
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 03 August 2023
Sec. Neurogenetics
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1187813
This article is part of the Research TopicNeurogenetics – Case Report Collection 2022View all 28 articles
 Lin Chen†‡
Lin Chen†‡ Yin Xu*†‡Ming-juan FangYong-guang ShiJie ZhangLiang-liang Zhang
Yin Xu*†‡Ming-juan FangYong-guang ShiJie ZhangLiang-liang Zhang Yu WangYong-zhu HanJi-yuan HuRen-min Yang
Yu WangYong-zhu HanJi-yuan HuRen-min Yang Xu-en Yu*
Xu-en Yu*Gerstmann-Sträussler-Scheinker syndrome (GSS) is a rare genetic prion disease caused by a mutation in the prion protein (PRNP) gene. It is typically characterized by progressive cerebellar ataxia and slowly progressive dementia. We present a case study of the GSS from China in which a 45-year-old male with a progressive gait and balance disorder developed cerebellar ataxia onset but was misdiagnosed as spinocerebellar ataxia (SCA) for 2 years. The patient's clinical, electrophysiological, and radiological data were retrospectively analyzed. Examination revealed ataxia, dysarthria, muscle weakness, areflexia in lower limbs, including a pyramidal sign, whereas cognitive decline was insignificant. His late mother had a similar unsteady gait. An electroencephalogram (EEG) showed normal findings, and 14-3-3 protein was negative. A brain MRI was performed for global brain atrophy and ventricular enlargement. Positron emission tomography–computed tomography (PET–CT) (18F-fluoro-2-deoxy-d-glucose, FDG) images showed mild to moderate decreased glucose metabolism in the left superior parietal lobe and left middle temporal lobe. According to genetic testing, his younger brother also had the P102L variant in the PRNP gene. This single case adds to the clinical and genetic phenotypes of GSS.
Gerstmann-Sträussler-Scheinker syndrome (GSS) is a rare genetic fatal prion disease with clinical heterogeneity where the prevalence ranges from 1 to 10 per 100 million individuals and is characterized by progressive cerebellar dysfunction and cognitive decline (1). GSS was initially described as a rare familial disease of the central nervous system. In 1995, a proline-to-leucine mutation at codon 102 (P102L) in the PRNP gene was identified in a family (2). Although the P102L mutation has been reported in several Chinese GSS cases, it may not be a common mutation in China (3). GSS syndrome with P102L mutation was first reported in China in 2006, and only 20 cases with P102L-associated GSS have been reported so far (Table 1) (7, 8, 12, 13, 15, 17, 18, 21, 23).
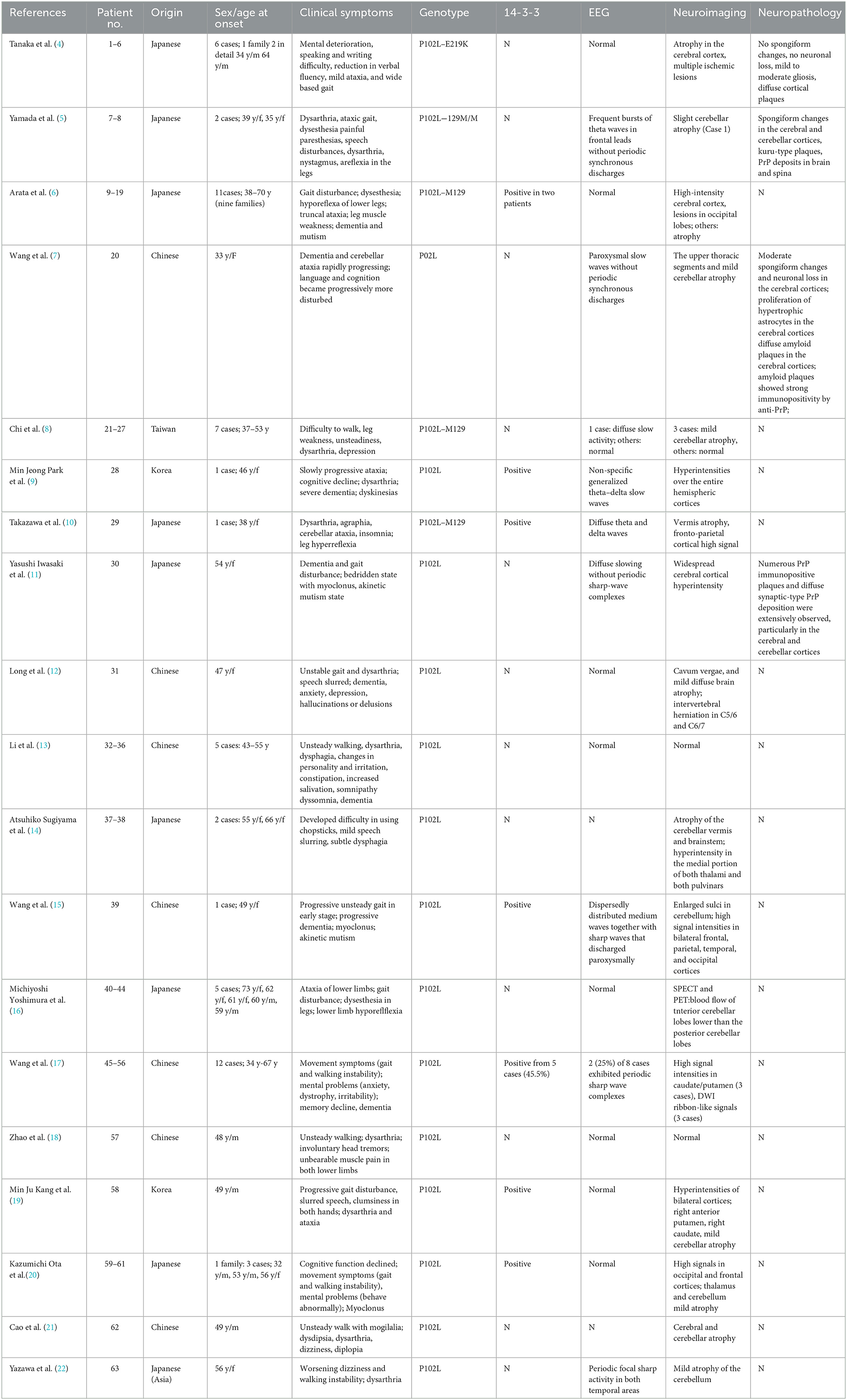
Table 1. Comparison of basic features of GSS cases with P102L mutation previously reported in Asian region.
We described a Chinese patient with GSS and a heterozygous mutation in the PRNP gene with progressive ataxia, pyramidal signs, and areflexia. The patient had a few cognitive declines previously misdiagnosed as spinocerebellar ataxia (SCA). This case report describes an unusual clinical condition with a positive family history confirmed by gene testing. Our patient and his younger brother both had heterozygous mutations in exon 2 of PRNP, located on chromosome 20. A pathogenic mutation causes the P102L mutation at codon 102 in PRNP, the most common variant associated with GSS.
A 41-year-old Chinese man was referred for an abnormal gait suggestive of ataxia. The patient's physical and intellectual level in early life was normal, but his family noticed decreased language fluency at the age of 40 years. One year later, he was 41-years-old, he often fell due to progressive aggravation of walking instability and decreased muscle strength in his lower limbs. He was treated at hospital at the age of 42 years for ataxia, and he was given buspirone. He deteriorated over time, when he was 44-years-old, he could not walk, and began using a wheelchair. There was no further decline in cognitive status over time.
He had a family history of similar symptoms in his mother. She presented to medical attention at the age of 55 years with an unsteady gait. She required a wheelchair by age 58 years, owing to progressive walking instability and decreased muscle strength in her lower limbs. She was subsequently bedbound but did not attend the hospital for a physical examination and finally died at the age of 60 years. During this time, her family did not realize significant cognitive difficulties. The cause of death was unknown, and her family could not provide further details.
Meanwhile, the results of SCA genetic sequencing were found negative. He was referred to our hospital in April 2022. The physical examination revealed mild dysarthria, gait ataxia, bilateral lower extremity weakness, and areflexia but with present Babinski responses bilaterally. The finger-to-nose and rapid alternating movement tests were both abnormal. Orientation, attention, calculation, comprehension, and memory were normal. Laboratory tests and cerebrospinal fluid evaluation were found normal, including the screening for paraneoplastic syndromes-related antibodies and evaluation of 14-3-3 protein levels. Blood and cerebrospinal fluid (CSF) tests were negative for neuromyelitis optica (NMO)-IgG, aquaporin 4 antibodies (AQP4-Ab), and paraneoplastic antibodies. His cognitive function was slightly impaired, and a Mini-Mental State Examination (MMSE) score of 27/30 was obtained during a neuropsychological examination. The interictal electroencephalogram (EEG) showed normal findings (Figure 1). Evoked potential: increase in the binaural threshold. The lower extremity deep sensory path revealed prolonged bilateral P40 latency with amplitude decrease. Brain MRI exhibited T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences, as well as global brain atrophy, ventricular enlargement and cerebellar atrophy. Diffusion-weighted imaging (DWI) revealed no other abnormalities (Figure 2). PET-CT (18F-fluoro-2-deoxy-d-glucose, FDG) images showed that the left superior parietal lobe and left middle temporal lobe had mild to moderate decreased glucose metabolism, with reductions of 10 and 19%, respectively (Figure 3). We questioned the possible diagnosis of autosomal-recessive cerebellar ataxia (ARCA) before hospitalization, but not exclude a dominant ataxia. Our case was initially diagnosed with SCA. However, the genes responsible for common subtypes of SCA (including SCA1/2/3/6/7/8/12/17, FRDA, and DRPLA) were sequenced for this proband, revealing no pathogenic mutations. The patient was then suspected of having spastic paraplegia; however, areflexia was inexplicable, although later autosomal dominant spastic paraplegia type 4 had a suspected pathogenic site on chromosome 17 (c.1786G>A). The whole-exome sequencing (WES) analysis identified pathogenic heterozygous missense mutations of the PRNP gene, c.305C>T (p.Pro102Leu). The Sanger sequencing confirmed that his younger brother inherited the same mutations from his parents (Figure 4). The codon 129 genotype of the patient and his young brother were both P102L-129M/M. His younger brother inherited the same mutations from his parents at the age of 39 years. Up to now, his younger brother still has no symptoms. Then, we diagnosed a case of P102L-associated GSS. We suggested a brain biopsy before making a final diagnosis, but the patient refused. There are currently no approved treatments for GSS. He was treated with buspirone (30 mg/day). The patient's limb weakness worsened rapidly. One year after onset, he often fell due to progressive aggravation of walking instability and decreased muscle strength in his lower limbs. Then, 2 years after onset, he began using a wheelchair and was completely paralyzed in bed most of the time.
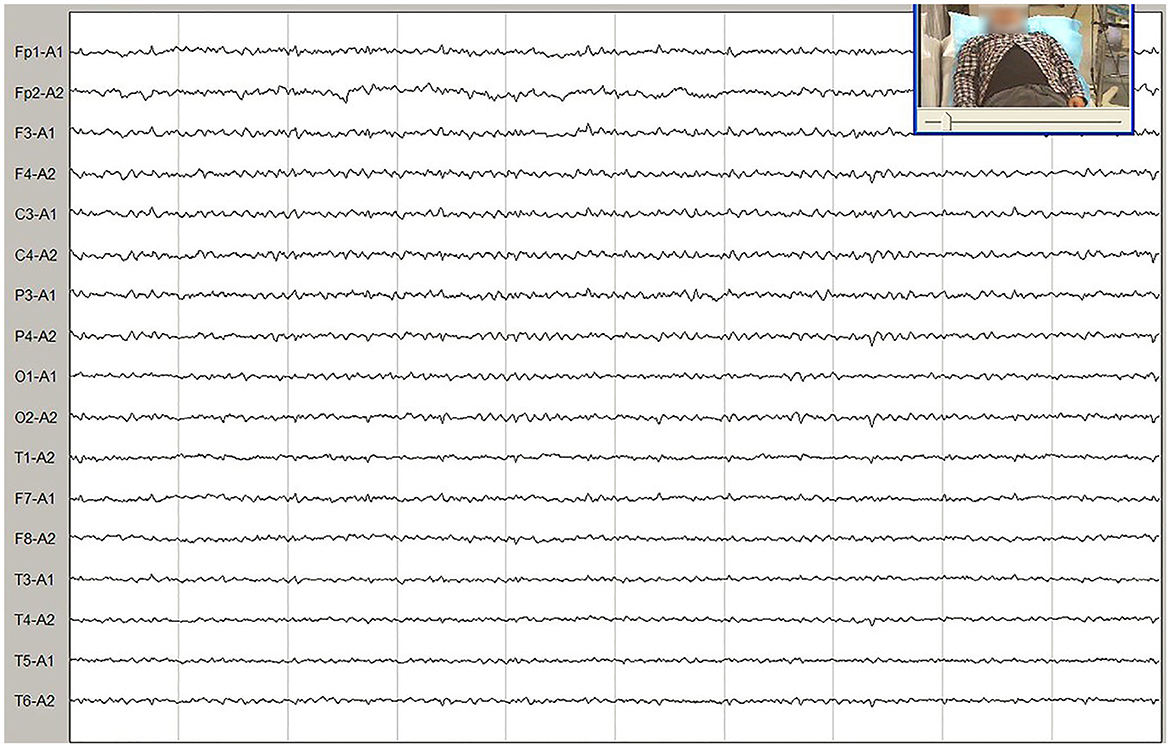
Figure 1. Video electroencephalogram (EEG) showed normal findings.

Figure 2. Magnetic resonance imaging (MRI) of the brain. Axial T2-weighted (A, B) and sagittal T2-weighted scan (C) revealed enlarged sulci in the cerebrum. Fluid-attenuated inversion recovery (FLAIR) sequences (D–F) revealed global brain atrophy, ventricular enlargement.
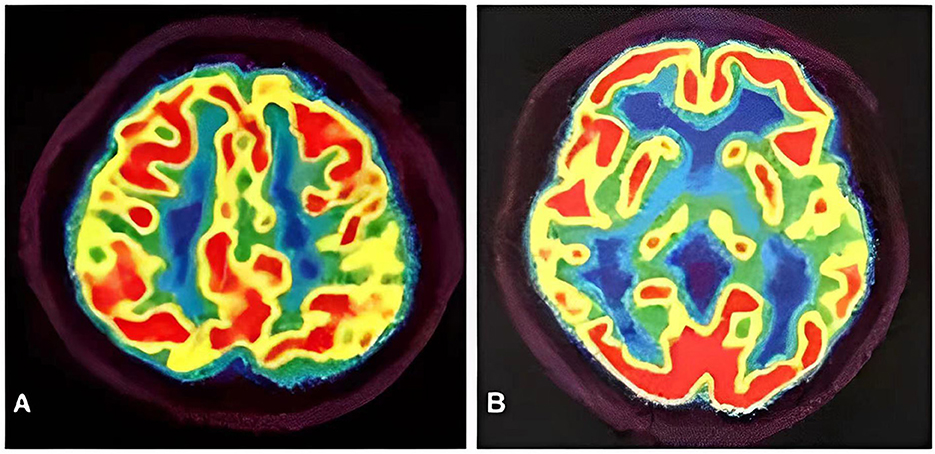
Figure 3. PET-CT images showed the left superior parietal lobe (A) and left middle temporal lobe (B) had mild to moderate decreased glucose metabolism, with reductions of 10 and 19%, respectively.
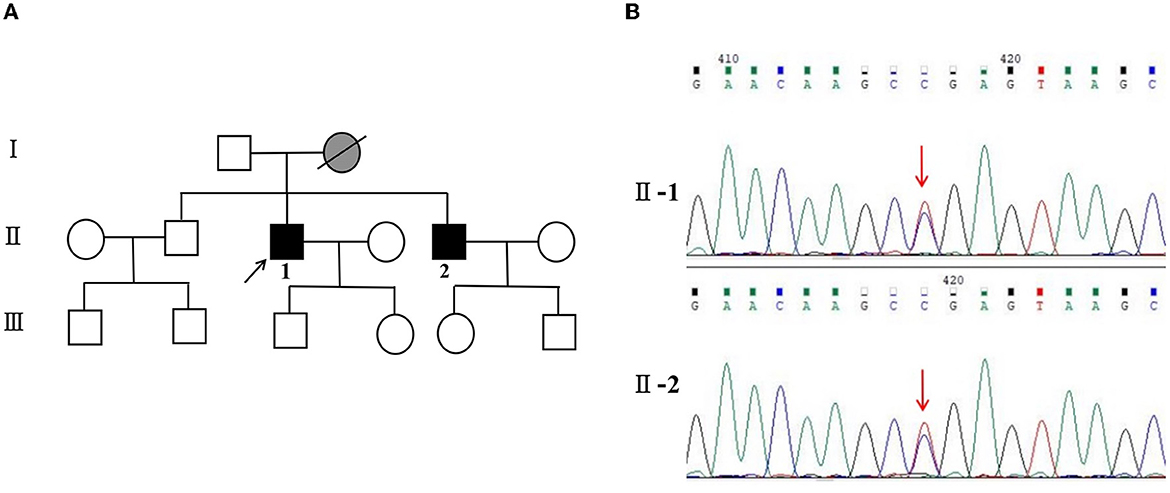
Figure 4. (A) Pedigree and PRNP sequences of the proband and his brother. Squares indicate men, circles indicate women, black symbols indicate affected individuals, gray indicates symptoms of presumed GSS, diagonal lines across symbols indicate deceased individuals, and the arrow indicates the proband.  for GSS, and
for GSS, and  for symptoms of presumed GSS. (B) II-1: PRNP sequence of the patient reveals a heterozygous substitution from C to T at position 305 of PRNP cDNA, resulting in an amino acid change from proline to leucine at position 102 (P102L mutation). II-2: PRNP sequence of his little brother confirms the P102L mutation. The arrow indicates the mutation.
for symptoms of presumed GSS. (B) II-1: PRNP sequence of the patient reveals a heterozygous substitution from C to T at position 305 of PRNP cDNA, resulting in an amino acid change from proline to leucine at position 102 (P102L mutation). II-2: PRNP sequence of his little brother confirms the P102L mutation. The arrow indicates the mutation.
We described a case of GSS with unusual clinical and genetic features. Since GSS is an autosomal dominant inherited disease, a single allele mutation can increase the risk of developing the disease. The duration of the disease ranges from 1 to 10 years. GSS has a relatively longer survival duration than other prion diseases. GSS with the P102L mutation is a rare genetic prion disease caused by a pathogenic mutation at codon 102 in the prion protein gene, with diverse clinical variability (7). GSS clinical symptoms include cerebellar ataxia and gait disturbance (72%), cognitive decline (80%), extrapyramidal damage (36%), psychiatric symptoms (21%), and myoclonus (15%) (24, 25). A high positivity rate (83.3%) for the family history was found in the present Chinese case of P102L-associated GSS, with slowly progressive cerebellar ataxia in 90% of patients. In contrast, visual disturbances, dystonia, and myoclonus are uncommon in patients with GSS (18). Ufkes et al. have reported a member of the GSS Indiana Kindred with supranuclear palsy, a less common feature in GSS (26). Li et al. reported five patients from China with progressive ataxia with age at onset ranging from 48 to 52 years (49.5 ± 4.51). All these patients were found to have the p.P102L mutation within PRNP (13). Of course, the vast majority of GSS cases are due to a missense mutation in the PRNP gene although there are a few other reports such as OPRI (27). From 1992 to the present, not much has been reported about Chinese cases of P102L-associated GSS (Tables 1, 2).
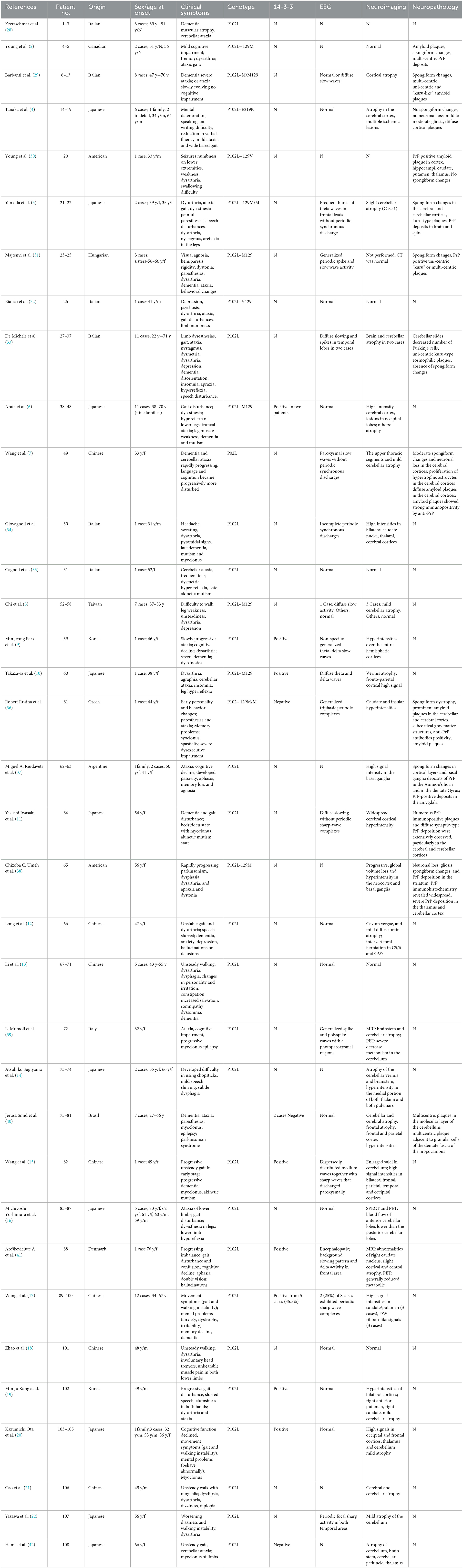
Table 2. Comparison of basic features of GSS cases with P102L mutation previously reported.
Genetic testing should be recommended for patients with rapidly progressing paralysis, including gait and balance disorders. Cluster analysis suggests the existence of four clinical phenotypes: typical GSS, GSS with areflexia and paresthesia, pure dementia GSS, and Creutzfeldt-Jakob disease-like GSS (43). The patient had GSS with areflexia. The symptoms at the early stage of the disease should be distinguished from those of hereditary ataxia and spastic paraplegia. Since the patient only presented with ataxia, muscle weakness, and positive family history, hereditary ataxia, such as spinocerebellar ataxia (SCA), should be distinguished.
Non-specific clinical presentation causes delays in diagnosis. Therefore, rare genetic diseases should be paid more attention especially when common causes have been excluded. The patient had no myoclonus, seizures, psychiatric symptoms, parkinsonism, and dementia. We also focused on EEG and 14-3-3 protein in the CSF because typical triphasic complexes and positivity for 14-3-3 protein in patients were useful in confirming the clinical diagnosis of prion disease. In this context, based on the analysis of 12 Chinese patients with P102L-associated GSS disease, Wang et al. found that only one-quarter and less than half of the Chinese patients had periodic sharp wave complexes (PSWC) in EEG and positivity for 14-3-3 protein in the CSF, respectively (17). Coincidental PSWC in EEG and 14-3-3 positivity in the CSF were observed in 50 and 31% of Caucasian GSS patients, respectively (24). Yazawa et al. reported a woman who developed GSS symptoms and was diagnosed with GSS due to the P102L mutation at the age of 58 years. There were no significant EEG findings during the early stage. Bilateral independent periodic discharges (BIPDs) in both temporal areas appeared at the age of 64 years (22), whereas 14-3-3 protein and EEG reports were normal for our patient, making the diagnosis more difficult.
The neuroimaging examination is an essential component in the differential diagnosis. For our patient, the MRI findings did not provide a clear diagnosis. The main imaging features of GSS are cortical atrophy (55.07%), cerebellar atrophy (42.03%), cortical hyperintensities (32.32%), and basal ganglia hyperintensities (21.54%) (43). However, an investigation based on data from the EuroCJD study found FLAIR or DWI hyperintensities in the basal ganglia in 30% of the P102L-associated GSS cases (24). Our patient revealed cortical atrophy and cerebellar atrophy, despite the absence of FLAIR or DWI hyperintensities consistent with GSS. Yoshimura et al. examined five patients from four Japanese families, and predominant abnormalities were found in the occipital and frontal lobes on SPECT and PET analyses, respectively. In SPECT analysis, the blood flow of the anterior cerebellar lobes was lower than that of the posterior cerebellar lobes (44). Hama et al. reported that a Japanese patient with 18F-2-fluorodeoxy-D-glucose (18F-FDG) PET demonstrated hypometabolism of the cerebral cortex, especially in the frontal lobes and thalamus (42). In contrast, we found reduced presynaptic dopamine transporter uptake in the left superior parietal lobe and left medial temporal lobe on PET-CT images. Thus, the significance of MRI findings in P102L-associated GSS needs further evaluation.
Among Japanese P102L-associated GSS cases, 21% presented with early and prominent dementia (45). Another study found that 40% of cases showed cognitive symptoms at the onset (18). However, unlike his mother, our patient had mild cognitive decline. More research in case studies is required to determine whether Chinese P102-associated GSS patients have a higher or lower proportion of cognitive problems. The presence of multicentric prion protein amyloid plaques in neuropathology remains the key feature of GSS that differentiates it from most other genetic prion diseases. There was no diagnosis for 3 years in the present case. Therefore, we do not have the pathological information of the patient. Nonno et al. demonstrated that GSS is a genuine prion disease characterized by both transmissibility and strain variation, expanding our understanding of the heterogeneous clinic-pathological phenotypes of GSS (46).
Our case highlights the clinical heterogeneity of GSS with the most common p.P102L mutation in the family screening. His younger brother showed no symptoms despite carrying the same P102L mutation in the PRNP gene. His mother walked unsteadily, eventually unable to walk until her death. Therefore, we inferred that his mother suffered from GSS, although the genetic screening was unavailable. His onset began earlier when he and his family refused to do a brain biopsy. His son and daughter were unaffected but did not consent to PRNP gene analysis. Therefore, we do not have full access to the genetic information of the entire family. Penetrance, age of onset, and duration of illness have been systematically characterized across PRNP variants in a global cohort. A genetic counseling session may be triggered by a symptomatic case within the family and may occur either before or after the patient has been tested. Other members of the family, including children need to be able to access clinical services for genetic counseling and testing (47). Several limitations are included in the study. Firstly, we were unable to obtain neuropathological data since the patient did not consent to brain biopsy. Secondly, we have not fully obtained the genetic information of the entire family due to the patient's compliance.
In summary, PRNP sequencing is an indispensable tool for diagnosing GSS due to the complexity of the clinical manifestations of GSS patients. The weakness of the patient's lower limbs developed rapidly, and he arrived at our hospital in a wheelchair. The patient was recently followed up, the strength of his upper limbs was still weak, and he is currently bedridden. However, the patient's younger brother remains asymptomatic.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by Ethics Committee of the Affiliated Hospital of the Institute of Neurology of Anhui University of Chinese Medicine. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
LC and YX wrote the manuscripts with input from all authors. All authors contributed to data acquisition and analysis. All authors contributed to the article and approved the submitted version.
This work was supported by the Key Project of Natural Science Research Project of Universities in Anhui Province (KJ2021A0551) and Research Fund of Anhui University of Chinese Medicine (2020sjzd05).
We thank the patient and her family for placing their trust in us. We also acknowledge TopEdit LLC for linguistic editing and proofreading during the preparation of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1187813/full#supplementary-material
1. Farlow MR, Yee RD, Dlouhy SR, Conneally PM, Azzarelli B, Ghetti B. Gerstmann-Sträussler-Scheinker disease. I. Extending the clinical spectrum. Neurology. (1989) 39:1446–52. doi: 10.1212/WNL.39.11.1446
2. Young K, Jones CK, Piccardo P, Lazzarini A, Golbe LI, Zimmerman TR, et al. Gerstmann-Sträussler-Scheinker disease with mutation at codon 102 and methionine at codon 129 of PRNP in previously unreported patients. Neurology. (1995) 45:1127–34. doi: 10.1212/WNL.45.6.1127
3. Kim DY, Shim KH, Bagyinszky E, An SSA. Prion Mutations in Republic of Republic of Korea, China, and Japan. Int J Mol Sci. (2022) 24:625. doi: 10.3390/ijms24010625
4. Tanaka Y, Minematsu K, Moriyasu H, Yamaguchi T, Yutani C, Kitamoto T, et al. A Japanese family with a variant of Gerstmann-Sträussler-Scheinker disease. J Neurol Neurosurg Psychiatr. (1997) 62:454–7. doi: 10.1136/jnnp.62.5.454
5. Yamada M, Tomimitsu H, Yokota T, Tomi H, Sunohara N, Mukoyama M, et al. Involvement of the spinal posterior horn in Gerstmann-Sträussler-Scheinker disease (PrP P102L). Neurology (1999) 52:260–5. doi: 10.1212/wnl.52.2.260
6. Arata H, Takashima H, Hirano R, Tomimitsu H, Machigashira K, Izumi K, et al. Early clinical signs and imaging findings in Gerstmann-Sträussler-Scheinker syndrome (Pro102Leu). Neurology (2006) 66:1672–8. doi: 10.1212/01.wnl.0000218211.85675.18
7. Wang Y, Qiao X-Y, Zhao C-B, Gao X, Yao Z-W, Qi L, et al. Report on the first Chinese family with Gerstmann-Sträussler-Scheinker disease manifesting the codon 102 mutation in the prion protein gene. Neuropathology. (2006) 26:429–32. doi: 10.1111/j.1440-1789.2006.00704.x
8. Chi N-F, Lee Y-C, Lu Y-C, Wu H-M, Soong B-W. Transmissible spongiform encephalopathies with P102L mutation of PRNP manifesting different phenotypes: clinical, neuroimaging, and electrophysiological studies in Chinese kindred in Taiwan. J Neurol. (2010) 257:191–7. doi: 10.1007/s00415-009-5290-4
9. Park MJ, Jo HY, Cheon S-M, Choi SS, Kim Y-S, Kim JW. A case of gerstmann-sträussler-scheinker disease. J Clin Neurol Seoul Korea. (2010) 6:46–50. doi: 10.3988/jcn.2010.6.1.46
10. Takazawa T, Ikeda K, Ito H, Aoyagi J, Nakamura Y, Miura K, et al. A distinct phenotype of leg hyperreflexia in a Japanese family with Gerstmann-Sträussler-Scheinker syndrome (P102L). Intern Med Tokyo Jpn. (2010) 49:339–42. doi: 10.2169/internalmedicine.49.2864
11. Iwasaki Y, Mori K, Ito M, Nokura K, Tatsumi S, Mimuro M, et al. Gerstmann-Straeussler-Scheinker disease with P102L prion protein gene mutation presenting with rapidly progressive clinical course. Clin Neuropathol. (2014) 33:344–53. doi: 10.5414/NP300733
12. Long L, Cai X, Shu Y, Lu Z. A family with hereditary cerebellar ataxia finally confirmed as Gerstmann-Straussler-Scheinker syndrome with P102L mutation in PRNP gene. Neurosci Riyadh Saudi Arab. (2017) 22:138–42. doi: 10.17712/nsj.2017.2.20160522
13. Li H-F, Liu Z-J, Dong H-L, Xie J-J, Zhao S-Y, Ni W, et al. Clinical features of Chinese patients with Gerstmann-Sträussler-Scheinker identified by targeted next-generation sequencing. Neurobiol Aging. (2017) 49:216.e1–e5. doi: 10.1016/j.neurobiolaging.2016.09.018
14. Sugiyama A, Sato N, Kimura Y, Maekawa T, Wakasugi N, Sone D, et al. Thalamic involvement determined using VSRAD advance on MRI and easy Z-score analysis of 99mTc-ECD-SPECT in Gerstmann- Sträussler-Scheinker syndrome with P102L mutation. J Neurol Sci. (2017) 373:27–30. doi: 10.1016/j.jns.2016.12.021
15. Wang J, Xiao K, Zhou W, Gao C, Chen C, Shi Q, et al. Chinese patient of P102L Gerstmann-Sträussler-Scheinker disease contains three other disease-associated mutations in SYNE1. Prion. (2018) 12:150–5. doi: 10.1080/19336896.2018.1447733
16. Yoshimura M, Yuan J-H, Higashi K, Yoshimura A, Arata H, Okubo R, et al. Correlation between clinical and radiologic features of patients with Gerstmann-Sträussler-Scheinker syndrome (Pro102Leu). J Neurol Sci. (2018) 391:15–21. doi: 10.1016/j.jns.2018.05.012
17. Wang J, Xiao K, Zhou W, Shi Q, Dong XP. Analysis of 12 Chinese patients with proline-to-leucine mutation at codon 102-associated Gerstmann-Sträussler-Scheinker disease. J Clin Neurol Seoul Korea. (2019) 15:184–90. doi: 10.3988/jcn.2019.15.2.184
18. Zhao M-M, Feng L-S, Hou S, Shen P-P, Cui L, Feng J-C. Gerstmann-Sträussler-Scheinker disease: a case report. World J Clin Cases. (2019) 7:389–95. doi: 10.12998/wjcc.v7.i3.389
19. Kang MJ, Suh J, An SS, Kim S, Park YH. Pearls & Oy-sters: Challenging diagnosis of Gerstmann-Sträussler-Scheinker disease: Clinical and imaging findings. Neurology (2019) 92:101–3. doi: 10.1212/WNL.0000000000006730
20. Ota K, Nakazato Y, Yokoyama R, Kawasaki H, Tamura N, Ohtake A, et al. A Japanese family with P102L Gerstmann-Sträussler-Scheinker disease with a variant Creutzfeldt-Jakob disease-like phenotype among the siblings: A case report. eNeurologicalSci. (2021) 25:100380. doi: 10.1016/j.ensci.2021.100380
21. Cao L, Feng H, Huang X, Yi J, Zhou Y. Gerstmann-Sträussler-Scheinker syndrome misdiagnosed as cervical spondylotic myelopathy: a case report with 5-year follow-up. Medicine. (2021) 100:e25687. doi: 10.1097/MD.0000000000025687
22. Yazawa S, Tsuruta K, Sugimoto A, Suzuki Y, Yagi K, Matsuhashi M, et al. Appearance of bitemporal periodic EEG activity in the last stage of Gerstmann-Sträussler-Scheinker syndrome (Pro102Leu): a case report. Clin Neurol Neurosurg. (2021) 204:106602. doi: 10.1016/j.clineuro.2021.106602
23. Shi Q, Chen C, Xiao K, Zhou W, Gao L-P, Chen D-D, et al. Genetic prion disease: insight from the features and experience of China National Surveillance for Creutzfeldt-Jakob Disease. Neurosci Bull. (2021) 37:1570–82. doi: 10.1007/s12264-021-00764-y
24. Webb TEF, Poulter M, Beck J, Uphill J, Adamson G, Campbell T, et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain J Neurol. (2008) 131:2632–46. doi: 10.1093/brain/awn202
25. Kovács GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, et al. Genetic prion disease: the EUROCJD experience. Hum Genet. (2005) 118:166–74. doi: 10.1007/s00439-005-0020-1
26. Ufkes NA, Woodard C, Dale ML, A. case of Gerstmann-Straussler-Scheinker (GSS) disease with supranuclear gaze palsy. J Clin Mov Disord. (2019) 6:7. doi: 10.1186/s40734-019-0082-1
27. Vital A, Laplanche J-L, Bastard J-R, Xiao X, Zou W-Q, Vital C, et al. case of Gerstmann-Sträussler-Scheinker disease with a novel six octapeptide repeat insertion. Neuropathol Appl Neurobiol. (2011) 37:554–9. doi: 10.1111/j.1365-2990.2011.01174.x
28. Kretzschmar HA, Kufer P, Riethmüller G, DeArmond S, Prusiner SB, Schiffer D. Prion protein mutation at codon 102 in an Italian family with Gerstmann-Sträussler-Scheinker syndrome. Neurology (1992) 42:809–10. doi: 10.1212/wnl.42.4.809
29. Barbanti P, Fabbrini G, Salvatore M, Petraroli R, Cardone F, Maras B, et al. Polymorphism at codon 129 or codon 219 of PRNP and clinical heterogeneity in a previously unreported family with Gerstmann-Sträussler-Scheinker disease (PrP-P102L mutation). Neurology (1996) 47:734–41. doi: 10.1212/wnl.47.3.734
30. Young K, Clark HB, Piccardo P, Dlouhy SR, Ghetti B. Gerstmann-Sträussler-Scheinker disease with the PRNP P102L mutation and valine at codon 129. Brain Res Mol Brain Res. (1997) 44:147–50. doi: 10.1016/s0169-328x(96)00251-3
31. Majtényi C, Brown P, Cervenáková L, Goldfarb LG, Tateishi J. A three-sister sibship of Gerstmann-Sträussler-Scheinker disease with a CJD phenotype. Neurology (2000) 54:2133–7. doi: 10.1212/wnl.54.11.2133
32. Bianca M, Bianca S, Vecchio I, Raffaele R, Ingegnosi C, Nicoletti F. Gerstmann-Sträussler-Scheinker disease with P102L-V129 mutation: a case with psychiatric manifestations at onset. Ann Genet. (2003) 46:467–9. doi: 10.1016/s0003-3995(03)00017-0
33. De Michele G, Pocchiari M, Petraroli R, Manfredi M, Caneve G, Coppola G, et al. Variable phenotype in a P102L Gerstmann-Sträussler-Scheinker Italian family. Can J Neurol Sci J Can Sci Neurol. (2003) 30:233–6. doi: 10.1017/s0317167100002651
34. Giovagnoli AR, Di Fede G, Aresi A, Reati F, Rossi G, Tagliavini F. Atypical frontotemporal dementia as a new clinical phenotype of Gerstmann-Straussler-Scheinker disease with the PrP-P102L mutation. Description of a previously unreported Italian family. Neurol Sci Off J Ital Neurol Soc Ital Soc Clin Neurophysiol. (2008) 29:405–10. doi: 10.1007/s10072-008-1025-z
35. Cagnoli C, Brussino A, Sbaiz L, Di Gregorio E, Atzori C, Caroppo P, et al. A previously undiagnosed case of Gerstmann-Sträussler-Scheinker disease revealed by PRNP gene analysis in patients with adult-onset ataxia. Mov Disord Off J Mov Disord Soc. (2008) 23:1468–71. doi: 10.1002/mds.21953
36. Rusina R, Fiala J, Holada K, Matejcková M, Nováková J, Ampapa R, et al. Gerstmann-Sträussler-Scheinker syndrome with the P102L pathogenic mutation presenting as familial Creutzfeldt-Jakob disease: a case report and review of the literature. Neurocase (2013) 19:41–53. doi: 10.1080/13554794.2011.654215
37. Riudavets MA, Sraka MA, Schultz M, Rojas E, Martinetto H, Begué C, et al. Gerstmann-Sträussler-Scheinker syndrome with variable phenotype in a new kindred with PRNP-P102L mutation. Brain Pathol Zurich Switz. (2014) 24:142–7. doi: 10.1111/bpa.12083
38. Umeh CC, Kalakoti P, Greenberg MK, Notari S, Cohen Y, Gambetti P, et al. Clinicopathological Correlates in a PRNP P102L Mutation Carrier with Rapidly Progressing Parkinsonism-dystonia. Mov Disord Clin Pract. (2016) 3:355–358. doi: 10.1002/mdc3.12307
39. Mumoli L, Labate A, Gambardella A. Gerstmann-Straussler-Scheinker disease with PRNP P102L heterozygous mutation presenting as progressive myoclonus epilepsy. Eur J Neurol. (2017) 24:e87–8. doi: 10.1111/ene.13447
40. Smid J, Studart A, Landemberger MC, Machado CF, Nóbrega PR, Canedo NHS, et al. High phenotypic variability in Gerstmann-Sträussler-Scheinker disease. Arq Neuropsiquiatr. (2017) 75:331–8. doi: 10.1590/0004-282X20170049
41. Areškeviciute A, Melchior LC, Broholm H, Krarup L-H, Lindquist SG, Johansen P, et al. Sporadic Creutzfeldt-Jakob disease in a woman married into a Gerstmann-Sträussler-Scheinker Family: An investigation of prions transmission via microchimerism. J Neuropathol Exp Neurol. (2018) 77:673–84. doi: 10.1093/jnen/nly043
42. Hama Y, Saitoh Y, Imabayashi E, Morimoto Y, Tsukamoto T, Sato K, et al. 18F-THK5351 positron emission tomography imaging for Gerstmann-Sträussler-Scheinker disease. J Neurol Sci. (2022) 441:120379. doi: 10.1016/j.jns.2022.120379
43. Krasnianski A, Heinemann U, Ponto C, Kortt J, Kallenberg K, Varges D, et al. Clinical findings and diagnosis in genetic prion diseases in Germany. Eur J Epidemiol. (2016) 31:187–96. doi: 10.1007/s10654-015-0049-y
44. Kepe V, Ghetti B, Farlow MR, Bresjanac M, Miller K, Huang S-C, et al. PET of brain prion protein amyloid in Gerstmann-Sträussler-Scheinker disease. Brain Pathol Zurich Switz. (2010) 20:419–30. doi: 10.1111/j.1750-3639.2009.00306.x
45. Higuma M, Sanjo N, Satoh K, Shiga Y, Sakai K, Nozaki I, et al. Relationships between clinicopathological features and cerebrospinal fluid biomarkers in Japanese patients with genetic prion diseases. PLoS ONE. (2013) 8:e60003. doi: 10.1371/journal.pone.0060003
46. Nonno R, Angelo Di Bari M, Agrimi U, Pirisinu L. Transmissibility of Gerstmann-Sträussler-Scheinker syndrome in rodent models: new insights into the molecular underpinnings of prion infectivity. Prion. (2016) 10:421–33. doi: 10.1080/19336896.2016.1239686
Keywords: Gerstmann-Sträussler-Scheinker syndrome, PRNP gene, P102L, spinocerebellar ataxia (SCA), prion disease
Citation: Chen L, Xu Y, Fang M-j, Shi Y-g, Zhang J, Zhang L-l, Wang Y, Han Y-z, Hu J-y, Yang R-m and Yu X-e (2023) Case report: A Chinese patient with spinocerebellar ataxia finally confirmed as Gerstmann-Sträussler-Scheinker syndrome with P102L mutation. Front. Neurol. 14:1187813. doi: 10.3389/fneur.2023.1187813
Received: 19 April 2023; Accepted: 13 July 2023;
Published: 03 August 2023.
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Ahmet Burak Caglayan, Istanbul Medipol University, TürkiyeCopyright © 2023 Chen, Xu, Fang, Shi, Zhang, Zhang, Wang, Han, Hu, Yang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yin Xu, Zm1yaXh1eUAxMjYuY29t; Xu-en Yu, eXV4dWVuMTc0NkAxNjMuY29t
†These authors share first authorship
‡ORCID: Lin Chen orcid.org/0009-0007-9449-2876
Yin Xu orcid.org/0000-0002-3123-561
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.