 Yilun Tao
Yilun Tao Chen Zhao2†
Chen Zhao2† Lihong Wang
Lihong Wang Xiaoze Li
Xiaoze Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 28 April 2023
Sec. Dementia and Neurodegenerative Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1170557
This article is part of the Research TopicDementia and Neurodegenerative Diseases – Case Report Collection 2022View all 25 articles
Pantothenate kinase-associated neurodegeneration (PKAN) is a rare genetic neurodegenerative disorder with brain iron accumulation characterized as dysarthria, spasticity, cognitive impairment, parkinsonism, and retinopathy. PKAN is caused by biallelic mutations in the mitochondrial pantothenate kinase 2 (PANK2) gene. Herein, we report a 4-year-old patient with PKAN from a Han Chinese family, who presented with developmental regression, progressive inability to walk, and limb tremors. Neuroimaging demonstrated “eye-of-the-tiger” sign. Whole exome sequencing (WES) identified compound heterozygous mutations of c.1213T>G (p.Tyr405Asp) and c.1502T>A (p.Ile501Asn) in PANK2 gene. In addition, a review of all known PANK2 variants observed in reported PKAN patients was conducted, to improve understanding of the genotype-phenotype associations that occur in PKAN patients.
Pantothenate kinase-associated neurodegeneration (PKAN, MIM 234200) is a rare autosomal recessive disorder characterized by progressive iron accumulation in the basal ganglia and other regions of the brain, resulting in extrapyramidal movements such as parkinsonism and dystonia (1). On brain magnetic resonance imaging (MRI), abnormalities are restricted to the globus pallidus and substantia nigra in most cases of PKAN, with almost 100% having an “eye of the tiger” sign (2, 3). PKAN incidence is extremely low, reaching ~1–3/1,00,00,000 globally (4, 5), but the accurate prevalence of PKAN is unclear, particularly in the Chinese population.
The disease was first described in 1922 by two German physicians, Hallervorden and Spatz (6). In 2001, the cause of PNAK was determined to be a homozygous or compound heterozygous mutation in PANK2 gene (MIM 606157), which is located on chromosome 20p13 and encodes a pantothenate kinase (7). The enzyme localizes to the mitochondria and phosphorylates pantothenate to synthesize coenzyme A (CoA) (8). Impaired activity of this pantothenate kinase may lead to increased levels of cysteine and its intermediate products in the basal ganglia. And meanwhile, iron accumulates with an unexplained mechanism. Cysteine can be chelated with iron and rapidly oxidizes itself. The resulting free cysteine disturbs energy metabolism and has a toxic effect on cell membrane synthesis, which can eventually result in central nervous system dysfunction (9, 10).
According to the age at onset, rate of progression, and severity of motor symptoms, PKAN can be classified into two subtypes: typical and atypical (2, 3). In typical PKAN, symptoms present within the first decade of life and usually progress rapidly, with loss of ambulation ~10–15 years later. In the atypical form, patients have an onset in the second decade, with slower progression and variable clinical features. Patients may still ambulate decades after disease onset.
In this study, we described the clinical phenotypes, biochemical features, and genetic findings of a Chinese patient with PKAN who had compound heterozygous mutations c.1213T>G (p.Tyr405Asp) and c.1502T>A (p.Ile501Asn) of PANK2. Furthermore, we review the clinical and genetic features of reported PKAN patients, to help understand the genotype-phenotype relationship of PKAN.
The proband, a 4-year-old female, was the first-born child of healthy and non-consanguineous Chinese parents. No history of genetic diseases exists in her family. The mother had no history of teratogenic pathogens or drug exposure during gestation. The birth weight was 3,770 g (75th percentile), and the birth length was 51 cm (50th percentile) at week 41 of gestation. At 18 months of age, the subject could walk with some aid but exhibited poor balance. At 3 years and 11 months of age, she exhibited developmental regression and could not walk, with progressive tremors in both the upper limbs and choreoathetosis. She had superimposed choreiform movements, mainly involving the distal upper limbs, and displayed involuntary self-injurious behavior (Figure 1A). Meanwhile, she exhibited progressive backward language formation and mental, intellectual disability retardation; she could not speak even simple words such as “baba” and “mama.” She seldom responded to language and was in a nearly semi-vegetative state. No feeding difficulties or dysphagia were found, and ophthalmological examination yielded normal results. Brain MRI at 4 years of age displayed a typical “eye-of-the-tiger” sign (Figure 1B). The child previously underwent rehabilitation therapy for several months; however, the effect was unsatisfactory. The couple underwent clinical genetic counseling and considered a prenatal diagnosis in a future pregnancy.
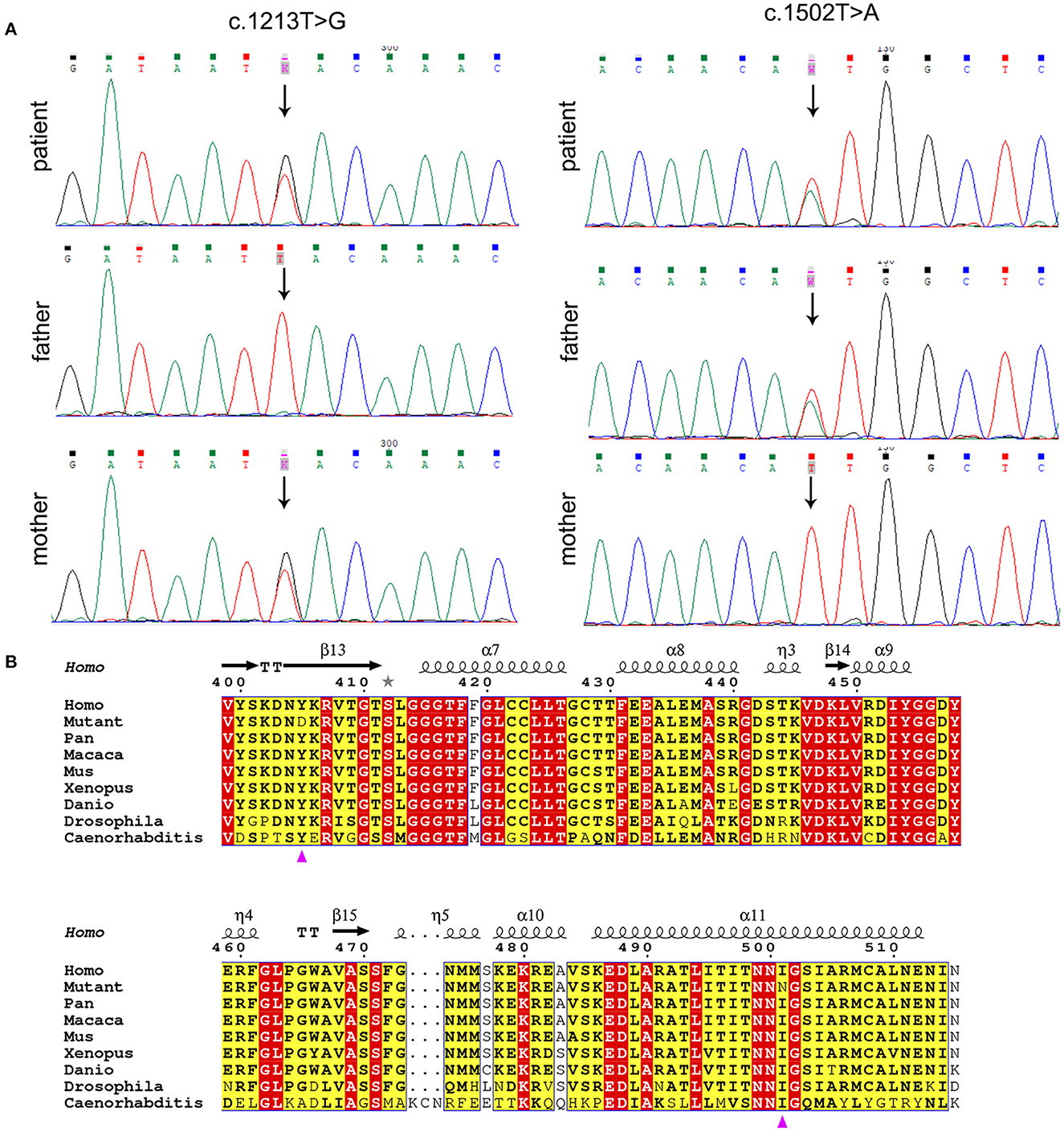
Figure 1. Clinical features and MRI examination of the proband. (A) Body scratches and bruise on the extremities by self-harm. (B) T2-weighted brain magnetic resonance image, showing the “eye-of-the-tiger” sign in the globus pallidus (arrow) at 4 years of age.
A peripheral blood sample of the patient was collected. Genomic DNA was extracted using QIAamp DNA Mini Kit (Qiagen, China). DNA library preparation was performed following Illumina protocols, which included end repair, adapter ligation, and PCR enrichment. The amplified DNA was then captured using a Whole Exome Capture Kit (MyGenostics Inc., Beijing, China). Biotinylated capture probes were designed to tile all exons without repeating regions. The enriched libraries were sequenced for paired-end reads of 150 bp using Illumina HiSeq × Ten platforms.
After sequencing, the clean reads were aligned to the UCSC hg19 human reference genome using the Burrows-Wheeler Alignment tool. Duplicate reads were removed using Picard (http://broadinstitute.github.io/picard). Insertions, deletions, and SNP variants were detected and filtered using the Genome Analysis Toolkit. The identified variants were annotated using ANNOVAR and associated with the following databases: 1,000 Genomes, Exome Aggregation Consortium (ExAC), GnomAD Database, and Human Gene Mutation Database (HGMD). In addition, the identified variants were predicted using Amino acid substitutions were studied in silico to predict the pathogenic effect of the change using VarSome (https://varsome.com/), which utilizes multiple bioinformatic algorithms. Pathogenicity of the mutations was explored following the American College of Medical Genetics and Genomics guidelines (ACMG). Candidate variable sites were confirmed using Sanger sequencing of the patient and his parents. Target sequences were sequenced on an ABI 3730 genetic analyzer (Applied Biosystems, Foster City Carlsbad, CA, USA) and identified using Chromas 2.6.5 (Technelysium Pty Ltd, Australia).
A literature search of PubMed, MEDLINE, and EMBASE databases for articles published on PKAN on August 1, 2022, was conducted. The following keywords were used in the literature search: “pantothenate kinase-associated neurodegeneration” or “PANK2.” Pertinent articles found using the keywords and with definite molecular genetic features of PKAN were screened, and the clinical and genetic findings and prognosis conditions of patients were summarized.
Whole exome sequencing (WES) data of the proband demonstrated two variants in PANK2, c.1213T>G (p.Tyr405Asp) and c.1502T>A (p.Ile501Asn) of transcript NM_153638.3, which were validated by Sanger sequencing (Figure 2A). The father was heterozygous for p.Ile501Asn and the mother was heterozygous for p.Tyr405Asp. Amino acid sequence alignment depicted that p.Tyr405Asp and p.Ile501Asn are highly conserved across different organisms, indicating that any non-synonymous change at positions 405 and 501 of PANK2 can be deleterious (Figure 2B). CADD values through VarSome were 26.7 for both two variants, also indicating the pathogenicity of both variants. According to ACMG classification, the two variants p.Tyr405Asp and p.Ile501Asn were predicted to be “likely pathogenic” (PP3_Strong+PM3+PM2_supporting) and “pathogenic” (PP3_Strong+PM3_strong+PM5+PM2_supporting). PAKN diagnosis in the proband was confirmed based on molecular genetics and available clinical findings.
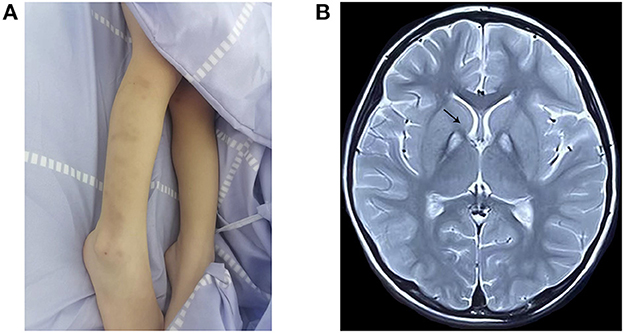
Figure 2. Molecular analysis of PANK2 variants. (A) PANK2 gene sequencing of the patient and his parents. The patient carried compound heterozygous PANK2 gene mutations of c.1213T>G and c.1502T>A; the father carried c.1502T>A, and the mother carried c.1213T>G. Arrow indicates the location of variants. (B) Multiple sequence alignment of PANK2 from different organisms. Residues are colored based on conservation. The white letters in red boxes represent the highest conservation-grade, black letters in yellow boxes represent the second-highest conservation grade, and no color indicates the least conserved grade. The variants p.Tyr405Asp and p.Ile501Asn are indicated by a purple triangle.
In this study, we report a case of typical PKAN in a patient who presented with developmental regression, dystonia, progressive inability to walk, limb tremors, cognitive impairment, and dysarthria. Two compound heterozygous variants in exons 3 (p.Tyr405Asp) and 5 (p.Ile501Asn) were identified. This novel genotype was found for the first time in this study, as far as we know. Although p.Tyr405Asp was first reported in 2005 (3), no more cases have been reported, and this is the first report of this variant in China. Bioinformatic analysis revealed that the two variants were damaging, and the clinical presentation of the patient was compatible with PKAN. These findings indicate that p.Tyr405Asp and p.Ile501Asn variants are associated with PKAN pathogenesis. However, further functional studies are required to validate the pathogenicity of these variants.
The comprehensive clinical features and genetic analysis of 270 reported cases of PKAN (including the present case) are summarized in Supplementary Table 1. The majority of patients were Asian (96, 35.56%) or European (46, 17.04%). Among 266 patients with clear onset age, 167 cases (62.78%) showed the typical form, and 99 (37.21%), the atypical one. The mean age at presentation was 9.87 years (range 0.5–48). There is no sex-related difference in prevalence of PKAN, which were 52.34% (134/256) for males and 47.66% (122/256) for males, as well as in survival of PKAN (p = 0.8355). Most of the patients presented with dystonia (238/251, 94.82%), “eye-of-the-tiger” sign on MRI (246/258, 95.35%), gait disturbance (205/225, 91.11%), and dysarthria (174/198, 87.88%). Other common phenotypes included pyramidal signs (113/166, 68.07%), cognitive impairment (110/177, 62.14%), choreoathetosis (22/41, 53.66%), parkinsonism (68/127, 53.54%), ocular abnormalities (86/174, 49.43%), tremor (45/85, 52.94%), and developmental delay (25/72, 34.72%). In some patients (59/132, 44.70%), behavioral abnormalities developed, including psychosis, depression, hyperactivity, or obsessive-compulsive disorder. Only a few cases (n = 8) presented with seizures (11–16).
A total of 163 distinct PANK2 variants were identified in these 270 patients (Supplementary Table 2), which were annotated based on transcript NM_153638.3. These 163 PANK2 variants corresponded to 104 missenses, 31 frameshifts, 12 splice-sites, 11 non-senses and 5 large deletions. Variants were unevenly distributed throughout PANK2 gene but were mainly concentrated in the exons (147/163, 90.18%), but particularly concentrated in exon 2 (27.61%), 3 (15.34%), and 4 (13.50%). One hundred and nine variants (66.87%) were observed only once or twice, indicating a high genetic heterogeneity among PKAN patients. The three most prevalent variants were p.Gly521Arg (5.91%), p.Asn404Ile (4.07%), and p.Thr528Met (3.88%), but have never been reported in the Chinese population. In addition, 114 out of 163 variants were absent in the Chinese population, and 40 variants were unique to the Chinese population. The six most prevalent variants in China, including p.Glu149Ter, p.Asp324Tyr, p.Asp378Gly, p.Asp452Gly, p.Ile501Thr and p.Phe519Leu, were only reported in Asia, including China, Korea, and Taiwan (4, 17–24). This finding indicates that the variants in PANK2 are population-specific.
The genotypes of the 270 PKAN patients were found to be highly heterogeneous, with more than 45% of the patients presenting with a unique genotype, and 56.67% (153/270) with homozygous genotypes. A high amount of consanguineous marriages (45/115, 39.13%) may have contributed to the homogeneity. However, in China, only 29.41% (15/51) of the patients carried homozygous mutations, with a lower rate of consanguineous marriage (1/21, 4.76%).
In the current study, null PANK2 alleles were defined as those containing non-sense, frameshift, and/or canonical splice site variants. Patients were divided into two groups with different genotypes: (i) M/M (missense/missense) (n = 161) and (ii) M/N (missense/null) or N/N (null/null) (n = 109). The age at onset was significantly earlier in “N/N” or “M/N” group than “M/M” group (median = 7.18 years vs. 11.24 years, p = 0.0003), and the ratio of typical early-onset patients in “N/N” or “M/N” group was larger than that in “M/M” group (80/107 vs. 87/159, 0.0009). Although there was no difference in survival of PKAN between patients with N/N or M/N and M/M genotypes (p = 0.4122), the death age was slight younger in those with null variants (median = 12.64 years vs. 14.80 years). Moreover, incidence of most phenotypes of the individuals with genotype “N/N” or “M/N” was larger compared with that of “M/M” group (Table 1). Taken together, these results indicate that patients with null variants might have a relatively poor survival. In this study, the proband carried compound heterozygous missense variants and was grouped in “M/M” genotype class. She appeared normal at birth, and there were no concerns about her development until over 3 years of age. The patient presented with the typical clinical manifestations of PKAN and a relatively poor prognosis. The patient also showed involuntary self-injurious behavior, which was not observed in any other PKAN patients, and poses challenges to the treatment and care of these patients.

Table 1. Clinical features of PKAN patients.
In summary, a novel genotype of PANK2, c.1213T>G and c.1502T>A, was identified in this study. These findings provide further information regarding the genetic variants that cause PKAN. The review section describes all known PANK2 variants, thus providing a basis for exploring genotype-phenotype correlations of PKAN.
The original contributions presented in the study are publicly available. This data can be found here: NCBI, accession number SRR23634966.
The studies involving human participants were reviewed and approved by Clinical Research Ethics Committee of Changzhi Maternal and Child Health Care Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
CZ and DH performed the experiment and patient follow-up. YT conceived and designed the experiment, conducted genetic data acquisition and interpretation, reviewed the published cases, and wrote the manuscript. CZ, LW, and XL performed in patient management. WS provided the clinical treatment guidance. YW analyzed the data and revised the manuscript. All authors have read and approved the final manuscript.
We are grateful to the patient and her families in our research. We express our gratitude to all the pediatricians who helped with this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1170557/full#supplementary-material
1. Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet. (2009) 46:73–80. doi: 10.1136/jmg.2008.061929
2. Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, Ching KHL, et al. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med. (2003) 348:33–40. doi: 10.1056/NEJMoa020817
3. Pellecchia MT, Valente EM, Cif L, Salvi S, Albanese A, Scarano V, et al. The diverse phenotype and genotype of pantothenate kinase-associated neurodegeneration. Neurology. (2005) 64:1810–2. doi: 10.1212/01.WNL.0000161843.52641.EC
4. Mak CM, Sheng B, Lee HH, Lau K, Chan W, Lam C, et al. Young-onset parkinsonism in a Hong Kong Chinese man with adult-onset Hallervorden-Spatz syndrome. Int J Neurosci. (2011) 121:224–7. doi: 10.3109/00207454.2010.542843
5. Brezavar D, Bonnen PE. Incidence of PKAN determined by bioinformatic and population-based analysis of ~140,000 humans. Mol Genet Metab. (2019) 128:463–9. doi: 10.1016/j.ymgme.2019.09.002
6. Hallervorden J, Spatz H. Eigenartige erkrankung im extrapyramidalen system mit besonderer beteiligung des globus pallidus und der substantia nigra. Zeitschrift Gesamte Neurol Psychiatr. (1922) 79:254–302. doi: 10.1007/BF02878455
7. Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. (2001) 28:345–9. doi: 10.1038/ng572
8. Alfonso-Pecchio A, Garcia M, Leonardi R, Jackowski S. Compartmentalization of mammalian pantothenate kinases. PLoS ONE. (2012) 7:e49509. doi: 10.1371/journal.pone.0049509
9. Tonekaboni SH, Mollamohammadi M. Neurodegeneration with brain iron accumulation: an overview. Iran J child Neurol. (2014) 8:1–8. doi: 10.22037/ijcn.v8i4.7574
10. Williams S, Gregory A, Hogarth P, Hayflick SJ, Gillingham MB. Metabolism and energy requirements in pantothenate kinase-associated neurodegeneration. Mol Genet Metab. (2013) 110:336–41. doi: 10.1016/j.ymgme.2013.06.017
11. Angural A, Singh I, Mahajan A, Pandoh P, Dhar MK, Kaul S, et al. A variation in PANK2 gene is causing Pantothenate kinase-associated Neurodegeneration in a family from Jammu and Kashmir - India. Sci Rep. (2017) 7:4834. doi: 10.1038/s41598-017-05388-9
12. Camargos ST, Gurgel-Giannetti J, Lees A, Hardy J, Singleton A, Cardoso F. Low prevalence of PANK2 mutations in Brazilian patients with early onset generalised dystonia and basal ganglia abnormalities on MRI. J Neurol Neurosurg Psychiatry. (2011) 82:1059–60. doi: 10.1136/jnnp.2009.200808
13. Dastsooz H, Nemati H, Fard MAF, Fardaei M, Faghihi MA. Novel mutations in PANK2 and PLA2G6 genes in patients with neurodegenerative disorders: two case reports. BMC Med Genet. (2017) 18:87. doi: 10.1186/s12881-017-0439-y
14. Li A, Paudel R, Johnson R, Courtney R, Lees AJ, Holton JL, et al. Pantothenate kinase-associated neurodegeneration is not a synucleinopathy. Neuropathol Appl Neurobiol. (2013) 39:121–31. doi: 10.1111/j.1365-2990.2012.01269.x
15. Dezfouli MA, Alavi A, Rohani M, Rezvani M, Nekuie T, Klotzle B, et al. PANK2 and C19orf12 mutations are common causes of neurodegeneration with brain iron accumulation. Mov Disord. (2013) 28:228–32. doi: 10.1002/mds.25271
16. Gatto E, Etcheverry JL, Converso DP, Bidinost C, Rosa A. Pantothenate kinase-associated neurodegeneration: novel mutations in the PANK2 gene in an Argentinean young woman. Mov Disord. (2010) 25:2262–64. doi: 10.1002/mds.23063
17. Wu YR, Chen C, Chao CY, Lyu RK, Lee-Chen GJ. Pantothenate kinase-associated neurodegeneration in two Taiwanese siblings: identification of a novel PANK2 gene mutation. Mov Disord. (2009) 24:940–1. doi: 10.1002/mds.22458
18. Lee JH, Park J, Ryu HS, Park H, Kim YE, Hong JY, et al. Clinical heterogeneity of atypical pantothenate kinase-associated neurodegeneration in Koreans. J Mov Disord. (2016) 9:20–7. doi: 10.14802/jmd.15058
19. Seo JH, Song SK, Lee PH. A novel PANK2 mutation in a patient with atypical pantothenate-kinase-associated neurodegeneration presenting with adult-onset parkinsonism. J Clin Neurol. (2009) 5:192–4. doi: 10.3988/jcn.2009.5.4.192
20. Zhang Y, Zhou D, Yang T. Novel PANK2 mutation in a Chinese boy with PANK2-associated neurodegeneration: a case report and review of Chinese cases. Medicine. (2019) 98:e14122. doi: 10.1097/MD.0000000000014122
21. Sakpichaisakul K, Saengow VE, Suwanpratheep P, Rongnoparat K, Panthan B, Trachoo O. Novel PANK2 mutation discovered among South East Asian children living in Thailand affected with pantothenate kinase associated neurodegeneration. J Clin Neurosci. (2019) 66:187–90. doi: 10.1016/j.jocn.2019.04.017
22. Ma LY, Wang L, Yang YM, Lu Y, Cheng FB, Wan XH. Novel gene mutations and clinical features in patients with pantothenate kinase-associated neurodegeneration. Clin Genet. (2015) 87:93–5. doi: 10.1111/cge.12341
23. Chang X, Zhang J, Jiang Y, Yao B, Wang J, Wu Y. Pilot trial on the efficacy and safety of pantethine in children with pantothenate kinase-associated neurodegeneration: a single-arm, open-label study. Orphanet J Rare Dis. (2020) 15:248. doi: 10.1186/s13023-020-01530-5
Keywords: PANK2 gene, pantothenate kinase-associated neurodegeneration, whole exome sequencing, case report, review
Citation: Tao Y, Zhao C, Han D, Wei Y, Wang L, Song W and Li X (2023) Typical pantothenate kinase-associated neurodegeneration caused by compound heterozygous mutations in PANK2 gene in a Chinese patient: a case report and literature review. Front. Neurol. 14:1170557. doi: 10.3389/fneur.2023.1170557
Received: 21 February 2023; Accepted: 04 April 2023;
Published: 28 April 2023.
Edited by:
Bruce Miller, University of California, San Francisco, United StatesReviewed by:
Anastasia Bougea, National and Kapodistrian University of Athens, GreeceCopyright © 2023 Tao, Zhao, Han, Wei, Wang, Song and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yilun Tao, eWx0YW8yMUAxNjMuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.