 Frederike Cosima Oertel
Frederike Cosima Oertel- 1Experimental and Clinical Research Center, Max-Delbrück-Centrum für Molekulare Medizin, Freie Universität Berlin and Humboldt-Universität zu Berlin, Charité – Universitätsmedizin Berlin, Berlin, Germany
- 2Neuroscience Clinical Research Center, Freie Universität Berlin and Humboldt-Universität zu Berlin, Charité – Universitätsmedizin Berlin, Berlin, Germany
- 3Department of Neurology, Freie Universität Berlin and Humboldt-Universität zu Berlin, Charité – Universitätsmedizin Berlin, Berlin, Germany
The first formal consensus diagnostic criteria for myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) were recently proposed. Yet, the distinction of MOGAD-defining characteristics from characteristics of its important differential diagnoses such as multiple sclerosis (MS) and aquaporin-4 antibody seropositive neuromyelitis optica spectrum disorder (NMOSD) is still obstructed. In preclinical research, MOG antibody-based animal models were used for decades to derive knowledge about MS. In clinical research, people with MOGAD have been combined into cohorts with other diagnoses. Thus, it remains unclear to which extent the generated knowledge is specifically applicable to MOGAD. Translational research can contribute to identifying MOGAD characteristic features by establishing imaging methods and outcome parameters on proven pathophysiological grounds. This article reviews suitable animal models for translational MOGAD research and the current state and prospect of translational imaging in MOGAD.
1 Introduction
Myelin oligodendrocyte glycoprotein (MOG) is a minor transmembrane glycoprotein located in the outermost membranes of the myelin sheath (1) that has long been an important target molecule for animal models of demyelinating diseases. Only in recent decades, antibodies against MOG (MOG-IgG) have been identified in people who were previously diagnosed with various other autoimmune-neurological diagnoses such as multiple sclerosis (MS), aquaporin-4-antibody (AQP4-IgG) seronegative neuromyelitis optica spectrum disorder (NMOSD), and acute disseminated encephalomyelitis (ADEM), as well as in isolated and recurrent optic neuritis (ON) and transverse myelitis (TM) (2–6). Furthermore, MOG-IgG can be discovered “false positively” in several other conditions, as demonstrated in several cases of peripheral neuropathy (7, 8) and tumor/lymphoma (9–11). Thus, care needs to be taken as to when MOG-IgG measurement should be performed as well as to the interpretation and consideration of possible differential diagnosis thereof, as has been pointed out in the formal consensus diagnostic criteria for MOG-IgG-associated diseases (MOGAD) that were recently established for the first time (12). Yet, clinical features of MOGAD partly overlap with its differential diagnoses, most importantly NMOSD and MS, delaying the time required until the correct treatment is applied, thus increasing relapse probability. Clinical and imaging studies until now also often included MOGAD patients grouped together with MOG-IgG seronegative patients (for example, as AQP4-IgG seronegative NMOSD), further limiting the discovery of MOGAD-specific features. This is not only true for clinical research: MOG-induced animal models, such as experimental autoimmune encephalomyelitis (EAE), have been widely used as models of demyelinating diseases in general and MS in particular. With the definition of MOGAD as a separate disease entity, it needs to be reevaluated to which extent the generated knowledge from MOG-induced models is specifically applicable to MOGAD versus what should be considered valid for its differential diagnosis (13).
Imaging can significantly aid differential diagnosis early in the disease course and guide the application of cell-based MOG-IgG assays (if available) (14–16). By using a back-translational approach to investigate disease-specific imaging features in preclinical models, imaging can also be used to improve the understanding of (A) distinct pathophysiology by using methods with single-cell resolution and (B) the pathophysiological basis of distinct imaging characteristics by using feature-specific histopathology. This article reviews translational imaging techniques in MOGAD and its animal models. It also discusses the current and potential future relevance of MOGAD-specific animal models and translational imaging for defining distinct pathophysiological features in MOGAD compared with important differential diagnoses, especially MS (17) and AQP4-IgG seropositive NMOSD (18).
2 The pathophysiology of MOGAD
There is very little autopsy and/or biopsy material that documents MOGAD pathology specifically (13, 19–22). Furthermore, these studies were conducted mostly on cerebral samples; there is only one case with spinal cord pathology reported (22). Optic nerves are missing in these evaluations. From the presented material, it can be deduced that there are clear histopathological differences discerning MOGAD from both NMOSD and MS, including a CD4+ dominated infiltrate, with fewer B cells, a moderate number of granulocytes (eosinophils and neutrophils), and many/abundant macrophages, some containing early myelin degradation products. While AQP4 and AQP1 were preserved in MOGAD, reactive astrogliosis and even scarring in and around the demyelinating lesions were observed. Axons and oligodendrocytes were unaffected or variably destructed, with a moderate number of axons showing disturbed fast axon transport and axonal spheroids, especially at the lesion rim. Demyelinating lesions occur usually in white matter in a mixed perivenous and confluent pattern of several perivenous lesions, with affection of cortico-medullary junctions and leptomeningeal areas of the cortex as well as the cerebral white matter. Furthermore, there are no “smoldering” radially expanding lesions with microglial/macrophage rim, as would be seen in progressive MS. A meningeal inflammation in 86% of biopsy cases could be seen. The studies, however, do not agree about complement deposition, one describing complement deposition/activation in white matter lesions (13) and the other describing only occasional perivascular-activated complement and IgG deposition (19). In the latter study and the study by Spadaro et al., MOG-dominated myelin loss with preserved oligodendrocytes was observed (20), whereas the previous one did not discern preferential loss of MOG (13). The pathology of one patient with a fulminant MOGAD-like disease including meningoencephalitis and leptomeningeal enhancement and positive MOG-IgG in the cerebrospinal fluid only showed relative axonal sparing, primary confluent demyelination, reactive gliosis, and CD4+ dominated inflammatory infiltrates (22).
There have also been attempts to define the cytokine profile in patients with MOGAD. A study by Nakajima et al. found elevated levels of serum IL-1ra, IL-5, and TGF-α as compared to MOG-negative patients (23). IL-6 was found to be elevated in the CSF of MOG-IgG seropositive children (24). In the study by Bauer et al., serum cytokine levels of MOG-IgG positive/AQP4-IgG positive NMOSD were compared to those measured in MS patients (25). They discovered 36 analytes being increased from MOGAD compared with MS (IL-8, SDF-1a, MCP-1, GRO-a, IL18, MIP-1b, Fractalkine, HGF, IP-10, SCF, VEGF-A, BAFF, IL-7, TWEAK, MIP-3a, M-CSF, CD40L, MMP-1, IL-27, MIG, LIF, MIP-1a, IL-17A, IL-23, TNF-β, IL-1a, IL-6, IL-21, IL-5, MDC, IL-9, FGF-2, Eotaxin-3, IL-10, Eotaxin-2, and IL-31). Only five cytokines differed between AQP4-IgG seropositive NMOSD and MOGAD, all being lower expressed in MOGAD (APRIL, TNFR2, TRAIL, MCP-2, and CD30). No differences were found in MOGAD/NMOSD with regard to disease activity (relapse/remission and amount of relapses), disease course (monophasic/relapsing), treatment modality, sex, or age; however, the availability of clinical data were incomplete.
3 Clinical features and clinical imaging in MOGAD
MOGAD affects pediatric and adult patients and shows no sex or ethnic predominance (26). Typical clinical attacks include ON, TM, and, to a lesser extent, cranial neuropathies, brainstem and cerebellar demyelinating attacks, tumefactive brain lesions, mono- and polyfocal CNS deficits, and white matter leukodystrophy-like damage, as well as encephalitis with seizures and neuropsychiatric symptoms (27–30). The most common first manifestation in adults is ON (>55%), whereas the most common first pediatric manifestation is ADEM (with or without ON, >45%) (31–33).
In contrast to the recurrent disease course in MS and NMOSD, MOGAD can be monophasic (~22–56%) (4, 13), preferentially in children (16, 34–36), or recurrent. The current estimation is limited by the short follow-up lengths of published studies, but only one in three MOGAD patients seems to have a relapse within a year after their initial manifestation (3, 4, 37). The risk is higher with steroid tapering and shortly after the initial attack (3, 4, 38, 39). Other longer studies with a small sample size suggest that the long-term risk for recurrent attacks is higher and that attacks can still occur up to >40 years after onset (40, 41). The risk of relapse is lower in pediatric patients; only one in five kids is affected (16, 31, 42, 43). In contrast to MS, clinical progression independent of attacks has not been widely reported in MOGAD so far (41, 44, 45). Histopathological analysis of autopsies/biopsies did not reveal “smoldering” (i.e., slowly expanding) lesions in patients with MOGAD, suggesting a different etiology, if there was a clinically progressive, meaning an attack-independent, disease course in MOGAD as compared to MS. Yet, the current state of research cannot shed light on the possibility of clinical or subclinical progression in MOGAD (46, 47). In a few cases in our outpatient clinic, we observed that patients experience relapse-free worsening of their symptoms over time; however, a thorough investigation on this matter is still needed.
3.1 Brain and brainstem
Cerebral manifestations and imaging findings in MOGAD are diverse. In adults with MOGAD, brain MRI findings are usually sparse and rarely occur in isolation without cerebral syndrome or concurrent optico-spinal lesions (3, 48). Silent lesions are seen in <5% of adult MOGAD patients and even those are usually associated with subsequent relapses (49). Cortical and infratentorial lesion locations are the most common, but large T2-hyperintense white matter lesions can occur (13, 48). In rare cases, tumefactive lesions with a risk for herniation are seen (50).
People with MOGAD have a higher frequency of cortical and juxtacortical lesions compared with people with AQP4-IgG seropositive NMOSD. Yet, the number of lesions in MOGAD is usually lower than in MS, especially at onset (3). Matthews, Juryńczyk and colleagues specifically proposed that lesions close to the lateral ventricle and/or in the inferior lobe, subcortical U-fiber lesions, and Dawson's finger-type lesions strongly suggest a diagnosis of MS vs. MOGAD (51, 52). For infratentorial lesions, the brainstem, especially the pons, close to the 4th ventricle and the middle cerebellar peduncle, are the most common locations in MOGAD — lesions can be found in up to 30% of patients (3, 48, 53, 54). Lesion demarcation is usually poor and lesions can disperse over time (4, 48). Particularly, lesions in the middle cerebellar peduncle can distinguish MOGAD from MS and AQP4-IgG seropositive NMOSD (54). Area postrema syndrome, however, is less common in MOGAD compared with AQP4-IgG seropositive NMOSD (55–57). In contrast to MS and AQP4-IgG seropositive NMOSD, the application of gadolinium rarely reveals a lesion enhancement pattern in MOGAD but can lead to unspecific leptomeningeal enhancement around the brainstem or in uni- or bilateral cortical areas, especially in MOGAD with cortical encephalitis.
In pediatric patients, the most common onset syndrome is ADEM, which typically presents on MRI with large asymmetric and diffuse, supra- and infratentorial T2-hyperintense white matter lesions (58–60). ADEM can also rarely occur in adults—with similar MRI features. Compared with MOG-IgG seronegative ADEM, MOGAD-ADEM more often involves the thalamus (61). MOG-IgG-associated autoimmune encephalitis, a second common pediatric manifestation, presents with large subcortical and/or cortical lesions (31, 62). In contrast to autoimmune encephalitis with other antibodies, normal MRI findings in MOG-IgG-associated autoimmune encephalitis are rare (63). A leukodystrophy-like phenotype of MOGAD, a rarer pediatric manifestation, also presents with large symmetric confluent white matter lesions, yet they are usually clinically progressive (47).
Advanced MRI techniques have been used for a limited number of MOGAD studies so far. Combining fluid-attenuated inversion recovery sequences (FLAIR) with traditional MRI metrics, hyperintense cortical lesions and numerous T2-hyperintense lesions in various locations were identified, respectively, in a subgroup of MOGAD referred to as FLAMES (FLAIR-hyperintense lesions in anti-MOG-associated encephalitis with seizures) (29, 64, 65). FLAMES can further be characterized by hyperperfusion of lesions on single photon emission computed tomography (SPECT) (56). Using diffusion-tensor imaging (DTI) and resting state functional MRI, reduced axial diffusivity in line with microstructural white matter damage, and interhemispheric functional connectivity changes of the motor, sensorimotoric and frontal lobe networks, respectively, were identified in MOGAD compared with healthy controls (66, 67). Applying volumetric analyses, no loss of gray or white matter was observed in adult MOGAD patients compared with healthy controls (66, 68). In pediatric ADEM, however, the brain volume as well as the expected brain growth were reduced (69). So far, no advanced MRI marker has been suggested to distinguish MOGAD from its differential diagnoses.
3.2 Spinal cord
TM in MOGAD can manifest as sensory, motor, and sphincter dysfunctions (70). It can occur in isolation or combined with other manifestations such as ADEM or ON. Despite often severe impairment in the acute stage, most patients have a good recovery. Yet, especially sexual, bladder, and bowel dysfunction can remain (44, 71). Persisting pain or spasms are uncommon and seen more often in AQP4-IgG seropositive NMOSD than in MOGAD. The MOGAD-associated spinal cord involvement in adult and pediatric patients is largely comparable (72).
Initial spinal cord MRI can be normal in 10% (73, 74). The most common finding on spinal cord MRI in MOGAD, however, is the so-called longitudinally extensive transverse myelitis (LETM) presenting as a hyperintense T2-lesion spanning over three or more segments and mainly affecting the cervical and/or thoracic cord (Figures 1A–D) (74–77). LETMs rarely occur in MS (78). While LETMs can also be seen in AQP4-IgG seropositive NMOSD, MOGAD patients present more often with multiple lesions and conus involvement (75, 79–81). Also, shorter TM, as typical for MS, can be seen in MOGAD and is more common compared with AQP4-IgG seropositive NMOSD (77, 79, 82).
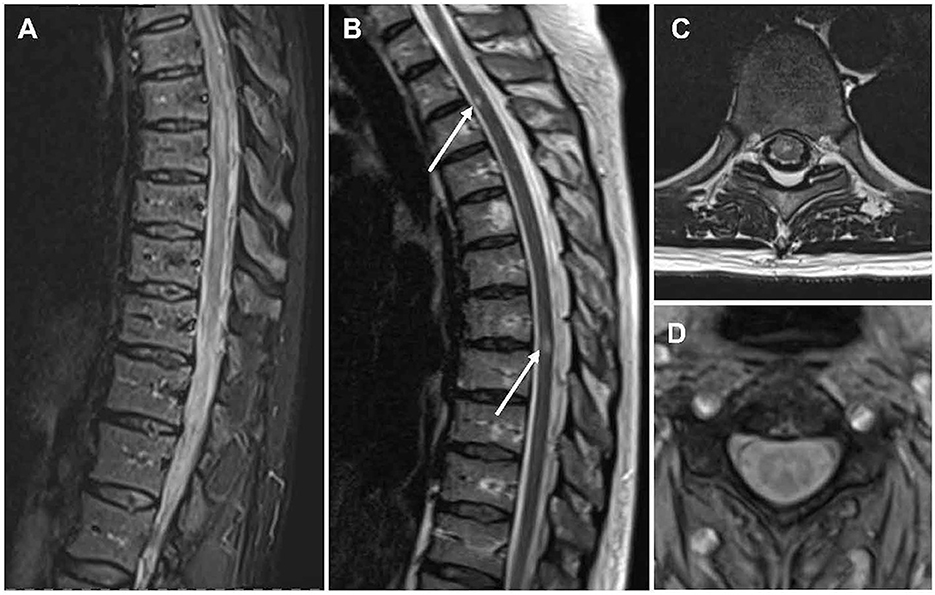
Figure 1. MR imaging of the spinal cord in people with MOGAD. Sagittal T2-weighted MRI showing hyperintense lesions in line with (A) an LETM and (B) shorter lesions. Axial T2-weighted MRI showing (C) a centrally located lesion and (D) the characteristic H-sign. LETM, longitudinally extensive transverse myelitis; MOGAD, myelin oligodendrocyte glycoprotein antibody associated disease; MRI, magnetic resonance imaging.
Up to 75% of lesions in MOGAD are centrally located and up to 50% of lesions are restricted to gray matter, which can often be identified as the characteristic H-sign on axial scans (Figures 1C, D) (73, 79, 81). This is particularly interesting since MOGAD is a highly inflammatory condition primed to the white matter. As discussed below, data from rodent models suggest that this severe white matter inflammation correlates with gray matter hypoxia and increased variation in oxygenation in the gray matter potentially leads to gray matter damage (83), as has been similarly suggested in MS (84, 85). Still the pathomechanism of gray matter damage remains to be elucidated and more autopsy/biopsy samples, especially in MOGAD, need to be analyzed to this end. In contrast to both MS and AQP4-IgG seropositive NMOSD, gadolinium-enhancement is less common in MOGAD (~50%) (72, 75). However, contrast enhancement of the pia and cauda as well as contrast enhancement and thickening of dorsal nerve roots can occur (28, 72).
The application of advanced spinal cord imaging in MOGAD has so far been very limited. Spinal cord atrophy as measured by volumetric MRI has been only seen after severe attacks (86, 87). Silent spinal cord lesions can occur during an attack of the brain or optic nerve but are extremely rare outside of attacks in MOGAD, making spinal cord involvement outside of acute attacks unlikely (49).
3.3 Retina and optic nerve
Optic neuritis (ON) is the most frequent onset feature in adults and one of the most common manifestations of MOGAD in general (88). Thus, imaging of the visual system is a promising approach for diagnosis and differential diagnosis (89, 90). In MOGAD, ON is often bilateral and mostly located in the anterior segment causing severe edema (39, 91, 92). Although single ON attacks often do not lead to tremendous retinal neurodegeneration, the high frequency of attacks in MOGAD can accumulate significant damage (92). Due to its severe symptoms, silent ON is uncommon in MOGAD, yet a bilateral ON can remain unrecognized due to stronger symptoms in one eye.
Lesions on optic nerve MRI usually show T2-hyperintensity and gadolinium enhancement on T1-weighted imaging (Figure 2A). In MOGAD, drastic nerve swelling and characteristic perineural/periorbital gadolinium enhancement are often seen (93, 94). Hemorrhages can occasionally occur, particularly in peripapillary regions. Optic nerve lesions are also extensive, involving more than half of the pre-chiasmic optic nerve, which distinguishes optic nerve lesions in MOGAD from shorter lesions in MS (95, 96). The optic nerve MRI can show the characteristic anterior involvement, which distinguishes optic nerve lesions in MOGAD from the also often extensive but mostly posterior lesions in AQP4-IgG seropositive NMOSD (96, 97). Simultaneous bilateral involvement is more common in MOGAD than in both MS and AQP4-IgG seropositive NMOSD (98).
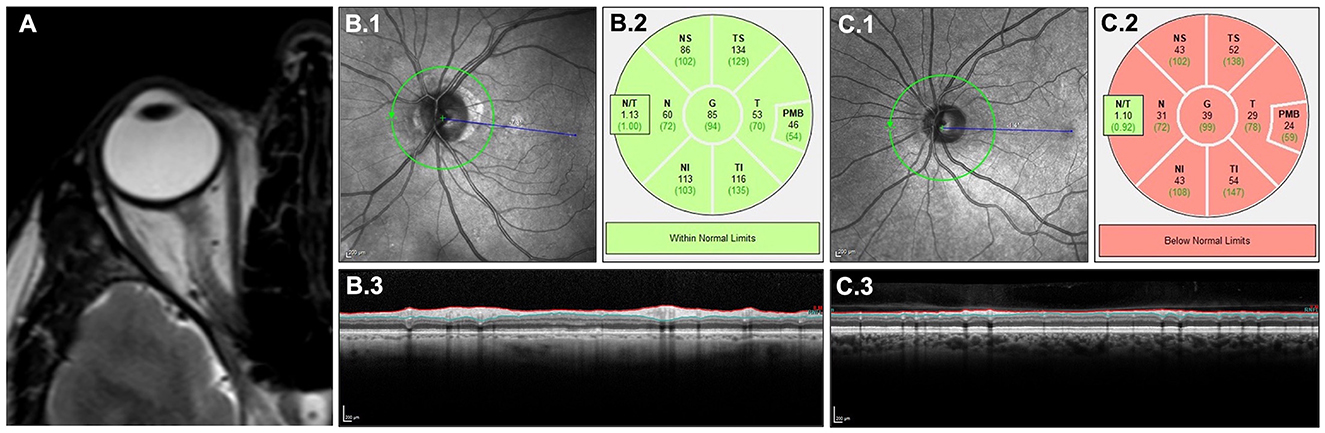
Figure 2. Clinical imaging of retina and optic nerve. T2-weighted MRI of the optic nerve (A) showing a longitudinal lesion with edema. OCT quantifying retinal neuroaxonal content measured by pRNFL around the optic nerve head in a retina without a history of ON (B) and with a history of ON (C) in MOGAD: scanning laser ophthalmoscopy (B.1, C.1), color-coded comparison with a healthy control cohort (B.2, C.2) and cross-sectional B-scans showing pRNFL atrophy in (C.3) compared with (B.3). MOGAD, myelin oligodendrocyte glycoprotein antibody associated disease; MRI, magnetic resonance imaging; OCT, optical coherence tomography; ON, optic neuritis; pRNFL, peripapillary retinal nerve fiber layer.
ON leads to retrograde retinal neurodegeneration, which can be monitored using spectral domain optical coherence tomography (OCT). OCT is a non-invasive imaging method using the interference of low coherent light to produce high-resolution images of the retina (99). Neurodegeneration after ON is quantified by OCT measuring the peripapillary retinal nerve fiber layer (pRNFL) and the combined ganglion cell and inner plexiform layer (GCIPL), which contain the axons and cell bodies of retinal ganglion cells, respectively (Figures 2B, C) (99, 100). Whereas, the pRNFL usually undergoes swelling during the acute phase before experiencing volume loss due to subsiding edema and concurrent degeneration; the GCIPL is less affected by swelling and undergoes a steadier volume loss due to neurodegeneration. According to the current consensus, the majority of retinal neurodegeneration happens within the first 6 months after the acute ON attack independent of the underlying disease. Yet, the acute pRNFL swelling in MOGAD is described to be more severe and is suggested as a diagnostic marker distinguishing MOGAD from MS (101). This might lead to a prolonged (more than 6 months) pRNFL reduction in MOGAD (102, 103).
To diagnose a history of ON, the use of the absolute or relative differences in pRNFL and GCIPL between both eyes of patients, the so-called inter-eye-difference (IED), has been suggested (104). Due to a higher frequency of unilateral ON, the diagnostic value of IED is very high in MS and reasonable in NMOSD (105–108). Yet, the use of IED has not been investigated in MOGAD and seems limited due to the high frequency of bilateral ON. When comparing absolute values of pRNFL and GCIPL after ON, MOGAD patients usually have more severe retinal neurodegeneration (ergo thinner pRNFL and GCIPL) than patients with MS. pRNFL and GCIPL after ON are comparable in people with MOGAD and AQP4-IgG seropositive NMOSD (109). Yet, several publications suggest that the neuronal loss per ON is lower in MOGAD, and only the higher frequency of ONs leads to damage that is comparable with AQP4-IgG seropositive NMOSD patients with less frequent but more severe ONs (109, 110). Despite the neuroaxonal loss being comparable, people with MOGAD often have a better long-term visual outcome compared with AQP4-IgG seropositive patients — the pathophysiological explanation for this difference is still pending (111–115).
Retinal and optic nerve damage independent of ON has been shown in MS, where it can also be used to predict disease activity (116–120) and, to a lesser extent, in AQP4-IgG seropositive NMOSD (121–126). Advanced OCT imaging suggests that ON-independent retinal changes in AQP4-IgG seropositive NMOSD are related to primary astrocytopathy (127). So far, no ON-independent neurodegeneration above aging-related standard and no primary and/or outer retinopathy has been shown in MOGAD, potentially aiding differential diagnosis (102, 103, 128). First applications of OCT angiography showed a significant decrease in vessel density after ON in MOGAD, which exceeded the changes in AQP4-IgG seropositive NMOSD (129, 130). A new generation of advanced OCT imaging methods including 3D-shape analyses and feature recognition can potentially contribute to a better understanding of ON-dependent and -independent changes in MOGAD and their use for differential diagnoses in the future (131–134).
4 Are MOG and MOG-IgG-induced animal models good models for MOGAD?
Animal models that induce encephalitis to mimic autoimmune-mediated disease in the CNS include approaches of active immunization, passive transfer, antibody (co-)mediated disease induction or exacerbation as well as transgenic/genetic modifications to mention the most common ones. MOG-mediated disease is one of the most commonly used to model MS and has been used in many variations that have been described and reviewed extensively elsewhere (135–164). However, with the emergence of MOGAD as a separate disease entity and considering that these models do present drawbacks in reproducing MS characteristics (mainly CD4+ mediated, no MOG-IgG present in any form of MS, etc.) (2, 12, 35, 165–168) the issues in their translation into new therapeutic modalities for MS could be viewed in a new light (169–171). In this chapter, we will discuss to what extent (some selected) MOG-induced animal models as well as some non-MOG-induced models resemble human MOGAD disease and to what extent they could be employed for diagnostic, prognostic, and therapeutic approaches (135).
4.1 MOG-induced animal models
The course and development of EAE are dependent on many different factors and their ratio to each other (172) including the conformation, concentration, solubility, specificity of the antigen used (135), age, species and genetic background of the experimental animals (139, 173–175), the adjuvant (176–179), and timing of immunizations/transfer to name just some variable instances. It has been shown, for instance, that the disease course—monophasic, relapsing, primary/secondary progressive, or chronic progressive (with disability accrual)—can be regulated by the immunization protocol of Lewis (LEW.1AV1) rats with MOG (135).
4.2 MOG-IgG-mediated models
In patients, MOG-IgG was shown to be present during very early stages of disease onset and to persist over long periods of time even during remission. The MOG-IgG titer is dependent on disease activity; however, the antibodies cannot independently induce the disease. In contrast, MOG-IgG has been found in early, intermediate, and late stages of EAE; however, the titer was not disease activity-dependent, being low at the beginning and higher in the end, with the amount being similar during the acute and remission phases (135). Complement-mediated pathology/demyelination could be induced in EAE (180, 181) in line with findings of complement deposition in MOGAD autopsy material. In a constitutively MOG-IgG-producing transgenic mouse model, EAE could be induced in the absence of B cells but required T cells (182).
Experimental studies suggest that MOG-IgG mediates a pathogenic effect in EAE (181, 183, 184). It seems, however, that circulating MOG-IgG require the presence of complement, cytokines, and/or a (T cell-induced) inflammatory milieu to trigger demyelination/enhance inflammation via CDC/antibody mediated cellular cytotoxicity (ADCC), as alone, they are not able to do so (152, 185–187). It was shown in naive recipient animals that primary demyelination restricted to CNS nerve fibers could be induced via injection of a monoclonal MOG-IgG (the 8-18C5) into their cerebrospinal fluid. In adult Sprague-Dawley rats, an association between antibody titer and degree of demyelination could be demonstrated after infusion of sera from Hartley guinea pigs previously immunized with homologous spinal cord lysate in adjuvant into their subarachnoid space. The presence of MOG-IgG in injected sera was demonstrated via an anti-MOG ELISA (185). The direct translational value of these experiments seems tenuous as the blood-brain barrier (BBB) was circumvented in these experiments. The demyelinating effect of antibodies directed against MOG was also demonstrated in a Sprague-Dawley animal model in which monoclonal MOG-IgG-producing B cell hybridomas were implanted into the right lateral ventricle (188). MOG-IgG titers could not be linked to disease outcome in MOGAD patients to this date (189); however, a longitudinally persistent MOG-IgG positivity seems to be associated with a higher risk for relapse (16, 34, 42, 190, 191). Furthermore, it was shown that children with monophasic ADEM lose MOG-IgG over time (192). This is mirrored in animals as high frequencies of relapses are associated with permanent damage. MOG-IgG injection was lethal when injected into SJL mice repeatedly challenged with passive MBP-specific T cell transfers (mimicking a relapsing disease course) that had not yet completely recovered from the previous relapse as opposed to no negative effect of the antibody if the disease score was zero (193). In another experiment with repeated passive transfer of T cells and subsequent antibody application, formation of large demyelinating lesions accompanied by lack of remyelination could be observed, with pronounced astrocytic scar formation traversed by “naked” axons, both characteristic of MS, and not described thus in the available MOGAD autopsy/biopsy cases (194). In mice engineered to produce high MOG-IgG titers, pathology could only be seen after immunization with MOG antigen, without regard to the genetically more (SJL) or less (C57BL/6) EAE-susceptible background (195). This is in line with experiments showing that B cells are not critical for the development of MOG-induced EAE (B cell-deficient muMT mice on C57BL/10 and DBA/1 genetic backgrounds and X-linked immunodeficiency (xid) mice on DBA/1 background) but contribute to the severity, i.e., demyelination rather than inflammation (196). However, the effect of the autoantibodies seems to differ regarding their enhancing characteristics of demyelination/inflammation depending on the agent EAE was induced with. Thus, MOG-specific T-cell-mediated inflammation can be enhanced via augmented antigen presentation (197), whereas in EAE induced by non-MOG-specific T-cells, demyelination is triggered but no enhancement of inflammation is observed (184).
4.3 Animal models targeting MBP
In the passive transfer EAE model (transfer of antigen-specific T cells propagated in vitro) with intravenous injection of MBP-specific T cells and subsequent intravenous (i.v.) MOG-IgG injection at the onset of the disease, a massive augmentation of clinical affection as well as primary demyelination could be observed in Lewis rats. Similarities to MOGAD include lesions located predominantly in the spinal cord and medulla oblongata at circumventricular organs (BBB is more transmissible at these points), predominantly mononuclear cell infiltrate with some granulocytes, perivascular, or focal confluent demyelinated lesion formation (75% of T cells infiltrate to the parenchyma), depending largely on the amount of injected T cells, extensive gliosis, preservation of axons, and remyelination of demyelinated lesions (20, 151, 198). There is a clear macrophage-dominated infiltrate seen in MBP EAE (macrophage: T cell ratio of approximately 6:1); in some cases of MOGAD histopathology, the amount of both cell types seems to be near to equal (1:1.2, respectively) (19), while in others, T cells seem to be somewhat outnumbered by macrophages, especially in the parenchyma [no ratios given, (13, 22)]. Furthermore, the relevance of complement involvement, in the form of membrane attack complex (MAC) formation as well as ADCC, was demonstrated in this model as well as MAC formation in PVG/c rats (±C6 complement component, immunized with guinea pig myelin basic protein (gpMBP) and Complete Freund's Adjuvants (CFA) containing Mycobacterium tuberculosis H37Ra) (199), which is in line with findings of complement deposition, to varying degrees, in MOGAD patients‘ biopsies/autopsies (200, 201).
Active EAE to MBP immunization has been induced in Lewis rats with subsequent MOG-IgG injection (MoAb 8-18C5) 10 days after sensitization. Antibody injection led to significant worsening of clinical and histopathological observations compared to the disease course without the addition of antibody (193, 202), granulocytic infiltrate, perivascular complement deposition, and inflammatory cuff formation, which could be observed similarly to histopathology found in MOGAD patients. The disease course after MBP immunization, with or without subsequent antibody injection, was monophasic; progression or relapse was not recorded after an observation period of 13 weeks (193).
4.4 Animal models induced by MOG-specific T cells
In animals (Lewis rat) with passive MOG-EAE (T cells raised against the MOG35−55 peptide, with and without MOG-IgG transfer), inflammatory changes were induced in the spinal cord without producing an according clinical correlate of typical EAE symptoms (tail tonus loss, gait instability, and severe weight loss) (173, 203). The macrophage: T cell ratio was clearly shifted toward T cells (1:6, respectively), and a few cells (7–20%) of the inflammatory perivascular infiltrate left the perivascular space toward parenchymal infiltration. Contrary to all previously analyzed passive EAE models [induced with MBP, S100β, PLP nicely reviewed in (140, 204, 205)], no peripheral affection was noted. Severe blood-brain barrier dysfunction was induced by passive MOG-EAE and subsequent intravenous injection of a demyelinating MOG-specific monoclonal antibody that induced severe clinical disease. Furthermore, it has been shown that the location of lesions was dependent on the antigen used to raise the T cells (204).
4.5 Animal models induced by MOG peptide immunization
Immunization (active MOG-EAE) in Lewis rats via a highly purified recombinant protein, mMOG, spanning its N-terminal domain (a.a 1–125 + CFA) failed to activate immunodominant T cell epitopes, producing an inflammatory non-demyelinating phenotype as seen previously with passive transfer EAE (206). No clinical symptoms could be observed, at least partly attributed to reduced macrophage recruitment as compared to immunization with MBP/PLP protein/peptide (173). Antibodies to MOG1−25 were induced by mMOG immunization and production could be enhanced by repeated immunization (booster) after 4 weeks; however, this epitope does not seem to produce a demyelinating phenotype. Again, extensive perivascular and subpial demyelination could be produced by co-injection of the MOG-specific mAb 8-18C5 on day 10 post-immunization. Thus, immunization with mMOG seems to reproduce MOGAD histopathology rather poorly. Contrary to these findings, immunization with MOG isolated from human/rat brain tissue as well as immunization with MOG35−55 peptide were able to induce a severe relapsing-remitting disease course in Lewis rats presenting with inflammatory demyelinating lesions and perivascular cuffs (mononuclear, including myelin debris) with accompanying MOG specific IgG production (in the former) (207). Different rat strains (BN, DA, Lewis.1N, Lew1AV1, and Lew1A) were challenged with different MOG compositions [soluble or precipitated in complete or incomplete Freund's Adjuvants (CFA/IFA)] and varying immunization protocols (205). This study shows a very good reproduction of core MOGAD characteristics, more or less expressed depending on strain/regime/immunogen composition, in all the animals. These include the development of a chronic relapsing disease course in 111/156 animals, 16/156 developed chronic progressive disease, 17/156 showed stable course with neurological deficit. Development of predominant or selective ON was seen in some animals. Neuropathology (in 133/156 animals) featuring perivenous inflammation, confluent demyelinating plaques with complement deposition at sites of active demyelination, relative axonal sparing, inflammatory perivenous infiltrates, and meninges with parenchymal infiltration adjacent to the pia mater with predominant T cell/macrophage infiltration as well as polymorphonuclear infiltrates (mostly in animals with ON/spinal cord affection) and frequent remyelination. Of the observed pathology, glial scar formation is not readily found in current reports of MOGAD histopathology. Acute disseminated leukoencephalomyelitis was seen in the other 23 animals, which is about ~15 of animals; in comparison, in human children ADEM occurs in >45% cases and in adults in ~10%, featuring severe perivenous inflammation and little/absent demyelination. Major patterns of lesion distribution across the CNS (optic nerve/spinal cord, isolated ON, spinal type, cerebellar type, periventricular type, acute disseminated leukoencephalomyelitis type, and destructive transverse myelitis) go along well with lesion distribution seen in MOGAD (classified by these authors at that time as neuromyelitis optica). In this study, the authors showed that optic nerve involvement was independent of MHC genes; in addition, it was shown by others that MHC haplotype seems to influence disease susceptibility to a certain amount (174, 208). Differences in these models compared to MOGAD were seen in relation to sex-associated characteristics, specifically in DA rats. It could be observed that female rats had a high incidence of ON, whereas none was seen in male rats. In a study by the Mayo Clinic, the authors observed that of the 87 MOGAD patients presenting with ON, 57% were female (92). Another clear sex difference was seen in eosinophilic granulocyte infiltration, which was seen only in female rats; however, this phenomenon was not mentioned by any of the MOGAD autopsy/biopsy studies cited above. In a transgenic mouse study with MHC II-restricted animals, immunodominant MOG epitopes were identified and EAE could be induced (209). This is in line with findings that CD4+ T cells (HLA class II) dominated cell infiltrates in MOGAD patients' lesions. In a Dutch and UK study, no negative association of MOGAD to an HLA subtype could be discerned to date, whereas a Chinese study suggested an association of DQB1*05:02-DRB1*16:02 alleles to pediatric-onset MOGAD (210–212). Notably, to this date, no definite genetic association could be shown in MOGAD; specifically, no strong HLA dependence, which is in contrast to what has been suggested in MS (210–212).
One of the most widely used EAE animal models to date is the C57BL/6 mouse MOG35−55 EAE (213, 214). Similar to MOG-induced EAE in Lewis rats, injection with only MOG35−55 peptide (and CFA, with and without pertussis toxin PT) was able to induce neurological impairment in C57BL/6J mice featuring a chronic, non-remitting disease course and mild clinical presentation usually restricted to paralysis in the tail and hind legs. Mice did not recover after immunization even after long-term observation (3 months), which, however, could not be observed in other studies (215). Lesions included perivascular infiltration of mononuclear cells and secondary demyelination. PT was observed to enhance EAE moderately and lead to earlier disease onset, however, PT is not needed to induce overt clinical disease per se (213).
It was shown in active C57BL/6 mouse MOG35−55 EAE (with CFA and PT) that natural killer cells (NK-cells) are involved in preventing EAE development, as Th1 response (including Th1 specific cytokine production, IFN-γ, and TNF-α) seemed to be elevated in NK-cell depleted animals (216). However, a reduced amount of NK-cells could only be seen in NMOSD but not MOGAD when compared to each other (217).
There have been some attempts to define the cytokine profile in patients with MOGAD (see above). In a study using actively induced MOG35−55-EAE in mice (induced with MOG35−55, CFA, PT) changes in cytokine production largely overlapping with MOGAD (IL-4, IL-6, IL-10, IL-12, IL-17, IL-23, TNF-α, IFN-γ and TGF-β) were observed (218). In EAE, the involvement of IL-6 has been extensively studied. It has been shown that IL-6 (conditionally) deficient mice are resistant to EAE (219–221), that IL-6 is involved in the induction phase of EAE (222) (MOG35−55 induced), that IL-6 inhibits T cell conversion to the Treg phenotype (Foxp3+) (223), and is (224, 225) or is not (223) involved in conversion to Th17 type T cells. It has been shown that tissue damage occurs preferentially at sites of IL-6 production (226, 227) and that induced antibodies against IL-6 are protective against EAE (228). Interestingly, PT which is often used to enhance EAE has been shown to induce IL-6 (229). In a mouse line deficient in the IL-6 gene (129/SvXC57BL/6), immunization with MOG35−55 peptide showed abrogated EAE induction (230). These findings are in line with the seemingly beneficial effect of Tocilizumab/Satralizumab (recombinant, monoclonal antil-IL6 receptor antibodies) on relapse prevention in MOGAD patients (231–236). IL-23 involvement was shown in EAE induction (237), as well as the development of Th1 and Th17 cells (238–240) but is not necessary in the effector phase of the disease.
It was demonstrated in different rodent animal models that IL-10 is involved in EAE via increased disease severity when deleted, and IL-10 contributed to disease course duration (shorter) and recovery (241, 242). In a passive transfer EAE with anti-MOG T cells into MyD88 animals, it was shown that resistance to EAE was mediated via the secretion of IL-10 by recipient T cells (243). Further, it was shown that immunization with MOG35−55 in susceptible (SJL and NOD) vs. resistant strains (B10.S or III) differed in the amount of cytokines produced, resistant strains secreting primarily IL-4/IL-10 and transforming growth factor (TGF)-β, vs. susceptible strains with predominant IFN-γ production (244). In contrast, 129/Sv mice knocked out for the gene coding for the ligand-binding chain of the IFN-γ receptor developed severe EAE (129/Sv are resistant to MOG-induced EAE), indicating that IFN-γ was involved in ameliorating EAE during both the effector and induction phase (245, 246). IFN-γ involvement in the determination of lesion location was shown in passive MOG-EAE induced in C57BL/6 mice lacking the IFN-γ receptor (IFNγR) (247) and it was shown that CFA/PT alone do not induce IFN-γ production, but immunization together with MOG is necessary (248).
The above-described patient cytokine profile points toward the direction of TH17 involvement (IL-17A, IL-23, IL-6, and IL-21) in the pathogenesis/disease course of MOGAD (249). The involvement of Th17 T cell subsets has been under discussion since their discovery in 2005 (250, 251), allocating a role for them in EAE induction/autoimmunity (252–256) or not (257) in different MOG-induced animal models [reviewed elsewhere (249)], going so far as to implicating the intestinal microbiome to EAE resistance of mice deficient in IL-17A and IL-17F (258). In a passive transfer model with MOG-specific T cells derived from 2D2 mice, it was shown that both Th1/Th17 cells are able to induce EAE; however, Th17 induce an atypical phenotype in half the cases (beginning with ataxia instead of paralysis, only developing paralysis later). Interestingly, histopathology [severe immune cells infiltration (CD4+ T cells and macrophages), astrogliosis, microglia activation, demyelination, and axonal damage] as well as lesion location (throughout the CNS as well as inflammatory infiltrates/demyelination in the PNS) were similar in both Th1 and Th17 recipients (259). A higher frequency of ataxia was found in children with ADEM positive for MOG-IgG compared to MOG-IgG negative cases (60). No involvement of IL-5 could be detected in the initiation or effector phases after immunization of C57BL/6J (or IL5−/−) mice with MOG35−55 (260). Likewise, IL-21 was found irrelevant for Th17 induction (261).
Cerebral cortical encephalitis is one of the core clinical demyelinating events suggested by Banwell et al. in the diagnostic criteria for MOGAD (12). Current models of EAE do not reflect cortical demyelination ideally. One model trying to recapitulate these lesions targeted the cerebral cortex by stereotactical injection of pro-inflammatory mediators into Lewis rats challenged with MOG1−125 (262). Inflammatory, demyelinating lesions were induced including complement deposition, and as seen in MOGAD autopsy cases, ready remyelination was observed. In a model of Dark Agouti rats immunized with MOG, inflammatory agents were injected into the subarachnoidal space to avoid parenchymal damage. Here as well, IgG and complement deposition were observed, the amount of inflammatory infiltrate was little and mostly limited to meninges, and as in the model described by Merkler et al., repair was rapid (263).
4.6 Transgenic animal models
There is a wealth of genetically modified/transgenic/humanized animal models that have been reviewed in more detail elsewhere (264–266). We will discuss some of those models in this review in regard to their similarities as models for MOGAD. In MOG35−55-induced active EAE in non-obese diabetic (NOD) mice, some groups showed that a switch from relapsing-remitting (RRMS) to secondary progressive (SPMS) can be induced and this model is considered to reflect the pathology of SPMS well (267, 268). Other groups could not observe the switch of clinical symptoms to a progressive disease course (269). When disease was induced in NOD mice via immunization with MOG35−55 and CFA/PT, inflammatory/demyelinating lesions developed preferentially in brain white matter (fimbria/internal capsule) and also in the spinal cord with macrophage infiltration. Microglia/astrocyte activation could be observed (268). Interestingly, disease development and progression could be prevented via anti-IL-12 antibodies in this model (270). NOD mice with transgenic TCR recognizing MOG35−55 were generated (1C6 TCR) (267) and showed development of spontaneous optic neuritis/EAE in around 1% of the animals, distributed similarly in both male and female animals. Upon passive transfer EAE, these mice developed preferentially spinal cord lesions and optic neuritis. When immunized with MOG35−55 and CFA, these mice developed chronic disease after the second relapse with CD4+ T cells predominating over CD8+ T cells at a ratio of 30:1 in the lesions, with elevated production of IFN-γ and IL-17. In following experiments, 1C6 TCR mice were crossed with Ig heavy-chain knock-in mice (IgHMOG or Th mice) on a C57BL/6 background (195). IgHMOG mice harbor autoreactive B cells producing anti-MOG antibodies with the heavy chain of the 8.18C5 demyelinating MOG-specific antibody; however, they do not develop spontaneous disease but were shown to both accelerate and exacerbate EAE irrespective of the inducing agent. The frequency of spontaneous disease was higher in 1C6 x IgHMOG mice (45% males, 79% females), CD4+ T cells still outnumbering CD8+ T cells 7:1, the amount of CD8+ T cells, however, being higher compared to 1C6 TCR mice. Lesions were located mostly in the spinal cord, with around 40% of the mice showing optic nerve lesions, and no formation of ectopic follicle-like structures was observed in the CNS of the animals. A large part (75%) of asymptomatic 1C6 × IgHMOG animals showed exclusively cerebellar lesions upon histopathological examination.
Also, in the Biozzi EAE model (271), chronic relapsing disease could be induced via subcutaneous injection at days 0 and 7 in both hind flanks with an emulsion spinal cord homogenate and CFA complemented with M. Butyricum. In these animals partial closing of the BBB, meningeal ectopic lymphoid tissue with adjacent subpial demyelinating lesions and a switch from T cell, to B cell, predominance and serum MOG-IgG generation in later chronic disease stages could be observed (272).
Another study in a transgenic mouse model, GFAPγR1Δ, induced EAE by active immunization with MOG35−55 to gain a progressive phenotype with sustained inflammation and increasing clinical disease. This study suggests that tumor necrosis factor (TNF) is predominantly produced by CNS infiltrating macrophages rather than microglia after the acute disease stage (273). Contrary to promising preclinical results of TNF blockade, however, the success of TNF suppression in MS patients did not yield uniformly positive results (274). For MOGAD in relation to TNF treatment, little is known and data from a small retrospective study (n = 5) is inconclusive regarding negative effects, however also no clear positive outcome is documented (275). Primary progressive-EAE (PP-EAE) was further established in A.SW mice sensitized with MOG92−106 and SJL/J mice sensitized with MOG92−106 and curdlan (276). A.SW mice develop large areas of demyelination, immunoglobulin deposition, and neutrophil infiltration in the absence of a T cell infiltrate (14, 16) while SJL mice show T cell infiltration and paralysis. Both models generated an anti-MOG antibody response (276).
Another model is the “genetic 2D2” EAE model (TCRMOG) in which mice were generated with a TCR that is directed against MOG35−55 (with a C57BL/6 background), about 5% of the animals develop EAE spontaneously with inflammatory/demyelinating lesions in brain, spinal cord, and optic nerves (277). Furthermore, a large proportion of non-clinically symptomatic mice showed ocular abnormalities, and around 15% of the 2D2 transgenic mice developed isolated optic neuritis in the absence of clinical/histological signs of EAE. These lesions showed macrophage infiltration, demyelination, and axonal damage. Interestingly, the challenge with PT alone was sufficient to induce clinical EAE in 39% and histological EAE in 56% of 2D2 mice. The GF-IL23 model, with astrocyte-specific IL-23 secretion on a 2D2 background (most CD4+ have TCR specific for MOG35−55), showed a spontaneous EAE induction with chronic disease course, clinical affection (ataxia/paraparesis), and a high proportion of B cells. A pronounced B cell accumulation and B cell follicle-like infiltrates have not been reported as such in MOGAD yet (160).
To generate double transgenic opticospinal EAE (OSE) mice (277–280), 2D2 mice were then crossed with IgHMOG (with a transgenic B cell receptor to MOG, described above). The offspring of these mice spontaneously develop ON and severe inflammatory spinal cord lesions, whereas the brain remains relatively spared, which is very similar to NMOSD/MOGAD disease in humans. A gene expression profiling study sought to discern whether spontaneous OSE or MOG-induced EAE reproduced the genetic contribution to MS pathogenesis more closely, and concluded that the OSE model is probably linked more closely to human MS risk genes due to differentially higher expressed Th1 genes (281). A thorough gene expression profile for MOGAD still needs to be generated; however, the cytokine profile (see above) is rather indicative of a predominant Th17 response in MOGAD, which needs to be verified.
It has been suggested that most axonal damage in MOGAD happens during the initial attack, measuring neuroinflammatory biomarkers (such as MBP, sNFL, GFAP, and Tau), and relapses are associated with increased myelin damage (282). It has been suggested that antineurofascin antibodies contribute to axonal pathology in a passive transfer MOG-EAE model (283). It has been shown in double-transgenic OSE mice that when MOG is knocked out, the autoimmune response of MOG TCR-specific T cells is redirected toward the medium-sized neurofilament (NF-M) (278). Subsequently, the same group was able to demonstrate that due to inefficient exposure to two self-antigens, these bi-specific T cells managed to escape tolerization (284). Interestingly, there are only few reports of MOG-IgG/AQP4-IgG double positivity in MOGAD/NMOSD patients (285–287), and peripheral involvement in MOGAD is rarely reported (288).
The major drawback of TCR transgenic 2D2 mice and double transgenic OSE mice is that there is no complement deposition or granulocyte recruitment present (277, 279). Several humanized models have been established (265). It was shown in a transgenic mouse line that was generated to express human fragment crystallizable gamma receptors (hFcgRs) that recognize Immunoglobulin G antibodies, nicely reviewed in (289), that FcgRs but not complement activation contribute to EAE and that the exacerbation is dependent on MOG recognition by the human-derived antibodies (290). However, it is currently unclear which disease should be mimicked with this model, as it was shown that MS does not harbor anti-MOG autoantibodies and MOGAD probably has a complement-activating component driving lesion formation (13, 35) although the extent of complement involvement in human pathology is under debate.
Other transgenic mouse models investigated the relevance of IL-6, TH17 cells, oligodendrocytes, Nrf2, and CXCR3 (225, 227, 240, 291–293). The presence of MOG-IgG in MOGAD patients suggests B cell involvement that could be mirrored in several EAE models (294–297). SJL/J mice expressing a MOG92−106-specific transgenic TCR1640 with high frequency (99% proportion of transgenic Vα8.3+/Vβ4+CD4+ T cells) spontaneously produced pathogenic MOG-specific IgG1 antibodies (162).
4.7 MOG induced EAE in non-human primates
Different EAE models in monkeys have been reviewed elsewhere (298–300). EAE models developed in the rhesus macaque (Macaca mulatta) and the cynomolgus monkey (Macaca fascicularis) tend to replicate acute disseminated (leuko)encephalomyelitis well (301). In all non-human primates (NHP) the disease course varies with a more acute/relapsing or chronic disease course depending on the adjuvant used, complete or incomplete Freund's Adjuvants, respectively.
In the common marmoset monkey (Callithrix jacchus), extensive cortical demyelination could be induced upon immunization with rMOG1−125 and CFA (302). Lesions were dominated by macrophage/microglia activation and T cell infiltration (mostly perivascular) with few B cells, the cellular infiltrate was generally lower than in the parenchyma. Furthermore, IgG infiltration and complement deposition were observed. No subpial demyelination could be observed which is in contrast to patients with MOGAD, as well as in another study that observed subpial lesions in all experimental animals (303). Another study with marmoset monkeys immunized with rMOG1−125 and CFA found inflammatory lesions in cerebral white matter with some animals being affected also in the spinal cord and optic nerve. Lesion composition was similar with activated macrophage/microglia, T cell infiltrate, few B cells, IgG and complement deposition, and large confluent demyelinating lesions with some perivascular preference. The authors mentioned some axonal damage and indications for early remyelination (304). The encephalitogenic epitope inducing EAE in marmosets in mixed human myelin and CFA-induced immunization was shown to be MOG14−36 and not MBP (305); however, it could be shown that EAE could also be induced with myelin (from both WT and MOG−/− C57BL/6 mice) but severity/disease progression were dependent on the presence of MOG-IgG (306). IL17-A production was found to be elevated compared to IFN-γ when marmoset monkeys were challenged with synthetic MOG34−56 peptide alone (307), which is in line with the cytokine profile suggested in MOGAD; however, although treatment with an anti-IL17-A antibody delayed onset of EAE, it did not abrogate its development (308). Another study found elevated levels of IL-6, G-CSF, IL-8, and IFN- γ in cynomolgus macaques immunized with rhMOG and IFA which was similar to CSF analyzed from children with acquired autoimmune disease positive for anti-MOG antibodies who had elevated levels of IL-6 and G-CSF (309).
4.8 Infection-induced animal models—Are they relevant models for MOGAD?
MOGAD has been associated with preceding infection or vaccination (310, 311) in ~20% of cases although a causal relationship to any specific agent has not been discerned yet. Recently, cases of MOGAD after infection or vaccination with COVID-19 vaccines (both mRNA and vector-based) were reported, some with detectable persistent long-term MOG-IgG (311–323). Different types of coronaviruses have been used extensively to induce EAE, resembling different aspects of MS/MOGAD in different species over the last six to seven decades to just give a few examples (205, 324–329). Biphasic disease with a short fulminant acute phase and a 1-month long chronic phase characterized by ongoing inflammatory demyelination can develop in mice infected with Theiler's murine encephalomyelitis virus (TMEV), which is not the case in all species (330–332). Similar to MOGAD, TMEV infection in mice features perivascular immune cell infiltrates, leptomeningeal and white matter mononuclear cell infiltrates in the spinal cord, and primary demyelination around day 15 after viral intracerebral inoculation (333–336). Spontaneously occurring ADEM-like disease could be observed in a Japanese macaque (JM) colony at the Oregon National Primate Research Center (ONPRC) that has been linked to infection by a gamma-herpesvirus, JM rhadinovirus (JMRV) (337). A case report from Japan with high titer MOG-IgG links influenza-A infection to longitudinally extensive TM (338).
Besides M. tuberculosis (339) and Pertussis toxin (induces IL-6 and reduces Treg compartment) (340) that are usually used for immune stimulation to induce EAE in mice, other infectious agents have been used prior or post-immunization with MOG33−35 like SEB (341) or LPS (342), exacerbation of MOG-induced EAE by intraperitoneal injections of a viral mimetic, polyinosinic-polycytidylic acid (PIC) (343), Cytomegalovirus infection (344) which induces susceptibility to EAE in resistant BALB/c mice (345), Influenza virus infection (346) by enhanced type I T cell infiltration. 2′-5′ oligoadenylate synthetase-like 1 (OASL1) deficient (Oasl1−/−) mice are resistant to viral infections, as OASL1 specifically inhibits the translation of interferon regulatory factor 7 (IRF7), the master transcription factor for interferon-1 (IFN-I). Thus, IFN-I production is negatively regulated upon viral infection and (Oasl1−/−) mice seem to have an enhanced resistance toward MOG-induced EAE (347). Protective effects toward EAE were also shown in a model of sepsis (348) and some malaria strains (349). Interestingly, it could be seen in a study by Nourbakhsh et al. that predominantly children seronegative for EBV presented with MOG-IgG (44%) compared to only 5.5% MOG+ in EBV+ children (350); likewise, no correlation between MOG+/EBNA+ was found in children in another study (351), suggesting that if infectious agents were involved/associated in the development of both diseases, they would be distinct. Cases linking LETM to M. tuberculosis infection have been reported (352, 353). Molecular mimicry between MOG18−32 and Semliki Forest Virus (SFV) could be demonstrated after demyelination-inducing immunization of C57Bl6/J mice (354). Infection with S. pneumoniae was shown to upregulate IL-6 and TNF-α in mice immunized with MOG35−55 (355).
In several animal models [Brown Norway rats challenged with MOG (356), rats challenged with replication-deficient adenovirus vector carrying IL-1β cDNA (AdIL-1β) (357)] a beneficial effect on EAE outcome was demonstrated with IFN beta-1a. It was also demonstrated in a mouse model (TMEV-infected SJL/J mice) that a shorter duration of treatment was associated with remyelination, whereas long-term treatment seemingly promoted demyelination (358).
5 Preclinical imaging in MOGAD animal models
Many clinical imaging methods can be applied to preclinical research in animal models with minimal adaptations. Additional methods beyond what is possible in clinical research allow imaging with higher, up to single-cell, resolution and better labeling of key players in pathophysiological processes. There are several applications for preclinical imaging: Firstly, comparing imaging features between MOGAD and its potential animal models can be used to validate the model's suitability. Secondly, imaging can be useful in traditional animal research investigating disease cause and pathophysiology by allowing longitudinal high-resolution analyses and the definition of time points based on imaging features, thereby reducing the number of needed animals. Thirdly, it can aid image marker development: New imaging methods can be tested in animal models for potential clinical application, especially regarding their safety, sensitivity, and correlation with histological features. When clinically established imaging methods are used to describe new distinct features in a disease, assumptions are often made about their pathophysiological origin. By back-translating these imaging methods and findings into an animal model, these assumptions can be tested using histology or molecular analyses. Finally, during drug development and testing, translatable methods can be extremely useful since future clinical trial endpoints can already be tested early on.
5.1 Brain and brainstem
As described above, actively induced MOG35−55-EAE (induced by MOG35−55, CFA, and PT) only has a low affection of the brainstem and cerebellum and mostly absent inflammation and tissue damage in the forebrain. Although this picture closely resembles the brain involvement of many MOGAD patients, it limits the use of this model for the investigation of MOGAD brain lesions. Using T1-weighted imaging with contrast enhancement, the brain involvement in 2D2+ mice was also shown to be little or non-existent (359). Only actively induced MOG35−55-EAE (induced by MOG35−55, CFA, and PT) in non-obese diabetic (NOD) mice, a model with relapsing-remitting disease course, leads to MRI gadolinium-enhanced lesions in T1-weighted imaging, located in corpus callosum, fimbria, and internal capsule (268). Although promising, this lesion pattern is more in line with MS pathology. In common marmoset monkeys, MOG1−125-induced EAE causes small T2 hyperintensities within the white matter with histopathologically confirmed demyelination, which can subsequently develop into expanding confluent lesions. This model might be suitable to model MOG-IgG seropositive ADEM, but further confirmatory research is warranted (268, 360).
Absent microstructural brain damage in actively induced MOG35−55-EAE in C57BL/6 mice (induced by MOG35−55, CFA, and PT) was confirmed by a DTI study, which did not detect differences in DTI parameters of anterior commissure, corpus callosum, cerebral peduncle, and external capsule between MOG35−55-EAE and controls (361). Similarly, the application of magnetization transfer ratio (MTR), which is suggested to be a sensitive method to detect demyelination, did not find any changes in actively induced MOG1−125-EAE in C57BL/6 mice (induced by MOG1−125, CFA, and PT) in line with absent histopathological findings, which is in contrast to results in monkeys described above (362). No in-depth diffusion-weighted MR studies in people with MOGAD exist yet. Lesion load, volumetric analyses, and diffusion-weighted imaging have also been applied in the preclinical testing of new and established therapeutic agents (363–366). Although easily translatable into clinical research, one has to be aware that preclinical MRI markers are not well-validated in distinct models so far.
However, some clinical imaging features of MOGAD patients can be reproduced: using serial post-contrast FLAIR (fluid-attenuated inversion recovery) sequences after gadolinium administration in actively induced MOG35−55-EAE (induced by MOG35−55, CFA, and PT) in C57BL/6 mice, Pol and colleagues showed leptomeningeal contrast enhancement in all mice that decreased during the chronic stage and correlated with the leptomeningeal invasion of macrophages as well as T- and B-cells in histology, which elucidates the leptomeningeal enhancement described in many MOGAD patients (367). Furthermore, two studies investigated the use of superparamagnetic iron oxide-enhanced MRI in MOG-EAE rats, which were actively induced by recombinant human MOG in 1AV1 congenic Lewis rats, and showed a demarcation of lesions in the cerebellum, brainstem, and periventricular regions, which were corresponding to lesional iron-laden macrophages in histology, suggesting that superparamagnetic iron oxide-enhanced MRI might be useful for the detection and demarcation of inflammatory CNS lesions (368, 369).
In the process of developing new imaging methods, preclinical research can help to establish the pathophysiological grounds. Especially when developing methods with potential side effects for patients, such as testing new positron emission tomography (PET) tracers, prior extensive preclinical research is warranted. In the CNS, translocator protein (TSPO) is thought to be mainly expressed in activated microglia cells, and TSPO ligands have been used to detect inflammatory CNS processes. Widespread accumulation of two different TSPO ligands was shown in actively induced MOG35−55-EAE in C57BL/6 mice (induced by MOG35−55, CFA, and PT) with and without additional cuprizone treatment including the spinal cord, cerebellum, cortex, striatum, and hippocampus (370, 371). Neuropathological analyses confirmed microglial activation and were correlated with tracer uptake, thereby validating the method. In a similar approach, tracers for CD19 and the cystine/glutamate antiporter were validated in actively induced MOG-based animal models in C57BL/6 mice (Hoehne et al.: MOG35−55, CFA, PT, Stevens et al.: MOG1−125, CFA, PT) (372, 373). Fluorinated molecules might be another promising and non-toxic option for MR-detectable tracers to study neuroinflammation in the near future (374–378).
Going one step further, preclinical research allows more invasive imaging approaches with up to single-cell resolution such as real-time confocal imaging and two-photon excitation microscopy. The latter uses the simultaneous non-linear excitation by two photons of fluorophores to report on the sequential order and interaction of different key players during a pathological process. Particularly interesting is the application in adoptive transfer models using autofluorescent lymphocytes, which can then be tracked longitudinally throughout the disease. By transferring MOG-sensitized lymphocytes isolated from green fluorescent protein (GFP)-transgenic mice to C57BL/6 mice, Yura et al. were able to track widespread invasion of these GFP-labeled CD4+ in the brain and spinal cord using confocal imaging and detected nearly exclusive production of T helper cell type 1 using real-time PCR (379). In a different approach, Siffrin and colleagues used the actively induced MOG35−55-EAE model (induced by MOG35−55, CFA, and PT) in mice with enhanced GFP (eGFP) expression in neurons and neuronal processes and red fluorescent protein in bone marrow-derived peripheral immune cells, as well as adoptive transfer models (stimulation performed with MOG35−55), to investigate neuron-immune cell interaction and to show that Th17 cells induce early neuronal damage (380).
5.2 Spinal cord
So far, only a few studies implemented preclinical spinal cord MRI: T1-weighted imaging with contrast enhancement was used to characterize spinal cord involvement in 2D2+ mice showing enhancement in half of the mice that correlated with histologically confirmed immune cell infiltration (359). Employing in vivo lumbar DTI, axial and radial diffusivity changes in line with microstructural axonal and myelin pathology in the spinal cord, respectively, have been shown in actively induced MOG35−55-EAE in C57BL/6 mice (induced by MOG35−55, CFA, only) and in an adoptive transfer model of MOG-reactive TH1 cells in C57BL/6 mice (stimulated with MOG35−55) (381, 382). In both models, exploratory treatments were suggested to improve DTI parameters toward control values, pointing toward a relative sensitivity of these metrics.
Spinal cord MRI has also been performed in two studies ex vivo post-fixation, potentially limiting morphometric analyses (383). Derdelinckx and colleagues treated actively induced MOG35−55-EAE in C57BL/6 mice (induced by MOG35−55, CFA, and PT) with myelin antigen-presenting tolerogenic dendritic cells and observed a stabilized EAE disability score and an inhibited T cell response (32). In this study, ex vivo gadolinium-enhanced spinal cord MRI was implemented post-fixation to confirm a reduced lesion load after treatment and to localize lesional and non-lesional tissue for histological analyses (32). Cahill and colleagues developed a new PPARαmut/WT 2D2+ animal model with a mild relapsing-remitting disease course and increasing hind limb clasping during the disease process (384). Apart from histological analyses showing T cell and microglial activation as well as axonal and myelin damage at several locations in the brain, brainstem, spinal cord, and optic nerve, they also applied ex vivo post-fixation MRI analyses after 9 months to confirm spinal cord atrophy compared with 2D2− littermates (384). Neither study generated imaging data that can easily be transferred/translated into clinical application.
In one recent study using advanced preclinical imaging, two-photon excitation microscopy was applied to the spinal cord in actively induced MOG35−55-EAE (induced by MOG35−55-EAE, CFA and PT) for the first time: Steudler et al. used ODCmitoGFP-Tomato mice, which have GFP-labeled mitochondria in tdTomato-labeled oligodendrocytes (385). They applied two-photon excitation microscopy to reveal the complex evolution of the mitochondrial redox state with increased and decreased oxidation at the preclinical and chronic stages, respectively, suggesting an early involvement of oligodendrocyte mitochondria in the inflammatory process in EAE (385).
5.3 Retina and optic nerve
Many techniques investigating the visual system in patients can directly be translated to their application in animals with only minimal technical adaptations, for example, to correct for differences in refraction. When back-translating OCT imaging to rodents, the inner retinal layer (IRL) is usually quantified, instead of separating pRNFL and GCIPL, due to the lower retinal neuroaxonal content and lower resolution in mice. Cruz-Herranz et al. performed comparative OCT in different neuroinflammatory mouse models: Actively induced MOG35−55-EAE (induced by MOG35−55, CFA, and PT) in C57BL/6 mice led to severe thickening of the IRL with subsequent thinning; a 32% retinal ganglion cell loss within 120 days (54% in 9 months) and T cell and microglia invasion were later confirmed by histopathology (386). In contrast, actively induced MBP-EAE (induced by MBP, CFA, and PT) led to a much milder disease course with stable IRL measurements and no retinal ganglion cell loss. Active MOG35−55 -EAE induction in TCR2D2 mice (induced by MOG35−55, CFA, and PT) led to an earlier IRL thinning without edema, yet the extent (49% within 120 days) was nearly comparable with C57BL/6 mice after MOG35−55 -EAE induction (386). Uninduced TCR2D2 mice also underwent IRL thinning and thereby neurodegeneration within a 120-day period suggesting an underlying process in the mouse line (386). In a similar fashion, actively induced PLP139−151-EAE in SJL/J mice (induced by PLP139−151, CFA, and PT) led to IRL atrophy, yet wild-type uninduced SJL/J mice also showed IRL thinning (386). This is most likely due to a homozygous Pde6brd1 mutation for retinopathy these mice carry (386). In marmoset monkeys actively induced with MOG1−125-EAE (induced by recombinant rat MOG1−125 and CFA), only 50% have an ON at all (387). Taken together, Cruz-Herranz and other independent studies imply a strong resemblance of actively induced MOG35−55-EAE in C57BL/6 with adult MOGAD-ON, describing features such as early edema and severe neuroaxonal loss over an extended period after ON, while other models might closer resemble the milder course in MS-ON (386).
Actively induced MOG-EAE models gained further stand as a MOGAD model by a recent study confirming bilateral ON in 70% of MOG1−125-EAE in Brown Norway (BN) rats (induced by MOG1−125, CFA only) using visually evoked potentials (VEPs) (388). In Dark Agouti rats actively induced with MOG1−125-EAE (induced by MOG1−125, CFA only), a VEP latency delay could be observed even before first motor deficits were present, i.e., during an inflammatory state, demyelination and axonal loss were observed at later disease stages (389). Severe ON was caused in BN rats actively induced with the same model (390). Induced apoptosis of retinal ganglion cells (RGCs) in this model in BN rats could be seen as independent of optic nerve involvement (391). Two additional studies employing OCT and histopathology measured early, inflammation-preceding, RNFL thickness reduction in this actively induced MOG1−125-EAE model in BN rats (induced by MOG1−125, CFA only) (392, 393), respectively. Later, an increase in oligodendrocyte alphaB-crystallin, a heat-shock protein induced by cellular stress, was observed during the preclinical stages, particularly in the optic nerve head in this actively induced MOG1−125-EAE model in BN rats (induced by MOG1−125, CFA only) (394). This is in line with measurements gained in an MS study (395, 396). Contrary to these observations, RGC loss induced in C57/B6 mice by actively induced MOG35−55 –EAE (induced by MOG35−55, CFA, and PT) occurred only in late stages of the disease (post-immunization day 42), whereas CD4+Tcell infiltration, demyelination, microglial, and astrocyte activation were induced in the optic nerve by PID 16 (397). Further late events include degeneration of retinal neurites and synapses as well as glial cell activation in the inner retina. Similarly, in actively induced PLP139−151-EAE in SJL/J mice (induced by PLP139−151, CFA, and PT), RGC loss was detected by PID14, which in this model was however after cell infiltrates had been detected in the optic nerve around PID 9, pointing toward inflammation preceding RGC loss in this model (398).
As a potentially promising development for pediatric MOGAD-ON, the OSE model shows good results: OCT in OSE mice with spontaneous encephalomyelitis starting on day 26 after birth showed retinal neurodegeneration, which was confirmed by histopathology as 38% loss at 6 weeks of age (399). The functional relevance of RGC loss was confirmed by electroretinogram (ERG) (399).
Due to the close correlation between structural and functional metrics, multimodal assessment including OCT and functional assessments is common in rodents. Functional metrics back-translated from clinical applications include ERGs and VEPs, usually performed as flash-VEP. As a metric for vision in mice, the optomotor response (OMR) is assessed, which quantifies the compensatory head movement when the mouse is exposed to a moving light-dark pattern. Despite being the current gold standard for vision in mice, the OMR was critiqued for (A) the interference of vision and motor function, (B) the overlay with the optokinetic response, and (C) not depicting the (retina–lateral geniculate nucleus—primary visual cortex)-pathway usually associated with vision in humans. Outputs of VEP, ERG, and OMR have been shown to correlate very well with the neuroaxonal content measured by OCT and by histopathology, for example, in actively induced MOG35−55-EAE in mice (induced with MOG35−55, CFA, and PT) (400, 401).
Applying visual outcome parameters in animal research currently serves two major purposes: Firstly, we can use animal models to better understand the pathophysiological basis of our functional metrics. Recently, VEP became an outcome parameter for myelin in clinical trials investigating potentially remyelinating agents. Although the measurement of conduction speed seems like a feasible metric for myelin, the pathophysiological basis of this assumption was never validated and the sensitivity of VEPs for myelin content was never shown. Using different demyelinating animal models including actively induced MOG35−55-EAE in C57BL/6J mice (induced with MOG35−55, CFA, PT), Cordano and colleagues now demonstrated that quantitative measurements of myelination and remyelination correspond well with VEP latency, thereby validating it as a tool (402). This VEP change also correlates well with the dysregulation of potassium channels around the nodes of Ranvier as shown during inflammatory demyelination in actively induced MOG35 − 55−EAE (induced by MOG35−55, CFA, and PT) (403, 404). So far, only one VEP study has been performed in actively induced MOG1−125-EAE in marmoset monkeys (induced by rat recombinant MOG1−125, CFA only). Unfortunately, this study only reports a loss of amplitudes in line with neurodegeneration in the later course of the disease but does not report potential latency delays (405).
Secondly, functional outcome parameters can be used in animal research to show functional relevance very early in the development and testing of new therapeutic agents. The visual system is especially suitable for early drug testing for neuroprotective agents due to the clear association of one localized lesion in the optic nerve with subsequent neurodegeneration in the retina and functional decline (406–411). A single study also used the rodent visual system in actively induced MOG35−55-EAE transgenic mice backcrossed to a C57BL/6 background (induced with MOG35−55, CFA, and PT) to investigate the functional effects of remyelinating with the agent chloroindazole using VEP and ERG, yet the structure-function correlation was less robust (412). The only study so far using the rodent visual system in actively induced MOG35−55-EAE in mice (induced with MOG35−55, CFA, PT) to investigate the effects of anti-inflammatory treatment with anti-IL-17 antibodies showed that retinal neurodegeneration as measured by OCT, but not motor symptoms, was completely prevented by neutralizing IL-17 (413). This is particularly interesting since MOGAD patients were shown to have more IL-17-positive central memory cells than healthy controls with a particular increase in IL-17-positive IFN- γ positive central memory cells during relapses, again suggesting important parallels between MOG-EAE and MOGAD (282).
Optic nerve MRI using T1- and T2-weighted imaging has been validated in actively induced MOG35−55-EAE in C57Bl/6 mice (induced with MOG35−55, CFA, and PT) but sparsely performed (414). Qi et al. were able to establish volumetric optic nerve analysis using T1-weighted 3D 4.7-tesla MRI (415). As suggested from clinical experience, they were able to show significant optic nerve swelling and subsequent volume loss in an EAE model induced by CFA and homologous spinal cord emulsion. Reducing mitochondrial reactive oxygen stress by increasing SOD2 gene expression using virally mediated gene transfer led to less edema and prevented significant volume loss, which the analysis was sensitive enough to detect (415). The involvement of the optic nerve, optic tract, and chiasm was also shown for 2D2+ mice by contrast-enhanced T1-weighted imaging.
Interestingly, DTI has been applied to the visual system in rodents but not yet specifically in MOGAD patients. Manogaran et al. performed a multimodal study including OCT, T2-weighted imaging, and DTI in actively induced MOG35 − 55−EAE in C57BL/6J mice (induced by MOG35−55, CFA, and PT). They confirmed signal increase around the optic nerve in T2-weighted MRI in line with significant inflammation. DTI showed a decrease in axial diffusivity and an increase in radial diffusivity in the optic nerve and optic tract compared with controls. These changes were correlated with neuroaxonal parameters from OCT (416). DTI changes were confirmed by other independent studies in actively induced MOG35 − 55−EAE (induced by MOG35−55, CFA, and PT) (361, 417). A newer diffusion MRI approach called diffusion basis spectrum imaging (DBSI) was specifically developed to separate axonal and inflammatory pathologies. In its first application in actively induced MOG-EAE in C57BL/6J mice (induced by unspecified MOG peptide, CFA, and PT), the DBSI data suggest that axonal loss in ON occurs early and in parallel to the optic nerve edema (417). The application of DTI to the visual system in people with MOGAD is still pending.
The possibilities of retinal imaging in rodents exceed the options in clinical research. One example is confocal scanning laser ophthalmoscopy (CSLO), which is a non-invasive technique for real-time imaging of autofluorescent targets in the retina. In actively induced MOG35 − 55−EAE (induced by MOG35−55, CFA, and PT), CSLO has been applied to track myeloid cells in CX3CR1GFP/− mice (expressing a green fluorescent protein under control of the endogenous CX3C locus chemokine receptor 1) (418, 419). CSLO was then used to characterize microglial activation longitudinally during the course of actively induced MOG35 − 55−EAE and to define time points of maximum microglial activation for further analyses (418). In the long term, this imaging method might be used with different targets and animal models. The more invasive alternative with better single-cell tracking is two-photon excitation microscopy, which can nowadays also be co-registered with OCT (420). Yet, it has been so far only applied to actively induced experimental autoimmune uveitis in CX3CR1eGFP/− mice [induced by IRBP1−20 (interphotoreceptor retinoid-binding protein), CFA, and PT], an inflammation localized in the iris and ciliary body (421). Uveitis also occurs in MOGAD patients (422) and MOGAD-depicting animal models (292). Histopathological findings in uveitis are comparable in actively induced MOG35−55-EAE in (C57BL/6 x SJL) F1 and C57BL/6 mice (induced by MOG35−55, CFA, and PT) and mice with passive transfer of T cells specific to MOG35−55, suggesting a T-cell-mediated origin of autoimmune uveitis in MOGAD (423). Translational imaging including co-registered OCT and two-photon excitation microscopy can help to further elucidate the cause.
6 Concluding remarks
Separating MOGAD as a disease entity presents a unique challenge since researchers have investigated MOG-IgG-based animal models and MOG-IgG seropositive patients for decades as models for or as part of other conditions. This review is a first step toward understanding how the generated knowledge is specifically applicable to MOGAD. Translational imaging in MOGAD has provided useful information on disease pathophysiology, commonalities between animal models and disease, and potential imaging markers. Yet, true translational imaging research including clinical and preclinical aspects within the same study is still warranted. Also, many open questions remain such as: (1) Is the histopathology of the optic nerve and spinal cord comparable between MOG animal models and MOGAD patients (due to the lack of human pathology studies), and which would be the closest to reflect human disease? (2) What causes the gray matter involvement in MOGAD? (3) Is there a relevant portion of MOGAD patients developing a clinically progressive disease course and do we need a disease-specific definition of neuropathological progression? and (4) Should current treatment regimens for MOGAD be reevaluated because (A) no adverse events to, e.g., Fingolimod/Natalizumab (as seen in AQP4-IgG seropositive NMOSD) were observed in MOG-IgG seropositive patients (217) and (B) many treatments have been shown to be beneficial in MOG-induced EAE that are less used in or have been unsuccessful in MS (160, 423–425). In the future, translational and advanced imaging might provide answers to these questions and support the development of biomarkers for the diagnosis and monitoring of MOGAD.
Author contributions
FO and MH participated in the original conceptualization and initial draft of the manuscript. FP contributed to substantial revisions of the manuscript. All authors contributed to the revisions of the manuscript and approved the submitted version.
Funding
We thank the Sumaira Foundation for supporting this article collection.
Acknowledgments
FO thanks the American Academy of Neurology (AAN), the National Multiple Sclerosis Society (NMSS), and the Hertie Foundation for fellowship support.
Conflict of interest
FP reports research support from Bayer, Novartis, Biogen, Teva, Sanofi-Aventis/Genzyme, Alexion, Roche and Merck Serono and research support from the German Research Council, Werth Stiftung of the City of Cologne, German Ministry of Education and Research, Arthur Arnstein Stiftung Berlin, EU FP7 Framework Program, Guthy-Jackson Charitable Foundation, and NMSS. He also reports receiving consultation fees as an associate editor for Neurology, Neuroimmunology, and Neuroinflammation and as an academic editor for PLoS ONE and consultant fees for Sanofi Genzyme, Biogen, MedImmune, Shire, and Alexion. He also reports receiving speaker honoraria from Bayer, Novartis, Biogen, Teva, Sanofi-Aventis/Genzyme, Merck Serono, Alexion, Chugai, MedImmune, and Shire. He is an advisory board member for Novartis and MedImmune Scientific and holds stocks of Nocturne GmbH—all outside the submitted work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Brunner C, Lassmann H, Waehneldt TV, Matthieu JM, Linington C. Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2′,3′-cyclic nucleotide 3′-phosphodiesterase in the CNS of adult rats. J Neurochem. (1989) 52:296–304. doi: 10.1111/j.1471-4159.1989.tb10930.x
2. Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol. (2019) 15:89–102. doi: 10.1038/s41582-018-0112-x
3. Cobo-Calvo A, Ruiz A, Maillart E, Audoin B, Zephir H, Bourre B, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: the MOGADOR study. Neurology. (2018) 90:e1858–69. doi: 10.1212/WNL.0000000000005560
4. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. (2017) 140:3128–38. doi: 10.1093/brain/awx276
5. Cobo-Calvo Á, d'Indy H, Ruiz A, Collongues N, Kremer L, Durand-Dubief F, et al. Frequency of myelin oligodendrocyte glycoprotein antibody in multiple sclerosis: a multicenter cross-sectional study. Neurol Neuroimmunol Neuroinflamm. (2020) 7:649. doi: 10.1212/NXI.0000000000000649
6. Held F, Kalluri SR, Berthele A, Klein A-K, Reindl M, Hemmer B. Frequency of myelin oligodendrocyte glycoprotein antibodies in a large cohort of neurological patients. Mult Scler J Exp Transl Clin. (2021) 7:20552173211022770. doi: 10.1177/20552173211022767
7. Dinoto A, Licciardi N, Reindl M, Chiodega V, Schanda K, Carta S, et al. Peripheral neuropathy and MOG-IgG: a clinical and neuropathological retrospective study. Eur J Neurol. (2022) 29:237. doi: 10.1016/j.msard.2022.104214
8. Dinoto A, Licciardi NM, Reindl M, Chiodega V, Schanda K, Carta S, et al. Myelin oligodendrocyte glycoprotein antibodies and peripheral neuropathies: a clinical and neuropathological retrospective study. J Peripher Nerv Syst. (2022) 27:S13.
9. Amin M, Mays M, Polston D, Flanagan EP, Prayson R, Kunchok A. Myelin oligodendrocyte glycoprotein (MOG) antibodies in a patient with glioblastoma: red flags for false positivity. J Neuroimmunol. (2021) 361:577743. doi: 10.1016/j.jneuroim.2021.577743
10. Uzura Y, Takeuchi H, Ashida S, Fujii C, Shishido-Hara Y, Inaba T, et al. A tumefactive anti-MOG antibody associated disorder heralding central nervous system B-cell lymphoma: case report on diagnostic challenge. J Neuroimmunol. (2022) 365:577823. doi: 10.1016/j.jneuroim.2022.577823
11. Cai MT, Lai QL, Tang JL, Du BQ, Shen CH, Zhang YX, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease preceding primary central nervous system lymphoma: causality or coincidence? Neurol Sci. (2023) doi: 10.1007/s10072-023-06919-1
12. Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: international MOGAD Panel proposed criteria. Lancet Neurol. (2023) 22:268–82. doi: 10.1016/S1474-4422(22)00431-8
13. Höftberger R, Guo Y, Flanagan EP, Lopez-Chiriboga AS, Endmayr V, Hochmeister S, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. (2020) 139:875–92. doi: 10.1007/s00401-020-02132-y
14. Reindl M, Schanda K, Woodhall M, Tea F, Ramanathan S, Sagen J, et al. International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm. (2020) 7:e674. doi: 10.1212/NXI.0000000000000674
15. Jarius S, Paul F, Aktas O, Asgari N, Dale RC, de Seze J, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation. (2018) 15:134. doi: 10.1186/s12974-018-1144-2
16. Waters P, Fadda G, Woodhall M, O'Mahony J, Brown RA, Castro DA, et al. Serial anti–myelin oligodendrocyte glycoprotein antibody analyses and outcomes in children with demyelinating syndromes. JAMA Neurol. (2020) 77:2940. doi: 10.1001/jamaneurol.2019.2940
17. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. (2018) 17:162–73. doi: 10.1016/S1474-4422(17)30470-2
18. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
19. Takai Y, Misu T, Kaneko K, Chihara N, Narikawa K, Tsuchida S, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain. (2020) 143:1431–46. doi: 10.1093/brain/awaa102
20. Spadaro M, Gerdes LA, Mayer MC, Ertl-Wagner B, Laurent S, Krumbholz M, et al. Histopathology and clinical course of MOG-antibody-associated encephalomyelitis. Ann Clin Transl Neurol. (2015) 2:295–301. doi: 10.1002/acn3.164
21. Hochmeister S, Gattringer T, Asslaber M, Stangl V, Haindl MT, Enzinger C, et al. A fulminant case of demyelinating encephalitis with extensive cortical involvement associated with anti-MOG antibodies. Front Neurol. (2020) 11:e00031. doi: 10.3389/fneur.2020.00031
22. Carta S, Höftberger R, Bolzan A, Bozzetti S, Bonetti B, Scarpelli M, et al. Antibodies to MOG in CSF only: pathological findings support the diagnostic value. Acta Neuropathol. (2021) 141:801–4. doi: 10.1007/s00401-021-02286-3
23. Nakajima H, Motomura M, Tanaka K, Ichinose K, Kawakami A, Tsujino A. Comprehensive cytokine profile in optic neuritis with antibodies to myelin oligodendrocyte glycoprotein. J Neurol Sci. (2017) 381:789–789. doi: 10.1016/j.jns.2017.08.2227
24. Horellou P, Wang M, Keo V, Chretien P, Serguera C, Waters P, et al. Increased interleukin-6 correlates with myelin oligodendrocyte glycoprotein antibodies in pediatric monophasic demyelinating diseases and multiple sclerosis. J Neuroimmunol. (2015) 289:1–7. doi: 10.1016/j.jneuroim.2015.10.002
25. Bauer A, Rudzki D, Berek K, Dinoto A, Lechner C, Wendel EM, et al. Increased peripheral inflammatory responses in myelin oligodendrocyte glycoprotein associated disease and aquaporin-4 antibody positive neuromyelitis optica spectrum disorder. Front Immunol. (2022) 13:1037812. doi: 10.3389/fimmu.2022.1037812
26. Kitley J, Waters P, Woodhall M, Leite MI, Murchison A, George J, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol. (2014) 71:276–83. doi: 10.1001/jamaneurol.2013.5857
27. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation. (2016) 13:280. doi: 10.1186/s12974-016-0718-0
28. Rinaldi S, Davies A, Fehmi J, Beadnall HN, Wang J, Hardy TA, et al. Overlapping central and peripheral nervous system syndromes in MOG antibody-associated disorders. Neurol Neuroimmunol Neuroinflamm. (2021) 8:924. doi: 10.1212/NXI.0000000000000924
29. Hamid SHM, Whittam D, Saviour M, Alorainy A, Mutch K, Linaker S, et al. Seizures and encephalitis in myelin oligodendrocyte glycoprotein IgG disease vs. aquaporin 4 IgG disease. JAMA Neurol. (2018) 75:65–71. doi: 10.1001/jamaneurol.2017.3196
30. Valencia-Sanchez C, Guo Y, Krecke KN, Chen JJ, Redenbaugh V, Montalvo M, et al. Cerebral cortical encephalitis in myelin oligodendrocyte glycoprotein antibody-associated disease. Ann Neurol. (2023) 93:297–302. doi: 10.1002/ana.26549
31. Armangue T, Olivé-Cirera G, Martínez-Hernandez E, Sepulveda M, Ruiz-Garcia R, Muñoz-Batista M, et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: a multicentre observational study. Lancet Neurol. (2020) 19:234–46. doi: 10.1016/S1474-4422(19)30488-0
32. Derdelinckx J, Mansilla MJ, De Laere M, Lee W-P, Navarro-Barriuso J, Wens I, et al. Clinical and immunological control of experimental autoimmune encephalomyelitis by tolerogenic dendritic cells loaded with MOG-encoding mRNA. J Neuroinflammation. (2019) 16:167. doi: 10.1186/s12974-019-1541-1
33. O'Connell K, Hamilton-Shield A, Woodhall M, Messina S, Mariano R, Waters P, et al. Prevalence and incidence of neuromyelitis optica spectrum disorder, aquaporin-4 antibody-positive NMOSD and MOG antibody-positive disease in Oxfordshire, UK. J Neurol Neurosurg Psychiatry. (2020) 91:1126–8. doi: 10.1136/jnnp-2020-323158
34. Hennes EM, Baumann M, Schanda K, Anlar B, Bajer-Kornek B, Blaschek A, et al. Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology. (2017) 89:900–8. doi: 10.1212/WNL.0000000000004312
35. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: Frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflammation. (2016) 13:279. doi: 10.1186/s12974-016-0717-1
36. Jitprapaikulsan J, Chen JJ, Flanagan EP, Tobin WO, Fryer JP, Weinshenker BG, et al. Aquaporin-4 and myelin oligodendrocyte glycoprotein autoantibody status predict outcome of recurrent optic neuritis. Ophthalmology. (2018) 125:1628–37. doi: 10.1016/j.ophtha.2018.03.041
37. Cobo-Calvo A, Ruiz A, Rollot F, Arrambide G, Deschamps R, Maillart E, et al. Clinical features and risk of relapse in children and adults with myelin oligodendrocyte glycoprotein antibody-associated disease. Ann Neurol. (2021) 89:30–41. doi: 10.1002/ana.25909
38. Levy M, Molazadeh N, Bilodeau PA, Vishnevetsky A, Lotan I, Salky R, et al. Multiple types of relapses in MOG antibody disease. Multiple Scler Relat Disord. (2023) 72:104613. doi: 10.1016/j.msard.2023.104613
39. Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. (2018) 89:127–37. doi: 10.1136/jnnp-2017-316880
40. Akaishi T, Misu T, Fujihara K, Takahashi T, Takai Y, Nishiyama S, et al. Relapse activity in the chronic phase of anti-myelin-oligodendrocyte glycoprotein antibody-associated disease. J Neurol. (2022) 269:3136–46. doi: 10.1007/s00415-021-10914-x
41. Akaishi T, Misu T, Takahashi T, Takai Y, Nishiyama S, Fujimori J, et al. Progression pattern of neurological disability with respect to clinical attacks in anti-MOG antibody-associated disorders. J Neuroimmunol. (2021) 351:577467. doi: 10.1016/j.jneuroim.2020.577467
42. López-Chiriboga AS, Majed M, Fryer J, Dubey D, McKeon A, Flanagan EP, et al. Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG-associated disorders. JAMA Neurol. (2018) 75:1355–63. doi: 10.1001/jamaneurol.2018.1814
43. Hacohen Y, Wong YY, Lechner C, Jurynczyk M, Wright S, Konuskan B, et al. Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol. (2018) 75:478–87. doi: 10.1001/jamaneurol.2017.4601
44. Deschamps R, Pique J, Ayrignac X, Collongues N, Audoin B, Zéphir H, et al. The long-term outcome of MOGAD: an observational national cohort study of 61 patients. Eur J Neurol. (2021) 28:1659–64. doi: 10.1111/ene.14746
45. Cobo-Calvo A, Sepúlveda M, d'Indy H, Armangué T, Ruiz A, Maillart E, et al. Usefulness of MOG-antibody titres at first episode to predict the future clinical course in adults. J Neurol. (2019) 266:806–15. doi: 10.1007/s00415-018-9160-9
46. Molazadeh N, Filippatou AG, Vasileiou ES, Levy M, Sotirchos ES. Evidence for and against subclinical disease activity and progressive disease in MOG antibody disease and neuromyelitis optica spectrum disorder. J Neuroimmunol. (2021) 360:577702. doi: 10.1016/j.jneuroim.2021.577702
47. Hacohen Y, Rossor T, Mankad K, Chong WK, Lux A, Wassmer E, et al. “Leukodystrophy-like” phenotype in children with myelin oligodendrocyte glycoprotein antibody-associated disease. Dev Med Child Neurol. (2018) 60:417–23. doi: 10.1111/dmcn.13649
48. Jurynczyk M, Geraldes R, Probert F, Woodhall MR, Waters P, Tackley G, et al. Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis. Brain. (2017) 140:617–27. doi: 10.1093/brain/aww350
49. Camera V, Holm-Mercer L, Ali AAH, Messina S, Horvat T, Kuker W, et al. Frequency of new silent MRI lesions in myelin oligodendrocyte glycoprotein antibody disease and aquaporin-4 antibody neuromyelitis optica spectrum disorder. JAMA Network Open. (2021) 4:e2137833. doi: 10.1001/jamanetworkopen.2021.37833
50. Sinha S, Banwell B, Tucker A, Storm PB, Huh J, Lang S-S. Hemicraniectomy and externalized ventricular drain placement in a pediatric patient with myelin oligodendrocyte glycoprotein-associated tumefactive demyelinating disease. Childs Nerv Syst. (2022) 38:185–9. doi: 10.1007/s00381-021-05139-2
51. Matthews L, Marasco R, Jenkinson M, Küker W, Luppe S, Leite MI, et al. Distinction of seropositive NMO spectrum disorder and MS brain lesion distribution. Neurology. (2013) 80:1330–7. doi: 10.1212/WNL.0b013e3182887957
52. Juryńczyk M, Tackley G, Kong Y, Geraldes R, Matthews L, Woodhall M, et al. Brain lesion distribution criteria distinguish MS from AQP4-antibody NMOSD and MOG-antibody disease. J Neurol Neurosurg Psychiatry. (2017) 88:132–6. doi: 10.1136/jnnp-2016-314005
53. Jarius S, Kleiter I, Ruprecht K, Asgari N, Pitarokoili K, Borisow N, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 3: Brainstem involvement - frequency, presentation and outcome. J Neuroinflammation. (2016) 13:281. doi: 10.1186/s12974-016-0719-z
54. Banks SA, Morris PP, Chen JJ, Pittock SJ, Sechi E, Kunchok A, et al. Brainstem and cerebellar involvement in MOG-IgG-associated disorder versus aquaporin-4-IgG and MS. J Neurol Neurosurg Psychiatry. (2020) jnnp-2020-325121. doi: 10.1136/jnnp-2020-325121
55. Salama S, Khan M, Shanechi A, Levy M, Izbudak I. MRI differences between MOG antibody disease and AQP4 NMOSD. Mult Scler. (2020) 26:1854–65. doi: 10.1016/j.msard.2019.11.041
56. Hyun J-W, Kwon YN, Kim S-M, Lee HL, Jeong WK, Lee HJ, et al. Value of area postrema syndrome in differentiating adults with AQP4 vs. MOG antibodies. Front Neurol. (2020) 11:e00396. doi: 10.3389/fneur.2020.00396
57. Kunchok A, Krecke KN, Flanagan EP, Jitprapaikulsan J, Lopez-Chiriboga AS, Chen JJ, et al. Does area postrema syndrome occur in myelin oligodendrocyte glycoprotein-IgG-associated disorders (MOGAD)? Neurology. (2020) 94:85–8. doi: 10.1212/WNL.0000000000008786
58. Chen L, Chen C, Zhong X, Sun X, Zhu H, Li X, et al. Different features between pediatric-onset and adult-onset patients who are seropositive for MOG-IgG: a multicenter study in South China. J Neuroimmunol. (2018) 321:83–91. doi: 10.1016/j.jneuroim.2018.05.014
59. Baumann M, Grams A, Djurdjevic T, Wendel E-M, Lechner C, Behring B, et al. MRI of the first event in pediatric acquired demyelinating syndromes with antibodies to myelin oligodendrocyte glycoprotein. J Neurol. (2018) 265:845–55. doi: 10.1007/s00415-018-8781-3
60. Baumann M, Sahin K, Lechner C, Hennes EM, Schanda K, Mader S, et al. Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein. J Neurol Neurosurg Psychiatry. (2015) 86:265–72. doi: 10.1136/jnnp-2014-308346
61. Cobo-Calvo Á, Ruiz A, D'Indy H, Poulat A-L, Carneiro M, Philippe N, et al. MOG antibody-related disorders: common features and uncommon presentations. J Neurol. (2017) 264:1945–55. doi: 10.1007/s00415-017-8583-z
62. Wegener-Panzer A, Cleaveland R, Wendel E-M, Baumann M, Bertolini A, Häusler M, et al. Clinical and imaging features of children with autoimmune encephalitis and MOG antibodies. Neurology Neuroimmunol Neuroinflamm. (2020) 7:731. doi: 10.1212/NXI.0000000000000731
63. Bartels F, Krohn S, Nikolaus M, Johannsen J, Wickström R, Schimmel M, et al. Clinical and magnetic resonance imaging outcome predictors in pediatric anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol. (2020) 88:148–59. doi: 10.1002/ana.25754
64. Ogawa R, Nakashima I, Takahashi T, Kaneko K, Akaishi T, Takai Y, et al. MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e322. doi: 10.1212/NXI.0000000000000322
65. Budhram A, Mirian A, Le C, Hosseini-Moghaddam SM, Sharma M, Nicolle MW. Unilateral cortical FLAIR-hyperintense Lesions in Anti-MOG-associated Encephalitis with Seizures (FLAMES): characterization of a distinct clinico-radiographic syndrome. J Neurol. (2019) 266:2481–7. doi: 10.1007/s00415-019-09440-8
66. Schmidt FA, Chien C, Kuchling J, Bellmann-Strobl J, Ruprecht K, Siebert N, et al. Differences in advanced magnetic resonance imaging in MOG-IgG and AQP4-IgG seropositive neuromyelitis optica spectrum disorders: a comparative study. Front Neurol. (2020) 11:499910. doi: 10.3389/fneur.2020.499910
67. Sun M, Zhou H, Xu Q, Yang M, Xu X, Zhou M, et al. Differential patterns of interhemispheric functional connectivity between AQP4-optic neuritis and MOG-optic neuritis: a resting-state functional MRI study. Acta Radiol. (2021) 62:776–83. doi: 10.1177/0284185120940250
68. Yang L, Li H, Xia W, Quan C, Zhou L, Geng D, et al. Quantitative brain lesion distribution may distinguish MOG-ab and AQP4-ab neuromyelitis optica spectrum disorders. Eur Radiol. (2020) 30:1470–9. doi: 10.1007/s00330-019-06506-z
69. Bartels F, Baumgartner B, Aigner A, Cooper G, Blaschek A, Wendel EM, et al. Impaired brain growth in myelin oligodendrocyte glycoprotein antibody-associated acute disseminated encephalomyelitis. Neurol Neuroimmunol Neuroinflamm. (2023) 10:e200066. doi: 10.1212/NXI.0000000000200066
70. Transverse Myelitis Consortium Working Group. Proposed diagnostic criteria and nosology of acute transverse myelitis. Neurology. (2002) 59:499–505. doi: 10.1212/WNL.59.4.499
71. Lopez-Chiriboga AS, Sechi E, Buciuc M, Chen JJ, Pittock SJ, Lucchinetti CF, et al. Long-term outcomes in patients with myelin oligodendrocyte glycoprotein immunoglobulin G–associated disorder. JAMA Neurol. (2020) 77:1575–7. doi: 10.1001/jamaneurol.2020.3115
72. Fadda G, Alves CA, O'Mahony J, Castro DA, Yeh EA, Marrie RA, et al. Comparison of spinal cord magnetic resonance imaging features among children with acquired demyelinating syndromes. JAMA Netw Open. (2021) 4:e2128871. doi: 10.1001/jamanetworkopen.2021.28871
73. Sechi E, Krecke KN, Pittock SJ, Dubey D, Lopez-Chiriboga AS, Kunchok A, et al. Frequency and characteristics of MRI-negative myelitis associated with MOG autoantibodies. Mult Scler. (2021) 27:303–8. doi: 10.1177/1352458520907900
74. Jitprapaikulsan J, Lopez Chiriboga AS, Flanagan EP, Fryer JP, McKeon A, Weinshenker BG, et al. Novel glial targets and recurrent longitudinally extensive transverse myelitis. JAMA Neurol. (2018) 75:892–5. doi: 10.1001/jamaneurol.2018.0805
75. Dubey D, Pittock SJ, Krecke KN, Morris PP, Sechi E, Zalewski NL, et al. Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol. (2019) 76:301. doi: 10.1001/jamaneurol.2018.4053
76. Cobo-Calvo Á, Sepúlveda M, Bernard-Valnet R, Ruiz A, Brassat D, Martínez-Yélamos S, et al. Antibodies to myelin oligodendrocyte glycoprotein in aquaporin 4 antibody seronegative longitudinally extensive transverse myelitis: clinical and prognostic implications. Mult Scler. (2016) 72:187–93. doi: 10.1177/1352458515591071
77. Ciccarelli O, Cohen JA, Reingold SC, Weinshenker BG International International Conference on Spinal Cord Involvement and Imaging in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorders. Spinal cord involvement in multiple sclerosis and neuromyelitis optica spectrum disorders. Lancet Neurol. (2019) 18:185–97. doi: 10.1016/S1474-4422(18)30460-5
78. Asnafi S, Morris PP, Sechi E, Pittock SJ, Weinshenker BG, Palace J, et al. The frequency of longitudinally extensive transverse myelitis in MS: a population-based study. Mult Scler Relat Disord. (2020) 37:101487. doi: 10.1016/j.msard.2019.101487
79. Mariano R, Messina S, Kumar K, Kuker W, Leite MI, Palace J. Comparison of clinical outcomes of transverse myelitis among adults with myelin oligodendrocyte glycoprotein antibody vs. aquaporin-4 antibody disease. JAMA Network Open. (2019) 2:e1912732. doi: 10.1001/jamanetworkopen.2019.12732
80. Etemadifar M, Salari M, Kargaran PK, Sigari AA, Nouri H, Etemadifar F, et al. Conus medullaris involvement in demyelinating disorders of the CNS: a comparative study. Mult Scler Relat Disord. (2021) 54:103127. doi: 10.1016/j.msard.2021.103127
81. ZhangBao J, Huang W, Zhou L, Wang L, Chang X, Lu C, et al. Myelitis in inflammatory disorders associated with myelin oligodendrocyte glycoprotein antibody and aquaporin-4 antibody: a comparative study in Chinese Han patients. Eur J Neurol. (2021) 28:1308–15. doi: 10.1111/ene.14654
82. Ciron J, Cobo-Calvo A, Audoin B, Bourre B, Brassat D, Cohen M, et al. Frequency and characteristics of short versus longitudinally extensive myelitis in adults with MOG antibodies: a retrospective multicentric study. Mult Scler. (2020) 26:936–44. doi: 10.1177/1352458519849511
83. Johnson TW, Wu Y, Nathoo N, Rogers JA, Yong VW, Dunn JF. Gray matter hypoxia in the brain of the experimental autoimmune encephalomyelitis model of multiple sclerosis. PLoS ONE. (2016) 11:e0167196. doi: 10.1371/journal.pone.0167196
84. Calabrese M, Magliozzi R, Ciccarelli O, Geurts JJ, Reynolds R, Martin R. Exploring the origins of grey matter damage in multiple sclerosis. Nat Rev Neurosci. (2015) 16:147–58. doi: 10.1038/nrn3900
85. Geurts JJ, Barkhof F. Grey matter pathology in multiple sclerosis. Lancet Neurol. (2008) 7:841–51. doi: 10.1016/S1474-4422(08)70191-1
86. Chien C, Scheel M, Schmitz-Hübsch T, Borisow N, Ruprecht K, Bellmann-Strobl J, et al. Spinal cord lesions and atrophy in NMOSD with AQP4-IgG and MOG-IgG associated autoimmunity. Mult Scler. (2019) 25:1926–36. doi: 10.1177/1352458518815596
87. Mariano R, Messina S, Roca-Fernandez A, Leite MI, Kong Y, Palace JA. Quantitative spinal cord MRI in MOG-antibody disease, neuromyelitis optica and multiple sclerosis. Brain. (2021) 144:198–212. doi: 10.1093/brain/awaa347
88. Lotan I, Oertel FC, Chien C, Asseyer S, Paul F, Stiebel-Kalish H. Practical recognition tools of immunoglobulin G serum antibodies against the myelin oligodendrocyte glycoprotein-positive optic neuritis and its clinical implications. Clin Exp Neuroimmunol. (2021) 12:42–53. doi: 10.1111/cen3.12623
89. Graves JS, Oertel FC, Van der Walt A, Collorone S, Sotirchos ES, Pihl-Jensen G, et al. Leveraging visual outcome measures to advance therapy development in neuroimmunologic disorders. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e1126. doi: 10.1212/NXI.0000000000001126
90. Ducloyer J-B, Caignard A, Aidaoui R, Ollivier Y, Plubeau G, Santos-Moskalyk S, et al. MOG-Ab prevalence in optic neuritis and clinical predictive factors for diagnosis. Br J Ophthalmol. (2020) 104:842–5. doi: 10.1136/bjophthalmol-2019-314845
91. Ramanathan S, Reddel SW, Henderson A, Parratt JDE, Barnett M, Gatt PN, et al. Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm. (2014) 1:40. doi: 10.1212/NXI.0000000000000040
92. Chen JJ, Flanagan EP, Jitprapaikulsan J, López-Chiriboga ASS, Fryer JP, Leavitt JA, et al. Myelin oligodendrocyte glycoprotein antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. (2018) 195:8–15. doi: 10.1016/j.ajo.2018.07.020
93. Rempe T, Tarhan B, Rodriguez E, Viswanathan VT, Gyang TV, Carlson A, et al. Anti-MOG associated disorder-Clinical and radiological characteristics compared to AQP4-IgG+ NMOSD-A single-center experience. Mult Scler Relat Disord. (2021) 48:102718. doi: 10.1016/j.msard.2020.102718
94. Ramanathan S, Fraser C, Curnow SR, Ghaly M, Leventer RJ, Lechner-Scott J, et al. Uveitis and optic perineuritis in the context of myelin oligodendrocyte glycoprotein antibody seropositivity. Eur J Neurol. (2019) 26:1137–e75. doi: 10.1111/ene.13932
95. Shor N, Aboab J, Maillart E, Lecler A, Bensa C, Le Guern G, et al. Clinical, imaging and follow-up study of optic neuritis associated with myelin oligodendrocyte glycoprotein antibody: a multicentre study of 62 adult patients. Eur J Neurol. (2020) 27:384–91. doi: 10.1111/ene.14089
96. Ramanathan S, Prelog K, Barnes EH, Tantsis EM, Reddel SW, Henderson AP, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler. (2016) 22:470–82. doi: 10.1177/1352458515593406
97. Tajfirouz D, Padungkiatsagul T, Beres S, Moss HE, Pittock S, Flanagan E, et al. Optic chiasm involvement in AQP-4 antibody–positive NMO and MOG antibody–associated disorder. Mult Scler. (2022) 28:149–53. doi: 10.1177/13524585211011450
98. Yang M, Wu Y, Lai M, Song H, Li H, Sun M, et al. Clinical predictive factors for diagnosis of MOG-IgG and AQP4-IgG related paediatric optic neuritis: a Chinese cohort study. Br J Ophthalmol. (2022) 106:262–6. doi: 10.1136/bjophthalmol-2020-317524
99. Oertel FC, Zimmermann H, Paul F, Brandt AU. Optical coherence tomography in neuromyelitis optica spectrum disorders: potential advantages for individualized monitoring of progression and therapy. EPMA J. (2018) 9:21–33. doi: 10.1007/s13167-017-0123-5
100. Petzold A, Balcer LJ, Calabresi PA, Costello F, Frohman TC, Frohman EM, et al. Retinal layer segmentation in multiple sclerosis: a systematic review and meta-analysis. Lancet Neurol. (2017) 16:797–812. doi: 10.1016/S1474-4422(17)30278-8
101. Chen JJ, Sotirchos ES, Henderson AD, Vasileiou ES, Flanagan EP, Bhatti MT, et al. OCT retinal nerve fiber layer thickness differentiates acute optic neuritis from MOG antibody-associated disease and multiple sclerosis: RNFL thickening in acute optic neuritis from MOGAD vs. MS. Mult Scler Relat Disord. (2022) 58:103525. doi: 10.1016/j.msard.2022.103525
102. Oertel FC, Sotirchos ES, Zimmermann HG, Motamedi S, Specovius S, Asseyer ES, et al. Longitudinal retinal changes in MOGAD. Ann Neurol. (2022) 92:476–85. doi: 10.1002/ana.26440
103. Oertel FC, Outteryck O, Knier B, Zimmermann H, Borisow N, Bellmann-Strobl J, et al. Optical coherence tomography in myelin-oligodendrocyte-glycoprotein antibody-seropositive patients: a longitudinal study. J Neuroinflammation. (2019) 16:154. doi: 10.1186/s12974-019-1521-5
104. Petzold A, Fraser CL, Abegg M, Alroughani R, Alshowaeir D, Alvarenga R, et al. Diagnosis and classification of optic neuritis. Lancet Neurol. (2022) 21:1120–34. doi: 10.1016/S1474-4422(22)00200-9
105. Coric D, Balk LJ, Uitdehaag BMJ, Petzold A. Diagnostic accuracy of optical coherence tomography inter-eye percentage difference for optic neuritis in multiple sclerosis. Eur J Neurol. (2017) 24:1479–84. doi: 10.1111/ene.13443
106. Bsteh G, Hegen H, Altmann P, Auer M, Berek K, Zinganell A, et al. Validation of inter-eye difference thresholds in optical coherence tomography for identification of optic neuritis in multiple sclerosis. Mult Scler Relat Disord. (2020) 45:102403. doi: 10.1016/j.msard.2020.102403
107. Nij Bijvank J, Uitdehaag BMJ, Petzold A. Retinal inter-eye difference and atrophy progression in multiple sclerosis diagnostics. J Neurol Neurosurg Psychiatry. (2022) 93:216–9. doi: 10.1136/jnnp-2021-327468
108. Oertel FC, Zimmermann HG, Motamedi S, Chien C, Aktas O, Albrecht P, et al. Diagnostic value of intereye difference metrics for optic neuritis in aquaporin-4 antibody seropositive neuromyelitis optica spectrum disorders. J Neurol Neurosurg Psychiatry. (2023) 94:560–66. doi: 10.1136/jnnp-2022-330608
109. Pache F, Zimmermann H, Mikolajczak J, Schumacher S, Lacheta A, Oertel FC, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 4: Afferent visual system damage after optic neuritis in MOG-IgG-seropositive versus AQP4-IgG-seropositive patients. J Neuroinflammation. (2016) 13:282. doi: 10.1186/s12974-016-0720-6
110. Stiebel-Kalish H, Lotan I, Brody J, Chodick G, Bialer O, Marignier R, et al. Retinal nerve fiber layer may be better preserved in MOG-IgG versus AQP4-IgG optic neuritis: a cohort study. PLoS ONE. (2017) 12:e0170847. doi: 10.1371/journal.pone.0170847
111. Gigengack NK, Oertel FC, Motamedi S, Bereuter C, Duchow A, Rust R, et al. Structure-function correlates of vision loss in neuromyelitis optica spectrum disorders. Sci Rep. (2022) 12:17545. doi: 10.1038/s41598-022-19848-4
112. Schmidt F, Zimmermann H, Mikolajczak J, Oertel FC, Pache F, Weinhold M, et al. Severe structural and functional visual system damage leads to profound loss of vision-related quality of life in patients with neuromyelitis optica spectrum disorders. Mult Scler Relat Disord. (2017) 11:45–50. doi: 10.1016/j.msard.2016.11.008
113. Sotirchos ES, Filippatou A, Fitzgerald KC, Salama S, Pardo S, Wang J, et al. Aquaporin-4 IgG seropositivity is associated with worse visual outcomes after optic neuritis than MOG-IgG seropositivity and multiple sclerosis, independent of macular ganglion cell layer thinning. Mult Scler. (2020) 26:1360–71. doi: 10.1177/1352458519864928
114. Zhao Y, Tan S, Chan TCY, Xu Q, Zhao J, Teng D, et al. Clinical features of demyelinating optic neuritis with seropositive myelin oligodendrocyte glycoprotein antibody in Chinese patients. Br J Ophthalmol. (2018) 102:1372–7. doi: 10.1136/bjophthalmol-2017-311177
115. Havla J, Pakeerathan T, Schwake C, Bennett JL, Kleiter I, Felipe-Rucián A, et al. Age-dependent favorable visual recovery despite significant retinal atrophy in pediatric MOGAD: how much retina do you really need to see well? J Neuroinflammation. (2021) 18:121. doi: 10.1186/s12974-021-02160-9
116. Zimmermann HG, Knier B, Oberwahrenbrock T, Behrens J, Pfuhl C, Aly L, et al. Association of retinal ganglion cell layer thickness with future disease activity in patients with clinically isolated syndrome. JAMA Neurol. (2018) 75:1071–9. doi: 10.1001/jamaneurol.2018.1011
117. Cordano C, Nourbakhsh B, Yiu HH, Papinutto N, Caverzasi E, Abdelhak AC, et al. Differences in age-related retinal and cortical atrophy rates in multiple sclerosis. Neurology. (2022) 99:e1685–93. doi: 10.1212/WNL.0000000000200977
118. Petracca M, Cordano C, Cellerino M, Button J, Krieger S, Vancea R, et al. Retinal degeneration in primary-progressive multiple sclerosis: a role for cortical lesions? Mult Scler. (2017) 23:43–50. doi: 10.1177/1352458516637679
119. Martinez-Lapiscina EH, Arnow S, Wilson JA, Saidha S, Preiningerova JL, Oberwahrenbrock T, et al. Retinal thickness measured with optical coherence tomography and risk of disability worsening in multiple sclerosis: a cohort study. Lancet Neurol. (2016) 15:574–84. doi: 10.1016/S1474-4422(16)00068-5
120. Cordano C, Nourbakhsh B, Devereux M, Damotte V, Bennett D, Hauser SL, et al. pRNFL as a marker of disability worsening in the medium/long term in patients with MS. Neurol Neuroimmunol Neuroinflamm. (2019) 6:533. doi: 10.1212/NXI.0000000000000533
121. Oertel FC, Specovius S, Zimmermann HG, Chien C, Motamedi S, Bereuter C, et al. Retinal optical coherence tomography in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. (2021) 8:e1068. doi: 10.1212/NXI.0000000000001068
122. Oertel FC, Havla J, Roca-Fernández A, Lizak N, Zimmermann H, Motamedi S, et al. Retinal ganglion cell loss in neuromyelitis optica: a longitudinal study. J Neurol Neurosurg Psychiatry. (2018) 83:1259–65. doi: 10.1136/jnnp-2018-318382
123. Oertel FC, Kuchling J, Zimmermann H, Chien C, Schmidt F, Knier B, et al. Microstructural visual system changes in AQP4-antibody–seropositive NMOSD. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e334. doi: 10.1212/NXI.0000000000000334
124. Motamedi S, Oertel FC, Yadav SK, Kadas EM, Weise M, Havla J, et al. Altered fovea in AQP4-IgG–seropositive neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm. (2020) 7:e805. doi: 10.1212/NXI.0000000000000805
125. Filippatou AG, Mukharesh L, Saidha S, Calabresi PA, Sotirchos ES. AQP4-IgG and MOG-IgG related optic neuritis-prevalence, optical coherence tomography findings, and visual outcomes: a systematic review and meta-analysis. Front Neurol. (2020) 11:540156. doi: 10.3389/fneur.2020.540156
126. Filippatou AG, Vasileiou ES, He Y, Fitzgerald KC, Kalaitzidis G, Lambe J, et al. Evidence of subclinical quantitative retinal layer abnormalities in AQP4-IgG seropositive NMOSD. Mult Scler. (2021) 27:1738–48. doi: 10.1177/1352458520977771
127. Roca-Fernández A, Oertel FC, Yeo T, Motamedi S, Probert F, Craner MJ, et al. Foveal changes in AQP4-Ab seropositive NMOSD are independent of optic neuritis and not overtly progressive. Eur J Neurol. (2021) 28:2280–93. doi: 10.1111/ene.14766
128. Lu A, Zimmermann HG, Specovius S, Motamedi S, Chien C, Bereuter C, et al. Astrocytic outer retinal layer thinning is not a feature in AQP4-IgG seropositive neuromyelitis optica spectrum disorders. J Neurol Neurosurg Psychiatry. (2022) 93:188–95. doi: 10.1136/jnnp-2021-327412
129. Yao Y, Li X, Xu Y, Liang X, Yang L, Shi F-D, et al. The difference of the retinal structural and microvascular characteristics in patients with MOGAD-ON and AQP4-ON. BMC Neurol. (2022) 22:323. doi: 10.1186/s12883-022-02848-2
130. Yu J, Huang Y, Quan C, Zhou L, ZhangBao J, Wu K, et al. Alterations in the retinal vascular network and structure in MOG antibody-associated disease: an optical coherence tomography angiography study. J Neuroophthalmol. (2021) 41:e424–32. doi: 10.1097/WNO.0000000000001116
131. Yadav SK, Motamedi S, Oberwahrenbrock T, Oertel FC, Polthier K, Paul F, et al. CuBe: parametric modeling of 3D foveal shape using cubic Bézier. Biomed Opt Express. (2017) 8:4181–99. doi: 10.1364/BOE.8.004181
132. Oertel FC, Zimmermann HG, Brandt AU, Paul F. Novel uses of retinal imaging with optical coherence tomography in multiple sclerosis. Expert Rev Neurother. (2019) 19:31–43. doi: 10.1080/14737175.2019.1559051
133. Petzold A, Albrecht P, Balcer L, Bekkers E, Brandt AU, Calabresi PA, et al. Artificial intelligence extension of the OSCAR-IB criteria. Ann Clin Transl Neurol. (2021) 8:1528–42. doi: 10.1002/acn3.51320
134. Motamedi S, Gawlik K, Ayadi N, Zimmermann HG, Asseyer S, Bereuter C, et al. Normative data and minimally detectable change for inner retinal layer thicknesses using a semi-automated OCT image segmentation pipeline. Front Neurol. (2019) 10:1117. doi: 10.3389/fneur.2019.01117
135. Sakuma H, Kohyama K, Park I-K, Miyakoshi A, Tanuma N, Matsumoto Y. Clinicopathological study of a myelin oligodendrocyte glycoprotein-induced demyelinating disease in LEW.1AV1 rats. Brain. (2004) 127:2201–13. doi: 10.1093/brain/awh260
136. Storch MK, Piddlesden S, Haltia M, Iivanainen M, Morgan P, Lassmann H. Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann Neurol. (1998) 43:465–71. doi: 10.1002/ana.410430409
137. Tsunoda I, Kuang L-Q, Fujinami RS. Induction of autoreactive CD8+ cytotoxic T cells during Theiler's murine encephalomyelitis virus infection: implications for autoimmunity. J Virol. (2002) 76:12834–44. doi: 10.1128/JVI.76.24.12834-12844.2002
138. Tsunoda I, Fujinami RS. Inside-Out versus Outside-In models for virus induced demyelination: axonal damage triggering demyelination. Springer Semin Immunopathol. (2002) 24:105–25. doi: 10.1007/s00281-002-0105-z
139. Iglesias A, Bauer J, Litzenburger T, Schubart A, Linington C. T- and B-cell responses to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis and multiple sclerosis. Glia. (2001) 36:220–34. doi: 10.1002/glia.1111
140. Wekerle H, Kojima K, Lannes-Vieira J, Lassmann H, Linington C. Animal models. Ann Neurol. (1994) 36(Suppl):S47–53. doi: 10.1002/ana.410360714
141. Bradl M, Linington C. Animal models of demyelination. Brain Pathol. (1996) 6:303–11. doi: 10.1111/j.1750-3639.1996.tb00857.x
142. Petry KG, Boullerne AI, Pousset F, Brochet B, Caillé JM, Dousset V. Experimental allergic encephalomyelitis animal models for analyzing features of multiple sclerosis. Pathol Biol. (2000) 48:47–53.
143. 't Hart BA, Amor S. The use of animal models to investigate the pathogenesis of neuroinflammatory disorders of the central nervous system. Curr Opin Neurol. (2003) 16:375–83. doi: 10.1097/01.wco.0000073940.19076.43
144. Levkovitch-Verbin H. Animal models of optic nerve diseases. Eye. (2004) 18:1066–74. doi: 10.1038/sj.eye.6701576
145. Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. (2006) 129:1953–71. doi: 10.1093/brain/awl075
146. Cassan C, Liblau RS. Immune tolerance and control of CNS autoimmunity: from animal models to MS patients. J Neurochem. (2007) 100:883–92. doi: 10.1111/j.1471-4159.2006.04270.x
147. Procaccini C, De Rosa V, Pucino V, Formisano L, Matarese G. Animal models of multiple sclerosis. Eur J Pharmacol. (2015) 759:182–91. doi: 10.1016/j.ejphar.2015.03.042
148. Yandamuri SS, Lane TE. Imaging axonal degeneration and repair in preclinical animal models of multiple sclerosis. Front Immunol. (2016) 7:189. doi: 10.3389/fimmu.2016.00189
149. Bjelobaba I, Begovic-Kupresanin V, Pekovic S, Lavrnja I. Animal models of multiple sclerosis: focus on experimental autoimmune encephalomyelitis. J Neurosci Res. (2018) 96:1021–42. doi: 10.1002/jnr.24224
150. Glatigny S, Bettelli E. Experimental autoimmune encephalomyelitis (EAE) as animal models of multiple sclerosis (MS). Cold Spring Harb Perspect Med. (2018) 8:a028977. doi: 10.1101/cshperspect.a028977
151. Lassmann H, Brunner C, Bradl M, Linington C. Experimental allergic encephalomyelitis: the balance between encephalitogenic T lymphocytes and demyelinating antibodies determines size and structure of demyelinated lesions. Acta Neuropathol. (1988) 75:566–76. doi: 10.1007/BF00686201
152. Linington C, Bradl M, Lassmann H, Brunner C, Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol. (1988) 130:443–54.
153. Schluesener HJ, Sobel RA, Weiner HL. Demyelinating experimental allergic encephalomyelitis (EAE) in the rat: treatment with a monoclonal antibody against activated T cells. J Neuroimmunol. (1988) 18:341–51. doi: 10.1016/0165-5728(88)90055-0
154. Xiao BG, Linington C, Link H. Antibodies to myelin-oligodendrocyte glycoprotein in cerebrospinal fluid from patients with multiple sclerosis and controls. J Neuroimmunol. (1991) 31:91–6. doi: 10.1016/0165-5728(91)90014-X
155. Weissert R. Actively induced experimental autoimmune encephalomyelitis in rats. Methods Mol Biol. (2016) 1304:161–9. doi: 10.1007/7651_2014_177
156. Hasselmann JPC, Karim H, Khalaj AJ, Ghosh S, Tiwari-Woodruff SK. Consistent induction of chronic experimental autoimmune encephalomyelitis in C57BL/6 mice for the longitudinal study of pathology and repair. J Neurosci Methods. (2017) 284:71–84. doi: 10.1016/j.jneumeth.2017.04.003
157. Giralt M, Molinero A, Hidalgo J. Active induction of experimental autoimmune encephalomyelitis (EAE) with MOG35-55 in the mouse. Methods Mol Biol. (2018) 1791:227–32. doi: 10.1007/978-1-4939-7862-5_17
158. Miyamura S, Matsuo N, Nagayasu K, Shirakawa H, Kaneko S. Myelin oligodendrocyte glycoprotein 35-55 (MOG 35-55)-induced experimental autoimmune encephalomyelitis: a model of chronic multiple sclerosis. Bio Protoc. (2019) 9:e3453. doi: 10.21769/BioProtoc.3453
159. Joly S, Mdzomba JB, Rodriguez L, Morin F, Vallières L, Pernet V, et al. cell-dependent EAE induces visual deficits in the mouse with similarities to human autoimmune demyelinating diseases. J Neuroinflammation. (2022) 19:54. doi: 10.1186/s12974-022-02416-y
160. Nitsch L, Petzinna S, Zimmermann J, Getts DR, Becker A, Müller M. MOG-specific T cells lead to spontaneous EAE with multilocular B cell infiltration in the GF-IL23 model. Neuromolecular Med. (2022) 24:415–23. doi: 10.1007/s12017-022-08705-2
161. Remlinger J, Madarasz A, Guse K, Hoepner R, Bagnoud M, Meli I, et al. Antineonatal Fc receptor antibody treatment ameliorates MOG-IgG-associated experimental autoimmune encephalomyelitis. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e1134. doi: 10.1212/NXI.0000000000001134
162. Salvador F, Deramoudt L, Leprêtre F, Figeac M, Guerrier T, Boucher J, et al. A spontaneous model of experimental autoimmune encephalomyelitis provides evidence of MOG-specific B cell recruitment and clonal expansion. Front Immunol. (2022) 13:755900. doi: 10.3389/fimmu.2022.755900
163. Triantafyllakou I, Clemente N, Khetavat RK, Dianzani U, Tselios T. Development of PLGA nanoparticles with a glycosylated myelin oligodendrocyte glycoprotein epitope (MOG35-55) against experimental autoimmune encephalomyelitis (EAE). Mol Pharm. (2022) 19:3795–805. doi: 10.1021/acs.molpharmaceut.2c00277
164. Voskuhl RR, MacKenzie-Graham A. Chronic experimental autoimmune encephalomyelitis is an excellent model to study neuroaxonal degeneration in multiple sclerosis. Front Mol Neurosci. (2022) 15:1024058. doi: 10.3389/fnmol.2022.1024058
165. Jarius S, Ruprecht K, Stellmann JP, Huss A, Ayzenberg I, Willing A, et al. MOG-IgG in primary and secondary chronic progressive multiple sclerosis: a multicenter study of 200 patients and review of the literature. J Neuroinflammation. (2018) 15:88. doi: 10.1186/s12974-018-1108-6
166. Lassmann H, Bradl M. Multiple sclerosis: experimental models and reality. Acta Neuropathol. (2017) 133:223–44. doi: 10.1007/s00401-016-1631-4
167. Aharoni R. New findings and old controversies in the research of multiple sclerosis and its model experimental autoimmune encephalomyelitis. Expert Rev Clin Immunol. (2013) 9:423–40. doi: 10.1586/eci.13.21
168. Peschl P, Bradl M, Höftberger R, Berger T, Reindl M. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. (2017) 8:529. doi: 10.3389/fimmu.2017.00529
169. 't Hart BA, Gran B, Weissert R. EAE: imperfect but useful models of multiple sclerosis. Trends Mol Med. (2011) 17:119–25. doi: 10.1016/j.molmed.2010.11.006
170. Friese MA, Montalban X, Willcox N, Bell JI, Martin R, Fugger L. The value of animal models for drug development in multiple sclerosis. Brain. (2006) 129:1940–52. doi: 10.1093/brain/awl083
171. Steinman L, Zamvil SS. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. (2005) 26:565–71. doi: 10.1016/j.it.2005.08.014
172. Wiedrick J, Meza-Romero R, Gerstner G, Seifert H, Chaudhary P, Headrick A, et al. Sex differences in EAE reveal common and distinct cellular and molecular components. Cell Immunol. (2021) 359:104242. doi: 10.1016/j.cellimm.2020.104242
173. Linington C, Berger T, Perry L, Weerth S, Hinze-Selch D, Zhang Y, et al. cells specific for the myelin oligodendrocyte glycoprotein mediate an unusual autoimmune inflammatory response in the central nervous system. Eur J Immunol. (1993) 23:1364–72. doi: 10.1002/eji.1830230627
174. Weissert R, Wallström E, Storch MK, Stefferl A, Lorentzen J, Lassmann H, et al. haplotype-dependent regulation of MOG-induced EAE in rats. J Clin Invest. (1998) 102:1265–73. doi: 10.1172/JCI3022
175. Khare M, Mangalam A, Rodriguez M, David CS. HLA DR and DQ interaction in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis in HLA class II transgenic mice. J Neuroimmunol. (2005) 169:1–12. doi: 10.1016/j.jneuroim.2005.07.023
176. Kummari E, Nichols JM, Yang E-J, Kaplan BLF. Neuroinflammation and B-cell phenotypes in cervical and lumbosacral regions of the spinal cord in experimental autoimmune encephalomyelitis in the absence of pertussis toxin. Neuroimmunomodulation. (2019) 26:198–207. doi: 10.1159/000501765
177. Heidari AR, Boroumand-Noughabi S, Nosratabadi R, Lavi Arab F, Tabasi N, Rastin M, et al. Acylated and deacylated quillaja saponin-21 adjuvants have opposite roles when utilized for immunization of C57BL/6 mice model with MOG35-55 peptide. Mult Scler Relat Disord. (2019) 29:68–82. doi: 10.1016/j.msard.2019.01.025
178. Huntemann N, Vogelsang A, Groeneweg L, Willison A, Herrmann AM, Meuth SG, et al. An optimized and validated protocol for inducing chronic experimental autoimmune encephalomyelitis in C57BL/6J mice. J Neurosci Methods. (2022) 367:109443. doi: 10.1016/j.jneumeth.2021.109443
179. Chase Huizar C, Ji N, Reddick R, Ostroff GR, Forsthuber TG. Glucan particles as a novel adjuvant for the induction of experimental autoimmune encephalomyelitis. Cell Immunol. (2021) 366:104383. doi: 10.1016/j.cellimm.2021.104383
180. Berg CT, Khorooshi R, Asgari N, Owens T. Influence of type I IFN signaling on anti-MOG antibody-mediated demyelination. J Neuroinflammation. (2017) 14:127. doi: 10.1186/s12974-017-0899-1
181. Peschl P, Schanda K, Zeka B, Given K, Böhm D, Ruprecht K, et al. Human antibodies against the myelin oligodendrocyte glycoprotein can cause complement-dependent demyelination. J Neuroinflammation. (2017) 14:208. doi: 10.1186/s12974-017-0984-5
182. Kinzel S, Lehmann-Horn K, Torke S, Häusler D, Winkler A, Stadelmann C, et al. Myelin-reactive antibodies initiate T cell-mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol. (2016) 132:43–58. doi: 10.1007/s00401-016-1559-8
183. Saadoun S, Waters P, Owens GP, Bennett JL, Vincent A, Papadopoulos MC. Neuromyelitis optica MOG-IgG causes reversible lesions in mouse brain. Acta Neuropathol Commun. (2014) 2:35. doi: 10.1186/2051-5960-2-35
184. Spadaro M, Winklmeier S, Beltran E, Macrini C, Hoftberger R, Schuh E, et al. Pathogenicity of human antibodies against myelin oligodendrocyte glycoprotein. Ann Neurol. (2018) 84:315–28. doi: 10.1002/ana.25291
185. Linington C, Lassmann H. Antibody-responses in chronic relapsing experimental allergic encephalomyelitis - correlation of serum demyelinating activity with antibody titer to the myelin oligodendrocyte glycoprotein (Mog). J Neuroimmunol. (1987) 17:61–9. doi: 10.1016/0165-5728(87)90031-2
186. Breithaupt C, Schafer B, Pellkofer H, Huber R, Linington C, Jacob U. Demyelinating myelin oligodendrocyte glycoprotein-specific autoantibody response is focused on one dominant conformational epitope region in rodents. J Immunol. (2008) 181:1255–63. doi: 10.4049/jimmunol.181.2.1255
187. Vass K, Heininger K, Schaefer B, Linington C, Lassmann H. Interferon-gamma potentiates antibody-mediated demyelination in vivo. Ann Neurol. (1992) 32:198–206. doi: 10.1002/ana.410320212
188. Zhou LX, Trapp BD, Miller RH. Demyelination in the central nervous system mediated by an anti-oligodendrocyte antibody. J Neurosci Res. (1998) 54:158–68. doi: 10.1002/(SICI)1097-4547(19981015)54:2<158::AID-JNR4>3.0.CO;2-D
189. Hoftberger R, Sepulveda M, Armangue T, Blanco Y, Rostasy K, Calvo AC, et al. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler J. (2015) 21:866–74. doi: 10.1177/1352458514555785
190. Oliveira LM, Apostolos-Pereira SL, Pitombeira MS, Torretta PHB, Callegaro D, Sato DK. Persistent MOG-IgG positivity is a predictor of recurrence in MOG-IgG-associated optic neuritis, encephalitis and myelitis. Mult Scler J. (2019) 25:1907–14. doi: 10.1177/1352458518811597
191. Wendel EM, Thonke HS, Bertolini A, Baumann M, Blaschek A, Merkenschlager A, et al. Temporal dynamics of MOG antibodies in children with acquired demyelinating syndrome. Neurol-Neuroimmunol. (2022) 9:e200035. doi: 10.1212/NXI.0000000000200035
192. Probstel AK, Dornmair K, Bittner R, Sperl P, Jenne D, Magalhaes S, et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology. (2011) 77:580–8. doi: 10.1212/WNL.0b013e318228c0b1
193. Schluesener HJ, Sobel RA, Linington C, Weiner HL. A monoclonal-antibody against a myelin oligodendrocyte glycoprotein induces relapses and demyelination in central-nervous-system autoimmune-disease. J Immunol. (1987) 139:4016–21. doi: 10.4049/jimmunol.139.12.4016
194. Linington C, Engelhardt B, Kapocs G, Lassman H. Induction of persistently demyelinated lesions in the rat following the repeated adoptive transfer of encephalitogenic T-cells and demyelinating antibody. J Neuroimmunol. (1992) 40:219–24. doi: 10.1016/0165-5728(92)90136-9
195. Litzenburger T, Fassler R, Bauer J, Lassmann H, Linington C, Wekerle H, et al. lymphocytes producing demyelinating autoantibodies: development and function in gene-targeted transgenic mice. J Exp Med. (1998) 188:169–80. doi: 10.1084/jem.188.1.169
196. Svensson L, Abdul-Majid KB, Bauer J, Lassmann H, Harris RA, Holmdahl R. A comparative analysis of B cell-mediated myelin oligodendrocyte glycoprotein-experimental autoimmune encephalomyelitis pathogenesis in B cell-deficient mice reveals an effect on demyelination. Eur J Immunol. (2002) 32:1939–46. doi: 10.1002/1521-4141(200207)32:7<1939::AID-IMMU1939>3.0.CO;2-S
197. Flach AC, Litke T, Strauss J, Haberl M, Gomez CC, Reindl M, et al. Autoantibody-boosted T-cell reactivation in the target organ triggers manifestation of autoimmune CNS disease. Proc Natl Acad Sci U S A. (2016) 113:3323–8. doi: 10.1073/pnas.1519608113
198. Meeson AP, Piddlesden S, Morgan BP, Reynolds R. The distribution of inflammatory demyelinated lesions in the central-nervous-system of rats with antibody-augmented demyelinating experimental allergic encephalomyelitis. Exp Neurol. (1994) 129:299–310. doi: 10.1006/exnr.1994.1172
199. Mead RJ, Singhrao SK, Neal JW, Lassmann H, Morgan BP. The membrane attack complex of complement causes severe demyelination associated with acute axonal injury. J Immunol. (2002) 168:458–65. doi: 10.4049/jimmunol.168.1.458
200. Linington C, Morgan BP, Scolding NJ, Wilkins P, Piddlesden S, Compston DA. The role of complement in the pathogenesis of experimental allergic encephalomyelitis. Brain. (1989) 112:895–911. doi: 10.1093/brain/112.4.895
201. Piddlesden SJ, Lassmann H, Zimprich F, Morgan BP, Linington C. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am J Pathol. (1993) 143:555–64.
202. Piddlesden S, Lassmann H, Laffafian I, Morgan BP, Linington C. Antibody-mediated demyelination in experimental allergic encephalomyelitis is independent of complement membrane attack complex-formation. Clin Exp Immunol. (1991) 83:245–50. doi: 10.1111/j.1365-2249.1991.tb05622.x
203. Pohl M, Kawakami N, Kitic M, Bauer J, Martins R, Fischer MT, et al. T cell-activation in neuromyelitis optica lesions plays a role in their formation. Acta Neuropathol Commun. (2013) 1:85. doi: 10.1186/2051-5960-1-85
204. Berger T, Weerth S, Kojima K, Linington C, Wekerle H, Lassmann H. Experimental autoimmune encephalomyelitis: the antigen specificity of T lymphocytes determines the topography of lesions in the central and peripheral nervous system. Lab Invest. (1997) 76:355–64.
205. Storch MK, Stefferl A, Brehm U, Weissert R, Wallstrom E, Kerschensteiner M, et al. Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol. (1998) 8:681–94. doi: 10.1111/j.1750-3639.1998.tb00194.x
206. Adelmann M, Wood J, Benzel I, Fiori P, Lassmann H, Matthieu JM, et al. The N-terminal domain of the myelin oligodendrocyte glycoprotein (MOG) induces acute demyelinating experimental autoimmune encephalomyelitis in the Lewis rat. J Neuroimmunol. (1995) 63:17–27. doi: 10.1016/0165-5728(95)00124-7
207. Johns TG, Derosbo NK, Menon KK, Abo S, Gonzales MF, Bernard CCA. Myelin oligodendrocyte glycoprotein induces a demyelinating encephalomyelitis resembling multiple-sclerosis. J Immunol. (1995) 154:5536–41. doi: 10.4049/jimmunol.154.10.5536
208. Stefferl A, Brehm U, Storch M, Lambracht-Washington D, Bourquin C, Wonigeit K, et al. Myelin oligodendrocyte glycoprotein induces experimental autoimmune encephalomyelitis in the “resistant” Brown Norway rat: disease susceptibility is determined by MHC and MHC-linked effects on the B cell response. J Immunol. (1999) 163:40–9. doi: 10.4049/jimmunol.163.1.40
209. Khare M, Rodriguez M, David C. Role of RLA class II molecules in myelin oligodendrocyte glycoprotein induced experimental autoimmune encepahlomyelitis. FASEB J. (2003) 17:C39–C39.
210. Bruijstens AL, Wong YYM, van Pelt DE, van der Linden PJE, Haasnoot GW, Hintzen RQ, et al. HLA association in MOG-IgG- and AQP4-IgG-related disorders of the CNS in the Dutch population. Neurol Neuroimmunol Neuroinflamm. (2020) 7:702. doi: 10.1212/NXI.0000000000000702
211. Sun X, Qiu W, Wang J, Wang S, Wang Y, Zhong X, et al. Myelin oligodendrocyte glycoprotein-associated disorders are associated with HLA subtypes in a Chinese paediatric-onset cohort. J Neurol Neurosurg Psychiatry. (2020) 91:733–9. doi: 10.1136/jnnp-2019-322115
212. Grant-Peters M, Passos GRD, Yeung H, Jacob A, Huda S, Leite MI, et al. No strong HLA association with MOG antibody disease in the UK population. Ann Clin Transl Neurol. (2021) 8:1502–7. doi: 10.1002/acn3.51378
213. Mendel I, Derosbo NK, Bennun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2(B) mice - fine specificity and T-cell receptor V-beta expression of encephalitogenic T-cells. Eur J Immunol. (1995) 25:1951–9. doi: 10.1002/eji.1830250723
214. Berard JL, Wolak K, Fournier S, David S. Characterization of relapsing-remitting and chronic forms of experimental autoimmune encephalomyelitis in C57BL/6 mice. Glia. (2010) 58:434–45. doi: 10.1002/glia.20935
215. Zhang GX, Yu S, Gran B, Li JF, Calida D, Ventura E, et al. Cell and antibody responses in remitting-relapsing experimental autoimmune encephalomyelitis in (C57BL/6xSJL) F1 mice. J Neuroimmunol. (2004) 148:1–10. doi: 10.1016/j.jneuroim.2003.10.057
216. Zhang BN, Yamamura T, Kondo T, Fujiwara M, Tabira T. Regulation of experimental autoimmune encephalomyelitis by natural killer (NK) cells. J Exp Med. (1997) 186:1677–87. doi: 10.1084/jem.186.10.1677
217. Chen B, Gui MC, Ji SQ, Xie Y, Tian DS, Bu BT. Distinct immunological features of inflammatory demyelinating diseases of the central nervous system. Neuroimmunomodulation. (2022) 29:220–30. doi: 10.1159/000519835
218. Xiong Y, Wu X, Qu X, Xie X, Ren Y. Functions of T-cell subsets and their related cytokines in the pathological processes of autoimmune encephalomyelitic mice. Int J Clin Exp Pathol. (2018) 11:4817–26.
219. Samoilova EB, Horton JL, Hilliard B, Liu TST, Chen YH. IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cell. J Immunol. (1998) 161:6480–6. doi: 10.4049/jimmunol.161.12.6480
220. Okuda Y, Sakoda S, Bernard CC, Fujimura H, Saeki Y, Kishimoto T, et al. IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int Immunol. (1998) 10:703–8. doi: 10.1093/intimm/10.5.703
221. Sanchis P, Fernandez-Gayol O, Comes G, Aguilar K, Escrig A, Giralt M, et al. new mouse model to study restoration of interleukin-6 (IL-6) expression in a Cre-dependent manner: microglial IL-6 regulation of experimental autoimmune encephalomyelitis. J Neuroinflammation. (2020) 17:304. doi: 10.1186/s12974-020-01969-0
222. Okuda Y, Sakoda S, Fujimura H, Saeki Y, Kishimoto T, Yanagihara T. IL-6 plays a crucial role in the induction phase of myelin oligodendrocyte glucoprotein 35-55 induced experimental autoimmune encephalomyelitis. J Neuroimmunol. (1999) 101:188–96. doi: 10.1016/S0165-5728(99)00139-3
223. Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. (2008) 105:18460–5. doi: 10.1073/pnas.0809850105
224. Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. (2008) 105:9041–6. doi: 10.1073/pnas.0802218105
225. Holz K, Prinz M, Brendecke SM, Hölscher A, Deng F, Mitrücker H-W, et al. Differing outcome of experimental autoimmune encephalitis in macrophage/neutrophil- and T cell-specific gp130-deficient mice. Front Immunol. (2018) 9:836. doi: 10.3389/fimmu.2018.00836
226. Quintana A, Muller M, Frausto RF, Ramos R, Getts DR, Sanz E, et al. Site-specific production of IL-6 in the central nervous system retargets and enhances the inflammatory response in experimental autoimmune encephalomyelitis. J Immunol. (2009) 183:2079–88. doi: 10.4049/jimmunol.0900242
227. Sanchis P, Fernández-Gayol O, Vizueta J, Comes G, Canal C, Escrig A, et al. Microglial cell-derived interleukin-6 influences behavior and inflammatory response in the brain following traumatic brain injury. Glia. (2020) 68:999–1016. doi: 10.1002/glia.23758
228. Galle P, Jensen L, Andersson C, Cuzzocrea S, Di Paola R, Nicoletti F, et al. Vaccination with IL-6 analogues induces autoantibodies to IL-6 and influences experimentally induced inflammation. Int Immunopharmacol. (2007) 7:1704–13. doi: 10.1016/j.intimp.2007.08.026
229. Chen X, Howard OM, Oppenheim JJ. Pertussis toxin by inducing IL-6 promotes the generation of IL-17-producing CD4 cells. J Immunol. (2007) 178:6123–9. doi: 10.4049/jimmunol.178.10.6123
230. Eugster HP, Frei K, Kopf M, Lassmann H, Fontana A. IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur J Immunol. (1998) 28:2178–87. doi: 10.1002/(SICI)1521-4141(199807)28:07<2178::AID-IMMU2178>3.0.CO;2-D
231. Novi G, Gastaldi M, Franciotta D, Pesce G, Benedetti L, Uccelli A. Tocilizumab in MOG-antibody spectrum disorder: a case report. Mult Scler Relat Dis. (2019) 27:312–4. doi: 10.1016/j.msard.2018.11.012
232. Rigal J, Pugnet G, Ciron J, Lepine Z, Biotti D. Off-label use of tocilizumab in neuromyelitis optica spectrum disorders and MOG-antibody-associated diseases: a case-series. Multiple Scler Relat Disord. (2020) 46:102483. doi: 10.1016/j.msard.2020.102483
233. Duchow A, Bellmann-Strobl J. Satralizumab in the treatment of neuromyelitis optica spectrum disorder. Neurodegener Dis Man. (2021) 11:46. doi: 10.2217/nmt-2020-0046
234. Elsbernd PM, Hoffman WR, Carter JL, Wingerchuk DM. Interleukin-6 inhibition with tocilizumab for relapsing MOG-IgG associated disorder (MOGAD): a case-series and review. Mult Scler Relat Dis. (2021) 48:102696. doi: 10.1016/j.msard.2020.102696
235. Ringelstein M, Ayzenberg I, Lindenblatt G, Fischer K, Gahlen A, Novi G, et al. Interleukin-6 receptor blockade in treatment-refractory MOG-IgG-associated disease and neuromyelitis optica spectrum disorders. Neurol-Neuroimmunol. (2022) 9:e1100. doi: 10.1212/NXI.0000000000001100
236. Yong KP, Kim HJ. Demystifying MOGAD and double seronegative NMOSD further with IL-6 blockade. Neurol-Neuroimmunol. (2022) 9:1110. doi: 10.1212/NXI.0000000000001110
237. Becher B, Durell BG, Noelle RJ. IL-23 produced by CNS-resident cells controls T cell encephalitogenicity during the effector phase of experimental autoimmune encephalomyelitis. J Clin Invest. (2003) 112:1186–91. doi: 10.1172/JCI200319079
238. Thakker P, Leach MW, Kuang W, Benoit SE, Leonard JP, Marusic S. IL-23 is critical in the induction but not in the effector phase of experimental autoimmune encephalomyelitis. J Immunol. (2007) 178:2589–98. doi: 10.4049/jimmunol.178.4.2589
239. Liu JB, Lin F, Strainic MG, An FQ, Miller RH, Altuntas CZ, et al. IFN-gamma and IL-17 production in experimental autoimmune encephalomyelitis depends on local APC-T cell complement production. J Immunol. (2008) 180:5882–9. doi: 10.4049/jimmunol.180.9.5882
240. Chung C-Y, Liao F. CXCR3 signaling in glial cells ameliorates experimental autoimmune encephalomyelitis by restraining the generation of a pro-Th17 cytokine milieu and reducing CNS-infiltrating Th17 cells. J Neuroinflammation. (2016) 13:76. doi: 10.1186/s12974-016-0536-4
241. Samoilova EB, Horton JL, Chen Y. Acceleration of experimental autoimmune encephalomyelitis in interleukin-10-deficient mice: roles of interleukin-10 in disease progression and recovery. Cell Immunol. (1998) 188:118–24. doi: 10.1006/cimm.1998.1365
242. Kennedy MK, Torrance DS, Picha KS, Mohler KM. Analysis of cytokine mRNA expression in the central nervous system of mice with experimental autoimmune encephalomyelitis reveals that IL-10 mRNA expression correlates with recovery. J Immunol. (1992) 149:2496–505. doi: 10.4049/jimmunol.149.7.2496
243. Cohen SJ, Cohen IR, Nussbaum G. IL-10 mediates resistance to adoptive transfer experimental autoimmune encephalomyelitis in MyD88(-/-) Mice. J Immunol. (2010) 184:212–21. doi: 10.4049/jimmunol.0900296
244. Maron R, Hancock WW, Slavin A, Hattori M, Kuchroo V, Weiner HL. Genetic susceptibility or resistance to autoimmune encephalomyelitis in MHC congenic mice is associated with differential production of pro- and anti-inflammatory cytokines. Int Immunol. (1999) 11:1573–80. doi: 10.1093/intimm/11.9.1573
245. Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. (1996) 157:3223–7. doi: 10.4049/jimmunol.157.8.3223
246. Willenborg DO, Fordham SA, Staykova MA, Ramshaw IA, Cowden WB. IFN-gamma is critical to the control of murine autoimmune encephalomyelitis and regulates both in the periphery and in the target tissue: a possible role for nitric oxide. J Immunol. (1999) 163:5278–86. doi: 10.4049/jimmunol.163.10.5278
247. Stoolman JS, Duncker PC, Huber AK, Giles DA, Washnock-Schmid JM, Soulika AM, et al. An IFNgamma/CXCL2 regulatory pathway determines lesion localization during EAE. J Neuroinflammation. (2018) 15:208. doi: 10.1186/s12974-018-1237-y
248. Evangelista MG, Castro SB, Alves CC, Dias AT, Souza VW, Reis LB, et al. Early IFN-gamma production together with decreased expression of TLR3 and TLR9 characterizes EAE development conditional on the presence of myelin. Autoimmunity. (2016) 49:258–67. doi: 10.3109/08916934.2016.1141898
249. Saravia J, Chapman NM, Chi H. Helper T cell differentiation. Cell Mol Immunol. (2019) 16:634–43. doi: 10.1038/s41423-019-0220-6
250. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. (2005) 6:1123–32. doi: 10.1038/ni1254
251. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. (2005) 6:1133–41. doi: 10.1038/ni1261
252. Uyttenhove C, Sommereyns C, Theate I, Michiels T, Van Snick J. Anti-IL-17A autovaccination prevents clinical and histological manifestations of experimental autoimmune encephalomyelitis. Ann N Y Acad Sci. (2007) 1110:330–6. doi: 10.1196/annals.1423.035
253. Sonobe Y, Jin S, Wang J, Kawanokuchi J, Takeuchi H, Mizuno T, et al. Chronological changes of CD4(+) and CD8(+) T cell subsets in the experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis. Tohoku J Exp Med. (2007) 213:329–39. doi: 10.1620/tjem.213.329
254. Zimmermann J, Nitsch L, Krauthausen M, Muller M. IL-17A facilitates entry of autoreactive T-cells and granulocytes into the CNS during EAE. Neuromolecular Med. (2023) doi: 10.1007/s12017-023-08739-0
255. Barthelmes J, Tafferner N, Kurz J, de Bruin N, Parnham MJ, Geisslinger G, et al. Induction of experimental autoimmune encephalomyelitis in mice and evaluation of the disease-dependent distribution of immune cells in various tissues. J Vis Exp. (2016) 8:53933. doi: 10.3791/53933
256. Steckner C, Weber A, Mausberg AK, Heininger M, Opdenhovel F, Kieseier BC, et al. Alteration of the cytokine signature by various TLR ligands in different T cell populations in MOG37-50 and MOG35-55-induced EAE in C57BL/6 mice. Clin Immunol. (2016) 170:22–30. doi: 10.1016/j.clim.2016.05.008
257. Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. (2009) 119:61–9. doi: 10.1172/JCI35997
258. Regen T, Isaac S, Amorim A, Nunez NG, Hauptmann J, Shanmugavadivu A, et al. IL-17 controls central nervous system autoimmunity through the intestinal microbiome. Sci Immunol. (2021) 6:aaz6563. doi: 10.1126/sciimmunol.aaz6563
259. Domingues HS, Mues M, Lassmann H, Wekerle H, Krishnamoorthy G. Functional and pathogenic differences of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. PLoS ONE. (2010) 5:e0015531. doi: 10.1371/journal.pone.0015531
260. Weir C, Bernard CCA, Backstrom BT. IL-5-deficient mice are susceptible to experimental autoimmune encephalomyelitis. Int Immunol. (2003) 15:1283–9. doi: 10.1093/intimm/dxg127
261. Sonderegger I, Kisielow J, Meier R, King C, Kopf M. IL-21 and IL-21R are not required for development of Th17 cells and autoimmunity in vivo. Eur J Immunol. (2008) 38:1833–8. doi: 10.1002/eji.200838511
262. Merkler D, Ernsting T, Kerschensteiner M, Bruck W, Stadelmann C. A new focal EAE model of cortical demyelination: multiple sclerosis-like lesions with rapid resolution of inflammation and extensive remyelination. Brain. (2006) 129:1972–83. doi: 10.1093/brain/awl135
263. Gardner C, Magliozzi R, Durrenberger PF, Howell OW, Rundle J, Reynolds R. Cortical grey matter demyelination can be induced by elevated pro-inflammatory cytokines in the subarachnoid space of MOG-immunized rats. Brain. (2013) 136:3596–608. doi: 10.1093/brain/awt279
264. Krishnamoorthy G, Holz A, Wekerle H. Experimental models of spontaneous in the central nervous system. J Mol Med. (2007) 85:1161–73. doi: 10.1007/s00109-007-0218-x
265. Ben-Nun A, Kaushansky N, Kawakami N, Krishnamoorthy G, Berer K, Liblau R, et al. From classic to spontaneous and humanized models of multiple sclerosis: impact on understanding pathogenesis and drug development. J Autoimmun. (2014) 54:33–50. doi: 10.1016/j.jaut.2014.06.004
266. Dash PK, Gorantla S, Poluektova L, Hasan M, Waight E, Zhang C, et al. Humanized mice for infectious and neurodegenerative disorders. Retrovirology. (2021) 18. doi: 10.1186/s12977-021-00557-1
267. Anderson AC, Chandwaskar R, Lee DH, Sullivan JM, Solomon A, Rodriguez-Manzanet R, et al. transgenic model of central nervous system autoimmunity mediated by CD4+ and CD8+ T and B cells. J Immunol. (2012) 188:2084–92. doi: 10.4049/jimmunol.1102186
268. Levy H, Assaf Y, Frenkel D. Characterization of brain lesions in a mouse model of progressive multiple sclerosis. Exp Neurol. (2010) 226:148–58. doi: 10.1016/j.expneurol.2010.08.017
269. Baker D, Nutma E, O'Shea H, Cooke A, Orian JM, Amor S. Autoimmune encephalomyelitis in NOD mice is not initially a progressive multiple sclerosis model. Ann Clin Transl Neurol. (2019) 6:1362–72. doi: 10.1002/acn3.792
270. Ichikawa M, Koh CS, Inoue A, Tsuyusaki J, Yamazaki M, Inaba Y, et al. Anti-IL-12 antibody prevents the development and progression of multiple sclerosis-like relapsing–remitting demyelinating disease in NOD mice induced with myelin oligodendrocyte glycoprotein peptide. J Neuroimmunol. (2000) 102:56–66. doi: 10.1016/S0165-5728(99)00153-8
271. Baker D, O'Neill JK, Gschmeissner SE, Wilcox CE, Butter C, Turk JL. Induction of chronic relapsing experimental allergic encephalomyelitis in Biozzi mice. J Neuroimmunol. (1990) 28:261–70. doi: 10.1016/0165-5728(90)90019-J
272. Nishri Y, Fainstein N, Goldfarb S, Hampton D, Macrini C, Meinl E, et al. Modeling compartmentalized chronic immune-mediated demyelinating CNS disease in the Biozzi ABH mouse. J Neuroimmunol. (2021) 356:577582. doi: 10.1016/j.jneuroim.2021.577582
273. Valentin-Torres A, Savarin C, Hinton DR, Phares TW, Bergmann CC, Stohlman SA. Sustained TNF production by central nervous system infiltrating macrophages promotes progressive autoimmune encephalomyelitis. J Neuroinflammation. (2016) 13:46. doi: 10.1186/s12974-016-0513-y
274. van Oosten BW, Barkhof F, Truyen L, Boringa JB, Bertelsmann FW, von Blomberg BM, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology. (1996) 47:1531–4. doi: 10.1212/WNL.47.6.1531
275. Redenbaugh V, Flanagan EP, Floris V, Zara P, Bhatti MT, Sanchez F, et al. Exposure to TNF inhibitors is rare at MOGAD presentation. J Neurol Sci. (2022) 432:120044. doi: 10.1016/j.jns.2021.120044
276. Omura S, Sato F, Martinez NE, Park A-M, Fujita M, Kennett NJ, et al. Bioinformatics analyses determined the distinct CNS and peripheral surrogate biomarker candidates between two mouse models for progressive multiple sclerosis. Front Immunol. (2019) 10:516. doi: 10.3389/fimmu.2019.00516
277. Bettelli E, Baeten D, Jäger A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest. (2006) 116:2393–402. doi: 10.1172/JCI28334
278. Krishnamoorthy G. Cumulative autoimmunity: myelin oligodendrocyte glycoprotein-specific T cells co-recognise neurofilament-M epitope in a spontaneous EAE of the C57BL/6 mouse. Mult Scler. (2009) 15:S28–9.
279. Krishnamoorthy G, Lassmann H, Wekerle H, Holz A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J Clin Invest. (2006) 116:2385–92. doi: 10.1172/JCI28330
280. Kalluri SR, Rothhammer V, Staszewski O, Srivastava R, Petermann F, Prinz M, et al. Functional characterization of aquaporin-4 specific T cells: towards a model for neuromyelitis optica. PLoS ONE. (2011) 6:e16083. doi: 10.1371/journal.pone.0016083
281. Faber H, Kurtoic D, Krishnamoorthy G, Weber P, Pütz B, Müller-Myhsok B, et al. Gene expression in spontaneous experimental autoimmune encephalomyelitis is linked to human multiple sclerosis risk genes. Front Immunol. (2020) 11:2165. doi: 10.3389/fimmu.2020.02165
282. Corbali O, Chitnis T. Pathophysiology of myelin oligodendrocyte glycoprotein antibody disease. Front Neurol. (2023) 14:1137998. doi: 10.3389/fneur.2023.1137998
283. Mathey EK, Derfuss T, Storch MK, Williams KR, Hales K, Woolley DR, et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med. (2007) 204:2363–72. doi: 10.1084/jem.20071053
284. Lucca LE, Axisa PP, Aloulou M, Perals C, Ramadan A, Rufas P, et al. Myelin oligodendrocyte glycoprotein induces incomplete tolerance of CD4(+) T cells specific for both a myelin and a neuronal self-antigen in mice. Eur J Immunol. (2016) 46:2247–59. doi: 10.1002/eji.201646416
285. Di Pauli F, Hoftberger R, Reindl M, Schanda K, Beer R, Sato D, et al. Antibodies to MOG and AQP4 in a patient with a fulminant demyelinating encephalomyelitis, clinical course and neuropathological examination: a case report. Mult Scler J. (2015) 21:131–131.
286. Spiezia AL, Carotenuto A, Iovino A, Moccia M, Gastaldi M, Iodice R, et al. AQP4-MOG double-positive neuromyelitis optica spectrum disorder: case report with central and peripheral nervous system involvement and review of literature. Int J Mol Sci. (2022) 23:559. doi: 10.3390/ijms232314559
287. Gou B, Yang P, Feng J, Li Y, Huang G, Shi J, et al. The case report of AQP4 and MOG IgG double positive NMOSD treated with subcutaneous Ofatumumab. J Neuroimmunol. (2023) 376:578035. doi: 10.1016/j.jneuroim.2023.578035
288. Gullapalli S, Ramesh R, Shanmugam S, Hazeena P. MOG antibody associated with central and peripheral demyelination. Prog Neurol Psychiat. (2023) 27:12–5. doi: 10.1002/pnp.775
289. Nimmerjahn F, Ravetch JV. Fc gamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
290. Khare P, Challa DK, Devanaboyina SC, Velmurugan R, Hughes S, Greenberg BM, et al. Myelin oligodendrocyte glycoprotein-specific antibodies from multiple sclerosis patients exacerbate disease in a humanized mouse model. J Autoimmun. (2018) 86:104–15. doi: 10.1016/j.jaut.2017.09.002
291. Pajoohesh-Ganji A, Karl M, Garrison E, Osei-Bonsu NA, Clarkson-Paredes C, Ahn J, et al. Developmental ablation of mature oligodendrocytes exacerbates adult CNS demyelination. Brain Behav Immun Health. (2020) 7:100110. doi: 10.1016/j.bbih.2020.100110
292. Larabee CM, Desai S, Agasing A, Georgescu C, Wren JD, Axtell RC, et al. Loss of Nrf2 exacerbates the visual deficits and optic neuritis elicited by experimental autoimmune encephalomyelitis. Mol Vis. (2016) 22:1503–13.
293. Li B, Wang X, Choi IY, Wang Y-C, Liu S, Pham AT, et al. miR-146a modulates autoreactive Th17 cell differentiation and regulates organ-specific autoimmunity. J Clin Invest. (2017) 127:3702–16. doi: 10.1172/JCI94012
294. Boyden AW, Brate AA, Karandikar NJ. Novel B cell-dependent multiple sclerosis model using extracellular domains of myelin proteolipid protein. Sci Rep. (2020) 10:5011. doi: 10.1038/s41598-020-61928-w
295. Häusler D, Häusser-Kinzel S, Feldmann L, Torke S, Lepennetier G, Bernard CCA, et al. Functional characterization of reappearing B cells after anti-CD20 treatment of CNS autoimmune disease. Proc Natl Acad Sci U S A. (2018) 115:9773–8. doi: 10.1073/pnas.1810470115
296. Lehmann-Horn K, Sagan SA, Winger RC, Spencer CM, Bernard CCA, Sobel RA, et al. accumulation of regulatory B cells is VLA-4-dependent. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e212. doi: 10.1212/NXI.0000000000000212
297. Bjarnadóttir K, Benkhoucha M, Merkler D, Weber MS, Payne NL, Bernard CCA, et al. cell-derived transforming growth factor-β1 expression limits the induction phase of autoimmune neuroinflammation. Sci Rep. (2016) 6:34594. doi: 10.1038/srep34594
298. Kap YS, Laman JD, 't Hart BA. Experimental autoimmune encephalomyelitis in the common marmoset, a bridge between rodent EAE and multiple sclerosis for immunotherapy development. J Neuroimmune Pharm. (2010) 5:220–30. doi: 10.1007/s11481-009-9178-y
299. 't Hart BA, Laman JD, Kap YS. Merits and complexities of modeling multiple sclerosis in non-human primates: implications for drug discovery. Expert Opin Drug Dis. (2018) 13:387–97. doi: 10.1080/17460441.2018.1443075
300. Stimmer L, Fovet CM, Serguera C. Experimental models of autoimmune demyelinating diseases in nonhuman primates. Vet Pathol. (2018) 55:27–41. doi: 10.1177/0300985817712794
301. 't Hart BA, Bauer J, Brok HPM, Amor S. Non-human primate models of experimental autoimmune encephalomyelitis: variations on a theme. J Neuroimmunol. (2005) 168:1–12. doi: 10.1016/j.jneuroim.2005.05.017
302. Merkler D, Boscke R, Schmelting B, Czeh B, Fuchs E, Bruck W, et al. Differential macrophage/microglia activation in neocortical EAE lesions in the marmoset monkey. Brain Pathol. (2006) 16:117–23. doi: 10.1111/j.1750-3639.2006.00004.x
303. Kramann N, Neid K, Menken L, Schlumbohm C, Stadelmann C, Fuchs E, et al. Increased meningeal T and plasma cell infiltration is associated with early subpial cortical demyelination in common marmosets with experimental autoimmune encephalomyelitis. Brain Pathol. (2015) 25:276–86. doi: 10.1111/bpa.12180
304. Merkler D, Schmelting B, Czeh B, Fuchs E, Stadelmann C, Bruck W. Myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis in the common marmoset reflects the immunopathology of pattern II multiple sclerosis lesions. Mult Scler. (2006) 12:369–74. doi: 10.1191/1352458506ms1290oa
305. Brok HP, Uccelli A, Kerlero De Rosbo N, Bontrop RE, Roccatagliata L, de Groot NG, et al. Myelin/oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis in common marmosets: the encephalitogenic T cell epitope pMOG24-36 is presented by a monomorphic MHC class II molecule. J Immunol. (2000) 165:1093–101. doi: 10.4049/jimmunol.165.2.1093
306. Jagessar SA, Smith PA, Blezer E, Delarasse C, Pham-Dinh D, Laman JD, et al. Autoimmunity against myelin oligodendrocyte glycoprotein is dispensable for the initiation although essential for the progression of chronic encephalomyelitis in common marmosets. J Neuropathol Exp Neurol. (2008) 67:326–40. doi: 10.1097/NEN.0b013e31816a6851
307. Jagessar SA, Kap YS, Heijmans N, van Driel N, van Straalen L, Bajramovic JJ, et al. Induction of progressive demyelinating autoimmune encephalomyelitis in common marmoset monkeys using MOG34-56 peptide in incomplete freund adjuvant. J Neuropathol Exp Neurol. (2010) 69:372–85. doi: 10.1097/NEN.0b013e3181d5d053
308. Kap YS, Jagessar SA, van Driel N, Blezer E, Bauer J, van Meurs M, et al. Effects of early IL-17A neutralization on disease induction in a primate model of experimental autoimmune encephalomyelitis. J Neuroimmune Pharm. (2011) 6:341–53. doi: 10.1007/s11481-010-9238-3
309. Serguera C, Stimmer L, Fovet CM, Horellou P, Contreras V, Tchitchek N, et al. Anti-MOG autoantibodies pathogenicity in children and macaques demyelinating diseases. J Neuroinflammation. (2019) 16:244. doi: 10.1186/s12974-019-1637-7
310. Frau J, Coghe G, Lorefice L, Fenu G, Cocco E. The role of microorganisms in the etiopathogenesis of demyelinating diseases. Life-Basel. (2023) 13:1309. doi: 10.3390/life13061309
311. Jarius S, Bieber N, Haas J, Wildemann B, MOG. encephalomyelitis after vaccination against severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2): case report and comprehensive review of the literature. J Neurol. (2022) 269:5198–212. doi: 10.1007/s00415-022-11194-9
312. Ahsan N, Jafarpour S, Santoro JD. Myelin oligodendrocyte glycoprotein antibody encephalitis following severe acute respiratory syndrome coronavirus 2 in a pediatric patient. Clin Exp Pediatr. (2021) 64:310–2. doi: 10.3345/cep.2020.01963
313. Colantonio M, Nwafor D, Shrestha A, Elkhooly M, Rollins S, Wen S, et al. Myelin oligodendrocyte glycoprotein antibody-associated optic neuritis and myelitis: a case report and literature review. Mult Scler J. (2022) 28:86. doi: 10.1186/s41983-022-00496-4
314. Gilardi M, Cortese A, Ferraro E, Rispoli M, Sadun R, Altavista MC, et al. MOG-IgG positive optic neuritis after SARS-CoV-2 infection. Eur J Ophthalmol. (2022) 33:NP87–90. doi: 10.1177/11206721221136319
315. Ide T, Kawanami T, Eriguchi M, Hara H. SARS-CoV-2-related myelin oligodendrocyte glycoprotein antibody-associated disease: a case report and literature review. Internal Med. (2022) 61:1253–8. doi: 10.2169/internalmedicine.8709-21
316. Khair AM, Nikam R, Husain S, Ortiz M, Kaur G. Para and post-COVID-19 CNS acute demyelinating disorders in children: a case series on expanding the spectrum of clinical and radiological characteristics. Cureus J Med Sci. (2022) 14:23405. doi: 10.7759/cureus.23405
317. Lambe J, McGinley MP, Moss BP, Mao-Draayer Y, Kassa R, Ciotti JR, et al. Myelin oligodendrocyte glycoprotein-IgG associated disorders (MOGAD) following SARS-CoV-2 infection: a case series. J Neuroimmunol. (2022) 370:577933. doi: 10.1016/j.jneuroim.2022.577933
318. Mariotto S, Carta S, Dinoto A, Lippi G, Salvagno GL, Masin L, et al. Is there a correlation between MOG-associated disorder and SARS-CoV-2 infection? Eur J Neurol. (2022) 29:1855–8. doi: 10.1111/ene.15304
319. Martins M, Pereira A, Teixeira A, Lima D, Lopes N, Amaral-Silva M, et al. Neurological complications associated with SARS-CoV-2 infection: a single-centre experience. Cureus J Med Sci. (2022) 14:32655. doi: 10.7759/cureus.32655
320. Matsumoto Y, Ohyama A, Kubota T, Ikeda K, Kaneko K, Takai Y, et al. MOG antibody-associated disorders following SARS-CoV-2 vaccination: a case report and literature review. Front Neurol. (2022) 13:e845755. doi: 10.3389/fneur.2022.845755
321. Sehgal V, Bansal P, Arora S, Kapila S, Bedi GS. Myelin oligodendrocyte glycoprotein antibody disease after COVID-19 vaccination - causal or incidental? Cureus J Med Sci. (2022) 14:27024. doi: 10.7759/cureus.27024
322. Wang JJ, Huang SM, Yu ZJ, Zhang S, Hou GH, Xu SY. Unilateral optic neuritis after vaccination against the coronavirus disease: two case reports. Doc Ophthalmol. (2022) 145:65–70. doi: 10.1007/s10633-022-09880-0
323. Uchino K, Soga K, Shinohara K, Imai T, Motohashi I, Okuma H, et al. Anti-myelin oligodendrocyte glycoprotein antibody-positive myelitis after coronavirus disease 2019. Internal Med. (2023) 62:1531–5. doi: 10.2169/internalmedicine.0394-22
324. Bailey OT, Pappenheimer AM, Cheever FS, Daniels JB. A murine virus (Jhm) causing disseminated encephalomyelitis with extensive destruction of myelin: Ii. Pathology J Exp Med. (1949) 90:195–212. doi: 10.1084/jem.90.3.195
325. Nagashima K, Wege H, Meyermann R, Meulen VT. Demyelinating encephalomyelitis induced by a long-term corona virus-infection in rats - preliminary-report. Acta Neuropathol. (1979) 45:205–13. doi: 10.1007/BF00702672
326. Stohlman SA, Weiner LP. Chronic central nervous system demyelination in mice after JHM virus infection. Neurology. (1981) 31:38–44. doi: 10.1212/WNL.31.1.38
327. Wege H, Koga M, Termeulen V. Corona virus-induced demyelination in rats. Neuropath Appl Neuro. (1981) 7:503–5.
328. Wu GF, Perlman S. Macrophage infiltration, but not apoptosis, is correlated with immune-mediated demyelination following murine infection with a neurotropic coronavirus. J Virol. (1999) 73:8771–80. doi: 10.1128/JVI.73.10.8771-8780.1999
329. Savarin C, Dutta R, Bergmann CC. Distinct gene profiles of bone marrow-derived macrophages and microglia during neurotropic coronavirus-induced demyelination. Front Immunol. (2018) 9:01325. doi: 10.3389/fimmu.2018.01325
330. Omura S, Kawai E, Sato F, Martinez NE, Minagar A, Al-Kofahi M, et al. Theiler's virus-mediated immunopathology in the CNS and heart: roles of organ-specific cytokine and lymphatic responses. Front Immunol. (2018) 9:2870. doi: 10.3389/fimmu.2018.02870
331. Omura S, Sato F, Martinez NE, Range T, Ekshyyan L, Minagar A, et al. Immunoregulation of Theiler's virus-induced demyelinating disease by glatiramer acetate without suppression of antiviral immune responses. Arch Virol. (2018) 163:1279–84. doi: 10.1007/s00705-018-3729-6
332. Sato F, Omura S, Kawai E, Martinez NE, Acharya MM, Reddy PC, et al. Distinct kinetics of viral replication, T cell infiltration, and fibrosis in three phases of myocarditis following Theiler's virus infection. Cell Immunol. (2014) 292:85–93. doi: 10.1016/j.cellimm.2014.10.004
333. Lipton HL. Theiler's virus infection in mice: an unusual biphasic disease process leading to demyelination. Infect Immun. (1975) 11:1147–55. doi: 10.1128/iai.11.5.1147-1155.1975
334. Dal Canto MC, Lipton HL. Primary demyelination in Theiler's virus infection. An ultrastructural study. Lab Invest. (1975) 33:626–37.
335. Croxford JL, Olson JK, Miller SD. Epitope spreading and molecular mimicry as triggers of autoimmunity in the Theiler's virus-induced demyelinating disease model of multiple sclerosis. Autoimmun Rev. (2002) 1:251–60. doi: 10.1016/S1568-9972(02)00080-0
336. Bieber AJ, Ure DR, Rodriguez M. Genetically dominant spinal cord repair in a murine model of chronic progressive multiple sclerosis. J Neuropathol Exp Neurol. (2005) 64:46–57. doi: 10.1093/jnen/64.1.46
337. Govindan AN, Fitzpatrick KS, Manoharan M, Tagge I, Kohama SG, Ferguson B, et al. Myelin-specific T cells in animals with Japanese macaque encephalomyelitis. Ann Clin Transl Neur. (2021) 8:456–70. doi: 10.1002/acn3.51303
338. Amano H, Miyamoto N, Shimura H, Sato DK, Fujihara K, Ueno S, et al. Influenza-associated MOG antibody-positive longitudinally extensive transverse myelitis: a case report. BMC Neurol. (2014) 14:224. doi: 10.1186/s12883-014-0224-x
339. Billiau A, Matthys P. Modes of action of Freund's adjuvants in experimental models of autoimmune diseases. J Leukoc Biol. (2001) 70:849–60. doi: 10.1189/jlb.70.6.849
340. Chen X, Winkler-Pickett RT, Carbonetti NH, Ortaldo JR, Oppenheim JJ, Howard OM. Pertussis toxin as an adjuvant suppresses the number and function of CD4+CD25+ T regulatory cells. Eur J Immunol. (2006) 36:671–80. doi: 10.1002/eji.200535353
341. Pakbaz Z, Sahraian MA, Noorbakhsh F, Salami SA, Pourmand MR. Staphylococcal enterotoxin B increased severity of experimental model of multiple sclerosis. Microb Pathog. (2020) 142:104064. doi: 10.1016/j.micpath.2020.104064
342. Mardiguian S, Ladds E, Turner R, Shepherd H, Campbell SJ, Anthony DC. The contribution of the acute phase response to the pathogenesis of relapse in chronic-relapsing experimental autoimmune encephalitis models of multiple sclerosis. J Neuroinflammation. (2017) 14:196. doi: 10.1186/s12974-017-0969-4
343. Petrisko TJ, Konat GW. Peripheral viral challenge exacerbates experimental autoimmune encephalomyelitis. Metab Brain Dis. (2019) 34:675–9. doi: 10.1007/s11011-019-0383-y
344. Vanheusden M, Broux B, Welten SPM, Peeters LM, Panagioti E, Van Wijmeersch B, et al. Cytomegalovirus infection exacerbates autoimmune mediated neuroinflammation. Sci Rep. (2017) 7:663. doi: 10.1038/s41598-017-00645-3
345. Milovanovic J, Popovic B, Milovanovic M, Kvestak D, Arsenijevic A, Stojanovic B, et al. Murine cytomegalovirus infection induces susceptibility to EAE in resistant BALB/c Mice. Front Immunol. (2017) 8:192. doi: 10.3389/fimmu.2017.00192
346. Chen Q, Liu Y, Lu A, Ni K, Xiang Z, Wen K, et al. Influenza virus infection exacerbates experimental autoimmune encephalomyelitis disease by promoting type I T cells infiltration into central nervous system. J Autoimmun. (2017) 77:1–10. doi: 10.1016/j.jaut.2016.10.006
347. Choi BY, Sim CK, Cho YS, Sohn M, Kim Y-J, Lee MS, et al. 2′-5′ oligoadenylate synthetase-like 1 (OASL1) deficiency suppresses central nervous system damage in a murine MOG-induced multiple sclerosis model. Neurosci Lett. (2016) 628:78–84. doi: 10.1016/j.neulet.2016.06.026
348. Jensen IJ, Jensen SN, Sjaastad FV, Gibson-Corley KN, Dileepan T, Griffith TS, et al. Sepsis impedes EAE disease development and diminishes autoantigen-specific naive CD4 T cells. Elife. (2020) 9:e55800. doi: 10.7554/eLife.55800
349. Hassan A, Wlodarczyk MF, Benamar M, Bassot E, Salvioni A, Kassem S, et al. Virus hosted in malaria-infected blood protects against T cell-mediated inflammatory diseases by impairing DC function in a type I IFN-dependent manner. MBio. (2020) 11:e03394–19. doi: 10.1128/mBio.03394-19
350. Nourbakhsh B, Cordano C, Asteggiano C, Ruprecht K, Otto C, Rutatangwa A, et al. Multiple sclerosis is rare in epstein-barr virus-seronegative children with central nervous system inflammatory demyelination. Ann Neurol. (2021) 89:1234–9. doi: 10.1002/ana.26062
351. Selter RC, Brilot F, Grummel V, Kraus V, Cepok S, Dale RC, et al. Antibody responses to EBV and native MOG in pediatric inflammatory demyelinating CNS diseases. Neurology. (2010) 74:1711–5. doi: 10.1212/WNL.0b013e3181e04096
352. Fang L, Gong YS, Han K, Lv YL, Li M, Wang J. Longitudinally extensive transverse myelitis with mycobacterium tuberculosis infection. Acta Neurol Belg. (2023) 123:243–6. doi: 10.1007/s13760-021-01723-0
353. Sahu SK, Giri S, Gupta N. Longitudinal extensive transverse myelitis due to tuberculosis: a report of four cases. J Postgrad Med. (2014) 60:409–12. doi: 10.4103/0022-3859.143977
354. Mokhtarian F, Zhang Z, Shi Y, Gonzales E, Sobel RA. Molecular mimicry between a viral peptide and a myelin oligodendrocyte glycoprotein peptide induces autoimmune demyelinating disease in mice. J Neuroimmunol. (1999) 95:43–54. doi: 10.1016/S0165-5728(98)00254-9
355. Herrmann I, Kellert M, Schmidt H, Mildner A, Hanisch UK, Bruck W, et al. Streptococcus pneumoniae Infection aggravates experimental autoimmune encephalomyelitis via Toll-like receptor 2. Infect Immun. (2006) 74:4841–8. doi: 10.1128/IAI.00026-06
356. Sattler MB, Demmer I, Williams SK, Maier K, Merkler D, Gadjanski I, et al. Effects of interferon-beta-1a on neuronal survival under autoimmune inflammatory conditions. Exp Neurol. (2006) 201:172–81. doi: 10.1016/j.expneurol.2006.04.015
357. Serres S, Bristow C, de Pablos RM, Merkler D, Soto MS, Sibson NR, et al. Magnetic resonance imaging reveals therapeutic effects of interferon-beta on cytokine-induced reactivation of rat model of multiple sclerosis. J Cerebr Blood F Met. (2013) 33:744–53. doi: 10.1038/jcbfm.2013.12
358. Njenga MK, Coenen MJ, DeCuir N, Yeh HY, Rodriguez M. Short-term treatment with interferon-alpha/beta promotes remyelination, whereas long-term treatment aggravates demyelination in a murine model of multiple sclerosis. J Neurosci Res. (2000) 59:661–70. doi: 10.1002/(SICI)1097-4547(20000301)59:5<661::AID-JNR9>3.0.CO;2-E
359. Bell JC, Liu Q, Gan Y, Liu Q, Liu Y, Shi F-D, et al. Visualization of inflammation and demyelination in 2D2 transgenic mice with rodent MRI. J Neuroimmunol. (2013) 264:35–40. doi: 10.1016/j.jneuroim.2013.09.008
360. 't Hart BA, Vogels J, Bauer J, Brok HPM, Blezer E. Non-invasive measurement of brain damage in a primate model of multiple sclerosis. Trends Mol Med. (2004) 10:85–91. doi: 10.1016/j.molmed.2003.12.008
361. Sun S-W, Liang H-F, Schmidt RE, Cross AH, Song S-K. Selective vulnerability of cerebral white matter in a murine model of multiple sclerosis detected using diffusion tensor imaging. Neurobiol Dis. (2007) 28:30–8. doi: 10.1016/j.nbd.2007.06.011
362. Fjær S, Bø L, Myhr K-M, Torkildsen Ø, Wergeland S. Magnetization transfer ratio does not correlate to myelin content in the brain in the MOG-EAE mouse model. Neurochem Int. (2015) 83–84:28–40. doi: 10.1016/j.neuint.2015.02.006
363. González-García C, Torres IM, García-Hernández R, Campos-Ruíz L, Esparragoza LR, Coronado MJ, et al. Mechanisms of action of cannabidiol in adoptively transferred experimental autoimmune encephalomyelitis. Exp Neurol. (2017) 298:57–67. doi: 10.1016/j.expneurol.2017.08.017
364. Levy Barazany H, Barazany D, Puckett L, Blanga-Kanfi S, Borenstein-Auerbach N, Yang K, et al. Brain MRI of nasal MOG therapeutic effect in relapsing-progressive EAE. Exp Neurol. (2014) 255:63–70. doi: 10.1016/j.expneurol.2014.02.010
365. Pol S, Liang S, Schweser F, Dhanraj R, Schubart A, Preda M, et al. Subcutaneous anti-CD20 antibody treatment delays gray matter atrophy in human myelin oligodendrocyte glycoprotein-induced EAE mice. Exp Neurol. (2021) 335:113488. doi: 10.1016/j.expneurol.2020.113488
366. Aharoni R, Sasson E, Blumenfeld-Katzir T, Eilam R, Sela M, Assaf Y, et al. Magnetic resonance imaging characterization of different experimental autoimmune encephalomyelitis models and the therapeutic effect of glatiramer acetate. Exp Neurol. (2013) 240:130–44. doi: 10.1016/j.expneurol.2012.11.004
367. Pol S, Schweser F, Bertolino N, Preda M, Sveinsson M, Sudyn M, et al. Characterization of leptomeningeal inflammation in rodent experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis. Exp Neurol. (2019) 314:82–90. doi: 10.1016/j.expneurol.2019.01.013
368. Linker RA, Kroner A, Horn T, Gold R, Mäurer M, Bendszus M. Iron particle-enhanced visualization of inflammatory central nervous system lesions by high resolution: preliminary data in an animal model. AJNR Am J Neuroradiol. (2006) 27:1225–9.
369. Chin C-L, Pai M, Bousquet PF, Schwartz AJ, O'Connor EM, Nelson CM, et al. Distinct spatiotemporal pattern of CNS lesions revealed by USPIO-enhanced MRI in MOG-induced EAE rats implicates the involvement of spino-olivocerebellar pathways. J Neuroimmunol. (2009) 211:49–55. doi: 10.1016/j.jneuroim.2009.03.012
370. Belloli S, Zanotti L, Murtaj V, Mazzon C, Di Grigoli G, Monterisi C, et al. 18F-VC701-PET and MRI in the in vivo neuroinflammation assessment of a mouse model of multiple sclerosis. J Neuroinflammation. (2018) 15:33. doi: 10.1186/s12974-017-1044-x
371. Nack A, Brendel M, Nedelcu J, Daerr M, Nyamoya S, Beyer C, et al. Expression of translocator protein and [18F]-GE180 ligand uptake in multiple sclerosis animal models. Cells. (2019) 8:94. doi: 10.3390/cells8020094
372. Hoehne A, James ML, Alam IS, Ronald JA, Schneider B, D'Souza A, et al. [18F]FSPG-PET reveals increased cystine/glutamate antiporter (xc-) activity in a mouse model of multiple sclerosis. J Neuroinflammation. (2018) 15:55. doi: 10.1186/s12974-018-1080-1
373. Stevens MY, Cropper HC, Lucot KL, Chaney AM, Lechtenberg KJ, Jackson IM, et al. Development of a CD19 PET tracer for detecting B cells in a mouse model of multiple sclerosis. J Neuroinflammation. (2020) 17:275. doi: 10.1186/s12974-020-01880-8
374. Waiczies H, Guenther M, Skodowski J, Lepore S, Pohlmann A, Niendorf T, et al. Monitoring dendritic cell migration using 19F/1H magnetic resonance imaging. J Vis Exp. (2013) 73:e50251. doi: 10.3791/50251
375. Ruiz-Cabello J, Barnett BP, Bottomley PA, Bulte JWM. Fluorine (19F) MRS and MRI in biomedicine. NMR Biomed. (2011) 24:114–29. doi: 10.1002/nbm.1570
376. Waiczies S, Millward JM, Starke L, Delgado PR, Huelnhagen T, Prinz C, et al. Enhanced fluorine-19 MRI sensitivity using a cryogenic radiofrequency probe: technical developments and ex vivo demonstration in a mouse model of neuroinflammation. Sci Rep. (2017) 7:9808. doi: 10.1038/s41598-017-09622-2
377. Waiczies S, Rosenberg JT, Kuehne A, Starke L, Delgado PR, Millward JM, et al. et al. Fluorine-19 MRI at 211 T: enhanced spin-lattice relaxation of perfluoro-15-crown-5-ether and sensitivity as demonstrated in ex vivo murine neuroinflammation. MAGMA. (2019) 32:37–49. doi: 10.1007/s10334-018-0710-z
378. Prinz C, Delgado PR, Eigentler TW, Starke L, Niendorf T, Waiczies S. Toward 19F magnetic resonance thermometry: spin-lattice and spin-spin-relaxation times and temperature dependence of fluorinated drugs at 94 T. MAGMA. (2019) 32:51–61. doi: 10.1007/s10334-018-0722-8
379. Yura M, Takahashi I, Serada M, Koshio T, Nakagami K, Yuki Y, et al. Role of MOG-stimulated Th1 type “light up” (GFP+) CD4+ T cells for the development of experimental autoimmune encephalomyelitis (EAE). J Autoimmun. (2001) 17:17–25. doi: 10.1006/jaut.2001.0520
380. Siffrin V, Radbruch H, Glumm R, Niesner R, Paterka M, Herz J, et al. In vivo imaging of partially reversible th17 cell-induced neuronal dysfunction in the course of encephalomyelitis. Immunity. (2010) 33:424–36. doi: 10.1016/j.immuni.2010.08.018
381. Wang X, Brieland JK, Kim JH, Chen Y-J, O'Neal J, O'Neil SR, et al. Diffusion tensor imaging detects treatment effects of FTY720 in experimental autoimmune encephalomyelitis mice. NMR Biomed. (2013) 26:3012. doi: 10.1002/nbm.3012
382. Cruz-Orengo L, Chen Y-J, Kim JH, Dorsey D, Song S-K, Klein RS. CXCR7 antagonism prevents axonal injury during experimental autoimmune encephalomyelitis as revealed by in vivo axial diffusivity. J Neuroinflammation. (2011) 8:170. doi: 10.1186/1742-2094-8-170
383. de Guzman AE, Wong MD, Gleave JA, Nieman BJ. Variations in post-perfusion immersion fixation and storage alter MRI measurements of mouse brain morphometry. Neuroimage. (2016) 142:687–95. doi: 10.1016/j.neuroimage.2016.06.028
384. Cahill LS, Zhang MA, Ramaglia V, Whetstone H, Sabbagh MP, Yi TJ, et al. Aged hind-limb clasping experimental autoimmune encephalomyelitis models aspects of the neurodegenerative process seen in multiple sclerosis. Proc Natl Acad Sci U S A. (2019) 116:22710–20. doi: 10.1073/pnas.1915141116
385. Steudler J, Ecott T, Ivan DC, Bouillet E, Walthert S, Berve K, et al. Autoimmune neuroinflammation triggers mitochondrial oxidation in oligodendrocytes. Glia. (2022) 70:2045–61. doi: 10.1002/glia.24235
386. Cruz-Herranz A, Dietrich M, Hilla AM, Yiu HH, Levin MH, Hecker C, et al. Monitoring retinal changes with optical coherence tomography predicts neuronal loss in experimental autoimmune encephalomyelitis. J Neuroinflammation. (2019) 16:203. doi: 10.1186/s12974-019-1583-4
387. Boretius S, Schmelting B, Watanabe T, Merkler D, Tammer R, Czéh B, et al. Monitoring of EAE onset and progression in the common marmoset monkey by sequential high-resolution 3D MRI. NMR Biomed. (2006) 19:41–9. doi: 10.1002/nbm.999
388. Weissert R, Hugosson T, Petzold A. Upregulated retinal neurofilament expression in experimental optic neuritis. Neuroophthalmology. (2022) 46:215–9. doi: 10.1080/01658107.2022.2025852
389. Castoldi V, Marenna S, d'Isa R, Huang S-C, Battista DD, Chirizzi C, et al. Non-invasive visual evoked potentials to assess optic nerve involvement in the dark agouti rat model of experimental autoimmune encephalomyelitis induced by myelin oligodendrocyte glycoprotein. Brain Pathol. (2020) 30:137–50. doi: 10.1111/bpa.12762
390. Meyer R, Weissert R, Diem R, Storch MK, de Graaf KL, Kramer B, et al. Acute neuronal apoptosis in a rat model of multiple sclerosis. J Neurosci. (2001) 21:6214–20. doi: 10.1523/JNEUROSCI.21-16-06214.2001
391. Hobom M, Storch MK, Weissert R, Maier K, Radhakrishnan A, Kramer B, et al. Mechanisms and time course of neuronal degeneration in experimental autoimmune encephalomyelitis. Brain Pathol. (2004) 14:148–57. doi: 10.1111/j.1750-3639.2004.tb00047.x
392. Hein K, Gadjanski I, Kretzschmar B, Lange K, Diem R, Sättler MB, et al. An optical coherence tomography study on degeneration of retinal nerve fiber layer in rats with autoimmune optic neuritis. Invest Ophthalmol Vis Sci. (2012) 53:157–63. doi: 10.1167/iovs.11-8092
393. Fairless R, Williams SK, Hoffmann DB, Stojic A, Hochmeister S, Schmitz F, et al. Preclinical retinal neurodegeneration in a model of multiple sclerosis. J Neurosci. (2012) 32:5585–97. doi: 10.1523/JNEUROSCI.5705-11.2012
394. Stojic A, Bojcevski J, Williams SK, Bas-Orth C, Nessler S, Linington C, et al. Preclinical stress originates in the rat optic nerve head during development of autoimmune optic neuritis. Glia. (2019) 67:512–24. doi: 10.1002/glia.23560
395. Green AJ, McQuaid S, Hauser SL, Allen IV, Lyness R. Ocular pathology in multiple sclerosis: retinal atrophy and inflammation irrespective of disease duration. Brain. (2010) 133:1591–601. doi: 10.1093/brain/awq080
396. Fisher JB, Jacobs DA, Markowitz CE, Galetta SL, Volpe NJ, Nano-Schiavi ML, et al. Relation of visual function to retinal nerve fiber layer thickness in multiple sclerosis. Ophthalmology. (2006) 113:324–32. doi: 10.1016/j.ophtha.2005.10.040
397. Jin J, Smith MD, Kersbergen CJ, Kam T-I, Viswanathan M, Martin K, et al. Glial pathology and retinal neurotoxicity in the anterior visual pathway in experimental autoimmune encephalomyelitis. Acta Neuropathol Commun. (2019) 7:125. doi: 10.1186/s40478-019-0767-6
398. Shindler KS, Guan Y, Ventura E, Bennett J, Rostami A. Retinal ganglion cell loss induced by acute optic neuritis in a relapsing model of multiple sclerosis. Mult Scler. (2006) 12:526–32. doi: 10.1177/1352458506070629
399. Petrikowski L, Reinehr S, Haupeltshofer S, Deppe L, Graz F, Kleiter I, et al. Progressive retinal and optic nerve damage in a mouse model of spontaneous opticospinal encephalomyelitis. Front Immunol. (2021) 12:759389. doi: 10.3389/fimmu.2021.759389
400. Marenna S, Huang S-C, Castoldi V, d'Isa R, Costa GD, Comi G, et al. Functional evolution of visual involvement in experimental autoimmune encephalomyelitis. Mult Scler J Exp Transl Clin. (2020) 6:2055217320963474. doi: 10.1177/2055217320963474
401. Dietrich M, Hecker C, Hilla A, Cruz-Herranz A, Hartung H-P, Fischer D, et al. Using optical coherence tomography and optokinetic response as structural and functional visual system readouts in mice and rats. JoVE. (2019) 142:e58571. doi: 10.3791/58571
402. Cordano C, Sin JH, Timmons G, Yiu HH, Stebbins K, Guglielmetti C, et al. Validating visual evoked potentials as a preclinical, quantitative biomarker for remyelination efficacy. Brain. (2022) 145:3943–52. doi: 10.1093/brain/awac207
403. Kapell H, Fazio L, Dyckow J, Cruz-Herranz A, Mayer C, Campos J, et al. Paranodal potassium channels as neuroprotective targets in inflammatory demyelination. Mult Scler J. (2022) 28:22–3.
404. Schirmer L, Möbius W, Zhao C, Cruz-Herranz A, Ben Haim L, Cordano C, et al. Oligodendrocyte-encoded Kir4.1 function is required for axonal integrity. eLife. (2018) 7:e36428. doi: 10.7554/eLife.36428
405. Diem R, Demmer I, Boretius S, Merkler D, Schmelting B, Williams SK, et al. Autoimmune optic neuritis in the common marmoset monkey: comparison of visual evoked potentials with MRI and histopathology. Invest Ophth Vis Sci. (2008) 49:3707–14. doi: 10.1167/iovs.08-1896
406. Talla V, Porciatti V, Chiodo V, Boye SL, Hauswirth WW, Guy J. Gene therapy with mitochondrial heat shock protein 70 suppresses visual loss and optic atrophy in experimental autoimmune encephalomyelitis. Invest Ophthalmol Vis Sci. (2014) 55:5214–26. doi: 10.1167/iovs.14-14688
407. Talla V, Yu H, Chou T-H, Porciatti V, Chiodo V, Boye SL, et al. NADH-dehydrogenase type-2 suppresses irreversible visual loss and neurodegeneration in the EAE animal model of MS. Mol Ther. (2013) 21:1876–88. doi: 10.1038/mt.2013.104
408. Talla V, Koilkonda R. Targeted Krüppel-like factor 4 gene knock-out in retinal ganglion cells improves visual function in multiple sclerosis mouse model. eNeuro. (2020) 7:ENEURO.0320-19.2020. doi: 10.1523/ENEURO.0320-19.2020
409. Talla V, Porciatti V, Chiodo V, Boye SL, Hauswirth WW, Guy J. Gene therapy with mitochondrial heat shock protein 70 suppresses visual loss and optic atrophy in experimental autoimmune encephalomyelitis. Invest Ophthalmol Vis Sci. (2014) 55:5214–26. doi: 10.1167/iovs.14-14688
410. Dietrich M, Helling N, Hilla A, Heskamp A, Issberner A, Hildebrandt T, et al. Early alpha-lipoic acid therapy protects from degeneration of the inner retinal layers and vision loss in an experimental autoimmune encephalomyelitis-optic neuritis model. J Neuroinflammation. (2018) 15:71. doi: 10.1186/s12974-018-1111-y
411. Liu P, Huang H, Fang F, Liu L, Li L, Feng X, et al. Neuronal NMNAT2 overexpression does not achieve significant neuroprotection in experimental autoimmune encephalomyelitis/optic neuritis. Front Cell Neurosci. (2021) 15:754651. doi: 10.3389/fncel.2021.754651
412. Sekyi MT, Lauderdale K, Atkinson KC, Golestany B, Karim H, Feri M, et al. Alleviation of extensive visual pathway dysfunction by a remyelinating drug in a chronic mouse model of multiple sclerosis. Brain Pathol. (2021) 31:312–32. doi: 10.1111/bpa.12930
413. Knier B, Rothhammer V, Heink S, Puk O, Graw J, Hemmer B, et al. Neutralizing IL-17 protects the optic nerve from autoimmune pathology and prevents retinal nerve fiber layer atrophy during experimental autoimmune encephalomyelitis. J Autoimmun. (2015) 56:34–44. doi: 10.1016/j.jaut.2014.09.003
414. Herrera SL, Palmer VL, Whittaker H, Smith BC, Kim A, Schellenberg AE, et al. Damage to the optic chiasm in myelin oligodendrocyte glycoprotein-experimental autoimmune encephalomyelitis mice. Magn Reson Insights. (2014) 7:23–31. doi: 10.4137/MRI.S19750
415. Qi X, Lewin AS, Sun L, Hauswirth WW, Guy J. Suppression of mitochondrial oxidative stress provides long-term neuroprotection in experimental optic neuritis. Invest Ophthalmol Vis Sci. (2007) 48:681–91. doi: 10.1167/iovs.06-0553
416. Manogaran P, Walker-Egger C, Samardzija M, Waschkies C, Grimm C, Rudin M, et al. Exploring experimental autoimmune optic neuritis using multimodal imaging. Neuroimage. (2018) 175:327–39. doi: 10.1016/j.neuroimage.2018.04.004
417. Nishioka C, Liang H-F, Barsamian B, Sun S-W. Sequential phases of RGC axonal and somatic injury in EAE mice examined using DTI and OCT. Mult Scler Relat Disord. (2019) 27:315–23. doi: 10.1016/j.msard.2018.11.010
418. Cruz-Herranz A, Oertel FC, Kim K, Cantó E, Timmons G, Sin JH, et al. Distinctive waves of innate immune response in the retina in experimental autoimmune encephalomyelitis. JCI Insight. (2021) 6:149228. doi: 10.1172/jci.insight.149228
419. Frenger MJ, Hecker C, Sindi M, Issberner A, Hartung H-P, Meuth SG, et al. Semi-automated live tracking of microglial activation in CX3CR1GFP mice during experimental autoimmune encephalomyelitis by confocal scanning laser ophthalmoscopy. Front Immunol. (2021) 12:761776. doi: 10.3389/fimmu.2021.761776
420. Rakhymzhan A, Reuter L, Raspe R, Bremer D, Günther R, Leben R, et al. Coregistered spectral optical coherence tomography and two-photon microscopy for multimodal near-instantaneous deep-tissue imaging. Cytometry Part A. (2020) 97:515–27. doi: 10.1002/cyto.a.24012
421. Bremer D, Pache F, Günther R, Hornow J, Andresen V, Leben R, et al. Longitudinal intravital imaging of the retina reveals long-term dynamics of immune infiltration and its effects on the glial network in experimental autoimmune uveoretinitis, without evident signs of neuronal dysfunction in the ganglion cell layer. Front Immunol. (2016) 7:642. doi: 10.3389/fimmu.2016.00642
422. Vosoughi AR, Ling J, Tam KT, Blackwood J, Micieli JA. Ophthalmic manifestations of myelin oligodendrocyte glycoprotein-IgG-associated disorder other than optic neuritis: a systematic review. Br J Ophthalmol. (2021) 105:1591–8. doi: 10.1136/bjophthalmol-2020-317267
423. Shao H, Sun SL, Kaplan HJ, Sun D. Induction of autoimmune encephalomyelitis and uveitis in B6 and (B6 x SJL) mice by peptides derived from myelin/oligodendrocyte glycoprotein. J Neuroimmunol. (2002) 132:117–22. doi: 10.1016/S0165-5728(02)00318-1
424. Papadopoulos D, Rundle J, Patel R, Marshall I, Stretton J, Eaton R, et al. FTY720 ameliorates MOG-induced experimental autoimmune encephalomyelitis by suppressing both cellular and humoral immune responses. J Neurosci Res. (2010) 88:346–59. doi: 10.1002/jnr.22196
Keywords: myelin oligodendrocyte glycoprotein associated disease, imaging, translational research, EAE, animal models
Citation: Oertel FC, Hastermann M and Paul F (2023) Delimiting MOGAD as a disease entity using translational imaging. Front. Neurol. 14:1216477. doi: 10.3389/fneur.2023.1216477
Received: 03 May 2023; Accepted: 23 August 2023;
Published: 07 December 2023.
Edited by:
Sasitorn Siritho, Bumrungrad International Hospital, ThailandReviewed by:
Yoshiki Takai, Tohoku University Hospital, JapanChristian Cordano, University of California, San Francisco, United States
Copyright © 2023 Oertel, Hastermann and Paul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frederike Cosima Oertel, frederike-cosima.oertel@charite.de
†These authors have contributed equally to this work and share first authorship