 Yoshiki Takai
Yoshiki Takai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 21 July 2023
Sec. Multiple Sclerosis and Neuroimmunology
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1209749
This article is part of the Research TopicMOGAD, Current Knowledge and Future TrendsView all 13 articles
Myelin oligodendrocyte glycoprotein (MOG) is expressed on the outermost layer of the myelin sheath in the central nervous system. Recently, the clinical concept of MOG antibody-associated disease (MOGAD) was established based on the results of human MOG-transfected cell-based assays which can detect conformation-sensitive antibodies against MOG. In this review, we summarized the pathological findings of MOGAD and discussed the issues that remain unresolved. MOGAD pathology is principally inflammatory demyelination without astrocyte destruction, characterized by perivenous demyelination previously reported in acute disseminated encephalomyelitis and by its fusion pattern localized in both the white and gray matter, but not by radially expanding confluent demyelination typically seen in multiple sclerosis (MS). Some of demyelinating lesions in MOGAD show severe loss of MOG staining compared with those of other myelin proteins, suggesting a MOG-targeted pathology in the disease. Perivascular cuffings mainly consist of macrophages and T cells with CD4-dominancy, which is also different from CD8+ T-cell-dominant inflammation in MS. Compared to aquaporin 4 (AQP4) antibody-positive neuromyelitis optica spectrum disorders (NMOSD), perivenous complement deposition is less common, but can be seen on myelinated fibers and on myelin degradation products within macrophages, resembling MS Pattern II pathology. Thus, the pathogenetic contribution of complements in MOGAD is still debatable. Together, these pathological features in MOGAD are clearly different from those of MS and AQP4 antibody-positive NMOSD, suggesting that MOGAD is an independent autoimmune demyelinating disease entity. Further research is needed to clarify the exact pathomechanisms of demyelination and how the pathophysiology relates to the clinical phenotype and symptoms leading to disability in MOGAD patients.
Myelin oligodendrocyte glycoprotein (MOG) is a glycoprotein (consisting of 218 amino acids) expressed in oligodendrocytes and is characterized by its distribution in the outermost layer of the myelin sheath (1). MOG is composed of multiple splicing variants (2, 3), all of which have extracellular immunoglobulin variable domains and thus belong to the immunoglobulin superfamily (4). Because of these structural features, MOG has a long history of research as an autoantigen that can induce inflammatory demyelinating pathology in the central nervous system (CNS) (5–9), and is one of the best-studied antigens in experimental autoimmune encephalomyelitis (EAE) (10–12). Therefore, autoantibodies against MOG have long been considered a potential cause of human inflammatory demyelinating diseases, particularly multiple sclerosis (MS). However, the discovery of clinically relevant MOG antibodies in human disease has not been successful until recently. Previous results on the detection of MOG antibodies by enzyme-linked immunosorbent assay (ELISA) or Western blot were confusing due to the low specificity (13). This is because the antigen is linear in ELISA or denatured in Western blot such that the three-dimensional structure of native MOG was lost; the issue was resolved when the conformation-sensitive MOG antibody became detectable by human MOG-transfected cell-based assays (CBAs) (14–16). As a result, MOG antibodies have been found in patients with optic neuritis, acute myelitis, neuromyelitis optica spectrum disorders (NMOSD) without aquaporin 4 (AQP4) antibodies (17, 18), acute disseminated encephalomyelitis (ADEM) (19, 20), and brainstem (21–23) and cerebral cortical encephalitis (24–26). In contrast, typical MS patients are essentially negative for MOG antibodies (27, 28). Consequently, patients with MOG antibodies came to be recognized as belonging to a group with inflammatory demyelinating conditions distinct from MS, and the international diagnostic criteria of MOG antibody-associated disease (MOGAD) were recently published (29).
In this review, we summarized the histopathological findings of MOGAD in published studies. In particular, we outlined the pathologies typically found in MOGAD and the issues that remain unresolved because of inconsistent results in previous studies. We also discussed the unique pathogenesis of MOGAD by comparing it with MS and AQP4 antibody-positive NMOSD (AQP4 + NMOSD).
The pattern of demyelination seen in well-known inflammatory demyelinating diseases can be classified into “confluent demyelination” in MS, “perivenous demyelination” in ADEM and “concentric demyelination” in Balo’s disease (Figure 1) (30, 31). “Confluent demyelination” is characterized by fusion and enlargement of perivascular demyelinating lesions with well-defined borders, resulting in the formation of large plaques, and the lesions may occasionally exhibit a map-like morphology (Figure 1A). On the other hand, “perivenous demyelination” is the one with indistinct borders around a single small vessel with inflammatory cell infiltration, and often multifocal (Figure 1B). Perivenous demyelination is considered useful in the pathological differentiation of ADEM from MS (32). However, it should be noted that we may miss “perivenous demyelination” because it can be very small, and the activity of myelin phagocytosis by macrophages is sometimes scarce, requiring careful observation (Figure 2).
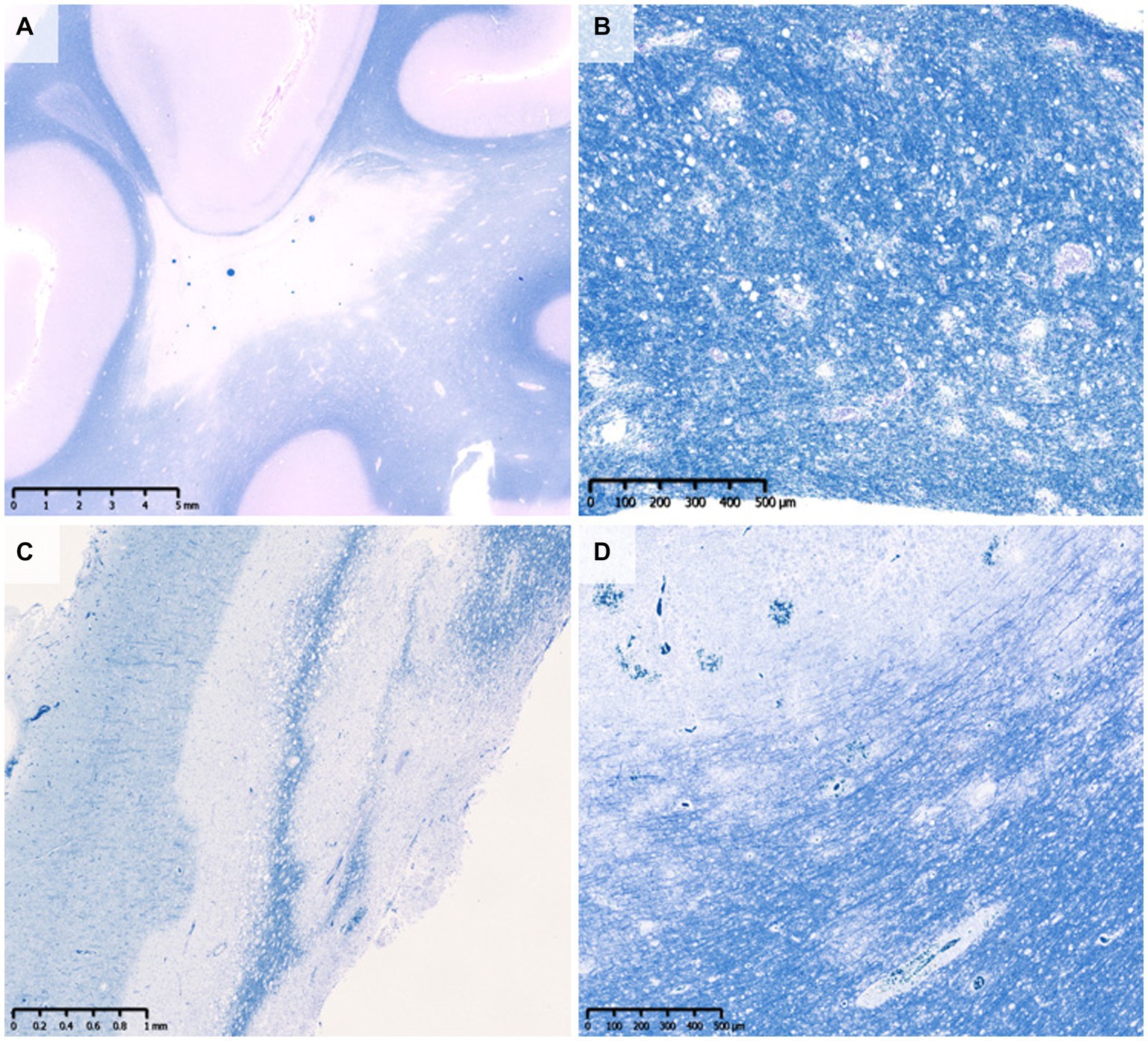
Figure 1. Various types of demyelination. (A) Confluent demyelination (SPMS). (B) Perivenous demyelination (ADEM). (C) Concentric demyelination (Balo’s disease). (D) Mixed pathology of confluent and perivenous demyelination (MOGAD). (A–D) Klüver-Barrera staining. ADEM, acute disseminated encephalomyelitis; MOGAD, myelin oligodendrocyte glycoprotein antibody-associated disease; SPMS, secondary progressive multiple sclerosis.
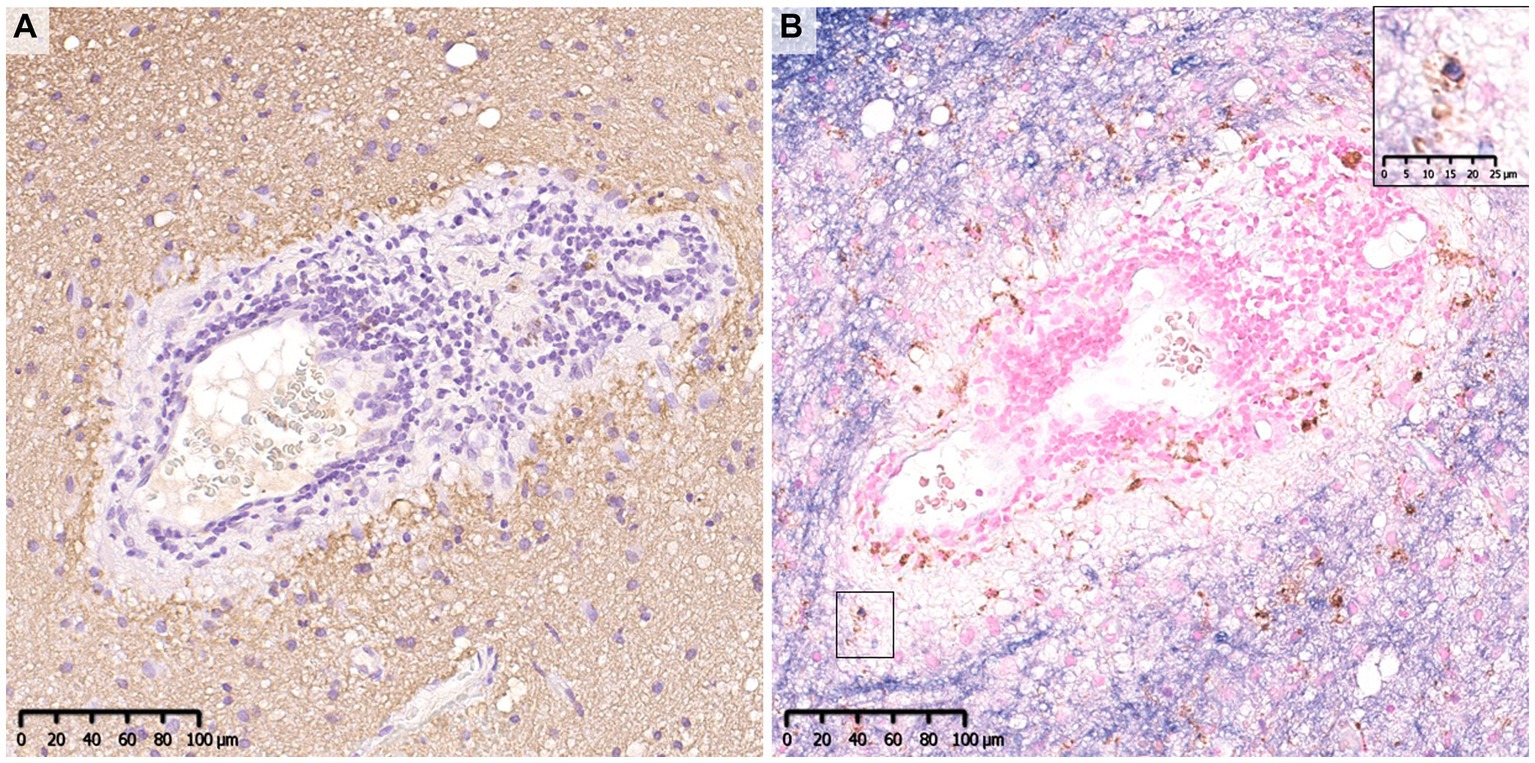
Figure 2. Perivenous demyelination in MOGAD. A demyelinating lesion was seen around small vessels with inflammatory cell infiltration. There were a small number of macrophages that phagocytosed myelin debris (insert in B). (A) MBP, (B) MOG (blue)/CD68 (brown). MBP, myelin basic protein; MOG, myelin oligodendrocyte glycoprotein; MOGAD, MOG antibody-associated disease.
The pathology of MOGAD is characterized by a mixture of perivenous and confluent demyelinating lesions (Figure 1D) (33, 34), and their proportions may depend on the timing of tissue sampling and disease severity (Tables 1, 2) (24, 33–47). The median time to tissue sampling in our study (one month) (33) was shorter than that in Höftberger et al. (seven months) (34) (Table 1), and the demyelination patterns in the two studies were different (90% of the lesions in our cases had perivenous demyelination, while 50% of the lesions in Höftberger’s study had a transitional pattern [a combination of perivenous and confluent demyelinations]). However, it is important to mention that among confluent demyelinating lesions, slowly expanding lesions (SELs) typically seen in the subacute to chronic stage of MS (Figure 3) (48) are rarely observed in MOGAD patients (34) or AQP4 + NMOSD patients (49). SELs are characterized by the accumulation of activated macrophages/microglia at the lesion edge with iron deposition (50), which is thought to be involved in the progression of MS (51–53). In other words, demyelinating lesion formation in MOGAD patients is characterized by simultaneous development of multiple perivascular inflammatory demyelination and its fusion to form confluent demyelination, which is different from radial expansion of the lesions in MS.
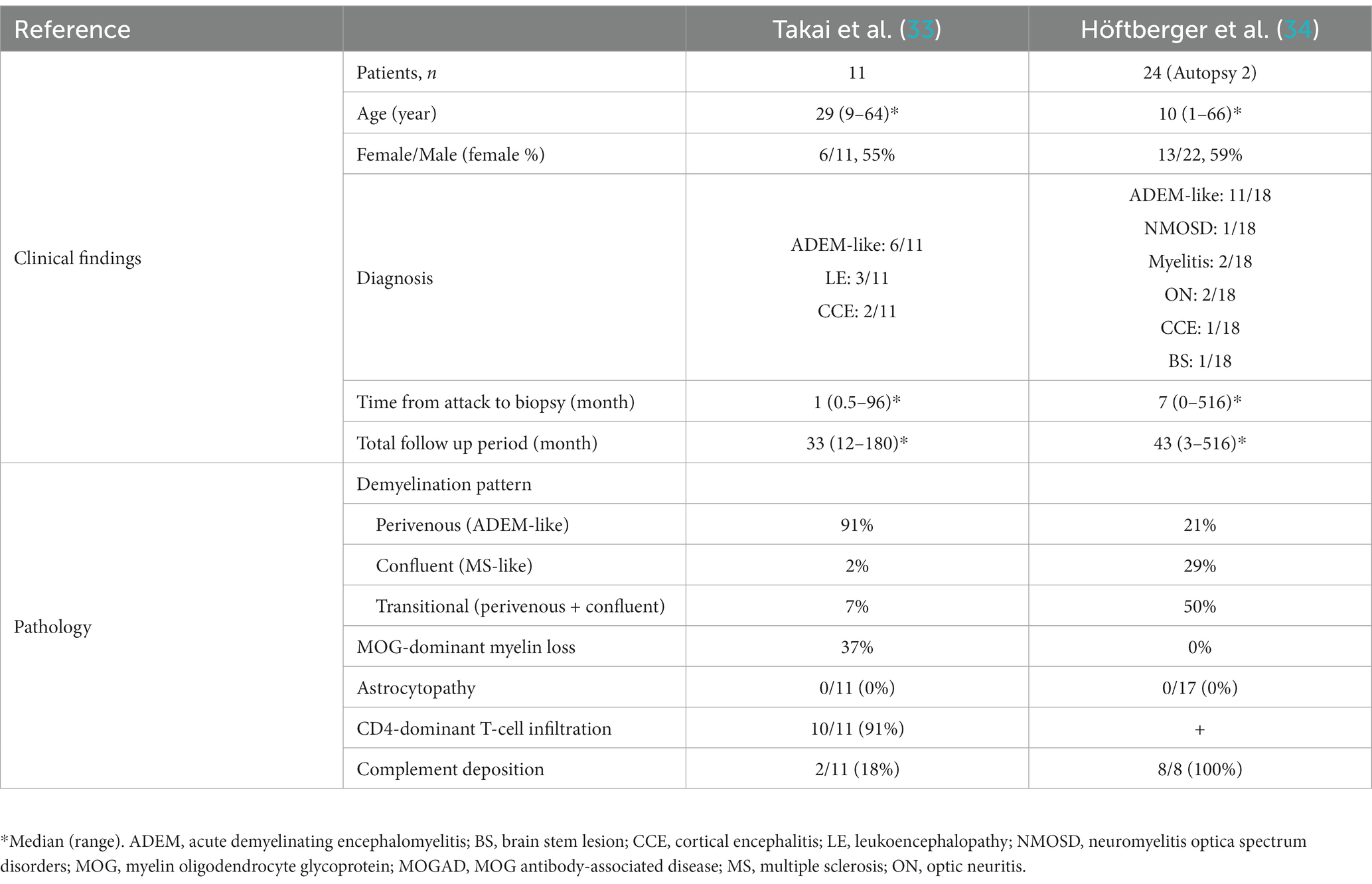
Table 1. Comparison of clinical findings and pathology of MOGAD in two studies of more than 10 patients.
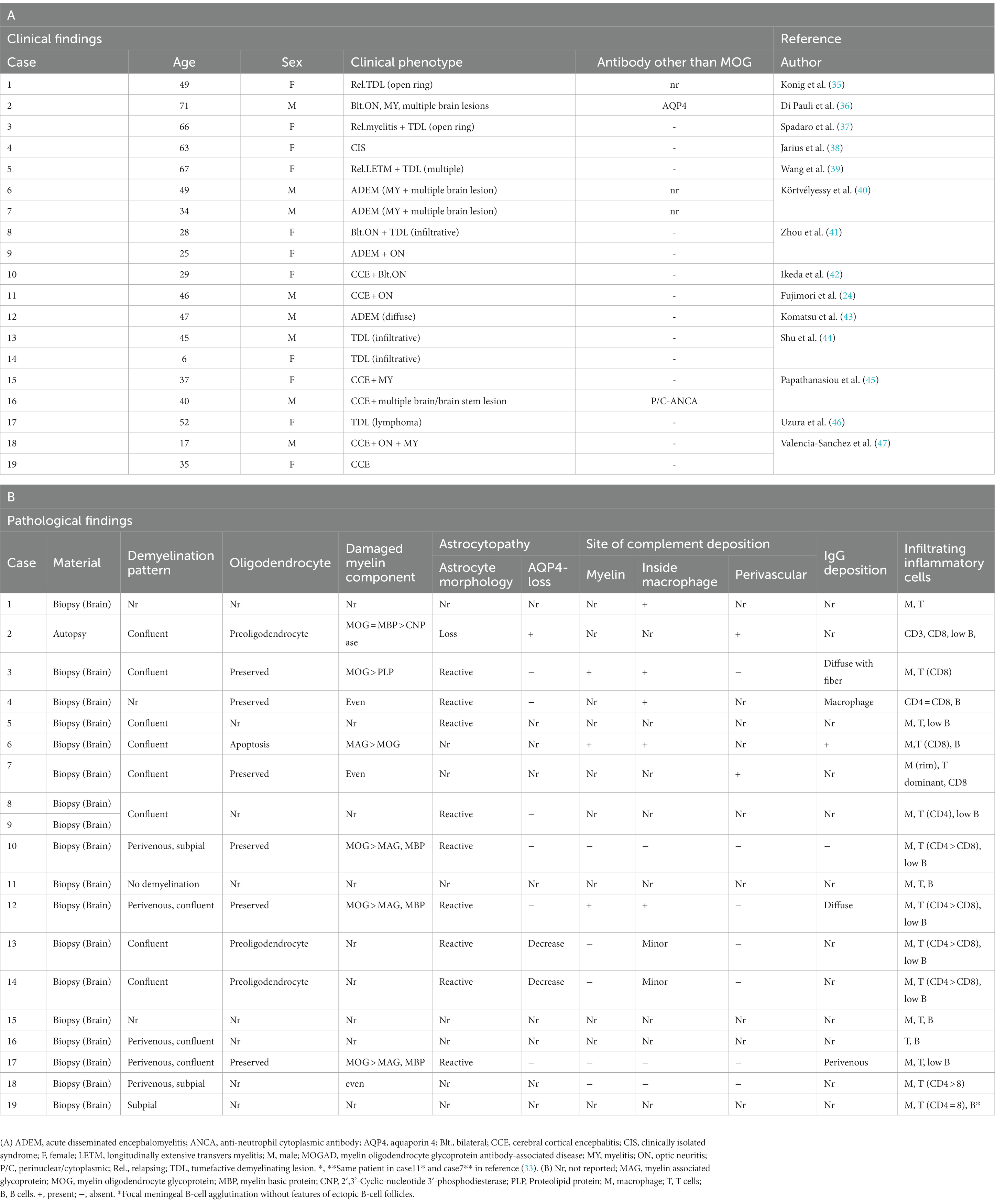
Table 2. Summary of the clinical and pathological findings of MOGAD in case reports.
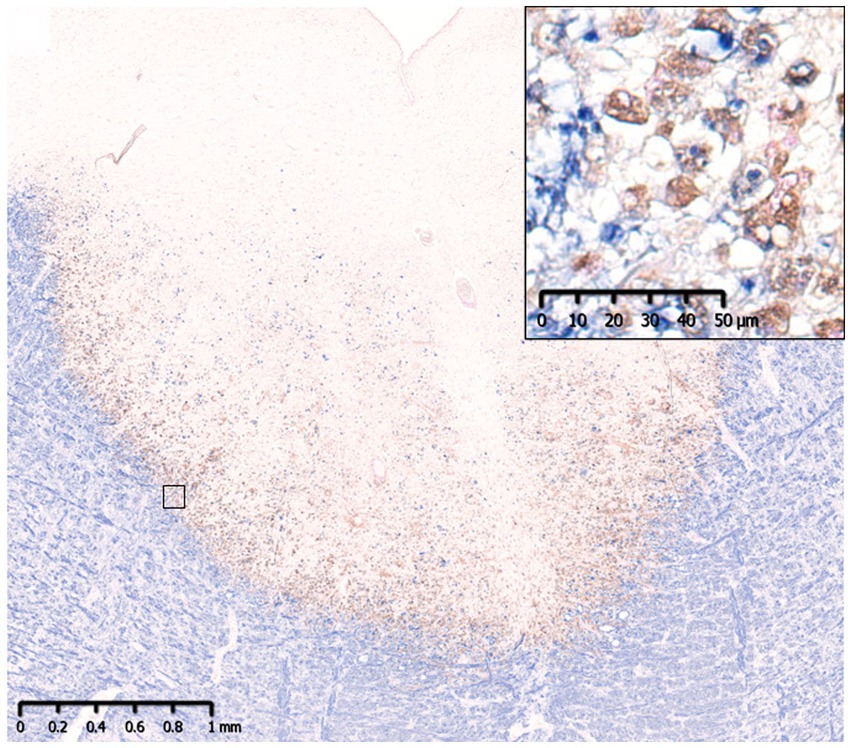
Figure 3. Slowly expanding lesion in SPMS. A large well-demarcated demyelinating lesion was seen with peripheral infiltration of myelin phagocytosed macrophages (insert). MBP (blue)/CD68 (brown). MBP, myelin basic protein; SPMS, secondary progressive multiple sclerosis.
Demyelinating lesions in MOGAD patients are found mainly in the white matter but also in the subpial cortex to cortico-medullary junction and deep gray matter (Figure 4) (33, 34, 47). Within these lesions, CD68-positive macrophages/microglia widely infiltrate the cortex (Figure 4). The frequency of cortical demyelination is reported to be higher in MOGAD patients with cerebral involvement than in MS patients (34), which is compatible with the high incidence of cortical involvement in MOGAD patients, evidenced by conditions such as ADEM and cerebral cortical encephalitis (29). In addition, inflammatory cells infiltrate around meningeal vessels adjacent to subpial demyelinating lesions (33, 34, 45, 47). In cerebral cortical encephalitis of MOGAD patients, brain MRI scans often show contrast enhancing effects in the meninges which may reflect such inflammation around the meningeal vessels (47).
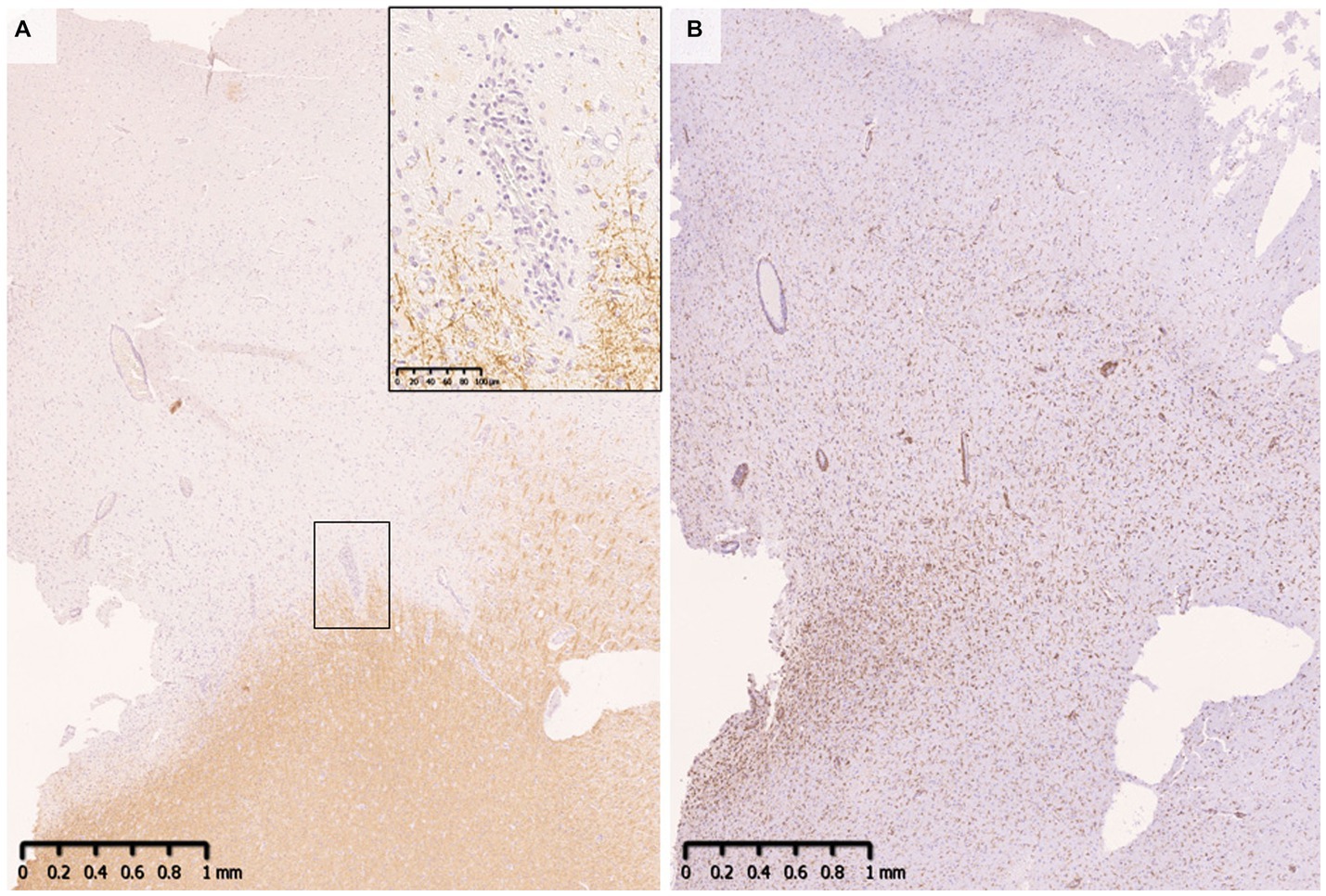
Figure 4. Subpial demyelination in MOGAD. (A) Myelin fibers were widely lost in the subpial cortex. Perivenous demyelination was observed at the cortical-medullary junction. (B) CD68 positive macrophages/microglia were diffusely infiltrated the demyelinated cortex. (A) MBP, (B) CD68. MBP, myelin basic protein; MOGAD, myelin oligodendrocyte glycoprotein antibody-associated disease.
When evaluating demyelinating lesions, it is important to identify primarily damaged myelin component(s) by immunohistochemistry to assess the type and stage of the disease (48, 54). Our study and several previous case reports indicated that some demyelinating lesions found in patients with MOGAD showed MOG-dominant myelin loss especially in the early-stage (Figure 5) (33, 37, 42, 46), and some myelin-laden macrophages localized at the perivascular space showed MOG-dominant phagocytosis (33). In addition, oligodendrocytes are relatively preserved in MOGAD demyelinating lesions (33, 37, 38, 44). These findings support that MOGAD actually targets MOG and that its demyelination process may initially occur on the surface of the myelin sheath. However, Höftberger et al. reported no MOG dominant myelin loss in their study (34), and it remains to be clarified what this difference originated from. On the other hand, some reports indicated that preferential loss of myelin associated glycoprotein (MAG) could occur in MOGAD patients although the incidence was very low (34, 40). Since MAG is expressed in the innermost layer of the myelin sheath and is most distant from the oligodendrocyte cell body, preferential loss of MAG is thought to reflect oligodendrocyte damage (distal oligodendrogliopathy) (55). This finding is seen in patients with MS pattern III lesions (56); Balo’s disease (57); AQP4 + NMOSD (58); and ischemic tissue damage such as cerebral infarction (59). Since MOG is also expressed at the surface of oligodendrocytes, depending on the concentration or characteristics of MOG antibodies, some oligodendrocytes may be damaged by MOG antibodies.
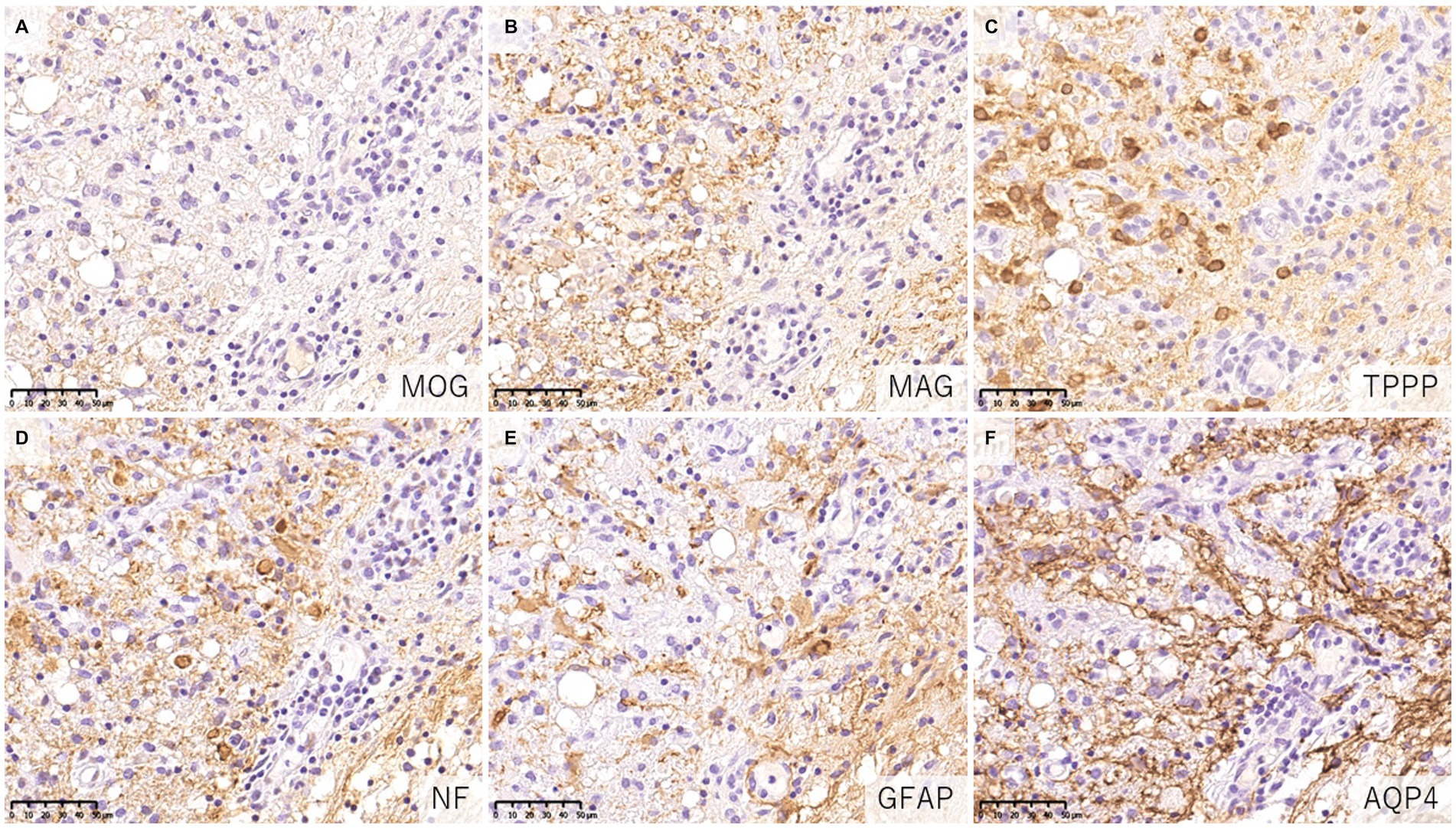
Figure 5. Characteristics of demyelinating lesions in MOGAD. (A,B) Loss of MOG staining was more evident than MAG staining. (C) Oligodendrocytes were well preserved in the demyelinating lesion. (D) Axonal enlargement was present, suggesting neuroaxonal alteration but axonal staining was relatively preserved compared to demyelination. (E,F) Activated astrocytes with dense AQP4 staining were observed. (A) MOG, (B) MAG, (C) TPPP, (D) NF, (E) GFAP, (F) AQP4. AQP4, aquaporin 4; GFAP, glial fibrillary acidic protein; MAG, myelin associated glycoprotein; MBP, myelin basic protein; MOG, myelin oligodendrocyte glycoprotein; MOGAD, MOG antibody-associated disease; NF, neurofilament; TPPP, tubulin polymerization promoting proteins.
The cellular infiltrate in inflammatory demyelinating lesions is composed mainly of myelin phagocytosing macrophages at the sites of demyelination and T-cell clusters in the perivascular space (perivascular cuffing). Infiltrating cells in the lesions of MOGAD are essentially similar (Tables 1, 2), but a characteristic feature of MOGAD is CD4+ T-cell-dominant infiltration in the demyelinating lesions (33, 34), which is different from the dominance of CD8+ T-cell infiltrates in MS lesions (60, 61). Some of those CD4+ T cells in MOGAD might be reactive to MOG epitopes. However, it should be noted that the timing of the sampling of specimens should be considered. In AQP4 + NMOSD, the main subpopulation of T cells infiltrated in the lesions changes from CD4 in the acute phase to CD8 in the chronic phase of the disease (49). Most of the CNS tissue specimens of MOGAD patients examined in the published studies were obtained in the acute phase, and the pathological findings in the chronic phase have not been examined. Therefore, the characteristics of T cells infiltrating the lesion may reflect differences in the stage of the disease rather than pathogenesis, and further detailed verification is required in the future. However, it is known that the levels of T helper 17 (Th17)-related cytokines are markedly elevated in the cerebrospinal fluid (CSF) of patients during the acute phase of MOGAD and AQP4 + NMOSD when compared with those of MS patients and control subjects (62). Thus, in the acute phase, T-cell subpopulations infiltrating the lesions are different between MOGAD and MS patients.
B cells are seen in small numbers in the perivascular space, but are less frequent than T cells (33, 34). Additionally, ectopic lymphoid follicles, as reported in MS (63, 64), have not been detected in MOGAD patients. However, occasionally B-cell aggregates may be seen in the leptomeninges (47). In MOGAD patients, intrathecal production of MOG antibodies seems to occur more frequently than in AQP4 + NMOSD patients (65–68), suggesting that B cells infiltrating the CNS produce MOG antibodies and contribute to the pathogenesis of the disease. In addition, CXCR4 is upregulated in B cells in patients with MOGAD (69), and its ligand, CXCL12, is known to be elevated in the CSF (70). Thus, CXCL12/CXCR4 may contribute to the chemotaxis of B cells and other inflammatory cells in MOGAD patients (71).
Since MOG antibodies are mainly composed of those in the IgG1 subclass (27, 72), complement-mediated cytotoxicity (CDC) has been considered to contribute to the pathogenesis of MOGAD. In fact, both in vitro and in vivo, it has been reported that MOG antibody-induced cytotoxicity and demyelination can occur in a complement-mediated manner (72–74). Indeed, some previous case reports on biopsied brain lesions in MOGAD patients showed the deposition of complement components on myelin fibers and myelin debris phagocytosed by macrophages, and the authors concluded that the lesions were probably caused by humoral immune-mediated demyelination, such as MS pattern II lesions (37, 38, 40, 56). This type of MS lesion is histologically characterized by extensive confluent demyelination with tissue deposition of humoral immune factors such as complements and immunoglobulins (22). However, the histopathological findings of complements in MOGAD patients remain debatable, as the reported results have been inconsistent (33, 34). In our study, only 2 of 11 MOGAD patients showed tissue deposition of complement, which was much less frequent and dense than in AQP4 + NMOSD patients with perivascular deposition of activated complements (C9neo) in all acute lesions (33). However, Höftberger et al. concluded that active complement deposition was observed in all 8 patients they evaluated (34) (Table 2). This difference may be due to the clinical severity, timing of tissue sampling or inter-individual variability in the severity of MOG-IgG-related cytotoxicity other than complement activation, such as antibody-dependent cellular phagocytosis (ADCP) and antibody-dependent cellular cytotoxicity (ADCC) (75, 76). However, a recent in vitro study demonstrated that MOG antibodies elicited much less complement activation than AQP4 antibodies (77). AQP4 has two isoforms, M1 and M23, that differ in their transcription start sites (78). AQP4-M1 and M23 are coexpressed in the CNS, and M23 is known to form large well-ordered assemblies called orthogonal arrays of particles (OAPs) (78) and is reported to be more highly expressed in the optic nerve and spinal cord, where NMOSD lesions are more likely to occur (79). The formation of OAPs allows AQP4 to be densely expressed on the cell surface, facilitating AQP4 antibody clustering on the cell membrane. The classical complement activation pathway is initiated by the binding of C1q to the Fc portion of IgG, but requires bivalent or multivalent binding (80). Thus, complement components are more likely to be activated when IgG is densely bound on the plasma membrane, and in fact, the presence of OAPs significantly enhanced complement-mediated cytotoxicity by the presence of AQP4 antibodies (81). On the other hand, MOG constitutes a quantitatively minor component (0·05%) of the myelin sheath (1), and MOG antibodies require bivalent binding when binding to MOG, making it difficult for them to assemble on the cell membrane, and resulting in low C1q binding ability (82). It is necessary to study in detail whether the amount and characteristics of MOG antibodies affect the degree of complement activation following binding to AQP4.
The dominant pattern of demyelinating lesions (perivenous demyelination) in MOGAD is similar to that of ADEM rather than MS. The characteristics of infiltrating T cells also differ in MOGAD and MS, as noted above (Table 3). However, we cannot rule out the possibility that some patients with MOGAD may have a pathology similar to MS since previous studies on MOG-EAE have demonstrated that the ratios of myelin antigen-specific lymphocytes and autoantibodies to myelin could influence the dominance of perivenous or confluent demyelination (8, 85). Additionally, there is a report that MOG antibodies purified from two MOGAD patients (whose MOG antibodies were capable of binding to rodent MOG) and administered intrathecally to EAE subjects induced by MOG-specific T cells did not produce demyelinating lesions with deposition of activated complement, but in the presence of MBP-specific T cells, demyelinating lesions similar to MS pattern II developed (86). These findings suggested that T cells in some cases of MOGAD may recognize myelin protein(s) other than MOG and activate complements. Thus, further investigations are needed to confirm whether this is the case in the human pathology of MOGAD. However, it should be noted that MS Pattern II is a pathological classification proposed before the discovery of conformation-sensitive MOG antibodies and includes many brain biopsy samples from cases with atypical or fulminant cerebral lesions for MS (56). Jarius et al. found that only one of the 13 cases with MS Pattern II pathology was positive for MOG antibodies and suggested its limited involvement (38). Therefore, it may include other inflammatory demyelinating pathologies than MS and MOGAD and require further verification.
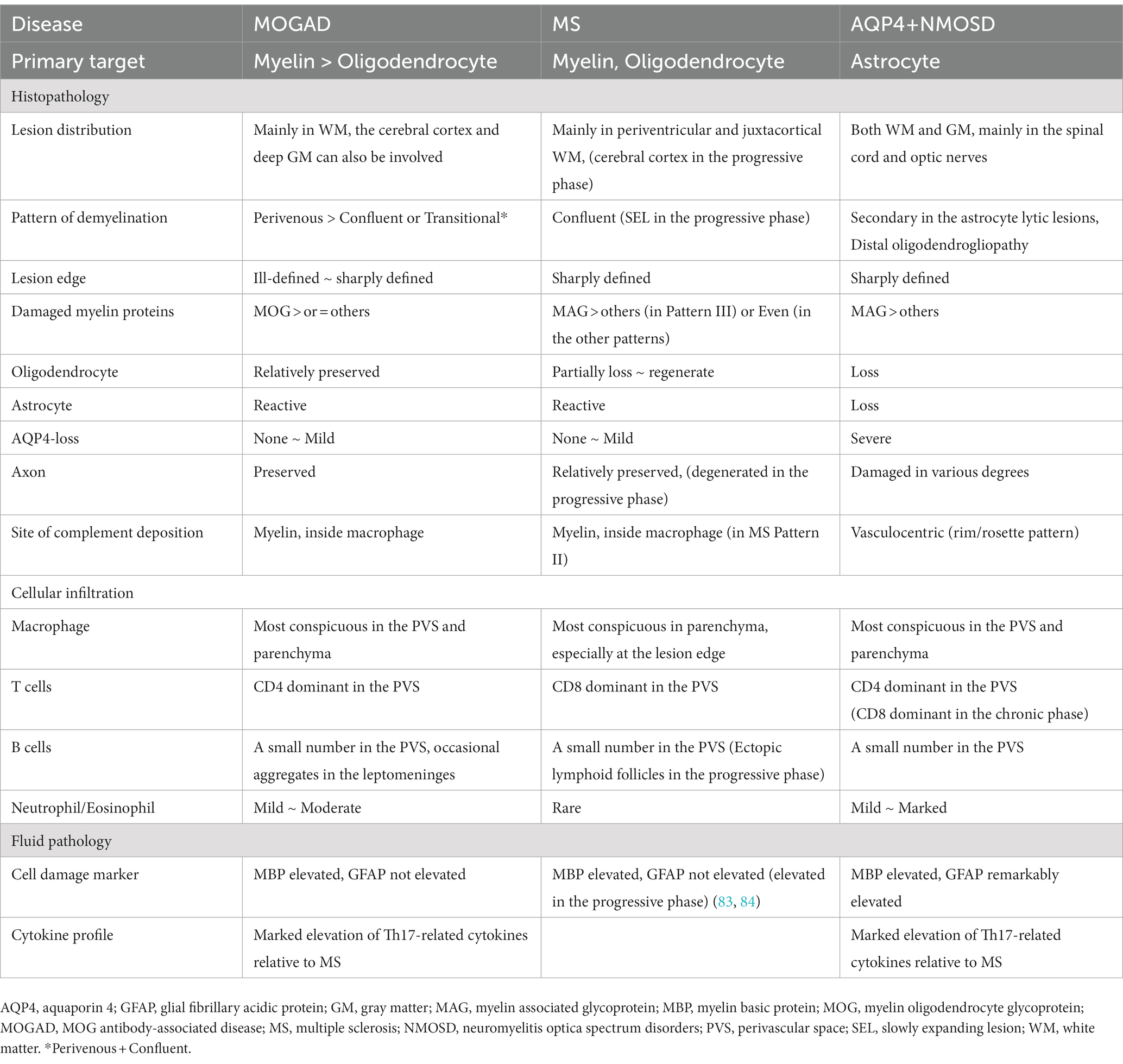
Table 3. Comparison of the major pathological findings of acute lesions in MOGAD, MS, and AQP4 + NMOSD.
The fundamental difference in the pathologies of the two diseases is that the main target of immune attack is myelin in MOGAD but is astrocytes in AQP4 + NMOSD (62). In previous pathological studies of MOGAD, there has been no astrocytic damage except in patient doubly positive for AQP4 and MOG antibodies (36). In the demyelinating lesions in MOGAD, astrocytes are essentially activated and AQP4 is also strongly stained on immunohistochemistry images (Figure 5), although two cases of partially decreased AQP4 expression in MOGAD with tumefactive brain lesions have been reported (Table 2) (44). The pathological process starts in the perivascular regions in both MOGAD and AQP4 + NMOSD, but they show distinct features of demyelinating lesions: in MOGAD, MOG is predominantly lost with relatively preserved oligodendrocytes, whereas in AQP4 + NMOSD, MAG is preferentially damaged, and oligodendrocytes are lost, but MOG is relatively preserved (Figure 6). The immunohistochemical staining pattern of activated complement deposition also differs: a rosette-like staining around blood vessels is seen in AQP4 + NMOSD (49, 87, 88), while in MOGAD, perivascular complement deposition is much less (Figure 7) but stained on myelinated fibers and in myelin degradation products within macrophages (Figure 7; Tables 2, 3) (37, 38, 40). Despite these different patterns of demyelination, MOGAD and AQP4 + NMOSD share some clinical features, such as optic neuritis and longitudinally extensive myelitis (18, 89), and cytokine profiles (upregulation of Th17-related cytokines) in the CSF (62) as autoantibody-associated CNS diseases.
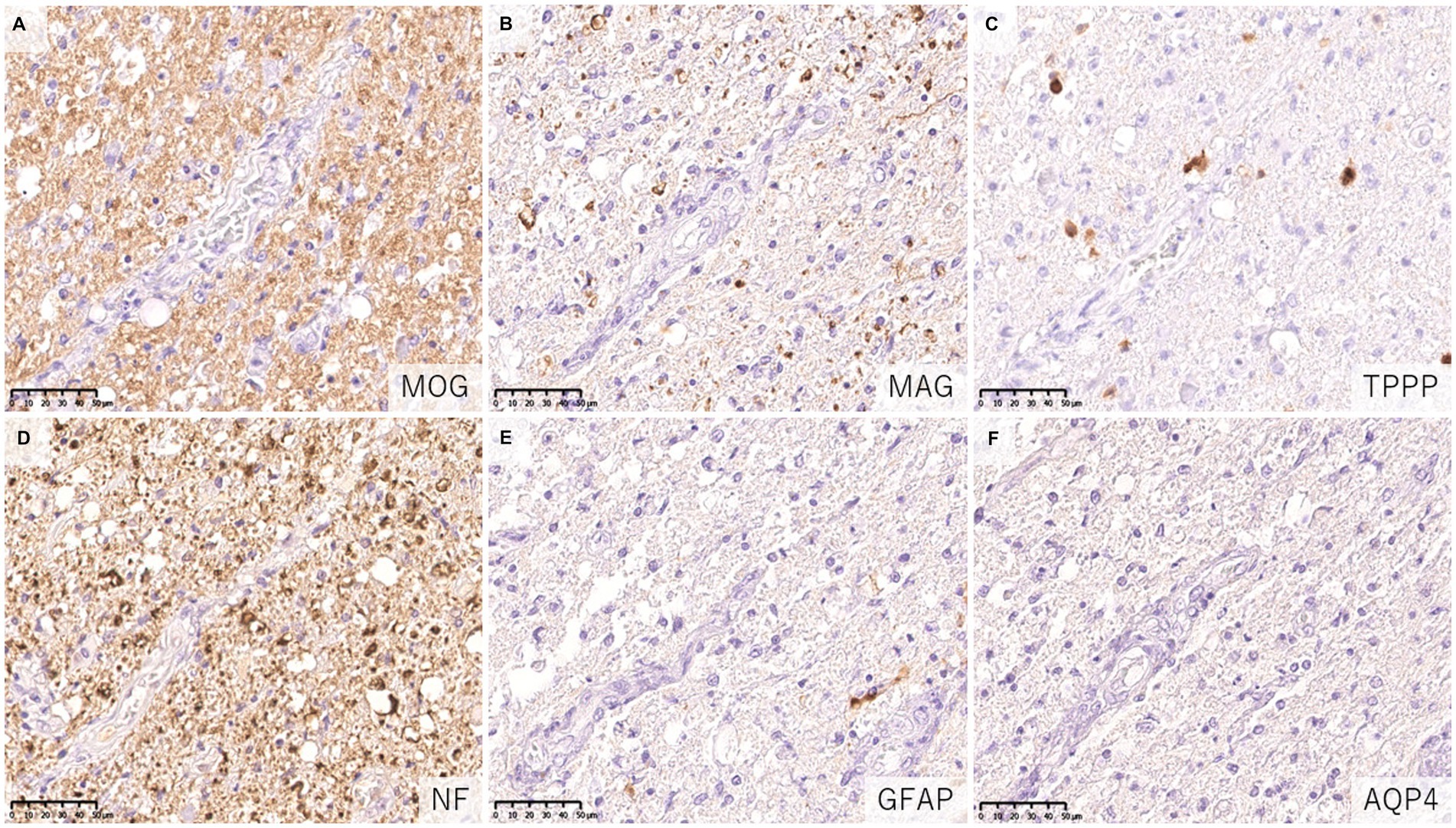
Figure 6. Characteristics of astrocytopathic lesions in AQP4 + NMOSD. (A,B) Loss of MAG staining was more evident than MOG staining. (C) Numerous oligodendrocytes were lost in the lesion. (D) Axonal enlargement was present, suggesting neuroaxonal alteration, but axonal staining was relatively preserved compared to demyelination and astrocyte loss. (D–F) Astrocytes were almost completely lost in the lesion. (A) MOG, (B) MAG, (C) TPPP, (D) NF, (E) GFAP, (F) AQP4. AQP4: aquaporin 4, GFAP, glial fibrillary acidic protein; MAG, myelin associated glycoprotein; MBP, myelin basic protein; MOG, myelin oligodendrocyte glycoprotein; MOGAD, MOG antibody-associated disease; NF, neurofilament; NMOSD, neuromyelitis optica spectrum disorders; TPPP, tubulin polymerization promoting proteins.
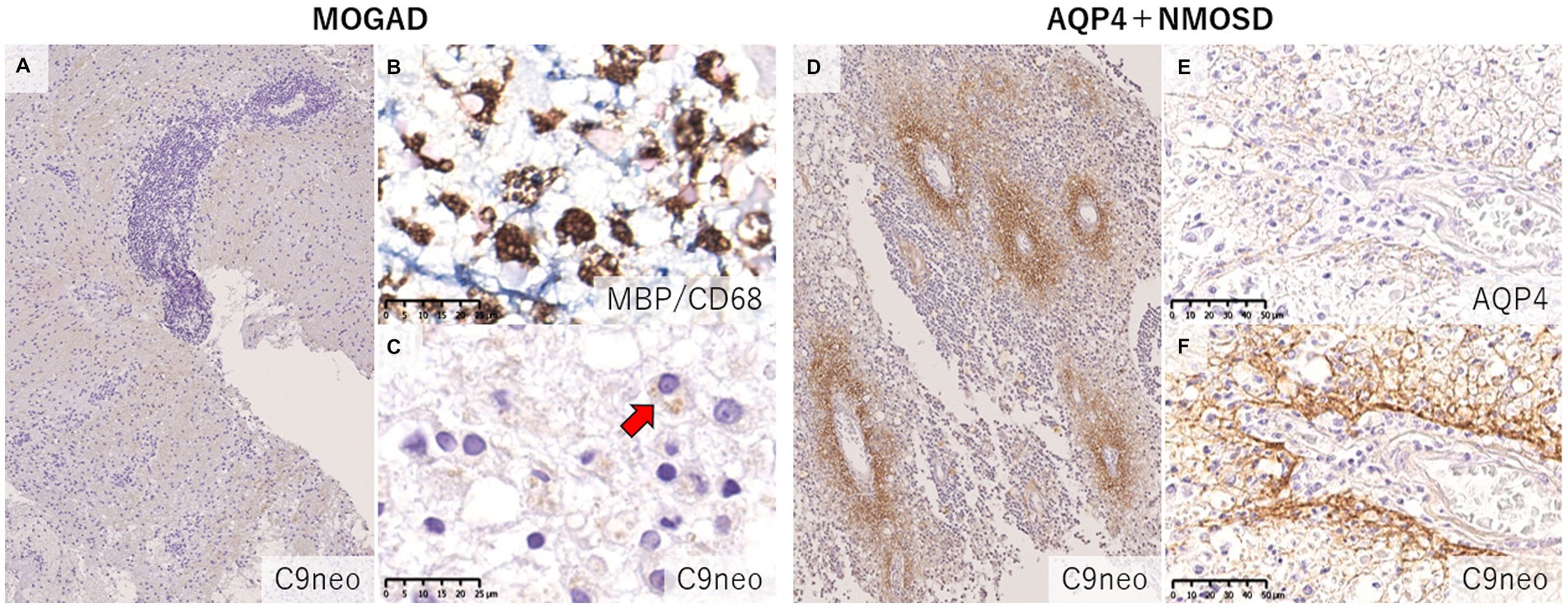
Figure 7. Comparison of the deposition pattern of activated complements in MOGAD and AQP4 + NMOSD. (A) Only mild perivascular depositions of complements were seen in MOGAD even in the active lesion where perivascular cuffing was evident. (B,C) In active demyelinating lesions, complement staining was detected on myelin debris phagocytosed by macrophages (red arrow). (D) Multiple rosette-like stainings of complement deposition were seen in the NMOSD lesion. (D–F) Perivascular complement depositions were seen within AQP4-loss lesions. (A,C,D,F) C9neo. (B) MBP/CD68, (E) AQP4. AQP4, aquaporin 4; MBP, myelin basic protein; MOGAD, myelin oligodendrocyte glycoprotein antibody-associated disease; NMOSD, neuromyelitis optica spectrum disorders.
Recently, the international diagnostic criteria for MOGAD have been proposed (29), and certain pathological features of the disease have been clarified (29), indicating that MOGAD is a disease entity distinct from MS and AQP4 + NMOSD. In fact, published articles on MOGAD have been rapidly increasing in recent years, and a few international clinical trials for relapsing MOGAD have already begun.
Considering the currently available data of the histopathological studies of MOGAD and some basic research with MOG antibodies, the immunopathological process in MOGAD may be summarized as follows. Initially, the breakdown of immune tolerance leads to the generation of MOG-reactive T cells that stimulate the production of MOG antibodies from B cells in the periphery. Triggered by infection, vaccination, or other stimuli, these MOG-reactive T cells are activated and penetrate the blood–brain barrier (BBB) into the CNS and aggregate at the perivascular space of the meninges and parenchyma (perivascular cuffing). MOG antigens in the CNS further activate these cells which are primarily CD4-positive T cells and promote a Th17-dominant cytokine milieu in the CNS and the BBB disruption. As a result, more MOG antibodies enter the CNS. Then the autoantibodies target myelins, especially MOG, to demyelinate the nerve fibers from the surface of myelin sheath via CDC (noted as deposition of activated complements), ADCC (in cooperation with infiltrating granulocytes), ADCP (seen as myelin phagocytosed macrophages), and other mechanisms. A fraction of the demyelinating lesions may exhibit MOG-dominant loss, suggesting a MOG-targeted pathology, and some oligodendrocytes may also be damaged. But compared to the remarkable CDC to cause astrocytolysis in AQP4 + NMOSD, the pathological role of CDC for demyelination may be less in some cases of MOGAD. These events probably occur simultaneously around multiple blood vessels (perivenous demyelination) in the white and gray matters. Subsequently, broken MOG and other myelin components are phagocytosed by macrophages (myelin-laden macrophages in the parenchyma and perivascular space), further enhancing antigen presentation and activating MOG-reactive CD4-positive T cells that induce the activation and infiltration of cytotoxic effector T cells against myelins and B cells that produce MOG antibodies intrathecally. These cellular and humoral immune responses are augmented through the interaction with proinflammatory cytokines/chemokines, which further exacerbates the disease state resulting in fusion of the lesions to form extensive demyelination (confluent demyelination). This confluent demyelination in MOGAD may develop by a different mechanism from that of the radial expansion of MS lesions.
However, we should investigate further details of the pathophysiology of MOGAD by means of various technologies including molecular immunology, omics, advanced imaging, neurophysiological tests, therapeutic response and artificial intelligence as well as conventional histopathological analyses. Furthermore, the histopathological studies in MOGAD to date have been derived from brain biopsies, and we should clarify whether the lesion characteristics are similar in other CNS regions, such as the optic nerve and spinal cord. Studies on how differences in the histopathologic findings may affect the severity and clinical phenotype in patients with MOGAD are also needed. These studies are expected to contribute to a better understanding and management of MOGAD.
YT, KF, and MA contributed to conception and design of the study. YT and TM organized the database. YT wrote the first draft of the manuscript, tables, and figures. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Johns, TG, and Bernard, CC. The structure and function of myelin oligodendrocyte glycoprotein. J Neurochem. (1999) 72:1–9. doi: 10.1046/j.1471-4159.1999.0720001.x
2. Boyle, LH, Traherne, JA, Plotnek, G, Ward, R, and Trowsdale, J. Splice variation in the cytoplasmic domains of myelin oligodendrocyte glycoprotein affects its cellular localisation and transport. J Neurochem. (2007) 102:1853–62. doi: 10.1111/j.1471-4159.2007.04687.x
3. Delarasse, C, Della Gaspera, B, Lu, CW, Lachapelle, F, Gelot, A, Rodriguez, D, et al. Complex alternative splicing of the myelin oligodendrocyte glycoprotein gene is unique to human and non-human primates. J Neurochem. (2006) 98:1707–17. doi: 10.1111/j.1471-4159.2006.04053.x
4. Pham-Dinh, D, Mattei, MG, Nussbaum, JL, Roussel, G, Pontarotti, P, Roeckel, N, et al. Myelin/oligodendrocyte glycoprotein is a member of a subset of the immunoglobulin superfamily encoded within the major histocompatibility complex. Proc Natl Acad Sci U S A. (1993) 90:7990–4. doi: 10.1073/pnas.90.17.7990
5. Lebar, R, Lubetzki, C, Vincent, C, Lombrail, P, and Boutry, JM. The M2 autoantigen of central nervous system myelin, a glycoprotein present in oligodendrocyte membrane. Clin Exp Immunol. (1986) 66:423–34.
6. Linington, C, and Lassmann, H. Antibody responses in chronic relapsing experimental allergic encephalomyelitis: correlation of serum demyelinating activity with antibody titre to the myelin/oligodendrocyte glycoprotein (MOG). J Neuroimmunol. (1987) 17:61–9. doi: 10.1016/0165-5728(87)90031-2
7. Schluesener, HJ, Sobel, RA, Linington, C, and Weiner, HL. A monoclonal antibody against a myelin oligodendrocyte glycoprotein induces relapses and demyelination in central nervous system autoimmune disease. J Immunol. (1987) 139:4016–21. doi: 10.4049/jimmunol.139.12.4016
8. Linington, C, Bradl, M, Lassmann, H, Brunner, C, and Vass, K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol. (1988) 130:443–54.
9. Lebar, R, Baudrimont, M, and Vincent, C. Chronic experimental autoimmune encephalomyelitis in the Guinea pig. Presence of anti-M2 antibodies in central nervous system tissue and the possible role of M2 autoantigen in the induction of the disease. J Autoimmun. (1989) 2:115–32. doi: 10.1016/0896-8411(89)90149-2
10. Bittner, S, Afzali, AM, Wiendl, H, and Meuth, SG. Myelin oligodendrocyte glycoprotein (MOG35-55) induced experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice. J Visual Exp. (2014) 15:51275. doi: 10.3791/51275-v
11. Lassmann, H, and Bradl, M. Multiple sclerosis: experimental models and reality. Acta Neuropathol. (2017) 133:223–44. doi: 10.1007/s00401-016-1631-4
12. Lassmann, H. The changing concepts in the neuropathology of acquired demyelinating central nervous system disorders. Curr Opin Neurol. (2019) 32:313–9. doi: 10.1097/WCO.0000000000000685
13. Lampasona, V, Franciotta, D, Furlan, R, Zanaboni, S, Fazio, R, Bonifacio, E, et al. Similar low frequency of anti-MOG IgG and IgM in MS patients and healthy subjects. Neurology. (2004) 62:2092–4. doi: 10.1212/01.WNL.0000127615.15768.AE
14. Reindl, M, Schanda, K, Woodhall, M, Tea, F, Ramanathan, S, Sagen, J, et al. International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm. (2020) 7:674. doi: 10.1212/NXI.0000000000000674
15. Gastaldi, M, Scaranzin, S, Jarius, S, Wildeman, B, Zardini, E, Mallucci, G, et al. Cell-based assays for the detection of MOG antibodies: a comparative study. J Neurol. (2020) 267:3555–64. doi: 10.1007/s00415-020-10024-0
16. Waters, PJ, Komorowski, L, Woodhall, M, Lederer, S, Majed, M, Fryer, J, et al. A multicenter comparison of MOG-IgG cell-based assays. Neurology. (2019) 92:e1255. doi: 10.1212/WNL.0000000000007096
17. Kitley, J, Woodhall, M, Waters, P, Leite, MI, Devenney, E, Craig, J, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology. (2012) 79:1273–7. doi: 10.1212/WNL.0b013e31826aac4e
18. Sato, DK, Callegaro, D, Lana-Peixoto, MA, Waters, PJ, de Haidar Jorge, FM, Takahashi, T, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. (2014) 82:474–81. doi: 10.1212/WNL.0000000000000101
19. Misu, T, Sato, DK, Nakashima, I, and Fujihara, K. MOG-IgG serological status matters in paediatric ADEM. J Neurol Neurosurg Psychiatry. (2015) 86:242. doi: 10.1136/jnnp-2014-308723
20. Di Pauli, F, Mader, S, Rostasy, K, Schanda, K, Bajer-Kornek, B, Ehling, R, et al. Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin Immunol. (2011) 138:247–54. doi: 10.1016/j.clim.2010.11.013
21. Matsumoto, Y, Misu, T, Mugikura, S, Takai, Y, Nishiyama, S, Kuroda, H, et al. Distinctive lesions of brain MRI between MOG-antibody-associated and AQP4-antibody-associated diseases. J Neurol Neurosurg Psychiatry. (2020) 92:682–4. doi: 10.1136/jnnp-2020-324818
22. Jurynczyk, M, Geraldes, R, Probert, F, Woodhall, MR, Waters, P, Tackley, G, et al. Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis. Brain. (2017) 140:617–27. doi: 10.1093/brain/aww350
23. Banks, SA, Morris, PP, Chen, JJ, Pittock, SJ, Sechi, E, Kunchok, A, et al. Brainstem and cerebellar involvement in MOG-IgG-associated disorder versus aquaporin-4-IgG and MS. J Neurol Neurosurg Psychiatry. (2020) 92:384–90. doi: 10.1136/jnnp-2020-325121
24. Fujimori, J, Takai, Y, Nakashima, I, Sato, DK, Takahashi, T, Kaneko, K, et al. Bilateral frontal cortex encephalitis and paraparesis in a patient with anti-MOG antibodies. J Neurol Neurosurg Psychiatry. (2017) 88:534–6. doi: 10.1136/jnnp-2016-315094
25. Ogawa, R, Nakashima, I, Takahashi, T, Kaneko, K, Akaishi, T, Takai, Y, et al. MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e322. doi: 10.1212/NXI.0000000000000322
26. Budhram, A, Mirian, A, Le, C, Hosseini-Moghaddam, SM, Sharma, M, and Nicolle, MW. Unilateral cortical FLAIR-hyperintense lesions in anti-MOG-associated encephalitis with seizures (FLAMES): characterization of a distinct clinico-radiographic syndrome. J Neurol. (2019) 266:2481–7. doi: 10.1007/s00415-019-09440-8
27. Waters, P, Woodhall, M, O'Connor, KC, Reindl, M, Lang, B, Sato, DK, et al. MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e89. doi: 10.1212/NXI.0000000000000089
28. Ketelslegers, IA, Van Pelt, DE, Bryde, S, Neuteboom, RF, Catsman-Berrevoets, CE, Hamann, D, et al. Anti-MOG antibodies plead against MS diagnosis in an acquired demyelinating syndromes cohort. Mult Scler. (2015) 21:1513–20. doi: 10.1177/1352458514566666
29. Banwell, B, Bennett, JL, Marignier, R, Kim, HJ, Brilot, F, Flanagan, EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: international MOGAD panel proposed criteria. Lancet Neurol. (2023) 22:268–82. doi: 10.1016/S1474-4422(22)00431-8
30. Popescu, BF, and Lucchinetti, CF. Pathology of demyelinating diseases. Annu Rev Pathol. (2012) 7:185–217. doi: 10.1146/annurev-pathol-011811-132443
31. Lucchinetti, CF. Multiple sclerosis and the spectrum of CNS inflammatory demyelinating diseases. Semin Neurol. (2008) 28:003–6. doi: 10.1055/s-2007-1019123
32. Young, NP, Weinshenker, BG, Parisi, JE, Scheithauer, B, Giannini, C, Roemer, SF, et al. Perivenous demyelination: association with clinically defined acute disseminated encephalomyelitis and comparison with pathologically confirmed multiple sclerosis. Brain. (2010) 133:333–48. doi: 10.1093/brain/awp321
33. Takai, Y, Misu, T, Kaneko, K, Chihara, N, Narikawa, K, Tsuchida, S, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain. (2020) 143:1431–46. doi: 10.1093/brain/awaa102
34. Hoftberger, R, Guo, Y, Flanagan, EP, Lopez-Chiriboga, AS, Endmayr, V, Hochmeister, S, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. (2020) 139:875–92. doi: 10.1007/s00401-020-02132-y
35. Konig, FB, Wildemann, B, Nessler, S, Zhou, D, Hemmer, B, Metz, I, et al. Persistence of immunopathological and radiological traits in multiple sclerosis. Arch Neurol. (2008) 65:1527–32. doi: 10.1001/archneur.65.11.1527
36. Di Pauli, F, Hoftberger, R, Reindl, M, Beer, R, Rhomberg, P, Schanda, K, et al. Fulminant demyelinating encephalomyelitis: insights from antibody studies and neuropathology. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e175. doi: 10.1212/NXI.0000000000000175
37. Spadaro, M, Gerdes, LA, Mayer, MC, Ertl-Wagner, B, Laurent, S, Krumbholz, M, et al. Histopathology and clinical course of MOG-antibody-associated encephalomyelitis. Ann Clin Translat Neurol. (2015) 2:295–301. doi: 10.1002/acn3.164
38. Jarius, S, Metz, I, Konig, FB, Ruprecht, K, Reindl, M, Paul, F, et al. Screening for MOG-IgG and 27 other anti-glial and anti-neuronal autoantibodies in 'pattern II multiple sclerosis' and brain biopsy findings in a MOG-IgG-positive case. Mult Scler. (2016) 22:1541–9. doi: 10.1177/1352458515622986
39. Wang, JJ, Jaunmuktane, Z, Mummery, C, Brandner, S, Leary, S, and Trip, SA. Inflammatory demyelination without astrocyte loss in MOG antibody-positive NMOSD. Neurology. (2016) 87:229–31. doi: 10.1212/WNL.0000000000002844
40. Kortvelyessy, P, Breu, M, Pawlitzki, M, Metz, I, Heinze, HJ, Matzke, M, et al. ADEM-like presentation, anti-MOG antibodies, and MS pathology: TWO case reports. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e335. doi: 10.1212/NXI.0000000000000335
41. Zhou, L, Huang, Y, Li, H, Fan, J, Zhangbao, J, Yu, H, et al. MOG-antibody associated demyelinating disease of the CNS: a clinical and pathological study in Chinese Han patients. J Neuroimmunol. (2017) 305:19–28. doi: 10.1016/j.jneuroim.2017.01.007
42. Ikeda, T, Yamada, K, Ogawa, R, Takai, Y, Kaneko, K, Misu, T, et al. The pathological features of MOG antibody-positive cerebral cortical encephalitis as a new spectrum associated with MOG antibodies: a case report. J Neurol Sci. (2018) 392:113–5. doi: 10.1016/j.jns.2018.06.028
43. Komatsu, T, Matsushima, S, Kaneko, K, and Fukuda, T. Perivascular enhancement in anti-MOG antibody demyelinating disease of the CNS. J Neurol Neurosurg Psychiatry. (2019) 90:111–2. doi: 10.1136/jnnp-2018-319235
44. Shu, Y, Long, Y, Wang, S, Hu, W, Zhou, J, Xu, H, et al. Brain histopathological study and prognosis in MOG antibody-associated demyelinating pseudotumor. Ann Clin Transl Neurol. (2019) 6:392–6. doi: 10.1002/acn3.712
45. Papathanasiou, A, Tanasescu, R, Davis, J, Rocha, MF, Singhal, S, O'Donoghue, MF, et al. MOG-IgG-associated demyelination: focus on atypical features, brain histopathology and concomitant autoimmunity. J Neurol. (2020) 267:359–68. doi: 10.1007/s00415-019-09586-5
46. Uzura, Y, Takeuchi, H, Ashida, S, Fujii, C, Shishido-Hara, Y, Inaba, T, et al. A tumefactive anti-MOG antibody associated disorder heralding central nervous system B-cell lymphoma: case report on diagnostic challenge. J Neuroimmunol. (2022) 365:577823. doi: 10.1016/j.jneuroim.2022.577823
47. Valencia-Sanchez, C, Guo, Y, Krecke, KN, Chen, JJ, Redenbaugh, V, Montalvo, M, et al. Cerebral cortical encephalitis in myelin oligodendrocyte glycoprotein antibody-associated disease. Ann Neurol. (2023) 93:297–302. doi: 10.1002/ana.26549
48. Kuhlmann, T, Ludwin, S, Prat, A, Antel, J, Brück, W, and Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. (2017) 133:13–24. doi: 10.1007/s00401-016-1653-y
49. Takai, Y, Misu, T, Suzuki, H, Takahashi, T, Okada, H, Tanaka, S, et al. Staging of astrocytopathy and complement activation in neuromyelitis optica spectrum disorders. Brain. (2021) 144:2401–15. doi: 10.1093/brain/awab102
50. Dal-Bianco, A, Grabner, G, Kronnerwetter, C, Weber, M, Kornek, B, Kasprian, G, et al. Long-term evolution of multiple sclerosis iron rim lesions in 7 T MRI. Brain. (2021) 144:833–47. doi: 10.1093/brain/awaa436
51. Prineas, JW, Kwon, EE, Cho, ES, Sharer, LR, Barnett, MH, Oleszak, EL, et al. Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol. (2001) 50:646–57. doi: 10.1002/ana.1255
52. Luchetti, S, Fransen, NL, van Eden, CG, Ramaglia, V, Mason, M, and Huitinga, I. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: a retrospective autopsy cohort analysis. Acta Neuropathol. (2018) 135:511–28. doi: 10.1007/s00401-018-1818-y
53. Frischer, JM, Weigand, SD, Guo, Y, Kale, N, Parisi, JE, Pirko, I, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol. (2015) 78:710–21. doi: 10.1002/ana.24497
54. Lassmann, H, Raine, CS, Antel, J, and Prineas, JW. Immunopathology of multiple sclerosis: report on an international meeting held at the Institute of Neurology of the University of Vienna. J Neuroimmunol. (1998) 86:213–7. doi: 10.1016/S0165-5728(98)00031-9
55. Lassmann, H, Brück, W, and Lucchinetti, C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends Mol Med. (2001) 7:115–21. doi: 10.1016/S1471-4914(00)01909-2
56. Lucchinetti, C, Bruck, W, Parisi, J, Scheithauer, B, Rodriguez, M, and Lassmann, H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. (2000) 47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
57. Stadelmann, C, Ludwin, S, Tabira, T, Guseo, A, Lucchinetti, CF, Leel-Ossy, L, et al. Tissue preconditioning may explain concentric lesions in Balo's type of multiple sclerosis. Brain. (2005) 128:979–87. doi: 10.1093/brain/awh457
58. Bruck, W, Popescu, B, Lucchinetti, CF, Markovic-Plese, S, Gold, R, Thal, DR, et al. Neuromyelitis optica lesions may inform multiple sclerosis heterogeneity debate. Ann Neurol. (2012) 72:385–94. doi: 10.1002/ana.23621
59. Aboul-Enein, F, Rauschka, H, Kornek, B, Stadelmann, C, Stefferl, A, Bruck, W, et al. Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol. (2003) 62:25–33. doi: 10.1093/jnen/62.1.25
60. Hohlfeld, R, Dornmair, K, Meinl, E, and Wekerle, H. The search for the target antigens of multiple sclerosis, part 2: CD8+ T cells, B cells, and antibodies in the focus of reverse-translational research. Lancet Neurol. (2016) 15:317–31. doi: 10.1016/S1474-4422(15)00313-0
61. Machado-Santos, J, Saji, E, Troscher, AR, Paunovic, M, Liblau, R, Gabriely, G, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain. (2018) 141:2066–82. doi: 10.1093/brain/awy151
62. Kaneko, K, Sato, DK, Nakashima, I, Nishiyama, S, Tanaka, S, Marignier, R, et al. Myelin injury without astrocytopathy in neuroinflammatory disorders with MOG antibodies. J Neurol Neurosurg Psychiatry. (2016) 87:1257–9. doi: 10.1136/jnnp-2015-312676
63. Magliozzi, R, Howell, O, Vora, A, Serafini, B, Nicholas, R, Puopolo, M, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. (2007) 130:1089–104. doi: 10.1093/brain/awm038
64. Howell, OW, Reeves, CA, Nicholas, R, Carassiti, D, Radotra, B, Gentleman, SM, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. (2011) 134:2755–71. doi: 10.1093/brain/awr182
65. Akaishi, T, Takahashi, T, Misu, T, Kaneko, K, Takai, Y, Nishiyama, S, et al. Difference in the source of anti-AQP4-IgG and anti-MOG-IgG antibodies in CSF in patients with Neuromyelitis Optica Spectrum disorder. Neurology. (2021) 97:e1–e12. doi: 10.1212/WNL.0000000000012175
66. Matsumoto, Y, Kaneko, K, Takahashi, T, Takai, Y, Namatame, C, Kuroda, H, et al. Diagnostic implications of MOG-IgG detection in sera and cerebrospinal fluids. Brain. (2023). doi: 10.1093/brain/awad122
67. Kwon, YN, Kim, B, Kim, JS, Mo, H, Choi, K, Oh, SI, et al. Myelin oligodendrocyte glycoprotein-immunoglobulin G in the CSF: clinical implication of testing and association with disability. Neurol Neuroimmunol Neuroinflamm. (2022) 9:1095. doi: 10.1212/NXI.0000000000001095
68. Mariotto, S, Gajofatto, A, Batzu, L, Delogu, R, Sechi, G, Leoni, S, et al. Relevance of antibodies to myelin oligodendrocyte glycoprotein in CSF of seronegative cases. Neurology. (2019) 93:e1867–72. doi: 10.1212/WNL.0000000000008479
69. Liu, J, Yang, X, Pan, J, Wei, Z, Liu, P, Chen, M, et al. Single-Cell transcriptome profiling unravels distinct peripheral blood immune Cell signatures of RRMS and MOG antibody-associated disease. Front Neurol. (2021) 12:807646. doi: 10.3389/fneur.2021.807646
70. Kothur, K, Wienholt, L, Tantsis, EM, Earl, J, Bandodkar, S, Prelog, K, et al. Th17, and neutrophil related cerebrospinal fluid cytokine/chemokines are elevated in MOG antibody associated demyelination. PLoS One. (2016) 11:e0149411. doi: 10.1371/journal.pone.0149411
71. Corbali, O, and Chitnis, T. Pathophysiology of myelin oligodendrocyte glycoprotein antibody disease. Front Neurol. (2023) 14:1137998. doi: 10.3389/fneur.2023.1137998
72. Mader, S, Gredler, V, Schanda, K, Rostasy, K, Dujmovic, I, Pfaller, K, et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation. (2011) 8:184. doi: 10.1186/1742-2094-8-184
73. Peschl, P, Schanda, K, Zeka, B, Given, K, Bohm, D, Ruprecht, K, et al. Human antibodies against the myelin oligodendrocyte glycoprotein can cause complement-dependent demyelination. J Neuroinflammation. (2017) 14:208. doi: 10.1186/s12974-017-0984-5
74. Kohyama, K, Nishida, H, Kaneko, K, Misu, T, Nakashima, I, and Sakuma, H. Complement-dependent cytotoxicity of human autoantibodies against myelin oligodendrocyte glycoprotein. Front Neurosci. (2023) 17:1014071. doi: 10.3389/fnins.2023.1014071
75. Yandamuri, SS, Filipek, B, Obaid, AH, Lele, N, Thurman, JM, Makhani, N, et al. MOGAD patient autoantibodies induce complement, phagocytosis, and cellular cytotoxicity. JCI Insight. (2023) 8:5373. doi: 10.1172/jci.insight.165373
76. Mader, S, Ho, S, Wong, HK, Baier, S, Winklmeier, S, Riemer, C, et al. Dissection of complement and fc-receptor-mediated pathomechanisms of autoantibodies to myelin oligodendrocyte glycoprotein. Proc Natl Acad Sci U S A. (2023) 120:e2300648120. doi: 10.1073/pnas.2300648120
77. Lerch, M, Schanda, K, Lafon, E, Würzner, R, Mariotto, S, Dinoto, A, et al. More efficient complement activation by anti-Aquaporin-4 compared with anti-myelin oligodendrocyte glycoprotein antibodies. Neurol Neuroimmunol Neuroinflamm. (2023) 10:59. doi: 10.1212/NXI.0000000000200059
78. Furman, CS, Gorelick-Feldman, DA, Davidson, KG, Yasumura, T, Neely, JD, Agre, P, et al. Aquaporin-4 square array assembly: opposing actions of M1 and M23 isoforms. Proc Natl Acad Sci U S A. (2003) 100:13609–14. doi: 10.1073/pnas.2235843100
79. Matiello, M, Schaefer-Klein, J, Sun, D, and Weinshenker, BG. Aquaporin 4 expression and tissue susceptibility to neuromyelitis optica. JAMA Neurol. (2013) 70:1118–25. doi: 10.1001/jamaneurol.2013.3124
80. Schumaker, VN, Hanson, DC, Kilchherr, E, Phillips, ML, and Poon, PH. A molecular mechanism for the activation of the first component of complement by immune complexes. Mol Immunol. (1986) 23:557–65. doi: 10.1016/0161-5890(86)90119-7
81. Phuan, PW, Ratelade, J, Rossi, A, Tradtrantip, L, and Verkman, AS. Complement-dependent cytotoxicity in neuromyelitis optica requires aquaporin-4 protein assembly in orthogonal arrays. J Biol Chem. (2012) 287:13829–39. doi: 10.1074/jbc.M112.344325
82. Macrini, C, Gerhards, R, Winklmeier, S, Bergmann, L, Mader, S, Spadaro, M, et al. Features of MOG required for recognition by patients with MOG antibody-associated disorders. Brain. (2021) 144:2375–89. doi: 10.1093/brain/awab105
83. Abdelhak, A, Huss, A, Kassubek, J, Tumani, H, and Otto, M. Serum GFAP as a biomarker for disease severity in multiple sclerosis. Sci Rep. (2018) 8:14798. doi: 10.1038/s41598-018-33158-8
84. Barro, C, Healy, BC, Liu, Y, Saxena, S, Paul, A, Polgar-Turcsanyi, M, et al. Serum GFAP and NfL levels differentiate subsequent progression and disease activity in patients with progressive multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2023) 10:52. doi: 10.1212/NXI.0000000000200052
85. Lassmann, H, Brunner, C, Bradl, M, and Linington, C. Experimental allergic encephalomyelitis: the balance between encephalitogenic T lymphocytes and demyelinating antibodies determines size and structure of demyelinated lesions. Acta Neuropathol. (1988) 75:566–76. doi: 10.1007/BF00686201
86. Spadaro, M, Winklmeier, S, Beltran, E, Macrini, C, Hoftberger, R, Schuh, E, et al. Pathogenicity of human antibodies against myelin oligodendrocyte glycoprotein. Ann Neurol. (2018) 84:315–28. doi: 10.1002/ana.25291
87. Lucchinetti, CF, Mandler, RN, McGavern, D, Bruck, W, Gleich, G, Ransohoff, RM, et al. A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain. (2002) 125:1450–61. doi: 10.1093/brain/awf151
88. Misu, T, Fujihara, K, Kakita, A, Konno, H, Nakamura, M, Watanabe, S, et al. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain. (2007) 130:1224–34. doi: 10.1093/brain/awm047
Keywords: myelin oligodendrocyte glycoprotein, antibody, acute disseminated encephalomyelitis, perivenous demyelination, confluent demyelination, multiple sclerosis lesion pattern-II
Citation: Takai Y, Misu T, Fujihara K and Aoki M (2023) Pathology of myelin oligodendrocyte glycoprotein antibody-associated disease: a comparison with multiple sclerosis and aquaporin 4 antibody-positive neuromyelitis optica spectrum disorders. Front. Neurol. 14:1209749. doi: 10.3389/fneur.2023.1209749
Edited by:
Lekha Pandit, Nitte University, IndiaReviewed by:
Tanuja Chitnis, Harvard Medical School, United StatesCopyright © 2023 Takai, Misu, Fujihara and Aoki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshiki Takai, eS10YWthaUBtZWQudG9ob2t1LmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.