 Haining Li1,2†
Haining Li1,2† Tingting Xuan
Tingting Xuan Zhenhai Wang
Zhenhai Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 13 September 2023
Sec. Neurogenetics
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1242472
This article is part of the Research Topic Case Reports in Neurogenetics, volume III - 2023 View all 20 articles
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterized by progressive degeneration of upper and lower motor neurons, with occasional involvement of the extrapyramidal system. Mutations in the sigma non-opioid intracellular receptor 1 (SIGMAR1) gene have been identified as one of the causes of ALS. Here, we present a case of a 49-year-old man diagnosed with ALS–Parkinson’s disease (PD) complex. The patient exhibited bradykinesia and tremor, and whole-exome sequencing revealed homozygous mutations in the SIGMAR1 gene (c.446-2A > T). In addition, we conducted an investigation into the clinical and molecular phenotype of previously reported variants of SIGMAR1 associated with ALS. This case report aims to raise awareness among physicians regarding atypical phenotypes of amyotrophic lateral sclerosis and to encourage further research on the factors leading to SIGMAR1 mutations in patients.
Amyotrophic lateral sclerosis (ALS) represents the most common form of motor neuron disease, characterized by the degeneration of upper and lower motor neurons. ALS is classified into two types: familial (fALS) and sporadic (sALS), with the latter accounting for 90–95% of cases, while fALS comprises only 5–10%. The etiology of sALS remains largely unknown although it is believed to involve both genetic and environmental factors. Genetic factors, in particular, play a significant role in the occurrence of sALS. To date, more than a hundred ALS-related genes have been identified, with approximately 30 genes primarily associated with ALS (1). Among Asians, Cu/Zn superoxide dismutase (SOD1) gene mutations are the most prevalent, whereas pathogenic mutations in the sigma non-opioid intracellular receptor 1 (SIGMAR1) gene are rare in Asian patients with familial or sporadic ALS. While mutations in the SIGMAR1 gene have been reported in association with ALS, with or without frontotemporal dementia or juvenile ALS, no instances of this mutation in the ALS–Parkinson’s disease (PD) complex have been described until now. Here, we present the case of a 49-year-old patient with ALS–PD complex, exhibiting slowly progressing motoneuron disease that may be linked to a homozygous SIGMAR1 mutation. Additionally, we conduct a comprehensive review of cases of ALS patients with mutations in this gene, as reported in the relevant scientific literature.
A 49-year-old man presented to our neurology department with complaints of involuntary shaking in both upper limbs for the past 3 years, along with slowness of movement for the past 2 years. He exhibited rest and action tremors in both upper limbs, along with simultaneous occurrence of bradykinesia and rigidity. Subsequently, he experienced unresponsiveness, memory decline, and choking while drinking, and his speaking rate began to slow down. Additionally, his facial expressions started to diminish, as noticed by his wife. In April 2021, the patient received a diagnosis of Parkinsonian syndrome at a local hospital. Initial treatment with levodopa at a daily dosage of 100 mg, gradually increased to 200 mg, resulted in partial improvement in involuntary shaking, but showed no significant overall improvement. Over the next year, his symptoms rapidly worsened, with progressive aggravation of stiffness and the appearance of mental irritability. Neurological examinations revealed decreased spontaneous facial expressions, poor eye movement in all directions, horizontal nystagmus, mildly increased muscle tone in the neck and limbs, and deep tendon reflexes in the biceps and triceps (1+). A positive Babinski sign was observed bilaterally. Symmetric muscle atrophy of the calves was also noted, which he reported experiencing for as long as he could remember (Figure 1A). Additionally, it was noted that he had planovalgus deformities of both feet since the age of 5 years (Figure 1B), a condition similar to that of his uncle’s feet. The strength of his upper and lower extremities, as well as proximal and distal muscles, was assessed as 5 on the Medical Research Council Muscle Scale. Brain magnetic resonance imaging revealed only mild atrophy, and his cognitive functions were deemed normal, scoring 28 on the standardized Mini-Mental State Examination and 23 points on the Montreal Cognitive Assessment. Further cervical and thoracic spine MRI showed degenerative changes, and electromyography revealed chronic denervation in both upper and lower extremities. Motor nerve conduction studies demonstrated reduced conduction velocity, amplitude, and distal latency in the left median nerve, as well as in the bilateral tibial and peroneal nerves. Sensory nerve conduction testing revealed normal sensory nerve action potential but showed delayed F-wave latencies in the left median and tibial nerves. However, anal sphincter electromyography was normal. The somatosensory-evoked potential showed abnormalities in the bilateral lower limbs, indicating a conduction block in the somatosensory pathway from the spinal cord to the cortex. Moreover, the bilateral visually evoked potential and bilateral auditory brainstem response were also found to be abnormal. The visually evoked potential showed prolonged P100 latency in both eyes. The auditory brainstem response suggested that bilateral ears were stimulated, but the waveform on both sides was relatively poor. The ambulatory electroencephalogram monitoring was normal. The routine cerebrospinal fluid (CSF) analysis showed normal pressure, cell counts, and levels of protein and glucose. Finally, whole-exome sequencing was performed using MyGenostics. In this study, four steps were employed to select potential pathogenic mutations for downstream analysis: (i) mutations with read counts less than 5 and mutation ratios below 30% were excluded; (ii) mutations with a frequency greater than 5% in 1,000 g, ESP6500, and Inhouse databases were removed; (iii) mutations present in the InNormal database (MyGenostics) were also discarded; (iv) synonymous mutations not listed in the HGMD database were excluded. The remaining mutations were considered potential pathogenic mutations for further analysis (Figure 2). Genomic DNA was extracted from the patient’s whole blood, and subsequent sequencing analysis identified a novel splice site mutation in intron 3 of SIGMAR1 gene (c.446-2A>T), which was further confirmed by Sanger sequencing (Figures 2, 3A). A review of the patient’s medical history revealed a longstanding presence of planovalgus deformities in both feet for over 40 years. Physical examination revealed muscle atrophy of both lower limbs at 10 years old, and he complained of mild discomfort while walking. However, his general condition was normal. The patient did not pursue further examination or treatment at that time. During the current clinical examination, upper and lower motor neuron damage was observed, and all the above findings were consistent with the diagnosis of ALS. At that time, the Unified Parkinson’s Disease Rating Scale-Part III motor score (in the morning without antiparkinsonian therapy) was 40. Next, we conducted a levodopa load test, and he showed a good response to levodopa. Based on the findings, the patient was eventually diagnosed with ALS–PD complex. Further exploration of the patient’s family history revealed that his parents were close relatives as they were second cousins. Unfortunately, his father was dead. Genetic testing was conducted on the mother, and it revealed that she has the same SIGMAR1 variant as detected in the proband (Figure 3B). Subsequently, a pedigree analysis was performed (Figure 3C). At the 3-month follow-up examination, the symptoms were observed to have remained relatively stable.
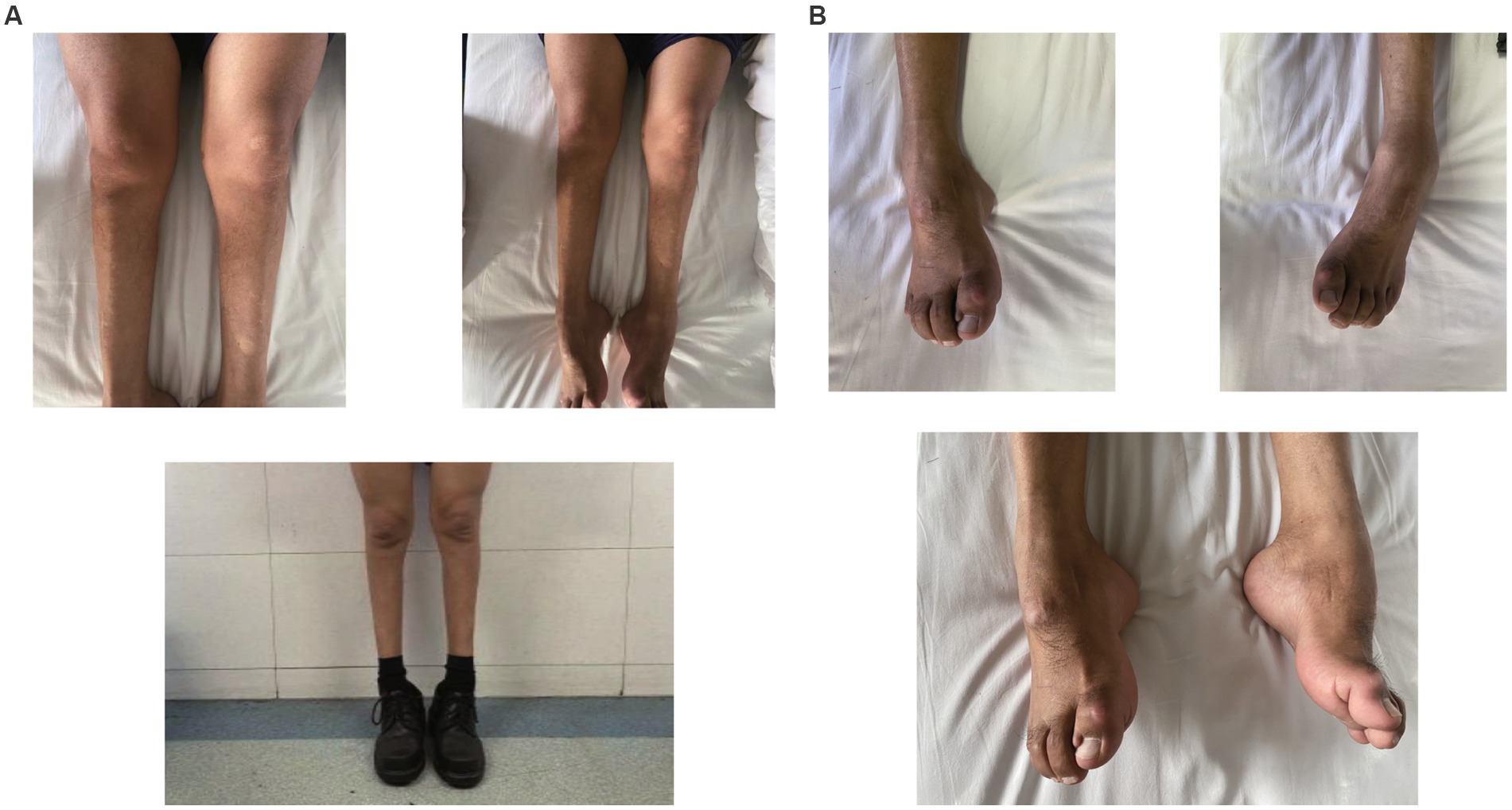
Figure 1. (A) Atrophy of bilateral calves more pronounced distally giving a “stork leg” appearance akin to Charcot-Marie-Tooth disease. (B) Planovalgus deformities of both feet.
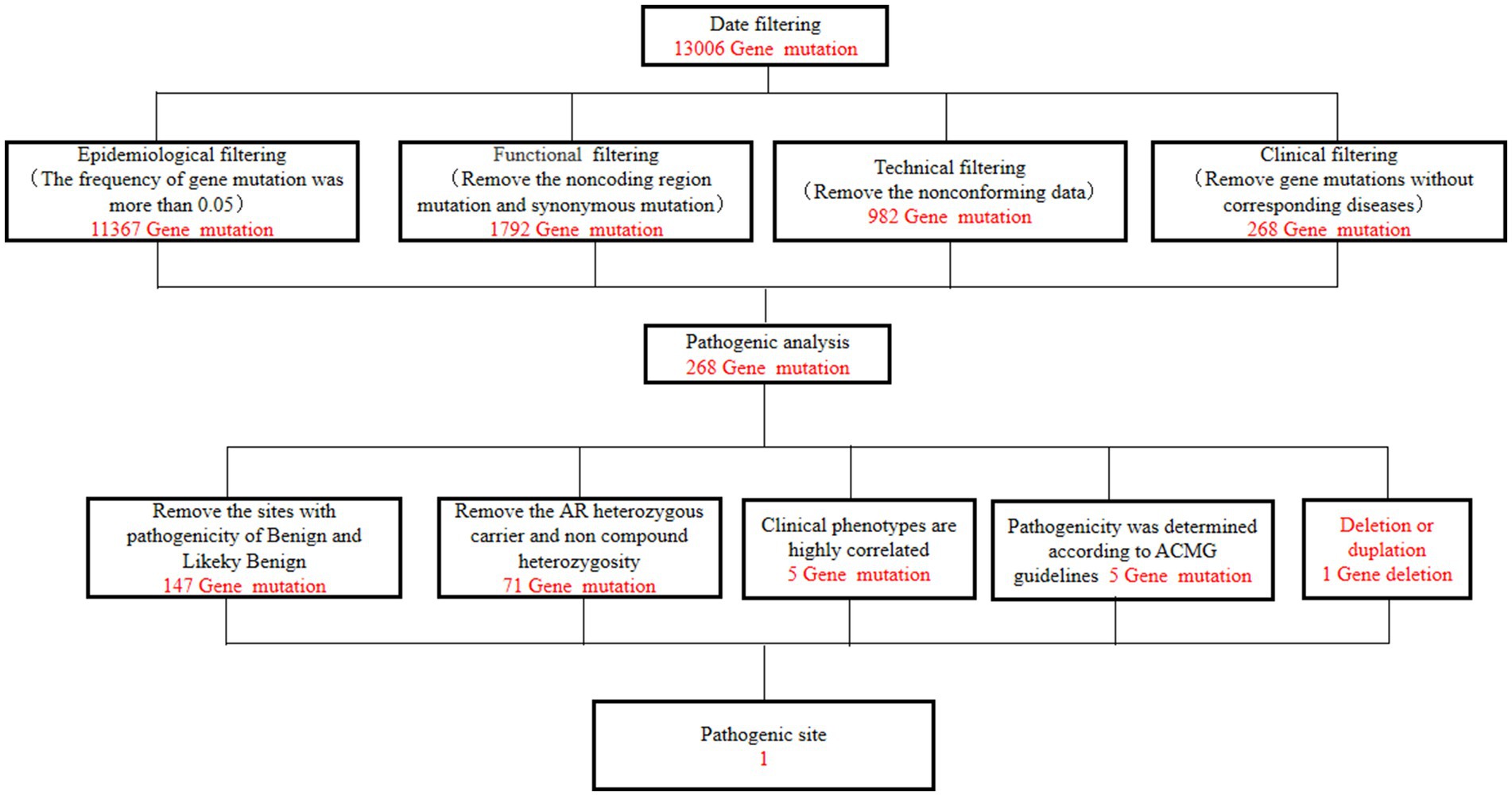
Figure 2. Filtering steps for the variant calls in whole-exome sequencing.
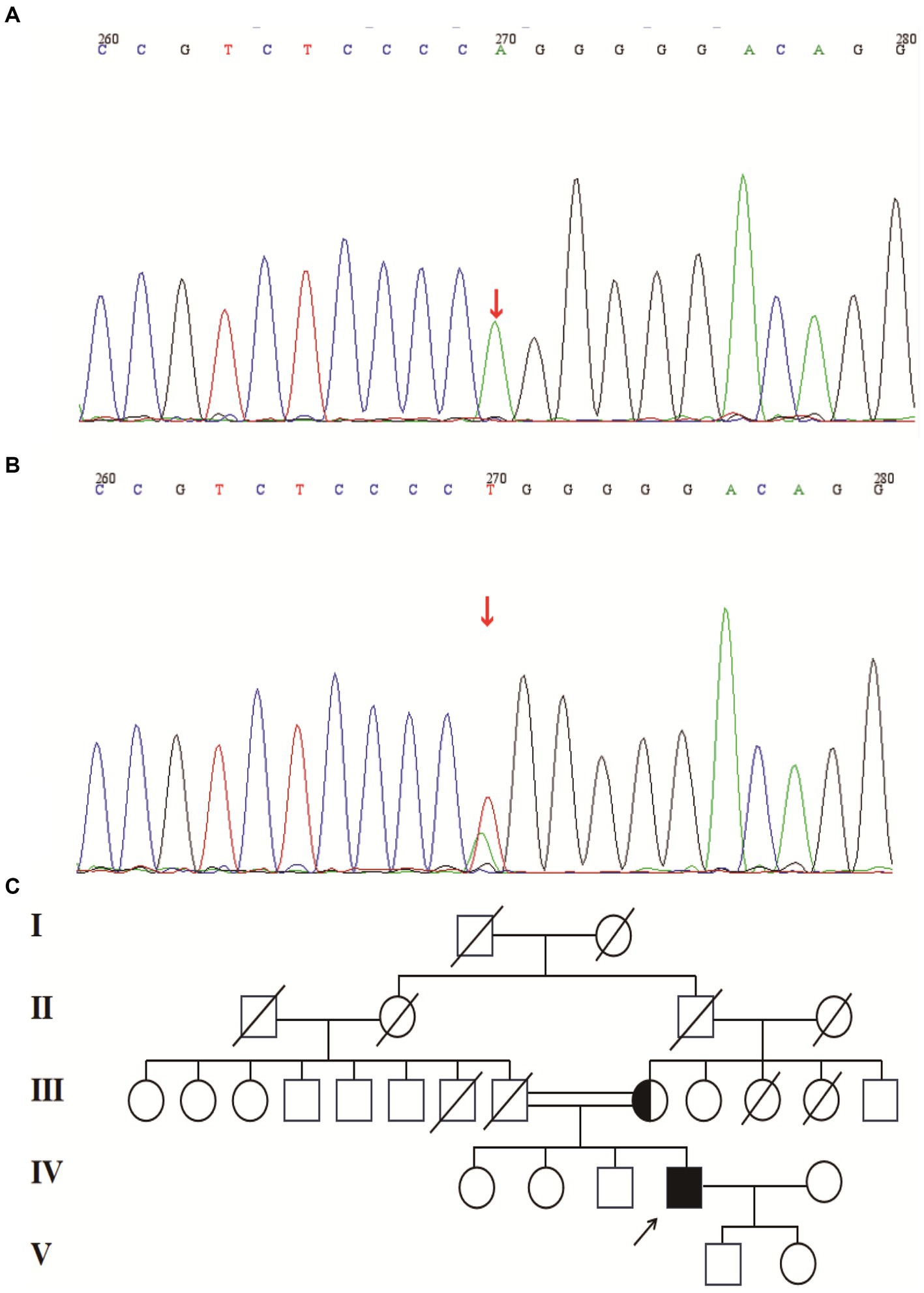
Figure 3. (A) Splice site mutation in the SIGMAR1 identified in our patient. Sanger sequencing was performed using cDNA generated from the patient. (B) Mutation in the SIGMAR1 identified in our patient’s mother. (C) Mutant pedigree map of familial mutations. Circles = females; squares = males; and slashes = deceased.
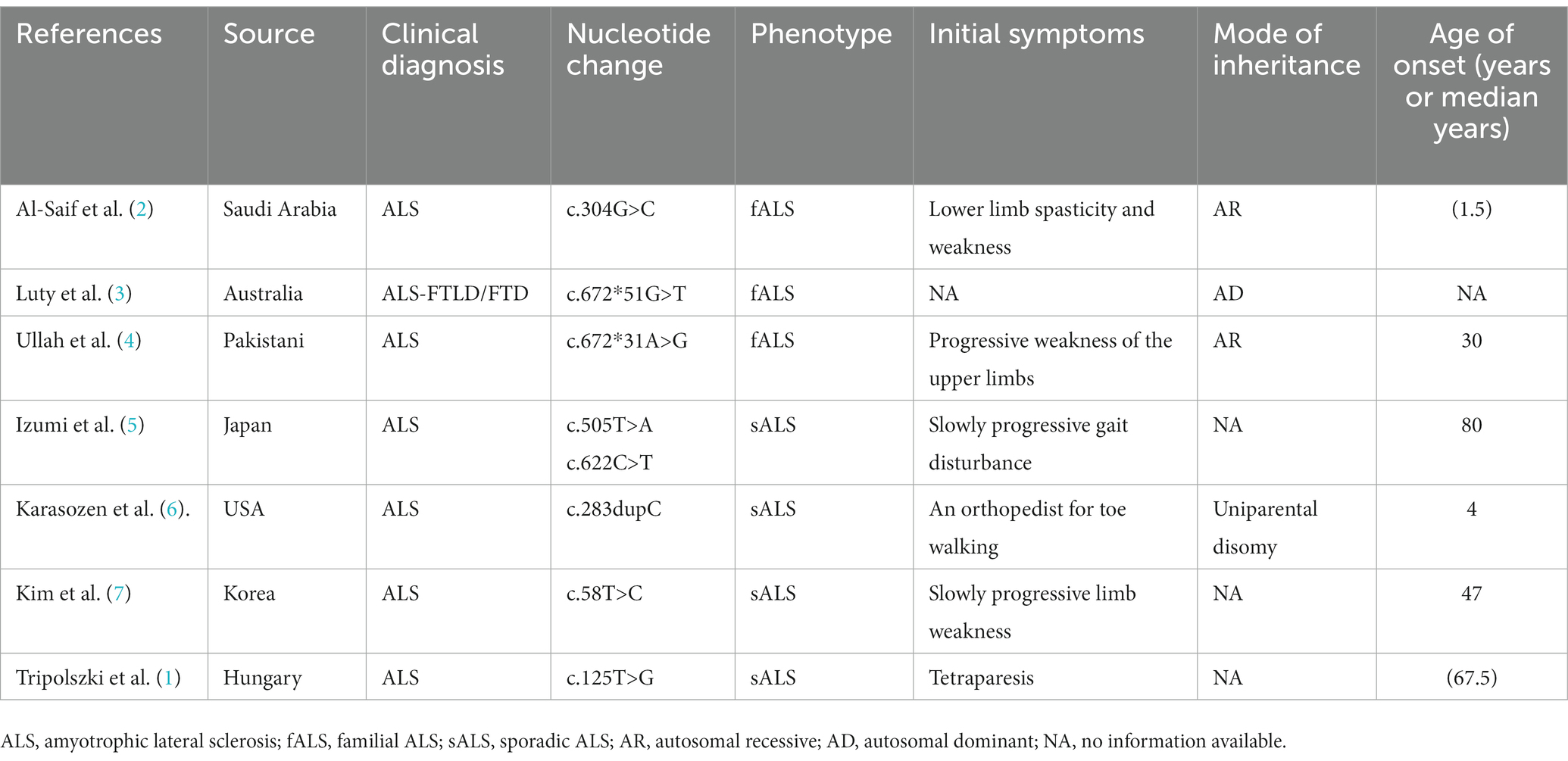
Table 1. Clinical phenotypes of patients with SIGMAR1 mutations.
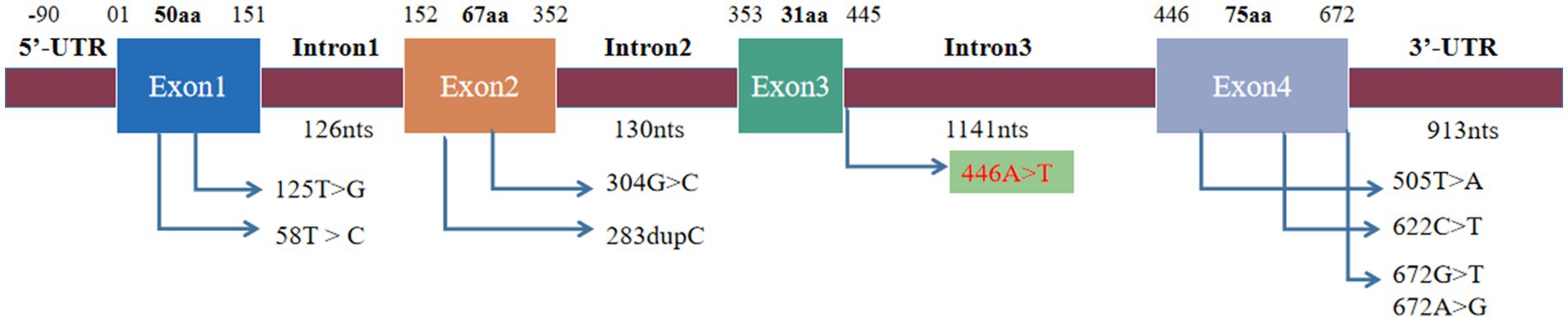
Figure 4. Mutation site of SIGMR1. The previously reported mutation site was located in exons and 3′-UTR, and the mutation site was located in intron.
A literature review was conducted by searching PubMed and China National Knowledge Infrastructure (CNKI) databases from their inception until May 2023 using the keywords “SIGMAR1,” “ALS,” and “amyotrophic lateral sclerosis.” Relevant articles describing cases of ALS with SIGMAR1 mutations were selected. Among the articles, eight described studies of interest, three reported cases of familial ALS (fALS), and five reported cases of sporadic ALS (sALS). Various mutations in the SIGMAR1 gene were identified in affected individuals, including a mutation (c.67251G > T) in the 3′-untranslated region in the FTLD-MND pedigree and mutations (c.304G > C, c.67231A > G, c.505 T > A, c.622C > T, c.283dupC, c.58 T > C, c.125 > G) in the SIGMAR1 gene in affected ALS patients. The phenotypes of each of these cases are presented in Table 1 and Figure 4.
According to the revised El Escorial criteria, patients with ALS may exhibit extrapyramidal involvement in addition to upper and lower motor neuron symptoms and signs (8). When ALS is associated with PD, it is known as Brait–Fahn–Schwartz syndrome or ALS–PD complex. Parkinsonism in these patients typically resembles PD and shows a response to levodopa. ALS–parkinsonism is more common than ALS–PD complex and refers to the presence of extrapyramidal findings that do not respond to levodopa treatment in ALS patients (8). Mutations in the SIGMAR1 gene have previously been linked to different forms of ALS, including juvenile (onset age < 20 years old) and adult-onset (early onset within 20–60 years and late onset >60 years) cases (9). Most SIGMAR1-related ALS cases present with a typical ALS phenotype, but the SIGMAR1 c.672*51G > T variant was identified in an Australian familial FTLD cohort with ALS, presenting as an ALS-frontotemporal dementia (FTD) phenotype (3). In this report, we present a case of ALS–PD complex in a patient with juvenile onset. We identified a potentially new pathogenic variant (c.446-2A > T) located in intron 3 of the SIGMAR1 gene. The sequencing data was saved in FASTQ format, and MGI sequencing adapters and low-quality reads (<80 bp) were filtered using the cutadapt software.1 The clean reads were then mapped to the UCSC hg19 human reference genome using the BWA parameter of the Sentieon software.2 Next, we removed duplicated reads using the driver parameter of Sentieon software, which also corrected the base to improve the quality of the output BAM file reads, making them closer to the real probability of mismatch with the reference genome. The mapped reads were used for variation detection. Variants of SNP and InDel were identified using the driver parameter of the Sentieon software. The data were then transformed into VCF format. To further analyze the variants, we used ANNOVAR software3 to annotate and cross-reference multiple databases, including 1,000 genome, ESP6500, dbSNP, EXAC, Inhouse (MyGenostics), HGMD, SIFT, PolyPhen-2, MutationTaster, and GERP++. Based on the ACMG guidelines, this variant was predicted to be likely pathogenic (PVS1+PP3+PM2). To the best of our knowledge, this is the first reported case of SIGMAR1-related ALS presenting as ALS–PD complex.
Compared to previously reported cases, the patient in this report exhibited distinct clinical phenotypes. While muscle weakness is the most commonly observed clinical feature in patients with SIGMAR1 mutations (10), the patient in our case presented with no symptoms of muscle weakness but instead displayed extrapyramidal symptoms, including bradykinesia, rigidity, and tremor. It is noteworthy that in the majority of ALS patients, motor nerve conduction velocities and terminal latencies are normal. However, this particular patient showed decreased conduction velocity, amplitude, and distal latency in the left median nerve, bilateral tibial, and peroneal nerves, which may be particularly relevant to the long-term atrophy of the lower limbs. Through our literature review, we found cases of ALS patients with SIGMAR1 mutations presenting with ALS either with or without frontotemporal dementia as well as cases of juvenile ALS. Nevertheless, this is the first reported case, where an association between ALS–PD phenotype and SIGMAR1 mutations has been observed.
SIGMAR1 is comprised of four exons and three introns, located on chromosome 9p13.3. This gene exhibits ubiquitous expression in various human tissues, including the brain (cerebellum), retinal ganglion cells, astrocytes, liver, and placenta. Particularly, it is prominently expressed in motor neurons found in the brainstem and spinal cord. The SIGMAR1 gene plays diverse roles in different cells and organs, encompassing ion channel regulation, chaperone function, regulation of mitochondrial morphology, dynamics, and function, as well as involvement in autophagy and endoplasmic reticulum (ER) stress response (10, 11). A common pathological feature in ALS is the disruption of the ER-associated membrane, where the ER-resident chaperone protein is predominantly localized (12, 13). Consequently, changes in SIGMAR1 function may alter ER morphology and impact ER stress responses, resulting in abnormal mitochondrial damage and the initiation of cellular autophagy, thereby contributing to the pathogenesis of ALS. PD, another prevalent neurodegenerative disease, has also been associated with alterations in SIGMAR1 functions. Studies by Finsterer et al. (9) demonstrated that SIGMAR1 agonist treatment in mice with 6-hydroxydopamine lesions reduced neuroinflammation, increased the density of dopaminergic fibers in denervated striatal regions, and elevated the levels of neurotrophic factors. Furthermore, Hong et al. (14) revealed that SIGMAR1 deficiency reduced 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced death of dopaminergic neurons and parkinsonism. Thus, pharmacological activation/inhibition of SIGMAR1 may potentially slow down the progression of PD. Overall, SIGMAR1 activation has demonstrated protective effects in neurodegenerative diseases by modulating various cellular pathways, including the regulation of mitochondrial function, autophagy, calcium homeostasis, and chaperone function.
SIGMAR1 activation has been found to induce potent neuroprotective effects, promote neuronal survival, and restore neuronal plasticity, leading to a deceleration of disease progression in neurodegenerative conditions. Moreover, it has shown promise in ameliorating the clinical symptoms of these diseases. On the contrary, dysfunction of SIGMAR1 may exacerbate the advancement of neurodegenerative disorders. Positioned at the interface of two crucial organelles commonly implicated in the majority of neurodegenerative disorders—the mitochondria and the ER—SIGMAR1 emerges as a robust therapeutic target with significant potential for intervention.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the General Hospital of Ningxia Medical University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
TXua examined the patient clinically. JY performed and analyzed neuroradiologic imaging studies. TXu analyzed electromyography results. HL analyzed performed genetic analyses. HL and TXua wrote the manuscript. JC and ZW analyzed and interpreted the data and revised the manuscript. All authors contributed to the article and approved the submitted version.
This research study was supported by the Key Research and Development Program of Ningxia Hui Autonomous Region (grant number 2022BEG03130), the Natural Science Fund project in Ningxia (grant number 2022AAC03561), and the National Nature Science Foundation of China (grant number 81960245).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tripolszki, K, Gampawar, P, Schmidt, H, Nagy, ZF, Nagy, D, Klivényi, P, et al. Comprehensive genetic analysis of a Hungarian amyotrophic lateral sclerosis cohort. Front Genet. (2019) 10:732. doi: 10.3389/fgene.2019.00732
2. Al-Saif, A, Al-Mohanna, F, and Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann Neurol. (2011) 70:913–9. doi: 10.1002/ana.22534
3. Luty, AA, Kwok, JB, Dobson-Stone, C, Loy, CT, Coupland, KG, Karlström, H, et al. Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease. Ann Neurol. (2010) 68:639–49. doi: 10.1002/ana.22274
4. Ullah, MI, Ahmad, A, Raza, SI, Amar, A, Ali, A, Bhatti, A, et al. In silico analysis of SIGMAR1 variant (rs4879809) segregating in a consanguineous Pakistani family showing amyotrophic lateral sclerosis without frontotemporal lobar dementia. Neurogenetics. (2015) 16:299–306. doi: 10.1007/s10048-015-0453-1
5. Izumi, Y, Morino, H, Miyamoto, R, Matsuda, Y, Ohsawa, R, Kurashige, T, et al. Compound heterozygote mutations in the SIGMAR1 gene in an oldest-old patient with amyotrophic lateral sclerosis. Geriatr Gerontol Int. (2018) 18:1519–20. doi: 10.1111/ggi.13506
6. Karasozen, Y, Sheikh, KA, Mancias, P, and Nguyen, TP. Uniparental Disomy leading to a rare juvenile form of ALS. J Pediatr Perinatol Child Health. (2020) 4:107–10. doi: 10.26502/jppch.74050049
7. Kim, HJ, Kwon, MJ, Choi, WJ, Oh, KW, Oh, SI, Ki, CS, et al. Mutations in UBQLN2 and SIGMAR1 genes are rare in Korean patients with amyotrophic lateral sclerosis. Neurobiol Aging. (2014) 35:1957.e7–8. doi: 10.1016/j.neurobiolaging.2014.03.001
8. Swinnen, B, and Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. (2014) 10:661–70. doi: 10.1038/nrneurol.2014.184
9. Finsterer, J, and Burgunder, JM. Recent progress in the genetics of motor neuron disease. Eur J Med Genet. (2014) 57:103–12. doi: 10.1016/j.ejmg.2014.01.002
10. Aishwarya, R, Abdullah, CS, Morshed, M, Remex, NS, and Bhuiyan, MS. Sigmar1's molecular, cellular, and biological functions in regulating cellular pathophysiology. Front Physiol. (2021) 12:705575. doi: 10.3389/fphys.2021.705575
11. Su, TP, Hayashi, T, Maurice, T, Buch, S, and Ruoho, AE. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci. (2010) 31:557–66. doi: 10.1016/j.tips.2010.08.007
12. Sakai, S, Watanabe, S, Komine, O, Sobue, A, and Yamanaka, K. Novel reporters of mitochondria-associated membranes (MAM), MAMtrackers, demonstrate MAM disruption as a common pathological feature in amyotrophic lateral sclerosis. FASEB J. (2021) 35:e21688. doi: 10.1096/fj.202100137R
13. Watanabe, S, Ilieva, H, Tamada, H, Nomura, H, Komine, O, Endo, F, et al. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol Med. (2016) 8:1421–37. doi: 10.15252/emmm.201606403
Keywords: amyotrophic lateral sclerosis, Parkinson’s disease, SIGMAR1 , genotype, phenotype
Citation: Li H, Xuan T, Xu T, Yang J, Cheng J and Wang Z (2023) SIGMAR1 variants in ALS–PD complex cases: a case report of a novel mutation and literature review. Front. Neurol. 14:1242472. doi: 10.3389/fneur.2023.1242472
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Cigdem Koroglu Altok, National Institute of Diabetes and Digestive and Kidney Diseases (NIH), United StatesCopyright © 2023 Li, Xuan, Xu, Yang, Cheng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiang Cheng, Y2hlbmdqaWFuZ254QDE2My5jb20=; Zhenhai Wang, d2FuZ3poZW5oYWkxOTY4QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.