Esteban, JC , Lezcano, E , Ros, R , Ampuero, I, et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia: new α-Synuclein gene mutation. Ann Neurol. (2004) 55:164–73. doi: 10.1002/ana.10795
101. Kasten, M , and Klein, C . The many faces of alpha-synuclein mutations: the many faces of alpha-Synuclein mutations. Mov Disord. (2013) 28:697–701. doi: 10.1002/mds.25499
102. Klein, C , and Lohmann, K . Parkinson disease(s): is “Parkin disease” a distinct clinical entity? Neurology. (2009) 72:106–7. doi: 10.1212/01.wnl.0000333666.65522.8d
103. Klein, C , and Schlossmacher, MG . Parkinson disease, 10 years after its genetic revolution: multiple clues to a complex disorder. Neurology. (2007) 69:2093–104. doi: 10.1212/01.wnl.0000271880.27321.a7
104. Castelo Rueda, MP , Raftopoulou, A , Gögele, M , Borsche, M , Emmert, D , Fuchsberger, C, et al. Frequency of heterozygous Parkin (PRKN) variants and penetrance of Parkinson’s disease risk markers in the population-based CHRIS cohort. Front Neurol. (2021) 12:706145. doi: 10.3389/fneur.2021.706145
105. Zhu, W , Huang, X , Yoon, E , Bandres-Ciga, S , Blauwendraat, C , Cade, JH, et al. Heterozygous PRKN mutations are common but do not increase the risk of Parkinson’s disease [internet]. Neurology. (2021). doi: 10.1101/2021.08.11.21261928
106. Kitada, T , Asakawa, S , Hattori, N , Matsumine, H , Yamamura, Y , Minoshima, S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. (1998) 392:605–8. doi: 10.1038/33416
107. Klein, C , Schumacher, K , Jacobs, H , Hagenah, J , Kis, B , Garrels, J, et al. Association studies of Parkinson’s disease andparkin polymorphisms. Ann Neurol. (2000) 48:126–7. doi: 10.1002/1531-8249(200007)48:1<126::AID-ANA22>3.0.CO;2-K
108. Hedrich, K , Eskelson, C , Wilmot, B , Marder, K , Harris, J , Garrels, J, et al. Distribution, type, and origin ofParkin mutations: review and case studies. Mov Disord. (2004) 19:1146–57. doi: 10.1002/mds.20234
109. Lesage, S , Lunati, A , Houot, M , Romdhan, SB , Clot, F , Tesson, C, et al. Characterization of recessive Parkinson disease in a large multicenter study. Ann Neurol. (2020) 88:843–50. doi: 10.1002/ana.25787
110. Dawson, TM , and Dawson, VL . The role of parkin in familial and sporadic Parkinson’s disease: the role of Parkin in familial and sporadic PD. Mov Disord. (2010) 25:S32–9. doi: 10.1002/mds.22798
111. Ishikawa, A , and Tsuji, S . Clinical analysis of 17 patients in 12 Japanese families with autosomal-recessive type juvenile parkinsonism. Neurology. (1996) 47:160–6. doi: 10.1212/WNL.47.1.160
112. Grünewald, A , Kasten, M , Ziegler, A , and Klein, C . Next-generation phenotyping using the Parkin example: time to catch up with genetics. JAMA Neurol. (2013) 70:1186. doi: 10.1001/jamaneurol.2013.488
113. Valente, EM , Abou-Sleiman, PM , Caputo, V , Muqit, MMK , Harvey, K , Gispert, S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. (2004) 304:1158–60. doi: 10.1126/science.1096284
114. Petit, A , Kawarai, T , Paitel, E , Sanjo, N , Maj, M , Scheid, M, et al. Wild-type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J Biol Chem. (2005) 280:34025–32. doi: 10.1074/jbc.M505143200
115. Bonifati, V , Rohe, CF , Breedveld, GJ , Fabrizio, E , De Mari, M , Tassorelli, C, et al. Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology. (2005) 65:87–95. doi: 10.1212/01.wnl.0000167546.39375.82
116. Paisán Ruíz, C , Jain, S , Evans, EW , Gilks, WP , Simón, J , van der Brug, M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. (2004) 44:595–600. doi: 10.1016/j.neuron.2004.10.023
117. Rui, Q , Ni, H , Li, D , Gao, R , and Chen, G . The role of LRRK2 in neurodegeneration of Parkinson disease. Curr Neuropharmacol. (2018) 16:1348–57. doi: 10.2174/1570159X16666180222165418
118. Nuytemans, K , Theuns, J , Cruts, M , and Van Broeckhoven, C . Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat. (2010) 31:763–80. doi: 10.1002/humu.21277
119. Gehrke, S , Imai, Y , Sokol, N , and Lu, B . Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. (2010) 466:637–41. doi: 10.1038/nature09191
120. Iwaki, H , Blauwendraat, C , Makarious, MB , Bandrés-Ciga, S , Leonard, HL , Gibbs, JR, et al. Penetrance of Parkinson’s disease in LRRK2 p.G2019S carriers is modified by a polygenic risk score. Mov Disord. (2020 May) 35:774–80. doi: 10.1002/mds.27974
121. Correia Guedes, L , Ferreira, JJ , Rosa, MM , Coelho, M , Bonifati, V , and Sampaio, C . Worldwide frequency of G2019S LRRK2 mutation in Parkinson’s disease: a systematic review. Parkinsonism Relat Disord. (2010) 16:237–42. doi: 10.1016/j.parkreldis.2009.11.004
122. Deng, H , Le, W , Guo, Y , Hunter, CB , Xie, W , and Jankovic, J . Genetic and clinical identification of Parkinson’s disease patients withLRRK2 G2019S mutation. Ann Neurol. (2005) 57:933–4. doi: 10.1002/ana.20510
123. Alcalay, RN , Mejia-Santana, H , Mirelman, A , Saunders-Pullman, R , Raymond, D , Palmese, C, et al. Neuropsychological performance in LRRK2 G2019S carriers with Parkinson’s disease. Parkinsonism Relat Disord. (2015) 21:106–10. doi: 10.1016/j.parkreldis.2014.09.033
124. Pankratz, N , Pauciulo, MW , Elsaesser, VE , Marek, DK , Halter, CA , Wojcieszek, J, et al. Mutations in DJ-1 are rare in familial Parkinson disease. Neurosci Lett. (2006) 408:209–13. doi: 10.1016/j.neulet.2006.09.003
125. Bonifati, V , Rizzu, P , van Baren, MJ , Schaap, O , Breedveld, GJ , Krieger, E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. (2003) 299:256–9. doi: 10.1126/science.1077209
126. Meulener, MC , Xu, K , Thomson, L , Ischiropoulos, H , and Bonini, NM . Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc Natl Acad Sci. (2006) 103:12517–22. doi: 10.1073/pnas.0601891103
127. Anderson, PC , and Daggett, V . Molecular basis for the structural instability of human DJ-1 induced by the L166P mutation associated with Parkinson’s disease. Biochemistry. (2008) 47:9380–93. doi: 10.1021/bi800677k
128. Malgieri, G , and Eliezer, D . Structural effects of Parkinson’s disease linked DJ-1 mutations. Protein Sci. (2008) 17:855–68. doi: 10.1110/ps.073411608
129. Weissbach, A , Wittke, C , Kasten, M , and Klein, C . ‘Atypical’ Parkinson’s disease – genetic. (2019) 207–235. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0074774219300972
130. Vilariño-Güell, C , Wider, C , Ross, OA , Dachsel, JC , Kachergus, JM , Lincoln, SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. (2011) 89:162–7. doi: 10.1016/j.ajhg.2011.06.001
131. Ando, M , Funayama, M , Li, Y , Kashihara, K , Murakami, Y , Ishizu, N, et al. VPS35 mutation in Japanese patients with typical Parkinson’s disease: VPS35 mutation in Japanese PD. Mov Disord. (2012) 27:1413–7. doi: 10.1002/mds.25145
132. Sharma, M , Ioannidis, JPA , Aasly, JO , Annesi, G , Brice, A , Bertram, L, et al. A multi-Centre clinico-genetic analysis of the VPS35 gene in Parkinson disease indicates reduced penetrance for disease-associated variants. J Med Genet. (2012) 49:721–6. doi: 10.1136/jmedgenet-2012-101155
133. Cerri, S , Ghezzi, C , Ongari, G , Croce, S , Avenali, M , Zangaglia, R, et al. GBA mutations influence the release and pathological effects of small extracellular vesicles from fibroblasts of patients with Parkinson’s disease. Int J Mol Sci. (2021) 22:2215. doi: 10.3390/ijms22042215
134. Zhao, F , Bi, L , Wang, W , Wu, X , Li, Y , Gong, F, et al. Mutations of glucocerebrosidase gene and susceptibility to Parkinson’s disease: An updated meta-analysis in a European population. Neuroscience. (2016) 320:239–46. doi: 10.1016/j.neuroscience.2016.02.007
135. Beavan, M , McNeill, A , Proukakis, C , Hughes, DA , Mehta, A , and Schapira, AHV . Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation–positive cohort. JAMA Neurol. (2015) 72:201–8. doi: 10.1001/jamaneurol.2014.2950
136. Sidransky, E , and Lopez, G . The link between the GBA gene and parkinsonism. Lancet Neurol. (2012) 11:986–98. doi: 10.1016/S1474-4422(12)70190-4
137. Granek, Z , Barczuk, J , Siwecka, N , Rozpędek-Kamińska, W , Kucharska, E , and Majsterek, I . GBA1 gene mutations in α-Synucleinopathies—molecular mechanisms underlying pathology and their clinical significance. Int J Mol Sci. (2023) 24:2044. doi: 10.3390/ijms24032044
138. Avenali, M , Blandini, F , and Cerri, S . Glucocerebrosidase defects as a major risk factor for Parkinson’s disease. Front Aging Neurosci. (2020) 12:97. doi: 10.3389/fnagi.2020.00097
139. Velez-Pardo, C , Lorenzo-Betancor, O , Jimenez-Del-Rio, M , Moreno, S , Lopera, F , Cornejo-Olivas, M, et al. The distribution and risk effect of GBA variants in a large cohort of PD patients from Colombia and Peru. Parkinsonism Relat Disord. (2019) 63:204–8. doi: 10.1016/j.parkreldis.2019.01.030
140. Ryan, E , Seehra, G , Sharma, P , and Sidransky, E . GBA1-associated parkinsonism: new insights and therapeutic opportunities. Curr Opin Neurol. (2019) 32:589–96. doi: 10.1097/WCO.0000000000000715
141. Riboldi, GM , and Di Fonzo, AB . GBA, Gaucher disease, and Parkinson’s disease: from genetic to clinic to new therapeutic approaches. Cells. (2019) 8:364. doi: 10.3390/cells8040364
142. Tayebi, N . Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab. (2003) 79:104–9. doi: 10.1016/S1096-7192(03)00071-4
143. Lill, CM . Genetics of Parkinson’s disease. Mol Cell Probes. (2016) 30:386–96. doi: 10.1016/j.mcp.2016.11.001
144. Ramirez, A , Heimbach, A , Gründemann, J , Stiller, B , Hampshire, D , Cid, LP, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. (2006) 38:1184–91. doi: 10.1038/ng1884
145. Tan, J , Zhang, T , Jiang, L , Chi, J , Hu, D , Pan, Q, et al. Regulation of intracellular manganese homeostasis by Kufor-Rakeb syndrome-associated ATP13A2 protein. J Biol Chem. (2011) 286:29654–62. doi: 10.1074/jbc.M111.233874
146. Park, JS , Blair, NF , and Sue, CM . The role of ATP13A2 in Parkinson’s disease: clinical phenotypes and molecular mechanisms: ATP13A2 in Parkinson’s disease. Mov Disord. (2015) 30:770–9. doi: 10.1002/mds.26243
147. Vidyadhara, DJ , Somayaji, M , Wade, N , Yücel, B , Zhao, H , Shashaank, N, et al. Dopamine transporter and synaptic vesicle sorting defects underlie auxilin-associated Parkinson’s disease. Cell Rep. (2023) 42:112231. doi: 10.1016/j.celrep.2023.112231
148. Yim, YI , Sun, T , Wu, LG , Raimondi, A , De Camilli, P , Eisenberg, E, et al. Endocytosis and clathrin-uncoating defects at synapses of auxilin knockout mice. Proc Natl Acad Sci. (2010) 107:4412–7. doi: 10.1073/pnas.1000738107
149. Edvardson, S , Cinnamon, Y , Ta-Shma, A , Shaag, A , Yim, YI , Zenvirt, S, et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific Clathrin-Uncoating co-chaperone Auxilin, is associated with juvenile parkinsonism. PLoS One. (2012) 7:e36458 doi: 10.1371/journal.pone.0036458
150. Köroğlu, Ç , Baysal, L , Cetinkaya, M , Karasoy, H , and Tolun, A . DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat Disord. (2013) 19:320–4. doi: 10.1016/j.parkreldis.2012.11.006
151. Olgiati, S , Quadri, M , Fang, M , Rood, JPMA , Saute, JA , Chien, HF, et al. D NAJC 6 mutations associated with early-onset Parkinson’s disease: DNAJC6 mutations in Parkinson’s disease. Ann Neurol. (2016) 79:244–56. doi: 10.1002/ana.24553
152. Elsayed, LEO , Drouet, V , Usenko, T , Mohammed, IN , Hamed, AAA , Elseed, MA, et al. A novel nonsense mutation in DNAJC 6 expands the phenotype of autosomal-recessive juvenile-onset Parkinson’s disease. Ann Neurol. (2016) 79:335–7. doi: 10.1002/ana.24591
153. Ng, J , Cortès-Saladelafont, E , Abela, L , Termsarasab, P , Mankad, K , Sudhakar, S, et al. DNAJC6 mutations disrupt dopamine homeostasis in juvenile PARKINSONISM-DYSTONIA. Mov Disord. (2020) 35:1357–68. doi: 10.1002/mds.28063
154. Kipreos, ET , and Pagano, M . The F-box protein family. Genome Biol. (2000) 1:REVIEWS3002. doi: 10.1186/gb-2000-1-5-reviews3002
155. Wei, L , Ding, L , Li, H , Lin, Y , Dai, Y , Xu, X, et al. Juvenile-onset parkinsonism with pyramidal signs due to compound heterozygous mutations in the F-box only protein 7 gene. Parkinsonism Relat Disord. (2018) 47:76–9. doi: 10.1016/j.parkreldis.2017.11.332
156. Shojaee, S , Sina, F , Banihosseini, SS , Kazemi, MH , Kalhor, R , Shahidi, GA, et al. Genome-wide linkage analysis of a parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet. (2008) 82:1375–84. doi: 10.1016/j.ajhg.2008.05.005
157. Fonzo, AD , Dekker, MCJ , Montagna, P , Baruzzi, A , Yonova, EH , Guedes, LC, et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology. (2009) 72:240–5. doi: 10.1212/01.wnl.0000338144.10967.2b
158. Paisán-Ruiz, C , Guevara, R , Federoff, M , Hanagasi, H , Sina, F , Elahi, E, et al. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations: complex recessive Parkinsonisms. Mov Disord. (2010) 25:1791–800. doi: 10.1002/mds.23221
159. Keller Sarmiento, IJ , Afshari, M , Kinsley, L , Silani, V , Akhtar, RS , Simuni, T, et al. Novel bi-allelic FBXO7 variants in a family with early-onset typical Parkinson’s disease. Parkinsonism Relat Disord. (2022) 104:88–90. doi: 10.1016/j.parkreldis.2022.10.014
160. Gündüz, A , Eken, AG , Bilgiç, B , Hanagasi, HA , Bilgüvar, K , Günel, M, et al. FBXO7–R498X mutation: phenotypic variability from chorea to early onset parkinsonism within a family. Parkinsonism Relat Disord. (2014) 20:1253–6. doi: 10.1016/j.parkreldis.2014.07.016
161. Conedera, S , Apaydin, H , Li, Y , Yoshino, H , Ikeda, A , Matsushima, T, et al. FBXO7 mutations in Parkinson’s disease and multiple system atrophy. Neurobiol Aging. (2016) 40:192.e1–5. doi: 10.1016/j.neurobiolaging.2016.01.003
162. Lohmann, E , Coquel, AS , Honoré, A , Gurvit, H , Hanagasi, H , Emre, M, et al. A new F-box protein 7 GENE MUTATION CAUSING TYPICAL Parkinson’s disease: FBXO7 GENE MUTATION CAUSING TYPICAL PD. Mov Disord. (2015) 30:1130–3. doi: 10.1002/mds.26266
163. Lesage, S , Mangone, G , Tesson, C , Bertrand, H , Benmahdjoub, M , Kesraoui, S, et al. Clinical variability of SYNJ1-associated early-onset parkinsonism. Front Neurol. (2021) 12:648457. doi: 10.3389/fneur.2021.648457
164. Quadri, M , Fang, M , Picillo, M , Olgiati, S , Breedveld, GJ , Graafland, J, et al. Mutation in the SYNJ1 gene associated with autosomal recessive. Early-Onset Parkinsonism Hum Mutat. (2013) 34:1208–15. doi: 10.1002/humu.22373
165. Krebs, CE , Karkheiran, S , Powell, JC , Cao, M , Makarov, V , Darvish, H, et al. The Sac1 domain of SYNJ 1 identified mutated in a family with early-onset progressive P arkinsonism with generalized seizures. Hum Mutat. (2013) 34:1200–7. doi: 10.1002/humu.22372
166. Klatt-Schreiner, K , Valek, L , Kang, J , Khlebtovsky, A , Trautmann, S , Hahnefeld, L, et al. High glucosylceramides and low anandamide contribute to sensory loss and pain in Parkinson’s disease. Mov Disord. (2020) 35:1822–33. doi: 10.1002/mds.28186
167. Alcalay, RN , Levy, OA , Waters, CH , Fahn, S , Ford, B , Kuo, SH, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain. (2015) 138:2648–58. doi: 10.1093/brain/awv179
168. Buhidma, Y , Rukavina, K , Chaudhuri, KR , and Duty, S . Potential of animal models for advancing the understanding and treatment of pain in Parkinson’s disease. Npj Park Dis. (2020) 6:1. doi: 10.1038/s41531-019-0104-6
Keywords: Parkinson’s disease, monogenic, genetic, pain, inheritance
Citation: Alizadeh P, Terroba-Chambi C, Achen B and Bruno V (2023) Pain in monogenic Parkinson’s disease: a comprehensive review. Front. Neurol. 14:1248828. doi: 10.3389/fneur.2023.1248828
Edited by:
Cristian Falup-Pecurariu, Transilvania University of Brașov, RomaniaReviewed by:
Konstantin Senkevich, McGill University, CanadaŁukasz Milanowski, Medical University of Warsaw, Poland
Christelle Jacqueline Tesson, INSERM U1127 Institut du Cerveau et de la Moelle épinière (ICM), France
Copyright © 2023 Alizadeh, Terroba-Chambi, Achen, and Bruno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Veronica Bruno dmV1YnJ1QGdtYWlsLmNvbQ==; dmVyb25pY2EuYnJ1bm9AdWNhbGdhcnkuY2E=
†These authors have contributed equally to this work and share first authorship