 Stefano Brunelli
Stefano Brunelli Emilia Giannella2
Emilia Giannella2 Domenico De Angelis
Domenico De Angelis Giulia Maria Sancesario
Giulia Maria Sancesario- 1NeuroRehabilitation Unit 4, IRCCS Santa Lucia Foundation, Rome, Italy
- 2Clinical Neurochemistry Unit and Biobank, IRCCS Santa Lucia Foundation, Rome, Italy
- 3Radiology and Diagnostic Imaging Unit, IRCCS Santa Lucia Foundation, Rome, Italy
Stroke is one of the leading causes of death and the primary source of disability in adults, resulting in neuronal necrosis of ischemic areas, and in possible secondary degeneration of regions surrounding or distant to the initial damaged area. Secondary neurodegeneration (SNDG) following stroke has been shown to have different pathogenetic origins including inflammation, neurovascular response and cytotoxicity, but can be associated also to regenerative processes. Aside from focal neuronal loss, ipsilateral and contralateral effects distal to the lesion site, disruptions of global functional connectivity and a transcallosal diaschisis have been reported in the chronic stages after stroke. Furthermore, SNDG can be observed in different areas not directly connected to the primary lesion, such as thalamus, hippocampus, amygdala, substantia nigra, corpus callosum, bilateral inferior fronto-occipital fasciculus and superior longitudinal fasciculus, which can be highlighted by neuroimaging techniques. Although the clinical relevance of SNDG following stroke has not been well understood, the identification of specific biomarkers that reflect the brain response to the damage, is of paramount importance to investigate in vivo the different phases of stroke. Actually, brain-derived markers, particularly neurofilament light chain, tau protein, S100b, in post-stroke patients have yielded promising results. This review focuses on cerebral morphological modifications occurring after a stroke, on associated cellular and molecular changes and on state-of-the-art of biomarkers in acute and chronic phase. Finally, we discuss new perspectives regarding the implementation of blood-based biomarkers in clinical practice to improve the rehabilitation approaches and post stroke recovery.
1. Introduction
Stroke is a major global health burden, affecting more than 12 million of individuals each year worldwide (1). According to the latest data, stroke is the second leading cause of death worldwide and third leading cause of disability (2). The economic impact of stroke is also significant, with direct and indirect costs estimated to be, in 2017, around 861 billion of international dollars each year, i.e., 1.12% of the global Gross Domestic Product (GDP) (1). Despite progress in understanding the underlying mechanisms of stroke and the development of new treatments, much work remains to be done to reduce the impact of stroke on individuals and society, not only during the acute phase but also during long-lasting invalidity in the following years.
The most common type of stroke, the ischemic one, occurs when a blood clot blocks the flow in an artery in the brain. The less frequent, the hemorrhagic stroke, can result from a ruptured blood vessel or from a structural abnormality of a blood vessel in the brain. Both stroke conditions cause local hypoxia and reduction of supply of other nutrients, with consequent damage or death of neurons (3, 4). The location, extensiveness and the number of the lesions can determine the degree of disability.
Clinically, the evaluation of stroke severity is performed through clinical measures by an experienced stroke clinician. Clinical assessments can provide valuable information about the severity of a stroke, but they are often imprecise and have only a moderate correlation with actual brain tissue damage and long-term outcomes. To determine the site of stroke area and the extent of brain damage the current strategies include standard neuroimaging such as conventional brain magnetic resonance (MRI) and computed tomography (CT) of the brain. There is also the possibility in the follow-up after the stroke, to study the evolution of the disease using advanced neuroimaging techniques such as functional MRI, perfusion imaging, diffusion imaging, magnetic resonance spectroscopy, and dual-energy computerized tomography.
Based on the area involved, different clinical phenotypes can be observed, with overlapping signs, including motor impairment, speech or cognitive deficits, as contralateral hemi-spatial neglect, memory or executive dysfunctions, visual field defects, dysphagia, urinary incontinence and other symptoms.
Furthermore, functional disturbances in remote parts of the brain, connected with the area with neuronal damage, were reported (5). This phenomenon could be explained by the secondary neurodegeneration (SNDG) that gradually spreads to different brain structures, even if not directly affected by reduction in cerebral blood flow caused by the initial stroke (6).
The progressive death of neurons, axonal degeneration and gliosis in distal regions of the brain have been observed in several experimental (7–9) and in clinical neuroimaging studies (10–14). The site of the infarct significantly influences the spreading of secondary changes (10), and the progressive involvement of these areas can lead to a further worsening of deficits and disability.
Actually, investigating the effect of SNDG following stroke on patients’ outcome, disability and recovery has reached increasing interest. Evidence suggests that measurement of circulating brain derived biomarkers in cerebrospinal fluid (CSF) or blood are a useful tool to evaluate the pathophysiological and biochemical modifications occurring during pathological processes (15, 16). Here, we describe: (i) the most frequent structural cerebral modifications occurring after a stroke; (ii) associated cellular and molecular changes; (iii) state-of-the-art and advances in the field of biomarkers in acute phase and in secondary neurodegeneration. In particular, we focus on markers of neuronal and astroglial damage, namely neurofilament light chain (Nf-L), tau proteins, neuron-specific enolase (NSE), S100 calcium binding protein B (S100b), as well as on marker of regeneration, as brain-derived neurotrophic factor (BDNF). Finally, we discuss new perspectives regarding the implementation of blood-based biomarkers in clinical practice to improve therapeutic intervention and neurorehabilitation.
1.1. Pathophysiology of Stroke and secondary neurodegeneration
After stroke, a cascade of inflammatory and degenerative mechanisms is triggered that ultimately determine the extent and severity of damage, the development of late secondary neurodegeneration, and the degree of disability or recovery (17). In the first phase, injury of the central nervous system (CNS) results from the dysfunction and death of neurons and of multiple cell types, including astrocytes, pericytes, smooth muscle cells, endothelial cells, oligodendrocytes, microglia and neural and glial precursor cells, which together constitute the so-called neurovascular unit (NVU) (18, 19). The NVU has been hypothesized to regulate the balance of the neural–glial–vascular signaling in a biphasic mode, that is mainly oriented to the damage during the acute injury phase and to the regeneration in the chronic recovery (20–22). Thus, post-stroke recovery results from reversal of tissue dysfunction, by promoting neurogenesis, angiogenesis and vascular remodeling (23–27) (Figure 1).
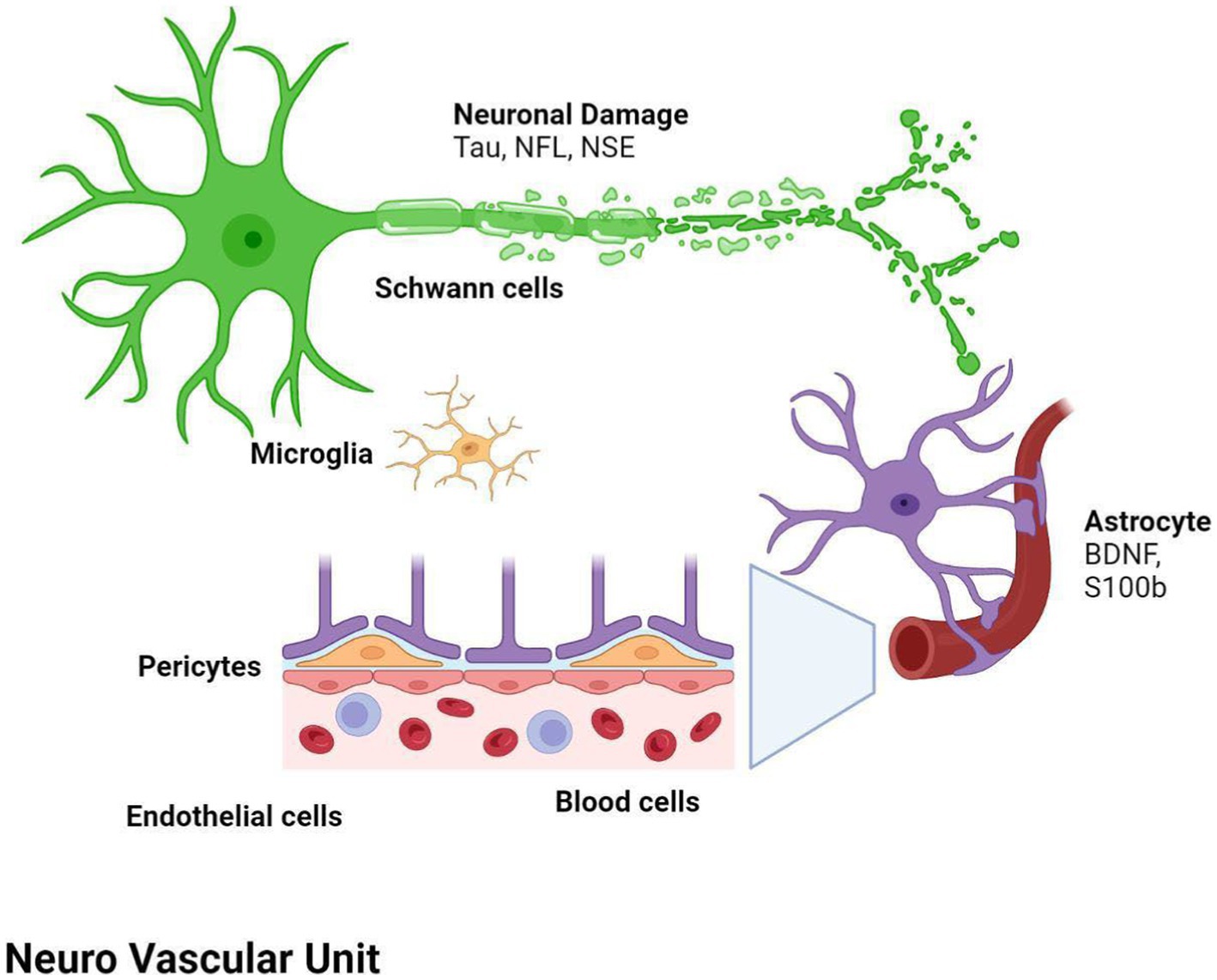
Figure 1. Both ischemic and hemorragic stroke cause local hypoxia and reduction of supply of other nutrients, with consequent damage or death of neurons and multiple cell types, glial, astrocytes, pericytes, smooth muscle cells, endothelial cells, oligodendrocytes, microglia and neural and glial precursor cells, which together constitute the neurovascular unit (NVU). Biomarker changes in CSF and blood reflect the extent of brain injury and development of pathological changes, and correlate with severity of damage and the activation of reparative mechanisms. BDNF, brain-derived neurotrophic factor; NF-L, neurofilament light proteins; NSE, neuron specific enolase; S100b, S100 calcium-binding protein B.
The imbalance between persistence of chronic inflammatory degenerative status and neurogenesis is considered responsible for persistent neurological dysfunction and impairment of functional recovery following damage of the CNS (28–30). Indeed, it is known that the occurrence of brain lesions, due to trauma or stroke injury, increases the incidence of late impairments and chronic neurodegenerative conditions, such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and chronic traumatic encephalopathy (31, 32).
SNDG refers to neurophysiological and histopathologic changes occurring in non-ischemic remote brain regions that have anterograde or retrograde synaptic connections with the primary lesion site (6); thus, SNDG has been hypothesized to be a potential modulator of post-stroke functional disorders (33).
Most of the knowledge on neurodegenerative processes associated with stroke is based on studies with neuroimaging and in particular brain MRI, providing an excellent anatomical detail and gray/white matter contrast. For this reason, structural MRI, using conventional sequences, especially T1, T2-weighted, in addition to contrast enhancement, has become the accepted standard for routine examination of the brain, offering high sensitivity to anatomical location and morphological characteristics of pathological processes.
Studying the SNDG with conventional MRI, disruption to global functional connectivity has been revealed in the ipsi-lesional corticospinal tract and in the inter-hemispheric connections (corpus callosum), in the bilateral inferior fronto-occipital fasciculus and in the bilateral superior longitudinal fasciculus (34–36), in the ipsilateral thalamus, in the substantia nigra, hippocampus and in amygdala (37–39).
The involvement of these areas distal to the primary lesion defines the onset of cognitive and behavioral symptoms different from those primarily related to stroke area (36, 40). For example, involvement of the thalamus can lead to hyperalgesia; damage in the substantia nigra provokes Parkinson-like symptoms (slow movements, tremor, stiffness and difficulty with walking and balance); involvement of amygdala results in difficulty with memory processing and emotional reactions, whereas an involvement of hippocampus to memory impairment (Table 1).
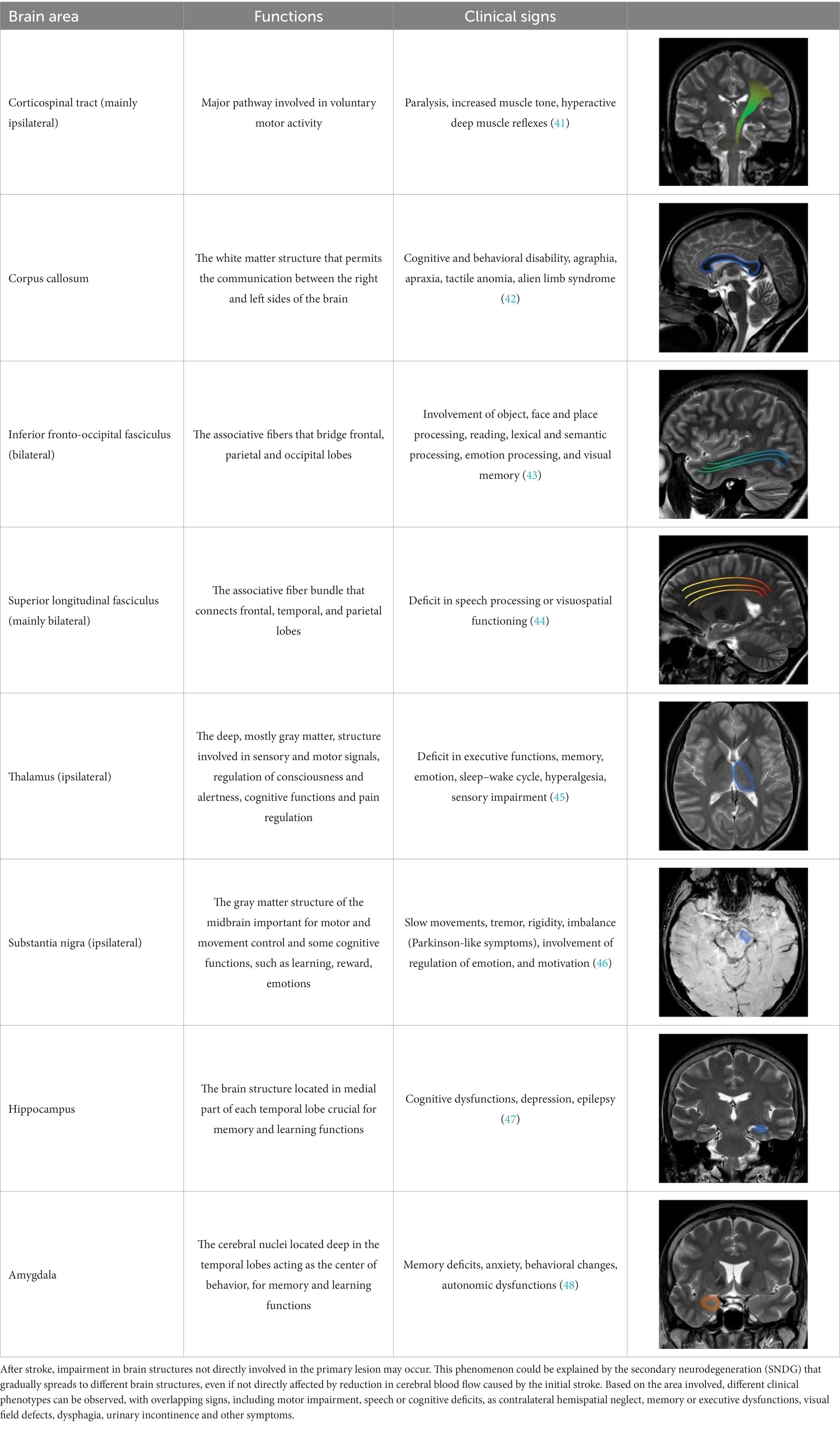
Table 1. Brain structures involved in secondary neurodegeneration after stroke and MRI: localization, functions, and clinical signs.
In addition, other pathogenic mechanisms occurring after stroke have been reported as potential factors leading to cognitive decline. Interestingly, some studies described alterations of functional connectivity measured with functional MRI in different parts of cortex that appear structurally normal, not contiguous to the stroke area (49, 50). This suggests that uninvolved areas of the brain, even if structurally normal, may have functional impairment. It was supposed that the stroke lesion can disrupt the mutual balanced inhibition between hemispheres by decreasing the inhibition of the contralateral non lesioned hemisphere and by reducing the number of cortical-spinal tract projections to the other brain areas, with the effect of a remote functional depression (51).
Following ischemic stroke, microglia and astrocytes are activated within hours, inducing the production of cytokines and chemokines and the infiltration of leukocytes (52, 53). Oxidative stress, a disturbance in the balance between the production of reactive oxygen species and antioxidant defenses, is induced in cerebral ischemia especially through inflammation and reperfusion, increasing the production of free radicals (54). After the activation of the peripheral immune response, macrophages and neutrophils are released by the spleen into the bloodstream, which can easily reach the brain due to the alteration of the blood–brain barrier (55). This immune response, associated with the activation of resident inflammatory cells such as microglia and astrocytes, has been demonstrated to contribute to the development of SNDG (56, 57).
On a cellular level, stroke lesions cause the disruption of the structure of the neuron’s cytoskeleton (58). Those changes in cytoskeletal structure and subsequent neuron instability and final neuronal death are associated with the formation of protein aggregates and the release of cytoplasmic proteins in the extracellular space, representing a valuable surrogate index of acute necrosis or slow
Among these, tau proteins, a microtubule associated protein, and the corresponding hyperphosphorylated forms, p-tau, are well-established biomarkers of neurodegeneration, and represent predictors of functional outcome (61) or development of cognitive dysfunction after cerebral ischemia (62–64). Further, increase of microtubule-associated protein (MAP2) reflects the fragmentation of neuronal dendrites, not only in the areas of stroke but even in other brain regions resulting in a widespread loss of synaptic plasticity (65, 66). Finally, stroke can also lead to accumulation of amyloid-beta (AB), a peptide that is the main component of amyloid plaques in the brains of subjects with Alzheimer’s disease (67).
The deposition of the neurotoxic aggregates of protein amyloid-beta could be further stimulated by the glial activation and pro-inflammatory cytokine release after cerebral ischemia, which persists for a long time and in some cases could determine the latter onset of Alzheimer’s type dementia (17).
2. Biomarkers of brain damage and Stroke
Brain atrophy patterns are recognized as signatures of neurodegenerative conditions, and have been included as topographical markers for AD and a number of other neurodegenerative diseases (68). Furthermore, regional atrophy rates have been shown to correlate with CSF and blood biomarkers of neurodegeneration. However, although the conventional neuroimaging techniques still represent the benchmark diagnostic tool, they may not have sufficient resolution to detect early changes in the brain at the cellular and molecular levels after stroke. Advanced neuroimaging may provide a better option to identify and follow up the biological processes involved in SNDG but not represent a real option because of the requirement of sophisticated and expensive instruments and trained personnel (69). Moreover, serial repetition of the neuroimaging is not feasible within a short interval to capture the evolution of the processes linked to SNDG. There is also to consider some general drawbacks of MRI such as long acquisition time, the possibility of movement artifacts, the contraindications in some patients with metallic surgical implants and patients’ claustrophobia.
Instead, blood-based biomarkers can reflect molecular and biochemical state in both normal and pathophysiologic processes, including neuronal and vascular injury, inflammation, oxidative stress, glial activation, etc. (70). Since stroke may induce blood brain barrier dysfunction (71), a progressive cross of brain-derived proteins into the bloodstream, and vice versa, may induce changes in concentration of several molecules and, accordingly, affect their clinical value. However, a great challenge is to understand whether assessment of biological markers after months or years after stroke, may be useful to early detect the occurrence of degenerative processes secondary to stroke.
The current knowledge on neurological biomarkers in stroke mainly takes advantage of findings in other neurodegenerative diseases, namely Alzheimer’s Disease (AD), Parkinson Disease (PD), dementia with Lewy bodies (LBD) and others. CSF biomarkers, including amyloid Beta 42 (Aβ42) and Aβ40, total tau (T-tau), phosphorylated tau (P-tau) have an added value in the differential diagnosis of AD and related disorders, including mixed pathologies, atypical presentations, and in case of ambiguous clinical dementia diagnosis (72–74).
Actually, a considerable number of published works have demonstrated that the blood level of some neuronal or glial proteins particularly increase after stroke and correlate with clinical features, severity and outcome, representing promising markers to evaluate the extent of brain injury (Table 2). In particular, we can differentiate: (1) markers of neuronal damage, namely neurofilament light chain (Nf-L), tau proteins, neuron-specific enolase (NSE); marker of astroglial damage, the S100 calcium binding protein B (S100b); (2) marker of neuroregeneration, the brain-derived neurotrophic factor (BDNF). Thus, longitudinal assessment of such biomarkers in subjects may reveal the presence of secondary subclinical degenerative or regenerative processes, providing information complementary to findings from routine examinations and neuroimaging (Figure 2).
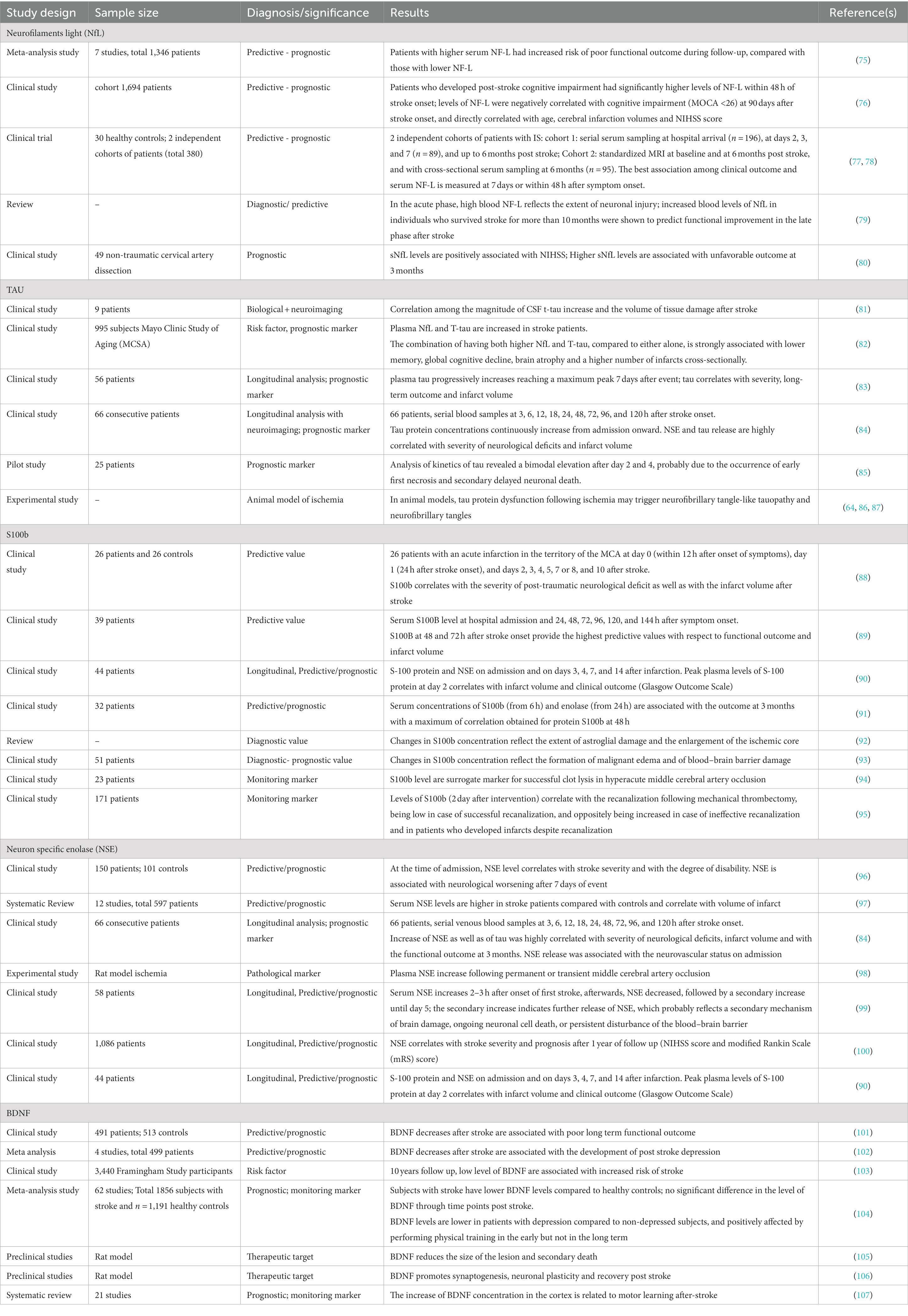
Table 2. Summary of CSF and blood biomarkers with potential diagnostic characteristics for SDNG after stroke.
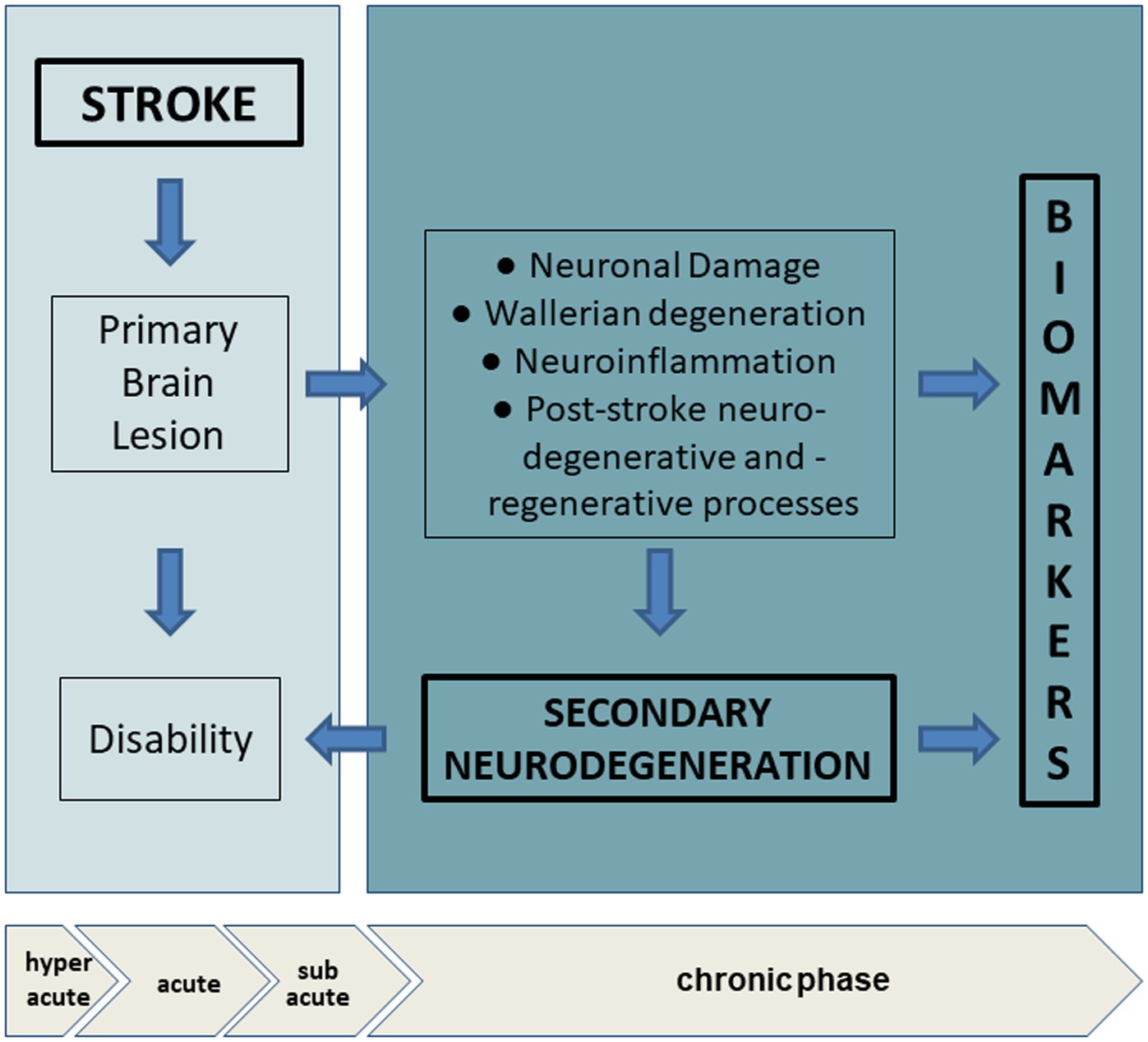
Figure 2. Biomarkers in chronic phase after stroke and secondary neurodegeneration. In hyperacute and acute phase, stroke provoques a primary brain lesion with consequent activation of a cascade of vascular, oxidative, inflammatory events. The location, extensiveness and the amount of the lesion can determine the degree of disability. In the chronic phase, adaptive neural plasticity and regenerative processes try to counteract wallerian degeneration, oxidative stress and neuroinflammation. Analysis of circulating biomarkers allow the evaluation of clinical silent changes, non-detectable with imaging technologies. Hyperacute (< 6 h), acute (6 to 72 h), subacute (after 7 days), chronic (after ~6 weeks).
2.1. Neurofilaments
Neurofilament proteins are components of the cytoskeleton of neurons, classified on their molecular weight in light (NF-L), middle (NF-M) and heavy (NF-H). As a subunit of neurofilament, serum NF-L has emerged in the last few years as the most promising biomarker of axonal injury and neurodegeneration, mainly in multiple sclerosis (MS), showing potential applications for both patient monitoring and for observational and interventional trials (78, 108).
Recently, numerous studies have been carried out on patients with stroke showing significant correlation with severity, according to National Institutes of Health Stroke Scale (NIHSS) upon admission (77, 78, 80, 109, 110) and after clinical outcome (111).
A meta-analysis on ischemic stroke or transient ischemic attack patients demonstrated that patients with higher serum NF-L, had increased risk of poor functional outcome during follow-up, compared with those with lower NF-L, strengthening the value of NF-L as predictive biomarker for ischemic stroke outcome (75). Furthermore, a study on a cohort of 1,694 patients with first-ever acute ischemic stroke investigated the correlation among NF-L and post-stroke cognitive impairment (PSCI). Among the cohort, 60.74% of patients developed cognitive impairment. Interestingly, authors found that the PSCI group exhibited significantly higher levels of NF-L within 48 h of stroke onset; levels of NF-L were negatively correlated with cognitive impairment defined by Montreal Cognitive Assessment (MOCA) (MOCA <26) at 90 days after stroke onset, and directly correlated with age, cerebral infarction volumes and NIHSS score (76).
Importantly, results from different studies showed a wide heterogeneity regarding the sampling time, reporting best association among clinical outcome and serum NF-L measured at 7 days or within 48 h after symptom onset (77, 78). Pending multicentric studies and large-scale validation, it is however suggested to perform the analysis of NF-L before the 7-day time point.
Converging evidence suggests that NF-L level has an added predictive value apart from stroke severity. This may be explained by the presence of SDNG outside the infarct area, such as white matter tracts connected to the infarct, which could contribute to the development of cognitive impairment after the stroke (76). A study in the acute and post-acute phase after stroke confirmed that high levels of NF-L are associated with poor clinical outcome, and later on, the concentration of NF-L positively correlates with the occurrence of SDNG, as assessed by MRI.
Surprisingly, increase of NF-L has shown to predict functional improvement in the late phase after stroke in patients who survived for more than 10 months. This could suggest that the kinetic of NF-L may reflect two concurrent but very distinct processes of NF-L release, namely neuroaxonal injury and synaptic damage, that are features of secondary neurodegeneration, and oppositely of late adaptive neural plasticity. Indeed, while in the acute phase high blood NF-L seem to reflect the extent of neuronal injury, in the late phase increase of NF-L may serve as a biomarker of adaptive neural plasticity and a positive predictor of functional improvement and, therefore, effectiveness of neurorehabilitation (79).
Thus, results from different studies suggest that at different time points, NF-L levels may be considered a measure of structural brain lesion, complementary to MRI (80) but in the late phase after stroke, the interpretation of elevated blood levels of NF-L should not be therefore limited to the extent of injury or neurodegeneration (79).
2.2. Tau
Tau, a microtubule-associated protein that regulates stability and dynamics of axons, is considered a well-recognized marker of neuronal degeneration by a huge amount of both clinical and preclinical studies. In the research framework of AD, CSF tau is one of the core markers of the so called “ATN system,” where “A” refers to the value of an amyloid β biomarker (amyloid PET or CSF Aβ42); “T,” the value of a tau pathology biomarker (CSF p-tau or tau PET); and “N,” a quantitative or topographic biomarker of neurodegeneration or neuronal injury (112, 113).
So far, the assessment of total tau level in the CSF or blood is thought to reflect the extent and intensity of neuronal damage of any etiology. Oppositely, the presence of post translationally modified tau species, i.e., truncated or phosphorylated, is considered a typical feature of some tauopathies. Blood and CSF levels of tau, but not p-tau, increases after ischemic stroke and mild head trauma, being highly increased in case of prion disease as Creutzfeldt-Jakob disease (114–116). Conversely, in tauopathies such as AD or other dementia, the production of p-tau, C- or N terminal truncated tau species are specifically increased, reflecting underlying neurofibrillary pathology and the formation of post translationally modified species which accumulate in brain and can be also detected in biological fluid (117–119).
Tau proteins have been proposed as candidate markers of stroke and SDNG for evaluating the extent of neurodegeneration, and potentially discriminating neuronal damage with the presence of degenerative processes of other etiology, such as Alzheimer’s pathology. Studies combining CSF analysis and neuroimaging demonstrate a correlation among the magnitude of t-tau increase and the volume of tissue damage following ischemic stroke (81). A recent study compared levels of plasma tau and NfL as cross-sectional and longitudinal markers of cognitive decline and neuroimaging changes, on a cohort of 995 subjects from the community-based Mayo Clinic Study of Aging (MCSA) (82). Both plasma NfL and T-tau have been found to be elevated in stroke patients, and among those with other cardiovascular conditions; the combination of having both higher NfL and T-tau, compared to either alone, was more strongly associated with lower memory, global cognitive decline, brain atrophy and a higher number of infarcts cross-sectionally (82).
Interestingly, in stroke patients, concentration of plasma tau progressively increases from admission reaching a maximum peak 7 days after event, showing a correlation with severity, long-term outcome, as well as infarct volume (83, 84). Analysis of kinetics of tau after global brain ischemia revealed a bimodal elevation after day 2 and 4, probably due to the occurrence of early first necrosis and secondary delayed neuronal death (85, 120). This is also confirmed by a recent retrospective study on head trauma and remote injury, involving a total of 164 subjects, 94 PD patients and 70 healthy controls, in which the levels of CSF tau were found to be increased in a subgroup of PD patients who reported lifetime head trauma preceding diagnosis, probably due to extended degeneration or occurrence of tauopathy (121).
Evidence from experimental models also suggest that tau has a key role in regulating neuronal damage and SDNG after stroke, up to the development of Alzheimer’s-type dementia (64). In animal models, tau protein dysfunction following ischemia may trigger neurofibrillary tangle-like tauopathy and neurofibrillary tangles (86, 87). In humans, history of ischemic stroke increases likelihood of developing AD-type dementia (122) and further, patients suffering of AD with previous brain ischemic injuries show more severe dementia phenotype (123). Certainly, tau in combination with other biomarkers is useful for clinical and research purposes in order to reveal the development of future or clinical subtle neurodegeneration.
2.3. S100b
S100b is a calcium-binding protein mainly expressed in astroglial and Schwann cells, myeloid-derived cells, and a few other cell types. Under physiological conditions, S100b is released in the extracellular space in response to hormonal or inflammatory stimuli and exerts both paracrine and autocrine effects on neurons and glia, but increases are observed in neuronal pathological conditions, such as brain trauma, ischemia and neurodegenerative, inflammatory and psychiatric diseases.
S100b is widely used in emergency medicine due to its high positive predictive value in cases of brain injury, showing a correlation with the severity of post-traumatic neurological deficit as well as with the infarct volume after stroke (88–90, 124).
A study on 32 consecutive patients evaluated changes of S100b and the neuron specific enolase (NSE) levels between the first 6 h, and in the 5 days after stroke. Serum concentrations of S100b from 6 h on and of NSE from 24 h on were associated with the outcome at 3 months with a maximum of correlation obtained for protein S100b at 48 h (91).
Moreover, the changes in S100b concentration reflect the extent of astroglial damage, the enlargement of the ischemic core, (92) as well as the formation of malignant edema and of blood–brain barrier damage (93). Conversely, in transient ischemia not associated with substantial tissue injury, S100b levels in serum are generally normal (94, 125). Recently, S100b has been proposed as a surrogate marker to monitor the stroke response after endovascular treatment. Indeed, levels of S100b are correlated with the recanalization following mechanical thrombectomy, being low in case of successful recanalization, and oppositely being increased in case of ineffective recanalization and in patients who developed infarcts despite recanalization (95).
So far, studies in patients with neurodegenerative diseases showed conflicting results. Some authors found that serum S100b concentrations were similar (126) or lower in CSF of AD patients compared with elderly controls, not correlating with brain atrophy (127), whereas others reported correlations between CSF S100b levels and AD brain atrophy or cognitive status as measured by the Mini Mental State Exam score (128, 129).
Notwithstanding, studies are still conducted on limited cohorts, therefore results need to be consolidated by larger cohorts and longitudinal studies, evaluating the association between S100b changes, volume of tissue damage and the functional or cognitive outcome over the post-stroke phases. Although S100B can be considered a useful biomarker in the acute phase after stroke to evaluate the damage of the NVU and astroglial cells, further investigations to understand its role in SDNG following stroke are needed.
2.4. Neuron specific enolase
Neuron specific enolase (NSE) is an enzyme involved in glycolytic energy metabolism in the brain, released from neurons during injury as a nonspecific marker of neuronal damage. Studies have investigated the potential of NSE as a predictor of outcome in patients in the early phase after stroke. In 150 cases of patients with stroke and 101 controls, NSE level showed a positive correlation with stroke severity at the time of admission and with the degree of disability, categorized into mild, moderate and severe according to NIHSS; further, NSE was associated with neurological worsening after 7 days of event (96).
Data obtained from 12 studies including 597 patients, found that serum NSE levels were higher in stroke patients compared with controls and correlated with volume of infarct; however, results do not support a correlation among NSE and functional outcome and further the relationship to stroke severity is unclear (97). Increase of NSE as well as of tau was highly correlated with severity of neurological deficits and infarct volume, and with the functional outcome at 3 months. Interestingly, NSE release was associated with the neurovascular status on admission (84).
Findings on CSF in patients with primary neurodegenerative diseases, such as AD, PD, LBD, suggest that NSE may be used to evaluate the presence and extent of axonal and glial degeneration (130). NSE in combination with tau may predict secondary damage after stroke, with specific windows that reflect different release mechanisms (84). In fact, the NSE concentration in serum increase 2–3 h after onset of first stroke, afterwards, NSE decreased, followed by a secondary increase until day 5, that is the last measurement in the observation period; the secondary increase indicates further release of NSE, which probably reflects a secondary mechanism of brain damage, ongoing neuronal cell death (98, 99), or persistent disturbance of the blood–brain barrier (131). A recent study found a correlation between NSE and both stroke severity and prognosis after 1 year of follow up documented by NIHSS score and modified Rankin Scale (mRS) score, respectively, on 1,086 patients grouped as hypertension and non-hypertension (100). However, studies mainly focus on the role of NSE in the acute and subacute phase after stroke and its potential predictive or prognostic value. Longitudinal studies on large cohorts are needed to evaluate the association between NSE levels and the development of brain morphological changes in secondary neurodegeneration.
2.5. BDNF
The brain derived neurotrophic factor (BDNF) is the most abundant neurotrophin in the adult brain, and has a remarkable capability to repair brain damage and maintain synaptic plasticity by inducing neuronal proliferation, survival and differentiation (132). However, unlike synaptic plasticity involved in normal cognitive function, post-stroke and rehabilitation neuroplasticity primarily refers to the brain’s ability to recover from injury to restore its normal structure and function. In this context, the significant role of BDNF in the regulation and maintenance of synaptic plasticity after stroke has been extensively investigated in both clinical and experimental studies, including the potential use of BDNF as a direct therapeutic agent for the stroke treatment (133).
After a stroke event, reactive astrocytes upregulate the expression of BDNF and other neurotrophic factors, and the resulting levels have been demonstrated to be associated with the clinical and functional outcome. For instance, BDNF decrease after stroke is associated with poor long term functional outcome (101) and development of post stroke depression (102); further, low level of BDNF is associated with increased risk of stroke (103).
A recent meta-analysis of data from 62 studies showed that subjects with stroke (n = 1856) had lower BDNF levels compared to healthy controls (n = 1,191), but there was no significant difference in the level of BDNF through time points post stroke. Furthermore, BDNF levels were lower in the patients with depression compared to non-depressed subjects, and positively affected by performing physical training in the early but not in the long term (104).
Preclinical studies demonstrated that BDNF induces anti apoptotic mechanisms, reducing the size of the lesion and secondary death (105), promoting synaptogenesis, neuronal plasticity and recovery post stroke (105, 106); moreover, increase of BDNF concentration in the cortex is related to motor learning after-stroke (107). Experimental studies demonstrated clinically positive outcomes reached by administration of stroke treatments that modulate BDNF expression, leading to consider BDNF as potential therapeutic target (133). Together, these studies suggest that BDNF exerts favorable effects in post-stroke recovery due to its attenuation of cell death and promotion of neurogenesis. However, longitudinal studies investigating the correlation between BDNF levels and other biomarkers of neurodegeneration, as well as morphological and clinical changes, are needed to better understand the development of SDNG and recovery in the chronic phase after stroke.
3. Future perspectives
Current therapeutic strategies for post-stroke patients are based on multidisciplinary approaches that include neuropsychological rehabilitation, physical, occupational, speech therapy and neuromodulation techniques, as the recent evidence of effectiveness of Transcranial Direct Current and Transcranial Magnetic Stimulations. Moreover, the prevention of disability must also take into account the presence of a possible secondary brain detriment that is driven by neuroinflammatory cascades and dysfunction of the NVU (39, 134) often leading to the development of SDNG. The early identification of degenerative pathological processes, also not clinically evident, is crucial for the pharmacological and rehabilitative treatment of patients. Neuroimaging techniques, which offer a complete view of the anatomical location and morphological characteristics of pathological processes within the brain, cannot be used as a screening and monitoring tool due to high costs and possible insufficient resolution to detect the early changes in the brain at a cellular and molecular level.
The possibility that biomarkers measured in blood may be predictive of future outcome and association with SDNG is appealing. Several potential markers of neurodegeneration have been identified, which can help capture a range of brain changes and pathologies. Plasma biomarkers, rather than CSF and imaging markers, provide a low-cost, non-invasive tool to evaluate neurodegeneration and to assess rate of disease progression, given the feasibility of repeat blood draws. Notwithstanding, it is crucial to understand what information each blood marker provides to know how they can best be applied for clinical and research purposes (82). Numerous studies have highlighted the correlation between changes in the levels of some biomarkers, such as NfL, tau, and clinical worsening in the chronic phases in stroke patients. Moreover, the levels of biomarkers also correlate with the morphological changes of the brain, suggesting a potential use in clinical settings. However, studies on the significance of changes in biomarker levels in the chronic phase and recovery, or on neurodegenerative mechanisms secondary to stroke, remain elusive and will require further investigation.
Most of the studies have been conducted on limited cohorts of patients and in the hyper, acute or subacute phases. Therefore, larger and longitudinal studies are needed to evaluate the association between biomarkers along the acute and chronic phases after stroke and the functional outcomes, as well as the role of biomarkers in late phase, over a period of years, to evaluate the effect of neuronal degeneration and rehabilitation therapy. For example, the biological efficacy of a rehabilitation method could be monitored by evaluation of specific SDNG biomarkers, e.g., NF-L and BDNF; therefore, in the future, blood biomarkers could be integrated into a tool for defining personalized rehabilitation approaches.
Nevertheless, the interpretation of biomarkers results in a clinical context needs expertise and caution. A limit of biomarkers analysis is that any changes in the levels can reflect acute or progressive pathological brain changes, but do not allow to recognize the etiology or identify where the degeneration is occurring. Therefore, a careful clinical evaluation is always important, and further integration of biochemical markers with neuroimaging is essential to reveal which brain areas are involved.
Moreover, the mechanisms participating in the development of SDNG after stroke have not been fully elucidated. A better understanding of the interlinks between inflammation, oxidative stress and degeneration may help to identify the appropriate biomarkers to be assessed to monitor the therapeutic and rehabilitation treatments. Last, since recent studies suggest that neurogenesis and angiogenesis processes are activated within brain areas after stroke events, it would be interesting to identify novel potential biomarkers associated with regeneration in stroke recovery.
4. Conclusion
Stroke care has been revolutionized in the last three decades by improved reperfusion treatments and rehabilitation therapies. The evaluation of neurodegenerative biomarkers in blood shows promising results for clinical and research purposes, especially in the evaluation of acute stroke patients. Further research is needed to better understand the pathophysiology of SDNG after stroke, to develop a useful tool to monitor and detect the occurrence of molecular and cellular pathological changes to finally predict the disability in patients. More rigorous studies should be conducted to validate the potential use of biomarkers in clinical settings, in order to define personalized pharmacological and neurorehabilitative treatments for stroke patients.
Author contributions
GS, SB, and DD contributed to the conception and design of the manuscript. MB contributed to the figures and neuroimagings. EG contributed to the first draft of the manuscript, the figures, and analysis. All the authors wrote sections of the manuscript and contributed to manuscript revision, read, and approved the submitted version.
Funding
This study was supported by the Italian Ministry of Health— Ricerca Corrente anno 2023.
Acknowledgments
Figure 1 was created using BioRender.com (Accessed on 30 March 2023).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. GBD 2019 Stroke Collaborators. Global, regional, and national burden of stroke and its risk factors, 1990-2019: a systematic analysis for the global burden of disease study 2019. Lancet Neurol. (2021) 20:795–820. doi: 10.1016/S1474-4422(21)00252-0
2. Owolabi, MO, Thrift, AG, Mahal, A, Ishida, M, Martins, S, Johnson, WD, et al. Primary stroke prevention worldwide: translating evidence into action. Lancet Public Health. (2022) 7:e74–85. doi: 10.1016/S2468-2667(21)00230-9
3. Al-Qazzaz, NK, Ali, SH, Ahmad, SA, Islam, S, and Mohamad, K. Cognitive impairment and memory dysfunction after a stroke diagnosis: a post-stroke memory assessment. Neuropsychiatr Dis Treat. (2014) 10:1677–91. doi: 10.2147/NDT.S67184
4. Sveinsson, OA, Kjartansson, O, and Valdimarsson, EM. Cerebral ischemia/infarction - epidemiology, causes and symptoms. Laeknabladid. (2014) 100:271–9. doi: 10.17992/lbl.2014.05.543
5. Pekny, M, Wilhelmsson, U, Tatlisumak, T, and Pekna, M. Astrocyte activation and reactive gliosis-a new target in stroke? Neurosci Lett. (2019) 689:45–55. doi: 10.1016/j.neulet.2018.07.021
6. Zhang, J, Zhang, Y, Xing, S, Liang, Z, and Zeng, J. Secondary neurodegeneration in remote regions after focal cerebral infarction: a new target for stroke management? Stroke. (2012) 43:1700–5. doi: 10.1161/STROKEAHA.111.632448
7. Butler, TL, Kassed, CA, Sanberg, PR, Willing, AE, and Pennypacker, KR. Neurodegeneration in the rat hippocampus and striatum after middle cerebral artery occlusion. Brain Res. (2002) 929:252–60. doi: 10.1016/S0006-8993(01)03371-6
8. Fujie, W, Kirino, T, Tomukai, N, Iwasawa, T, and Tamura, A. Progressive shrinkage of the thalamus following middle cerebral artery occlusion in rats. Stroke. (1990) 21:1485–8. doi: 10.1161/01.str.21.10.1485
9. Holmberg, P, Liljequist, S, and Wägner, A. Secondary brain injuries in thalamus and hippocampus after focal ischemia caused by mild, transient extradural compression of the somatosensori cortex in the rat. Curr Neurovasc Res. (2009) 6:1–11. doi: 10.2174/156720209787466073
10. Baudat, C, Maréchal, B, Corredor-Jerez, R, Kober, T, Meuli, R, Hagmann, P, et al. Automated MRI-based volumetry of basal ganglia and thalamus at the chronic phase of cortical stroke. Neuroradiology. (2020) 62:1371–80. doi: 10.1007/s00234-020-02477-x
11. Fernández-Andújar, M, Doornink, F, Dacosta-Aguayo, R, Soriano-Raya, JJ, Miralbell, J, Bargalló, N, et al. Remote thalamic microstructural abnormalities related to cognitive function in ischemic stroke patients. Neuropsychology. (2014) 28:984–96. doi: 10.1037/neu0000087
12. Nakane, M, Tamura, A, Sasaki, Y, and Teraoka, A. MRI of secondary changes in the thalamus following a cerebral infarct. Neuroradiology. (2002) 44:915–20. doi: 10.1007/s00234-002-0846-3
13. Stebbins, GT, Nyenhuis, DL, Wang, C, Cox, JL, Freels, S, Bangen, K, et al. Gray matter atrophy in patients with ischemic stroke with cognitive impairment. Stroke. (2008) 39:785–93. doi: 10.1161/STROKEAHA.107.507392
14. Veldsman, M, Curwood, E, Pathak, S, Werden, E, and Brodtmann, A. Default mode network neurodegeneration reveals the remote effects of ischaemic stroke. J Neurol Neurosurg Psychiatry. (2018) 89:318–20. doi: 10.1136/jnnp-2017-315676
15. Sancesario, G, and Bernardini, S. AD biomarker discovery in CSF and in alternative matrices. Clin Biochem. (2019) 72:52–7. doi: 10.1016/j.clinbiochem.2019.08.008
16. Schirinzi, T, Zenuni, H, Grillo, P, Bovenzi, R, Guerrera, G, Gargano, F, et al. Tau and amyloid-β peptides in serum of patients with Parkinson's disease: correlations with CSF levels and clinical parameters. Front Neurol. (2022) 13:748599. doi: 10.3389/fneur.2022.748599
17. Lin Kooi, O, Frederick Rohan, W, and Michael, N. Is Stroke a neurodegenerative condition? A critical review of secondary neurodegeneration and amyloid-beta accumulation after Stroke. AIMS Med Sci. (2017) 4:1–16. doi: 10.3934/medsci.2017.1.1
18. Lo, EH, Broderick, JP, and Moskowitz, MA. tPA and proteolysis in the neurovascular unit. Stroke. (2004) 35:354–6. doi: 10.1161/01.STR.0000115164.80010.8A
19. Lo, EH, Dalkara, T, and Moskowitz, MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. (2003) 4:399–414. doi: 10.1038/nrn1106
20. Lo, EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. (2008) 14:497–500. doi: 10.1038/nm1735
21. Moskowitz, MA, Lo, EH, and Iadecola, C. The science of stroke: mechanisms in search of treatments. Neuron. (2010) 67:181–98. doi: 10.1016/j.neuron.2010.07.002
22. Tiedt, S, Buchan, AM, Dichgans, M, Lizasoain, I, Moro, MA, and Lo, EH. The neurovascular unit and systemic biology in stroke - implications for translation and treatment. Nat Rev Neurol. (2022) 18:597–612. doi: 10.1038/s41582-022-00703-z
23. Li, L, Lundkvist, A, Andersson, D, Wilhelmsson, U, Nagai, N, Pardo, AC, et al. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab. (2008) 28:468–81. doi: 10.1038/sj.jcbfm.9600546
24. Pekna, M, Pekny, M, and Nilsson, M. Modulation of neural plasticity as a basis for stroke rehabilitation. Stroke. (2012) 43:2819–28. doi: 10.1161/STROKEAHA.112.654228
25. Pekny, M, and Nilsson, M. Astrocyte activation and reactive gliosis. Glia. (2005) 50:427–34. doi: 10.1002/glia.20207
26. Pekny, M, and Pekna, M. (2016) reactive gliosis in the pathogenesis of CNS diseases. Biochim Biophys Acta. (1862) 1862:483–91. doi: 10.1016/j.bbadis.2015.11.014
27. Wieloch, T, and Nikolich, K. Mechanisms of neural plasticity following brain injury. Curr Opin Neurobiol. (2006) 16:258–64. doi: 10.1016/j.conb.2006.05.011
28. Anwar, MA, Al Shehabi, TS, and Eid, AH. Inflammogenesis of secondary spinal cord injury. Front Cell Neurosci. (2016) 10:98. doi: 10.3389/fncel.2016.00098
29. Feng, Y, Liao, S, Wei, C, Jia, D, Wood, K, Liu, Q, et al. Infiltration and persistence of lymphocytes during late-stage cerebral ischemia in middle cerebral artery occlusion and photothrombotic stroke models. J Neuroinflammation. (2017) 14:248. doi: 10.1186/s12974-017-1017-0
30. Xiong, Y, Mahmood, A, and Chopp, M. Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chin J Traumatol. (2018) 21:137–51. doi: 10.1016/j.cjtee.2018.02.003
31. Johnson, VE, Stewart, W, Arena, JD, and Smith, DH. Traumatic brain injury as a trigger of neurodegeneration. Adv Neurobiol. (2017) 15:383–400. doi: 10.1007/978-3-319-57193-5_15
32. Sundman, MH, Hall, EE, and Chen, NK. Examining the relationship between head trauma and neurodegenerative disease: a review of epidemiology, pathology and neuroimaging techniques. J Alzheimers Dis Parkinsonism. (2014) 4:137. doi: 10.4172/2161-0460.1000137
33. Aamodt, EB, Lydersen, S, Alnæs, D, Schellhorn, T, Saltvedt, I, Beyer, MK, et al. Longitudinal brain changes after Stroke and the association with cognitive decline. Front Neurol. (2022) 13:856919. doi: 10.3389/fneur.2022.856919
34. Egorova, N, Dhollander, T, Khlif, MS, Khan, W, Werden, E, and Brodtmann, A. Pervasive white matter fiber degeneration in ischemic Stroke. Stroke. (2020) 51:1507–13. doi: 10.1161/STROKEAHA.119.028143
35. Gupta, RK, Saksena, S, Hasan, KM, Agarwal, A, Haris, M, Pandey, CM, et al. Focal Wallerian degeneration of the corpus callosum in large middle cerebral artery stroke: serial diffusion tensor imaging. J Magn Reson Imaging. (2006) 24:549–55. doi: 10.1002/jmri.20677
36. Koyama, T, and Domen, K. Diffusion tensor fractional anisotropy in the superior longitudinal fasciculus correlates with functional Independence measure cognition scores in patients with cerebral infarction. J Stroke Cerebrovasc Dis. (2017) 26:1704–11. doi: 10.1016/j.jstrokecerebrovasdis.2017.03.034
37. Forno, LS. Reaction of the substantia nigra to massive basal ganglia infarction. Acta Neuropathol. (1983) 62:96–102. doi: 10.1007/BF00684925
38. Ogawa, T, Yoshida, Y, Okudera, T, Noguchi, K, Kado, H, and Uemura, K. Secondary thalamic degeneration after cerebral infarction in the middle cerebral artery distribution: evaluation with MR imaging. Radiology. (1997) 204:255–62. doi: 10.1148/radiology.204.1.9205256
39. Stuckey, SM, Ong, LK, Collins-Praino, LE, and Turner, RJ. Neuroinflammation as a key driver of secondary neurodegeneration following Stroke? Int J Mol Sci. (2021) 22:13101. doi: 10.3390/ijms222313101
40. Corbetta, M, Ramsey, L, Callejas, A, Baldassarre, A, Hacker, CD, Siegel, JS, et al. Common behavioral clusters and subcortical anatomy in stroke. Neuron. (2015) 85:927–41. doi: 10.1016/j.neuron.2015.02.027
41. Ludwig, PE, Reddy, V, and Varacallo, M, Neuroanatomy, Central Nervous System (CNS). StatPearls. Treasure Island, FL: StatPearls Publishing (2022).
42. Friese, SA, Bitzer, M, Freudenstein, D, Voigt, K, and Küker, W. Classification of acquired lesions of the corpus callosum with MRI. Neuroradiology. (2000) 42:795–802. doi: 10.1007/s002340000430
43. Herbet, G, Zemmoura, I, and Duffau, H. Functional anatomy of the inferior longitudinal fasciculus: from historical reports to current hypotheses. Front Neuroanat. (2018) 12:77. doi: 10.3389/fnana.2018.00077
44. Janelle, F, Iorio-Morin, C, D'amour, S, and Fortin, D. Superior longitudinal fasciculus: a review of the anatomical descriptions with functional correlates. Front Neurol. (2022) 13:794618. doi: 10.3389/fneur.2022.794618
45. Maeshima, S, and Osawa, A. Thalamic lesions and aphasia or neglect. Curr Neurol Neurosci Rep. (2018) 18:39. doi: 10.1007/s11910-018-0844-4
46. Linck, PA, Kuchcinski, G, Munsch, F, Griffier, R, Lopes, R, Okubo, G, et al. Neurodegeneration of the substantia Nigra after ipsilateral infarct: MRI R2* mapping and relationship to clinical outcome. Radiology. (2019) 291:438–48. doi: 10.1148/radiol.2019182126
47. Shetty, AK. Hippocampal injury-induced cognitive and mood dysfunction, altered neurogenesis, and epilepsy: can early neural stem cell grafting intervention provide protection? Epilepsy Behav. (2014) 38:117–24.
48. Nikolenko, VN, Oganesyan, MV, Rizaeva, NA, Kudryashova, VA, Nikitina, AT, Pavliv, MP, et al. Amygdala: neuroanatomical and Morphophysiological features in terms of neurological and neurodegenerative diseases. Brain Sci. (2020) 10:502. doi: 10.3390/brainsci10080502
49. Carter, AR, Patel, KR, Astafiev, SV, Snyder, AZ, Rengachary, J, Strube, MJ, et al. Upstream dysfunction of somatomotor functional connectivity after corticospinal damage in stroke. Neurorehabil Neural Repair. (2012) 26:7–19. doi: 10.1177/1545968311411054
50. He, BJ, Shulman, GL, Snyder, AZ, and Corbetta, M. The role of impaired neuronal communication in neurological disorders. Curr Opin Neurol. (2007) 20:655–60. doi: 10.1097/WCO.0b013e3282f1c720
51. Auriat, AM, Neva, JL, Peters, S, Ferris, JK, and Boyd, LA. A review of transcranial magnetic stimulation and multimodal neuroimaging to characterize post-Stroke neuroplasticity. Front Neurol. (2015) 6:226. doi: 10.3389/fneur.2015.00226
52. Iadecola, C, and Anrather, J. The immunology of stroke: from mechanisms to translation. Nat Med. (2011) 17:796–808. doi: 10.1038/nm.2399
53. Jin, R, Yang, G, and Li, G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. (2010) 87:779–89. doi: 10.1189/jlb.1109766
54. Sinning, C, Westermann, D, and Clemmensen, P. Oxidative stress in ischemia and reperfusion: current concepts, novel ideas and future perspectives. Biomark Med. (2017) 11:11031–40. doi: 10.2217/bmm-2017-0110
55. Emsley, HC, Smith, CJ, Gavin, CM, Georgiou, RF, Vail, A, Barberan, EM, et al. An early and sustained peripheral inflammatory response in acute ischaemic stroke: relationships with infection and atherosclerosis. J Neuroimmunol. (2003) 139:93–101. doi: 10.1016/S0165-5728(03)00134-6
56. Ajmo, CT, Vernon, DO, Collier, L, Hall, AA, Garbuzova-Davis, S, Willing, A, et al. The spleen contributes to stroke-induced neurodegeneration. J Neurosci Res. (2008) 86:2227–34. doi: 10.1002/jnr.21661
57. Kluge, MG, Abdolhoseini, M, Zalewska, K, Ong, LK, Johnson, SJ, Nilsson, M, et al. Spatiotemporal analysis of impaired microglia process movement at sites of secondary neurodegeneration post-stroke. J Cereb Blood Flow Metab. (2019) 39:2456–70.
58. Gutiérrez-Vargas, JA, Castro-Álvarez, JF, Zapata-Berruecos, JF, Abdul-Rahim, K, and Arteaga-Noriega, A. Neurodegeneration and convergent factors contributing to the deterioration of the cytoskeleton in Alzheimer's disease, cerebral ischemia and multiple sclerosis (review). Biomed Rep. (2022) 16:27. doi: 10.3892/br.2022.1510
59. Cao, L, Tan, L, Wang, HF, Jiang, T, Zhu, XC, and Yu, JT. Cerebral microinfarcts and dementia: a systematic review and Metaanalysis. Curr Alzheimer Res. (2017) 14:802–8. doi: 10.2174/1567205013666161201200429
60. McMurray, CT. Neurodegeneration: diseases of the cytoskeleton? Cell Death Differ. (2000) 7:861–5. doi: 10.1038/sj.cdd.4400764
61. Bitsch, A, Horn, C, Kemmling, Y, Seipelt, M, Hellenbrand, U, Stiefel, M, et al. Serum tau protein level as a marker of axonal damage in acute ischemic stroke. Eur Neurol. (2002) 47:45–51. doi: 10.1159/000047946
62. Guo, T, Noble, W, and Hanger, DP. Roles of tau protein in health and disease. Acta Neuropathol. (2017) 133:665–704. doi: 10.1007/s00401-017-1707-9
63. Kosik, KS, Joachim, CL, and Selkoe, DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A. (1986) 83:4044–8. doi: 10.1073/pnas.83.11.4044
64. Pluta, R, Ułamek-Kozioł, M, Januszewski, S, and Czuczwar, SJ. Tau protein dysfunction after brain ischemia. J Alzheimers Dis. (2018) 66:429–37. doi: 10.3233/JAD-180772
65. Gutiérrez-Vargas, JA, Moreno, H, and Cardona-Gómez, GP. Targeting CDK5 post-stroke provides long-term neuroprotection and rescues synaptic plasticity. J Cereb Blood Flow Metab. (2017) 37:2208–23. doi: 10.1177/0271678X16662476
66. Pérez-Corredor, PA, Gutiérrez-Vargas, JA, Ciro-Ramírez, L, Balcazar, N, and Cardona-Gómez, GP. High fructose diet-induced obesity worsens post-ischemic brain injury in the hippocampus of female rats. Nutr Neurosci. (2022) 25:122–36. doi: 10.1080/1028415X.2020.1724453
67. Ong, LK, Zhao, Z, Kluge, M, Walker, FR, and Nilsson, M. Chronic stress exposure following photothrombotic stroke is associated with increased levels of amyloid beta accumulation and altered oligomerisation at sites of thalamic secondary neurodegeneration in mice. J Cereb Blood Flow Metab. (2017) 37:1338–48. doi: 10.1177/0271678X16654920
68. Dubois, B, Feldman, HH, Jacova, C, Hampel, H, Molinuevo, JL, Blennow, K, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. (2014) 13:614–29. doi: 10.1016/S1474-4422(14)70090-0
69. Puhr-Westerheide, D, Froelich, MF, Solyanik, O, Gresser, E, Reidler, P, Fabritius, MP, et al. Cost-effectiveness of short-protocol emergency brain MRI after negative non-contrast CT for minor stroke detection. Eur Radiol. (2022) 32:1117–26. doi: 10.1007/s00330-021-08222-z
70. Liu, Z, Yang, C, Wang, X, and Xiang, Y. Blood-based biomarkers: a forgotten friend of Hyperacute ischemic Stroke. Front Neurol. (2021) 12:634717. doi: 10.3389/fneur.2021.634717
71. Yang, J, d'Esterre, C, Ceruti, S, Roversi, G, Saletti, A, Fainardi, E, et al. Temporal changes in blood-brain barrier permeability and cerebral perfusion in lacunar/subcortical ischemic stroke. BMC Neurol. (2015) 15:214. doi: 10.1186/s12883-015-0468-0
72. Hansson, O, Zetterberg, H, Buchhave, P, Londos, E, Blennow, K, and Minthon, L. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. (2006) 5:228–34. doi: 10.1016/S1474-4422(06)70355-6
73. Jack, CR, Bennett, DA, Blennow, K, Carrillo, MC, Dunn, B, Haeberlein, SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. (2018) 14:535–62. doi: 10.1016/j.jalz.2018.02.018
74. Molinuevo, JL, Blennow, K, Dubois, B, Engelborghs, S, Lewczuk, P, Perret-Liaudet, A, et al. The clinical use of cerebrospinal fluid biomarker testing for Alzheimer's disease diagnosis: a consensus paper from the Alzheimer's biomarkers standardization Initiative. Alzheimers Dement. (2014) 10:808–17. doi: 10.1016/j.jalz.2014.03.003
75. Liu, D, Chen, J, Wang, X, Xin, J, Cao, R, and Liu, Z. Serum Neurofilament light chain as a predictive biomarker for ischemic Stroke outcome: a systematic review and meta-analysis. J Stroke Cerebrovasc Dis. (2020) 29:104813. doi: 10.1016/j.jstrokecerebrovasdis.2020.104813
76. Wang, Z, Wang, R, Li, Y, Li, M, Zhang, Y, Jiang, L, et al. Plasma Neurofilament light chain as a predictive biomarker for post-stroke cognitive impairment: a prospective cohort study. Front Aging Neurosci. (2021) 13:631738. doi: 10.3389/fnagi.2021.631738
77. Al-Khaled, M. Serum neurofilament light as diagnostic biomarker for acute cerebral ischemia: a promising tool. Eur J Neurol. (2018) 25:609. doi: 10.1111/ene.13588
78. Tiedt, S, Duering, M, Barro, C, Kaya, AG, Boeck, J, Bode, FJ, et al. Serum neurofilament light: a biomarker of neuroaxonal injury after ischemic stroke. Neurology. (2018) 91:e1338–47. doi: 10.1212/WNL.0000000000006282
79. Pekny, M, Wilhelmsson, U, Stokowska, A, Tatlisumak, T, Jood, K, and Pekna, M. Neurofilament light chain (NfL) in blood-a biomarker predicting Unfavourable outcome in the acute phase and improvement in the late phase after Stroke. Cells. (2021) 10:1537. doi: 10.3390/cells10061537
80. Traenka, C, Disanto, G, Seiffge, DJ, Gensicke, H, Hert, L, Grond-Ginsbach, C, et al. Serum neurofilament light chain levels are associated with clinical characteristics and outcome in patients with cervical artery dissection. Cerebrovasc Dis. (2015) 40:222–7. doi: 10.1159/000440774
81. Hesse, C, Rosengren, L, Vanmechelen, E, Vanderstichele, H, Jensen, C, Davidsson, P, et al. Cerebrospinal fluid markers for Alzheimer's disease evaluated after acute ischemic stroke. J Alzheimers Dis. (2000) 2:199–206. doi: 10.3233/JAD-2000-23-402
82. Marks, JD, Syrjanen, JA, Graff-Radford, J, Petersen, RC, Machulda, MM, Campbell, MR, et al. Comparison of plasma neurofilament light and total tau as neurodegeneration markers: associations with cognitive and neuroimaging outcomes. Alzheimers Res Ther. (2021) 13:199. doi: 10.1186/s13195-021-00944-y
83. Bielewicz, J, Kurzepa, J, Czekajska-Chehab, E, Stelmasiak, Z, and Bartosik-Psujek, H. Does serum tau protein predict the outcome of patients with ischemic stroke? J Mol Neurosci. (2011) 43:241–5. doi: 10.1007/s12031-010-9403-4
84. Wunderlich, MT, Lins, H, Skalej, M, Wallesch, CW, and Goertler, M. Neuron-specific enolase and tau protein as neurobiochemical markers of neuronal damage are related to early clinical course and long-term outcome in acute ischemic stroke. Clin Neurol Neurosurg. (2006) 108:558–63. doi: 10.1016/j.clineuro.2005.12.006
85. Randall, J, Mörtberg, E, Provuncher, GK, Fournier, DR, Duffy, DC, Rubertsson, S, et al. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: results of a pilot study. Resuscitation. (2013) 84:351–6. doi: 10.1016/j.resuscitation.2012.07.027
86. Bi, M, Gladbach, A, van Eersel, J, Ittner, A, Przybyla, M, van Hummel, A, et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat Commun. (2017) 8:473. doi: 10.1038/s41467-017-00618-0
87. Kato, T, Hirano, A, Katagiri, T, Sasaki, H, and Yamada, S. Neurofibrillary tangle formation in the nucleus basalis of Meynert ipsilateral to a massive cerebral infarct. Ann Neurol. (1988) 23:620–3. doi: 10.1002/ana.410230617
88. Büttner, T, Weyers, S, Postert, T, Sprengelmeyer, R, and Kuhn, W. S-100 protein: serum marker of focal brain damage after ischemic territorial MCA infarction. Stroke. (1997) 28:1961–5. doi: 10.1161/01.str.28.10.1961
89. Foerch, C, Singer, OC, Neumann-Haefelin, T, du Mesnil de Rochemont, R, Steinmetz, H, and Sitzer, M. Evaluation of serum S100B as a surrogate marker for long-term outcome and infarct volume in acute middle cerebral artery infarction. Arch Neurol. (2005) 62:1130–4. doi: 10.1001/archneur.62.7.1130
90. Missler, U, Wiesmann, M, Friedrich, C, and Kaps, M. S-100 protein and neuron-specific enolase concentrations in blood as indicators of infarction volume and prognosis in acute ischemic stroke. Stroke. (1997) 28:1956–60. doi: 10.1161/01.STR.28.10.1956
91. Wunderlich, MT, Wallesch, CW, and Goertler, M. Release of neurobiochemical markers of brain damage is related to the neurovascular status on admission and the site of arterial occlusion in acute ischemic stroke. J Neurol Sci. (2004) 227:49–53. doi: 10.1016/j.jns.2004.08.005
92. Brunkhorst, R, Pfeilschifter, W, and Foerch, C. Astroglial proteins as diagnostic markers of acute intracerebral hemorrhage-pathophysiological background and clinical findings. Transl Stroke Res. (2010) 1:246–51. doi: 10.1007/s12975-010-0040-6
93. Foerch, C, Otto, B, Singer, OC, Neumann-Haefelin, T, Yan, B, Berkefeld, J, et al. Serum S100B predicts a malignant course of infarction in patients with acute middle cerebral artery occlusion. Stroke. (2004) 35:2160–4. doi: 10.1161/01.STR.0000138730.03264.ac
94. Foerch, C, du Mesnil de Rochemont, R, Singer, O, Neumann-Haefelin, T, Buchkremer, M, Zanella, FE, et al. S100B as a surrogate marker for successful clot lysis in hyperacute middle cerebral artery occlusion. J Neurol Neurosurg Psychiatry. (2003) 74:322–5. doi: 10.1136/jnnp.74.3.322
95. Luger, S, Koerbel, K, Martinez Oeckel, A, Schneider, H, Maurer, CJ, Hintereder, G, et al. Role of S100B serum concentration as a surrogate outcome parameter after mechanical Thrombectomy. Neurology. (2021) 97:e2185–94. doi: 10.1212/WNL.0000000000012918
96. Bharosay, A, Bharosay, VV, Varma, M, Saxena, K, Sodani, A, and Saxena, R. Correlation of brain biomarker neuron specific enolase (NSE) with degree of disability and neurological worsening in cerebrovascular Stroke. Indian J Clin Biochem. (2012) 27:186–90. doi: 10.1007/s12291-011-0172-9
97. Anand, N, and Stead, LG. Neuron-specific enolase as a marker for acute ischemic stroke: a systematic review. Cerebrovasc Dis. (2005) 20:213–9. doi: 10.1159/000087701
98. Barone, FC, Clark, RK, Price, WJ, White, RF, Feuerstein, GZ, Storer, BL, et al. Neuron-specific enolase increases in cerebral and systemic circulation following focal ischemia. Brain Res. (1993) 623:77–82. doi: 10.1016/0006-8993(93)90012-C
99. Wunderlich, MT, Ebert, AD, Kratz, T, Goertler, M, Jost, S, and Herrmann, M. Early neurobehavioral outcome after stroke is related to release of neurobiochemical markers of brain damage. Stroke. (1999) 30:1190–5. doi: 10.1161/01.STR.30.6.1190
100. Gao, L, Xie, J, Zhang, H, Zheng, H, Zheng, W, Pang, C, et al. Neuron-specific enolase in hypertension patients with acute ischemic stroke and its value forecasting long-term functional outcomes. BMC Geriatr. (2023) 23:294. doi: 10.1186/s12877-023-03986-z
101. Stanne, TM, Åberg, ND, Nilsson, S, Jood, K, Blomstrand, C, Andreasson, U, et al. Low circulating acute brain-derived neurotrophic factor levels are associated with poor long-term functional outcome after ischemic Stroke. Stroke. (2016) 47:1943–5. doi: 10.1161/STROKEAHA.115.012383
102. Xu, HB, Xu, YH, He, Y, Xue, F, Wei, J, Zhang, H, et al. Decreased serum brain-derived neurotrophic factor may indicate the development of Poststroke depression in patients with acute ischemic Stroke: a Meta-analysis. J Stroke Cerebrovasc Dis. (2018) 27:709–15. doi: 10.1016/j.jstrokecerebrovasdis.2017.10.003
103. Pikula, A, Beiser, AS, Chen, TC, Preis, SR, Vorgias, D, DeCarli, C, et al. Serum brain-derived neurotrophic factor and vascular endothelial growth factor levels are associated with risk of stroke and vascular brain injury: Framingham study. Stroke. (2013) 44:2768–75. doi: 10.1161/STROKEAHA.113.001447
104. Mojtabavi, H, Shaka, Z, Momtazmanesh, S, Ajdari, A, and Rezaei, N. Circulating brain-derived neurotrophic factor as a potential biomarker in stroke: a systematic review and meta-analysis. J Transl Med. (2022) 20:126. doi: 10.1186/s12967-022-03312-y
105. Schäbitz, WR, Steigleder, T, Cooper-Kuhn, CM, Schwab, S, Sommer, C, Schneider, A, et al. Intravenous brain-derived neurotrophic factor enhances poststroke sensorimotor recovery and stimulates neurogenesis. Stroke. (2007) 38:2165–72. doi: 10.1161/STROKEAHA.106.477331
106. Ploughman, M, Windle, V, MacLellan, CL, White, N, Doré, JJ, and Corbett, D. Brain-derived neurotrophic factor contributes to recovery of skilled reaching after focal ischemia in rats. Stroke. (2009) 40:1490–5. doi: 10.1161/STROKEAHA.108.531806
107. Alcantara, CC, García-Salazar, LF, Silva-Couto, MA, Santos, GL, Reisman, DS, and Russo, TL. Post-stroke BDNF concentration changes following physical exercise: a systematic review. Front Neurol. (2018) 9:637. doi: 10.3389/fneur.2018.00637
108. Yuan, A, and Nixon, RA. Neurofilament proteins as biomarkers to monitor neurological diseases and the efficacy of therapies. Front Neurosci. (2021) 15:689938. doi: 10.3389/fnins.2021.689938
109. Gattringer, T, Pinter, D, Enzinger, C, Seifert-Held, T, Kneihsl, M, Fandler, S, et al. Serum neurofilament light is sensitive to active cerebral small vessel disease. Neurology. (2017) 89:2108–14. doi: 10.1212/WNL.0000000000004645
110. Onatsu, J, Vanninen, R, Jäkälä, P, Mustonen, P, Pulkki, K, Korhonen, M, et al. Serum Neurofilament light chain concentration correlates with infarct volume but not prognosis in acute ischemic Stroke. J Stroke Cerebrovasc Dis. (2019) 28:2242–9. doi: 10.1016/j.jstrokecerebrovasdis.2019.05.008
111. Uphaus, T, Bittner, S, Gröschel, S, Steffen, F, Muthuraman, M, Wasser, K, et al. NfL (Neurofilament light chain) levels as a predictive marker for long-term outcome after ischemic Stroke. Stroke. (2019) 50:3077–84. doi: 10.1161/STROKEAHA.119.026410
112. Ebenau, JL, Timmers, T, Wesselman, LMP, Verberk, IMW, Verfaillie, SCJ, Slot, RER, et al. ATN classification and clinical progression in subjective cognitive decline: the science project. Neurology. (2020) 95:e46–58. doi: 10.1212/WNL.0000000000009724
113. Jack, CR, Bennett, DA, Blennow, K, Carrillo, MC, Feldman, HH, Frisoni, GB, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. (2016) 87:539–47.
114. Hesse, C, Rosengren, L, Andreasen, N, Davidsson, P, Vanderstichele, H, Vanmechelen, E, et al. Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neurosci Lett. (2001) 297:187–90. doi: 10.1016/S0304-3940(00)01697-9
115. Sancesario, GM, and Bernardini, S. How many biomarkers to discriminate neurodegenerative dementia? Crit Rev Clin Lab Sci. (2015) 52:314–26. doi: 10.3109/10408363.2015.1051658
116. Stoeck, K, Sanchez-Juan, P, Gawinecka, J, Green, A, Ladogana, A, Pocchiari, M, et al. Cerebrospinal fluid biomarker supported diagnosis of Creutzfeldt-Jakob disease and rapid dementias: a longitudinal multicentre study over 10 years. Brain. (2012) 135:3051–61. doi: 10.1093/brain/aws238
117. Amadoro, G, Corsetti, V, Sancesario, GM, Lubrano, A, Melchiorri, G, Bernardini, S, et al. Cerebrospinal fluid levels of a 20-22 kDa NH2 fragment of human tau provide a novel neuronal injury biomarker in Alzheimer's disease and other dementias. J Alzheimers Dis. (2014) 42:211–26. doi: 10.3233/JAD-140267
118. Parnetti, L, Lanari, A, Amici, S, Gallai, V, Vanmechelen, E, Hulstaert, F, et al. CSF phosphorylated tau is a possible marker for discriminating Alzheimer's disease from dementia with Lewy bodies. Neurol Sci. (2001) 22:77–8. doi: 10.1007/s100720170055
119. Vanderstichele, H, De Vreese, K, Blennow, K, Andreasen, N, Sindic, C, Ivanoiu, A, et al. Analytical performance and clinical utility of the INNOTEST PHOSPHO-TAU181P assay for discrimination between Alzheimer's disease and dementia with Lewy bodies. Clin Chem Lab Med. (2006) 44:1472–80.
120. Mörtberg, E, Zetterberg, H, Nordmark, J, Blennow, K, Catry, C, Decraemer, H, et al. Plasma tau protein in comatose patients after cardiac arrest treated with therapeutic hypothermia. Acta Anaesthesiol Scand. (2011) 55:1132–8. doi: 10.1111/j.1399-6576.2011.02505.x
121. Schirinzi, T, Grillo, P, Di Lazzaro, G, Zenuni, H, Salimei, C, Dams-O'Connor, K, et al. Effects of head trauma and sport participation in young-onset Parkinson's disease. J Neural Transm. (2021) 128:1185–93. doi: 10.1007/s00702-021-02370-8
122. Honig, LS, Tang, MX, Albert, S, Costa, R, Luchsinger, J, Manly, J, et al. Stroke and the risk of Alzheimer disease. Arch Neurol. (2003) 60:1707–12. doi: 10.1001/archneur.60.12.1707
123. Snowdon, DA, Greiner, LH, Mortimer, JA, Riley, KP, Greiner, PA, and Markesbery, WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study JAMA. (1997) 277:813–7. doi: 10.1001/jama.1997.03540340047031
124. Cervellin, G, Benatti, M, Carbucicchio, A, Mattei, L, Cerasti, D, Aloe, R, et al. Serum levels of protein S100B predict intracranial lesions in mild head injury. Clin Biochem. (2012) 45:408–11. doi: 10.1016/j.clinbiochem.2012.01.006
125. Wunderlich, MT, Wallesch, CW, and Goertler, M. Release of glial fibrillary acidic protein is related to the neurovascular status in acute ischemic stroke. Eur J Neurol. (2006) 13:1118–23. doi: 10.1111/j.1468-1331.2006.01435.x
126. Nooijen, PT, Schoonderwaldt, HC, Wevers, RA, Hommes, OR, and Lamers, KJ. Neuron-specific enolase, S-100 protein, myelin basic protein and lactate in CSF in dementia. Dement Geriatr Cogn Disord. (1997) 8:169–73. doi: 10.1159/000106627
127. Chaves, ML, Camozzato, AL, Ferreira, ED, Piazenski, I, Kochhann, R, Dall'Igna, O, et al. Serum levels of S100B and NSE proteins in Alzheimer's disease patients. J Neuroinflammation. (2010) 7:6. doi: 10.1186/1742-2094-7-6
128. Peskind, ER, Griffin, WS, Akama, KT, Raskind, MA, and Van Eldik, LJ. Cerebrospinal fluid S100B is elevated in the earlier stages of Alzheimer's disease. Neurochem Int. (2001) 39:409–13. doi: 10.1016/s0197-0186(01)00048-1
129. Petzold, A, Jenkins, R, Watt, HC, Green, AJ, Thompson, EJ, Keir, G, et al. Cerebrospinal fluid S100B correlates with brain atrophy in Alzheimer's disease. Neurosci Lett. (2003) 336:167–70. doi: 10.1016/S0304-3940(02)01257-0
130. Katayama, T, Sawada, J, Takahashi, K, Yahara, O, and Hasebe, N. Meta-analysis of cerebrospinal fluid neuron-specific enolase levels in Alzheimer's disease, Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy. Alzheimers Res Ther. (2021) 13:163. doi: 10.1186/s13195-021-00907-3
131. Marchi, N, Cavaglia, M, Fazio, V, Bhudia, S, Hallene, K, and Janigro, D. Peripheral markers of blood-brain barrier damage. Clin Chim Acta. (2004) 342:1–12. doi: 10.1016/j.cccn.2003.12.008
132. Binder, DK, and Scharfman, HE. Brain-derived neurotrophic factor. Growth Factors. (2004) 22:123–31. doi: 10.1080/08977190410001723308
133. Liu, W, Wang, X, O'Connor, M, Wang, G, and Han, F. Brain-derived neurotrophic factor and its potential therapeutic role in Stroke comorbidities. Neural Plast. (2020) 2020:1–13. doi: 10.1155/2020/1969482
Keywords: biomarkers, stroke, secondary neurodegeneration, neurofilaments light, rehabilitation, brain-derived neurotrophic factor (BDNF), neuron specific enolase (NSE), tau
Citation: Brunelli S, Giannella E, Bizzaglia M, De Angelis D and Sancesario GM (2023) Secondary neurodegeneration following Stroke: what can blood biomarkers tell us? Front. Neurol. 14:1198216. doi: 10.3389/fneur.2023.1198216
Edited by:
Daniela Basso, University of Padua, ItalyReviewed by:
Ge Dang, Jinan University, ChinaDaniel J. Beard, The University of Newcastle, Australia
Copyright © 2023 Brunelli, Giannella, Bizzaglia, De Angelis and Sancesario. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giulia Maria Sancesario, Z2l1bGlhLnNhbmNlc2FyaW9AZ21haWwuY29t