 Gina Perez-Giraldo
Gina Perez-Giraldo Natalia Gonzalez Caldito
Natalia Gonzalez Caldito Elena Grebenciucova
Elena Grebenciucova
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurol. , 06 July 2023
Sec. Multiple Sclerosis and Neuroimmunology
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1210972
This article is part of the Research Topic MOGAD, Current Knowledge and Future Trends View all 13 articles
Transverse myelitis (TM) is the second most common presentation of myelin oligodendrocyte antibody-associated disease (MOGAD), occurring in approximately 26% of affected patients. The diagnosis may be complicated by the lack of diagnostic specificity of low titers of MOG antibody in serum, fluctuation in seropositivity overtime, including initially normal MRI in up to 10% of patients, and in many instances complete resolution of radiological abnormalities when MRI is done in a significantly delayed fashion. The use of preventive disease modifying treatments is limited by the uncertainty whether the disease process will remain monophasic or become relapsing, as well as by the lack FDA approved treatments. In this review, we discuss clinical, radiological and cerebrospinal fluid (CSF) characteristics, including the significance of MOG titers and changes in the seropositivity status for the diagnosis of MOGAD-associated TM, its radiological features and management options, highlighting the data on the risk of relapses associated with TM at presentation and the need for further randomized clinical trials to empower effective treatment algorithms.
Transverse myelitis (TM) is the second most common presentation of myelin oligodendrocyte antibody-associated disease (MOGAD), occurring in approximately 26% of MOGAD (1). TM can occur in isolation, simultaneously with optic neuritis (less than 10% of patients) or as part of acute disseminated encephalomyelitis (ADEM) (2–4). Clinical presentation in children and adults may vary. TM in MOGAD can occur shortly after an infectious illness (1). It is therefore not surprising that rare cases of MOGAD TM have been reported both after SARS-CoV2 vaccination (ChAdOx1 nCoV-19) and after the infection itself (5, 6). The severity of the attack varies, typically moderate to severe with EDSS scores >4 in about half of patients (7) with up to one third being nonambulatory at the nadir of acute myelitis attack (8), and many requiring bladder catheterization (9). In a prospective UK cohort, transverse myelitis as part of MOGAD was found to be less common in children as compared to adults, and relapsing TM was only observed in adults (3). Optic neuritis is the most common relapsing phenotype of MOGAD, and patients with TM at onset have less risk of relapse as compared to those with other MOGAD phenotypes (3).
The clinical manifestations of MOGAD myelitis include sensory, motor or bowel-bladder symptoms, erectile dysfunction, typically with acute to subacute onset of paraparesis or quadriplegia (10). Motor and sensory symptoms are usually bilateral, and sphincter dysfunction is more common than in aquaporin 4 antibody (AQP 4 antibody) positive NMOSD (4). Patients may also present with acute flaccid myelitis (AFM) and may be initially thought to have post-viral or viral-induced AFM (8, 11). Tonic spasms and severe neuropathic pain are less common with MOGAD as compared to AQP4 positive NMOSD (12). Prognosis is generally good, with excellent motor recovery, but residual neurogenic bladder and sexual dysfunction can occur (2).
MOGAD myelitis is diagnosed when a patient has neurological deficits and tempo of symptomatic development compatible with myelitis, a clear positive serum MOG-IgG test, and supportive of diagnosis MRI features (2). The latest international expert consensus advocates towards the use of live cell-based assays to increase diagnostic specificity. If not available, fixed cell-based assays can be used, with a clear positive result being a titer >1:100 (2). Titers lower than 1:100 have a lower predictive value (number of true-positive results/total positive results) for MOGAD and may lead to misdiagnosis. For example, a titer of MOG of 1:20 to 1:40 carry a positive predictive value of 51%, meaning that nearly 50% of patients with this titer may have a different etiology of their clinical presentation (13). Serological testing should ideally occur before administration of corticosteroids, intravenous immunoglobulins, or plasma exchange, as these interventions can increase the risk of false negative result. In cases of a high clinical suspicion but a negative test, if done after initiation of immune therapies, testing should be repeated about 3 months or later. Screening for the presence of serum MOG-IgG routinely in patients with clear features of multiple sclerosis is not recommended, as false positives can occur, decreasing the positive predictive value of the test and leading to misdiagnosis (2, 10).
In addition, titers of MOG antibody can fluctuate with intermittent seroconversion to negativity and can become undetectable over time (14); this, and the fact that the MRI abnormalities can disappear (up to 72% of the brain lesions; 79% of spinal cord lesions) (13, 15, 16), make a retrospective diagnosis of MOGAD impossible in some scenarios, which warrants a continuity of high degree of suspicion if new neurological symptoms arise (17).
Cerebrospinal fluid (CSF) analysis typically demonstrates lymphocytic pleocytosis, with >50 WBC/mm3 seen in about 30% of patients (9). Oligoclonal bands are frequently absent, with positivity rates being less than 10%–15% (4, 18). MOG-IgG antibody concentrations in CSF are typically low, however in some cases MOG-IgG can be positive in CSF and not in serum. Intrathecal antibody production occurs more frequently in MOGAD than in AQP4-positive NMOSD (19). Caution is advised in interpretation of the MOG-IgG positivity when clinical features are atypical of MOGAD, as false positives can occur. Testing both CSF and serum is not recommended for routine evaluations (2). However, if the serum MOG antibody is negative in a patient presenting with clinical, radiological and CSF findings typical of MOGAD, CSF testing for MOG antibody may be considered, as well as re-testing serum several months later.
Current diagnostic criteria for MOGAD require exclusion of an alternative diagnosis. Differential diagnostic considerations include multiple sclerosis, neuromyelitis optica spectrum disorder, CRMP-5 antibody positive paraneoplastic myelopathy, GFAP antibody positive encephalomyelitis, neurosarcoidosis, SLE associated TM, ischemic myelopathy, metabolic myelopathy, infections and neoplastic causes. Specific findings pointing to other inflammatory diagnoses are summarized in Table 1.
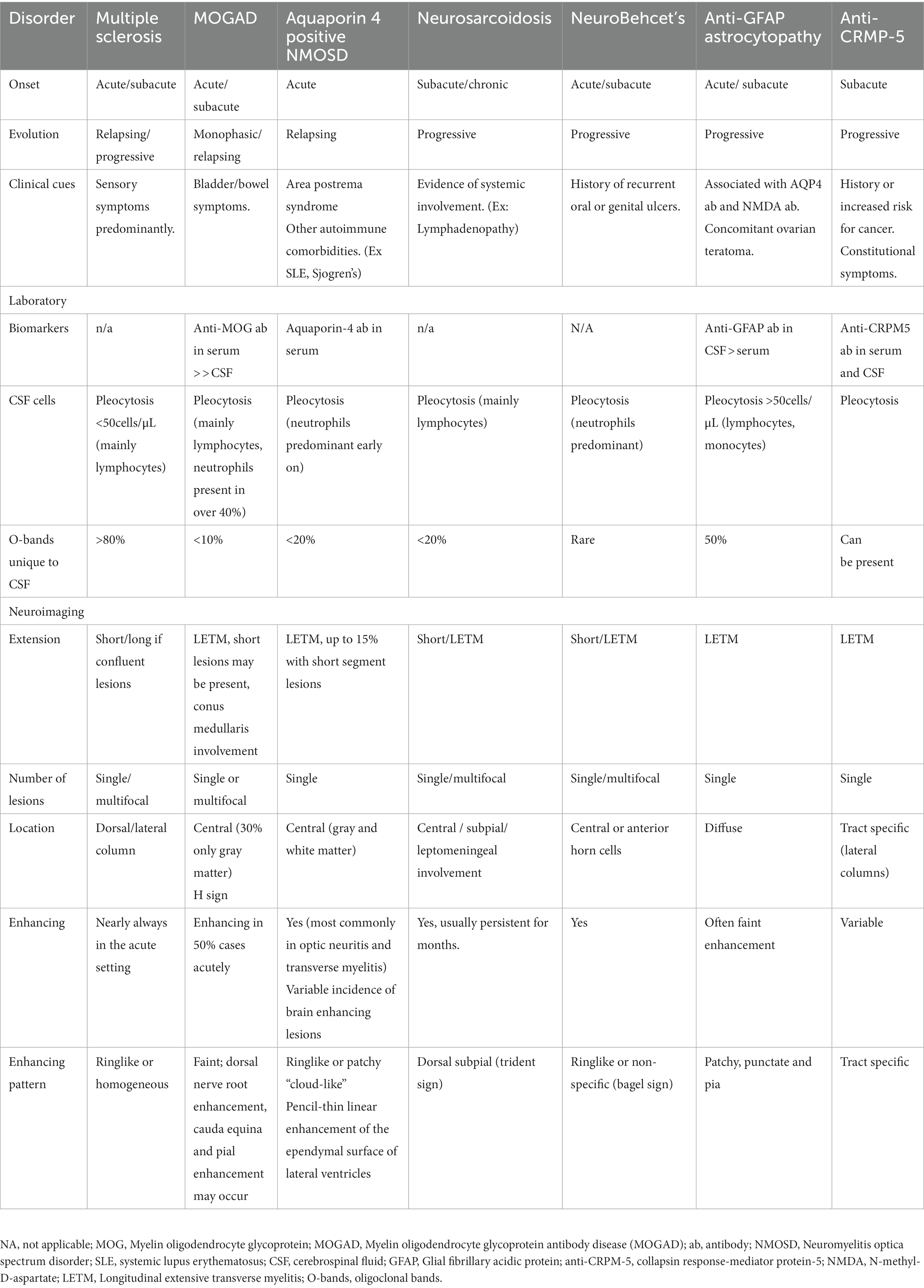
Table 1. Differential diagnosis of MOGAD associated-TM.
Acute myelitis due to MOGAD is typically longitudinally extensive involving 3 or more vertebral segments in length (Figure 1). Short lesions can occur in about 7% of patients, more commonly in those patients that are approximately 40 years of age (3). Multifocal spinal cord lesions can occur (2, 18). Spinal cord necrosis is typically not seen (20). Some T2 signal abnormalities may be initially subtle and contrast the severity of clinical presentation. In fact, in some cases linear T2 signal abnormalities may be difficult to distinguish from a prominent central canal. In these instances, carefully evaluating axial images may be helpful, as well as repeating the MRI at a later time to evaluate for the evolution of the lesion. MRI of the brain may also show MOGAD-associated lesions and should be considered in TM evaluation. Some patients with MOGAD TM may also have clinically silent spinal cord lesions (20). Although current data are limited by the retrospective nature, one study found silent spinal cord MRI lesions occurred in none of the patients during remission and in up to 7.3% of those during acute attack (21). Another study confirmed that finding new or enlarging spinal cord lesions inter-attack is rare and occurs in <1% of patients (22). Initial MRI of the spinal cord may be normal in up to 10% of patients, and repeat MRI within days would be warranted (13–15).
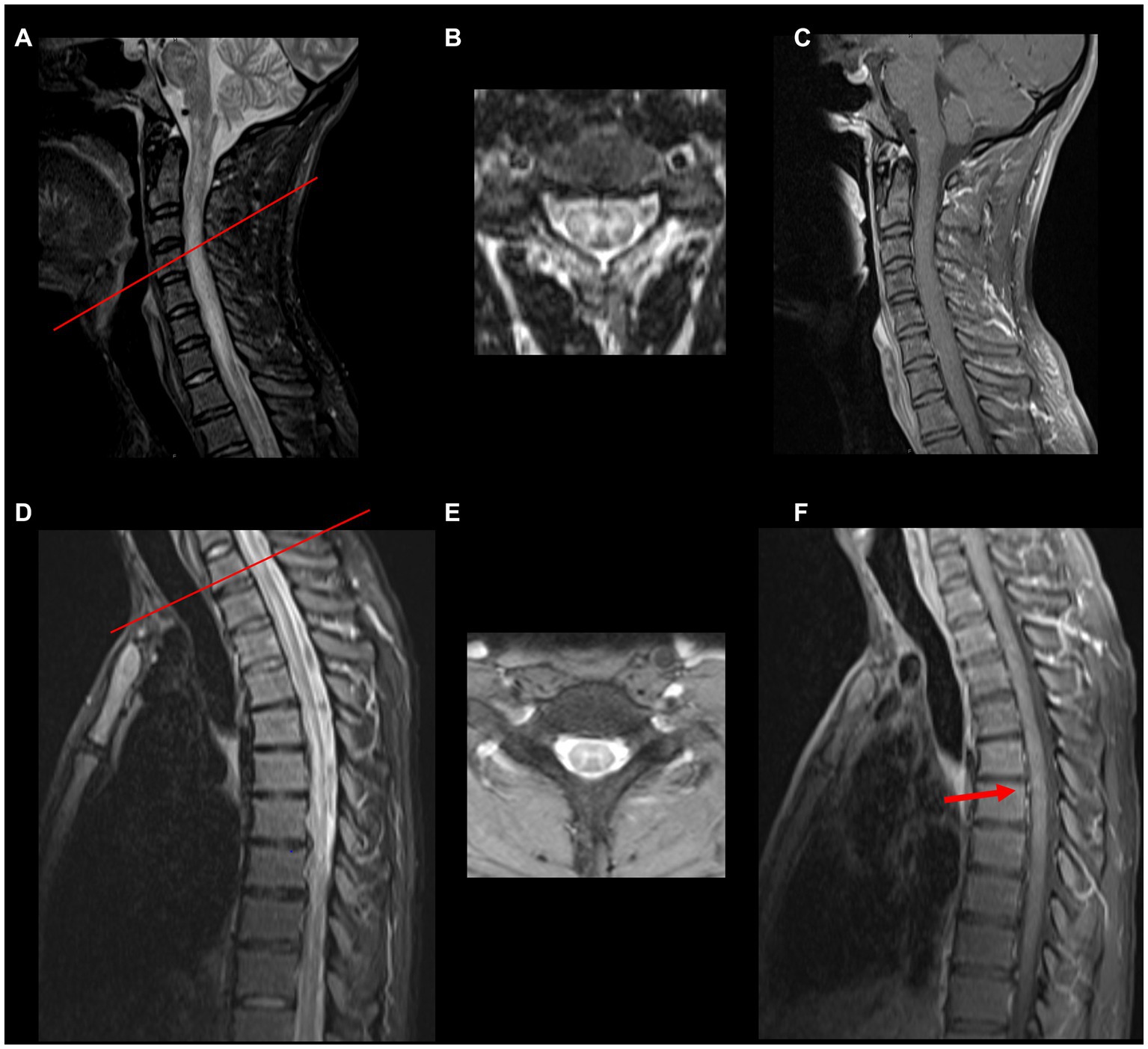
Figure 1. Spinal cord MRI in MOGAD myelitis. (A,D) Abnormal hyperintense T2/FLAIR signal abnormality with cord expansion from the cervicomedullary junction extending to the thoracic spinal cord. (B,E) Show axial T2 images (red bar indicates the spinal cord of the axial section) resembling H sign (grey matter involvement). (C,F) Show post gadolinium T1 sequences, respectively. No enhancement is noted in the cervical spinal cord, but a small focus of enhancement is noted at T9 and T10 (red arrow).
A negative MRI in a patient presenting acutely with clinical features of myelitis should not deter from testing for MOG antibody. The spinal cord lesions are typically located in the central cord, can involve both the gray and white matter, affect more than 50% of the axial section or can be restricted to the grey matter in about 50% of patients, which on axial imaging can be seen as the “H sign” (2, 23). T2 signal abnormality confined to gray matter (sagittal line and axial H sign) as well as lack of enhancement favor diagnosis of MOGAD over NMOSD or MS (8).
A straight T2 hyperintense line with surrounding hyperintense signal in the anterior and posterior gray matter horns on sagittal view can also be seen in MOGAD myelitis and is known as pseudo dilation of the central canal (23). The conus medullaris can be affected in about 26% of patients with MOGAD (24), which is a valuable radiographic clue, as the conus medullaris is less frequently affected in other demyelinating conditions, such as multiple sclerosis (MS) and neuromyelitis optica spectrum disorder (NMOSD), with rates of involvement as low as 1.3 and 6%, respectively, (8, 24–26). Gadolinium enhancement can occur in about half of patients with MOGAD myelitis and can be heterogeneous giving a cloud-like appearance (2, 9, 23). Interestingly, dorsal nerve root enhancement, cauda equina and pial enhancement can also occur (2).
Clinically silent new lesions on MRI are rare and occur in only 3% of patients with MOGAD (21, 27). Upon follow up and resolution of acute myelitis, complete resolution of spinal cord lesions can be seen in up to 79% of MOGAD cases (4). Spinal cord atrophy can be seen in severe cases, but this is rare (2).
In the acute setting, patients presenting with TM receive intravenous corticosteroids. For those with incomplete recovery or severe clinical picture overall, plasma exchange is typically used next (9, 28). Intravenous immunoglobulins (IVIG) can also be used following the plasma exchange completion, given that intravenous immunoglobulins can also serve as an effective preventive therapy for those patients who elect long-term preventive therapy option (29). Steroids are typically tapered slowly over at least a month or longer, with some data pointing to a lower risk of relapse in those with steroid use >1 month (16).
In about 40%–50% of cases, MOGAD appears to be monophasic long-term; however, 50%–60% patients presenting with their first attack of MOGAD will go on to develop the relapsing type of the disease. While the disappearance of MOG antibody serologically cannot be used as a definitive sign of the disease being monophasic (17), some data show that pediatric patients who seroconvert to negativity may have a somewhat lower risk of relapse (30). In the UK cohort, the final status of MOG antibody was not found to be associated with the relapsing disease overall, but the longitudinal analysis showed a reduction of 4–5% in monthly relapse risk in those who seroconverted negative for MOG IgG. Same study showed that the patients presenting with TM may have lower rates of relapsing disease: TM as the first attack was associated with a lower risk of relapse (OR, 0.03; 95% CI, 0.004–0.23; p = 0.001) and a longer time to first relapse (HR, 0.42; 95% CI, 0.22–0.82; p = 0.011) (16). In another study, TM at presentation alone or in combination with another syndrome (ON, ADEM, brainstem) was likewise associated with lower risk of relapse (HR, 0.41; 95% CI, 0.20–0.88; p = 0.01) (3).
A discussion whether a long-term disease modifying treatment needs to be started after the first attack or only after the disease course proves itself to be relapsing, risks versus benefits of both approaches should be carefully explored with every patient. Some clinicians may offer preventive disease modifying treatments as early as after the first attack, if the attack was severe with poor recovery leading to significant residual disability, thus if the second attack were to occur, it would be detrimental to the patient’s quality of life and independence.
Treatments commonly used in the prevention of MOGAD relapses are rituximab, azathioprine, mycophenolate mofetil, tocilizumab, and intravenous immunoglobulins. A recent meta-analysis of 41 studies (3 prospective, 1 ambispective, 37 retrospective) evaluating efficacy of MOGAD-associated treatments found that the proportions of patients free of relapse were 65% [95% confidence interval (CI): 49%–82%] on azathioprine, 73% (95% CI: 62%–84%) on mycophenolate mofetil, 66% (95% CI: 55%–77%) on rituximab, 79% (95% CI: 66%–91%) on IVIG, and 93% (95% CI: 54%–100%) on tocilizumab (31).
Patients with MOGAD transverse myelitis can go on to develop chronic neuropathic pain, weakness, and bladder dysfunction. Addressing these symptoms effectively and in a timely manner is critical to the patient’s quality of life. Medications utilized for the treatment of neuropathic pain such as gabapentin, pregabalin, and in some instances addition of duloxetine can be utilized. Physical and occupational therapy during recovery period and periodically in those without complete recovery are recommended. Bladder symptoms are best addressed and managed by a knowledgeable urologist or a neuro-urologist. Neuropsychiatric consultation may be warranted in patients with adjustment disorder, anxiety or depressive symptoms secondary to the medical condition.
Neurological outcomes of patients with TM due to MOGAD are typically more favorable than of those with anti-aquaporin4 antibody positive NMOSD. A recent study evaluating long-term outcomes of patients with TM due to MOGAD (n = 32) vs. NMOSD (n = 57) found that MOGAD TM patients on average had a lower EDSS score than patients with AQP4-Ab TM (1.8 [1.0–8.0] vs. 3.0 [1.0–8.0]), reflecting better outcomes. Due to higher predilection of MOG positive TM to conus localization, persistent bladder dysfunction was more common in patients with MOGAD (59% with MOGAD and 48% with AQP-4 positive NMOSD), with up to 23% requiring long-term catheterization in both groups. In addition, neuropathic pain was less common in patients with MOGAD TM vs. NMOSD TM (29% vs. 13%) (32).
Transverse myelitis is the second most common presentation of MOGAD, with a substantial number of patients presenting with para- or quadriplegia at nadir of the attack. Although generally purporting better outcomes than AQP 4 antibody positive NMOSD, some patients are left with persistent bladder dysfunction, erectile problems, and weakness. The diagnosis is often complicated by the lack of diagnostic specificity of low titers of MOG antibody in the serum, its serologic fluctuation from positive to negative and back to positive, disappearance in some cases when diagnostic workup is delayed by months, including initially normal MRI in up to 10% patients, and resolution of radiological abnormalities when MRI is done in a significantly delayed fashion. Moreover, many patients with low titer of MOG antibody in serum such as 1:20 or 1:40 are initially misdiagnosed with MOGAD and may go one to develop other conditions such as MS or NMOSD, among others. Long-term management of MOGAD-associated TM is complicated by the uncertainty as to whether the disease process will remain monophasic or become relapsing. Because MOG antibody titers can fluctuate and go from positive to negative to positive again, basing the risk of relapse purely on a decrease in titer or seroconversion to negativity does not appear to guarantee monophasic course of the disease. Recent data suggest that male gender and TM at onset may overall have a lower risk of relapse; however, small patient numbers, and the length of follow up continue to be important limiting factors. Because at least 40%–50% cases will remain monophasic, most clinicians will start preventive disease modifying treatment only if the disease proves to be of relapsing phenotype; others may choose to start disease modifying treatments after the first attack in those with poor response to initial treatment and significant residual disability, in fear that the second attack might be devastating to what may already be a significant neurological disability with poor quality of life and reduced independence. Currently there are no FDA approved drugs specifically targeting MOGAD. Despite several off label management options discussed above, some patients may relapse through multiple disease modifying therapies. Randomized controlled clinical trials are crucially needed to create an evidence-based treatment algorithm for those affected by MOGAD.
GP-G and EG made a substantial, direct, and intellectual contribution to the work. NC provided MRI imaging Figure 1 and Table 1 with legends. All authors approved the submitted version.
EG has served on advisory boards and received honoraria from Horizon Therapeutics, Alexion, Genentech, Prevail Therapeutics and has received research support from NIH and Genentech.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Longbrake, E. Myelin oligodendrocyte glycoprotein-associated disorders. Continuum (Minneap Minn). (2022) 28:1171–93. doi: 10.1212/CON.0000000000001127
2. Banwell, B, Bennett, JL, Marignier, R, Kim, HJ, Brilot, F, Flanagan, EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: international MOGAD panel proposed criteria. Lancet Neurol. (2023) 22:268–82. doi: 10.1016/S1474-4422(22)00431-8
3. Satukijchai, C, Mariano, R, Messina, S, Sa, M, Woodhall, MR, Robertson, NP, et al. Factors associated with relapse and treatment of myelin oligodendrocyte glycoprotein antibody-associated disease in the United Kingdom. JAMA Netw Open. (2022) 5:e2142780. doi: 10.1001/jamanetworkopen.2021.42780. Erratum in: JAMA Netw Open 2022;5(3):e225056
4. Fadda, G, Flanagan, EP, Cacciaguerra, L, Jitprapaikulsan, J, Solla, P, Zara, P, et al. Myelitis features and outcomes in CNS demyelinating disorders: comparison between multiple sclerosis, MOGAD, and AQP4-IgG-positive NMOSD. Front Neurol. (2022) 13:1011579. doi: 10.3389/fneur.2022.1011579
5. Dams, L, Kraemer, M, and Becker, J. MOG-antibody-associated longitudinal extensive myelitis after ChAdOx1 nCoV-19 vaccination. Mult Scler. (2022) 28:1159–62. doi: 10.1177/13524585211057512
6. Dias da Costa, M, Leal Rato, M, Cruz, D, Valadas, A, Antunes, AP, and Albuquerque, L. Longitudinally extensive transverse myelitis with anti-myelin oligodendrocyte glycoprotein antibodies following SARS-CoV-2 infection. J Neuroimmunol. (2021) 361:577739. doi: 10.1016/j.jneuroim.2021.577739
7. Mariano, R, Messina, S, Roca-Fernandez, A, Leite, MI, Kong, Y, and Palace, JA. Quantitative spinal cord MRI in MOG-antibody disease, neuromyelitis optica and multiple sclerosis. Brain. (2021) 144:198–212. doi: 10.1093/brain/awaa347
8. Dubey, D, Pittock, SJ, Krecke, KN, Morris, PP, Sechi, E, Zalewski, NL, et al. Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol. (2019) 76:301–9. doi: 10.1001/jamaneurol.2018.4053
9. Sechi, E, Cacciaguerra, L, Chen, JJ, Mariotto, S, Fadda, G, Dinoto, A, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD): a review of clinical and MRI features, diagnosis, and management. Front Neurol. (2022) 13:885218. doi: 10.3389/fneur.2022.885218
10. Ambrosius, W, Michalak, S, Kozubski, W, Kalinowska, A, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: current insights into the disease pathophysiology, diagnosis and management. Int J Mol Sci. (2020) 22:100. doi: 10.3390/ijms22010100
11. Wang, C, Narayan, R, and Greenberg, B. Anti-myelin oligodendrocyte glycoprotein antibody associated with gray matter predominant transverse myelitis mimicking acute flaccid myelitis: a presentation of two cases. Pediatr Neurol. (2018) 86:42–5. doi: 10.1016/j.pediatrneurol.2018.06.003
12. Kim, KH, Kim, SH, Hyun, JW, and Kim, HJ. Clinical and radiological features of myelin oligodendrocyte glycoprotein-associated myelitis in adults. J Clin Neurol. (2022) 18:280–9. doi: 10.3988/jcn.2022.18.3.280
13. Sechi, E, Buciuc, M, Pittock, SJ, Chen, JJ, Fryer, JP, Jenkins, SM, et al. Positive predictive value of myelin oligodendrocyte glycoprotein autoantibody testing. JAMA Neurol. (2021) 78:741–6. doi: 10.1001/jamaneurol.2021.0912
14. Huda, S, Whittam, D, Jackson, R, Karthikeayan, V, Kelly, P, Linaker, S, et al. Predictors of relapse in MOG antibody associated disease: a cohort study. BMJ Open. (2021) 11:e055392. doi: 10.1136/bmjopen-2021-055392
15. Sechi, E, Krecke, KN, Messina, SA, Buciuc, M, Pittock, SJ, Chen, JJ, et al. Comparison of MRI lesion evolution in different central nervous system demyelinating disorders. Neurology. (2021) 97:e1097–109. doi: 10.1212/WNL.0000000000012467
16. Sechi, E, Krecke, KN, Pittock, SJ, Dubey, D, Lopez-Chiriboga, AS, Kunchok, A, et al. Frequency and characteristics of MRI-negative myelitis associated with MOG autoantibodies. Mult Scler. (2021) 27:303–8. doi: 10.1177/1352458520907900
17. López-Chiriboga, AS, Majed, M, Fryer, J, Dubey, D, McKeon, A, Flanagan, EP, et al. Association of MOG-IgG Serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG-associated disorders. JAMA Neurol. (2018) 75:1355–63. doi: 10.1001/jamaneurol.2018.1814
18. Dos Passos, GR, Oliveira, LM, da Costa, BK, Apostolos-Pereira, SL, Callegaro, D, Fujihara, K, et al. MOG-IgG-associated optic neuritis, encephalitis, and myelitis: lessons learned from Neuromyelitis Optica Spectrum disorder. Front Neurol. (2018) 9:217. doi: 10.3389/fneur.2018.00217
19. Akaishi, T, Takahashi, T, Misu, T, Kaneko, K, Takai, Y, Nishiyama, S, et al. Difference in the source of anti-AQP4-IgG and anti-MOG-IgG antibodies in CSF in patients with Neuromyelitis Optica Spectrum disorder. Neurology. (2021) 97:e1–e12. doi: 10.1212/WNL.0000000000012175
20. Jarius, S, Ruprecht, K, Kleiter, I, Borisow, N, Asgari, N, Pitarokoili, K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation. (2016) 13:280. doi: 10.1186/s12974-016-0718-0
21. Camera, V, Holm-Mercer, L, Ali, AAH, Messina, S, Horvat, T, Kuker, W, et al. Frequency of new silent MRI lesions in myelin oligodendrocyte glycoprotein antibody disease and aquaporin-4 antibody neuromyelitis optica spectrum disorder. JAMA Netw Open. (2021) 4:e2137833. doi: 10.1001/jamanetworkopen.2021.37833
22. B Syc-Mazurek, S, Chen, JJ, Morris, P, Sechi, E, Mandrekar, J, Tillema, JM, et al. Frequency of new or enlarging lesions on MRI outside of clinical attacks in patients with MOG-antibody-associated disease. Neurology. (2022) 99:795–9. doi: 10.1212/WNL.0000000000201263
23. Shahriari, M, Sotirchos, ES, Newsome, SD, and Yousem, DM. MOGAD: how it differs from and resembles other neuroinflammatory disorders. AJR Am J Roentgenol. (2021) 216:1031–9. doi: 10.2214/AJR.20.24061
24. Etemadifar, M, Salari, M, Kargaran, PK, Sigari, AA, Nouri, H, Etemadifar, F, et al. Conus medullaris involvement in demyelinating disorders of the CNS: a comparative study. Mult Scler Relat Disord. (2021) 54:103127. doi: 10.1016/j.msard.2021.103127
25. Sato, DK, Callegaro, D, Lana-Peixoto, MA, Waters, PJ, de Haidar Jorge, FM, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. (2014) 82:474–81. doi: 10.1212/WNL.0000000000000101
26. Kitley, J, Waters, P, Woodhall, M, Leite, MI, Murchison, A, George, J, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol. (2014) 71:276–83. doi: 10.1001/jamaneurol.2013.5857
27. Fadda, G, Banwell, B, Waters, P, Marrie, RA, Yeh, EA, O'Mahony, J, et al. Canadian pediatric demyelinating disease network. Silent new brain MRI lesions in children with MOG-antibody associated disease. Ann Neurol. (2021) 89:408–13. doi: 10.1002/ana.25957
28. Whittam, DH, Karthikeayan, V, Gibbons, E, Kneen, R, Chandratre, S, Ciccarelli, O, et al. Treatment of MOG antibody associated disorders: results of an international survey. J Neurol. (2020) 267:3565–77. doi: 10.1007/s00415-020-10026-y
29. Chen, JJ, Huda, S, Hacohen, Y, Levy, M, Lotan, I, Wilf-Yarkoni, A, et al. Association of maintenance intravenous immunoglobulin with prevention of relapse in adult myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol. (2022) 79:518–25. doi: 10.1001/jamaneurol.2022.0489
30. Cobo-Calvo, A, Ruiz, A, Rollot, F, Arrambide, G, Deschamps, R, Maillart, E, et al. Clinical features and risk of relapse in children and adults with myelin oligodendrocyte glycoprotein antibody-associated disease. Ann Neurol. (2021) 89:30–41. doi: 10.1002/ana.25909
31. Chang, X, Zhang, J, Li, S, Wu, P, Wang, R, Zhang, C, et al. Meta-analysis of the effectiveness of relapse prevention therapy for myelin-oligodendrocyte glycoprotein antibody-associated disease. Mult Scler Relat Disord. (2023) 72:104571. doi: 10.1016/j.msard.2023.104571
Keywords: transverse myelitis, MOG myelitis, myelin oligodendrocyte glycoprotein associated disease, MOG antibody positive myelitis, MOGAD myelitis
Citation: Perez-Giraldo G, Caldito NG and Grebenciucova E (2023) Transverse myelitis in myelin oligodendrocyte glycoprotein antibody-associated disease. Front. Neurol. 14:1210972. doi: 10.3389/fneur.2023.1210972
Edited by:
Sasitorn Siritho, Bumrungrad International Hospital, ThailandReviewed by:
Yoshiki Takai, Tohoku University Hospital, JapanCopyright © 2023 Perez-Giraldo, Caldito and Grebenciucova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Grebenciucova, ZWxlbmEuZ3JlYmVuY2l1Y292YUBub3J0aHdlc3Rlcm4uZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.