 Jolien Perneel
Jolien Perneel Masood Manoochehri3
Masood Manoochehri3 Rosa Rademakers
Rosa Rademakers
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 03 April 2023
Sec. Neurogenetics
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1160248
This article is part of the Research Topic Neurogenetics – Case Report Collection 2022 View all 28 articles
Frontotemporal dementia (FTD) is the second-most common young-onset dementia. Variants in the TMEM106B gene have been proposed as modifiers of FTD disease risk, especially in progranulin (GRN) mutation carriers. A patient in their 50s presented to our clinic with behavioral variant FTD (bvFTD). Genetic testing revealed the disease-causing variant c.349 + 1G > C in GRN. Family testing revealed that the mutation was inherited from an asymptomatic parent in their 80s and that the sibling also carries the mutation. Genetic analyses showed that the asymptomatic parent and sibling carry two copies of the protective TMEM106B haplotype (defined as c.554C > G, p.Thr185Ser), whereas the patient is heterozygous. This case report illustrates that combining TMEM106B genotyping with GRN mutation screening may provide more appropriate genetic counseling on disease risk in GRN families. Both the parent and sibling were counseled to have a significantly reduced risk for symptomatic disease. Implementing TMEM106B genotyping may also promote the collection of biosamples for research studies to improve our understanding of the risk-and disease-modifying effect of this important modifier gene.
Frontotemporal dementia (FTD) is the second-most common presenile dementia after Alzheimer’s disease. Up to 40% of affected individuals have a family history of FTD or a related neurodegenerative disease, with 10–30% having an autosomal dominant pattern of inheritance (1). Pathologically, FTD is characterized by the accumulation of Tau, TDP-43 or FUS protein which leads to atrophy of the frontal and temporal regions of the brain referred to as frontotemporal lobar degeneration (FTLD) (2) Mutations in progranulin (GRN) account for 5–20% of familial FTLD-TDP and 1–5% of sporadic cases. The phenotype of symptomatic GRN carriers has considerable intra-and inter-familial variation, including age of onset, duration of disease, and manifested symptoms. Penetrance is age-related with 90% of carriers being affected by age 70 (3, 4).
TMEM106B was first identified as a risk-associated gene for FTD with TDP-43 pathology (FTLD-TDP) and since then has been recognized as an important modifier of disease risk in a variety of neurodegenerative disorders [reviewed in (5, 6)]. Multiple single nucleotide polymorphisms (SNPs) are present at the TMEM106B locus on chromosome 7p21 which are in strong linkage disequilibrium (LD). Consequently, it has been difficult to pinpoint the specific functional variant (s) modulating the disease risk. The genetic status of TMEM106B is therefore usually described as either a risk or protective haplotype with the most significant SNP from the original genome-wide association study, rs1990622, often stated as the sentinel SNP representing the haplotype (Figure 1A). Here, the major T allele is associated with an increased risk and the minor C allele with a reduced risk for developing disease (7). Alternatively, the two haplotypes can be differentiated by the only coding variant rs3173615 (c.554C > G) where the risk haplotype carries a Threonine and the protective haplotype carries a Serine at position 185.
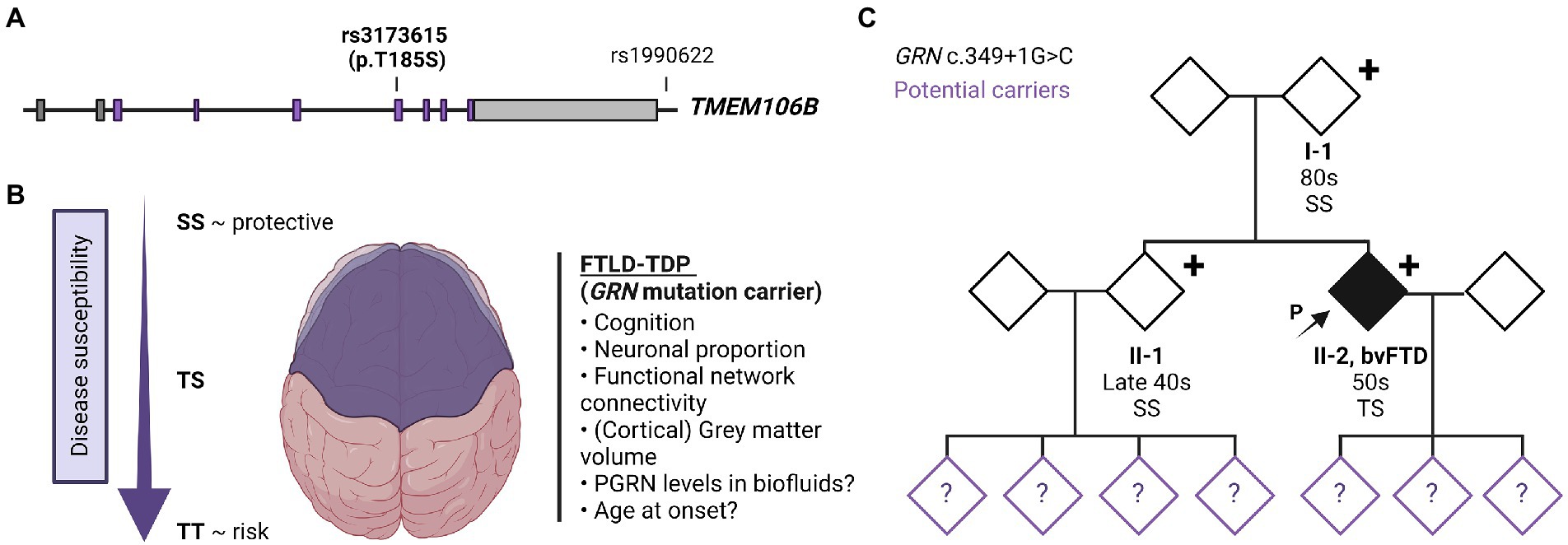
Figure 1. TMEM106B gene structure, TMEM106B haplotype-associated phenotypes and family pedigree. (A) Gene architecture and effect of risk/protective haplotype of TMEM106B. The TMEM106B gene is comprised of nine exons, coding regions are labeled in purple. The major SNPs associated with neurodegenerative diseases are indicated with sentinel SNP (rs1990622), located in the regulatory sequence downstream of TMEM106B, and coding variant rs3173615 (c.554C > G, p.Thr185Ser). (B) Current understanding of the effect of TMEM106B haplotype on brain health and disease susceptibility in FTLD-GRN. (C) Family pedigree. Patient II-2 presented in the clinic in their 50s with symptoms of bvFTD. Genetic testing revealed a disease-causing variant in GRN (c.349 + 1G > C, Splice donor). The patient’s asymptomatic parent (I-1) and younger sibling (II-1) who also carry the mutation were evaluated in their 80s and late 40s, respectively. TMEM106B genotyping showed I-1 and II-1 to have two copies of the protective TMEM106B allele (SS), while the patient is heterozygous (TS). The children of II-1 and II-2, were not genetically tested but are at risk of carrying the mutation, as represented with a question mark within the symbol. Figure was created using BioRender.com.
While the functional variant remains a topic of active discussion, the TMEM106B haplotype is proposed to alter TMEM106B expression, with an increased expression correlating with the risk haplotype. The non-coding variant, rs1990620, is proposed to modulate TMEM106B expression through transcriptional activation due to altered long-range chromatin-looping interactions (8) while the one coding variant (rs3173615, p.T185S), located in TMEM106B’s fourth N-X-T/S glycosylation motif, may affect TMEM106B protein levels by affecting the protein stability and degradation rate due to differences in N-glycosylation (9). As an integral lysosomal transmembrane protein, TMEM106B regulates several aspects of lysosomal functioning, and proper TMEM106B protein levels are crucial for maintaining lysosomal health.
Interestingly, the risk-modifying effect of TMEM106B is most prominent in FTLD-TDP patients harboring disease-associated GRN mutations (4) and the premise that the alteration in TMEM106B levels is the driver of the disease-modulating effect is further substantiated by the observation that TMEM106B mRNA and protein levels were significantly increased in GRN mutation carriers (10, 11). Considering, PGRN is cleaved within the lysosome into functional granulins and also affects several aspects of lysosomal function, it is likely that the exceptionally strong disease-modifying effect in GRN mutation carriers occurs within the endolysosomal system, but the precise mechanism remains unknown.
Functionally, the TMEM106B ‘risk’ haplotype has been associated with lower progranulin levels (12, 13), reduced volume of the superior temporal gyrus (especially in the left hemisphere) (14), decreased functional network connectivity (15), decreased neuronal proportion (16), and a faster cognitive decline (17) (Figure 1B). The association with cognition, neuronal proportion, and general brain health was also replicated in the absence of brain disease (18), suggesting that TMEM106B may modulate the susceptibility of an individual to the pathophysiology of FTLD and related disorders and functions as natural protection against neurodegeneration in general. However, despite the clear functional effect of the TMEM106B haplotype, TMEM106B genotyping is not routinely implemented in diagnostic testing.
A patient in their 50s presented to our clinic with progressive cognitive and behavioral impairment (Patient II-2; Figure 1C). The patient’s partner reported that the proband suffered from cognitive changes starting a year earlier with forgetfulness, difficulty with calculations and computers, impaired judgment while driving, behavioral changes, and lack of initiative in household chores. Concurrent with onset of cognitive symptoms, the patient had prominent dietary changes, including increased appetite and sweet cravings, resulting in a 30 pounds weight gain. Mood swings including episodes of crying were also noted, for which the patient was treated with Zoloft with some benefit. Psychotic symptoms included visual illusions, paranoia, and rare auditory hallucinations. The patient had partial insight into their cognitive and behavioral changes, with relative preservation of emotional range such as maintained interest in their family. The clinical impression was behavioral variant frontotemporal dementia (bvFTD), which the neuropsychological testing and neuroimaging workup supported (Table 1; Figure 2).
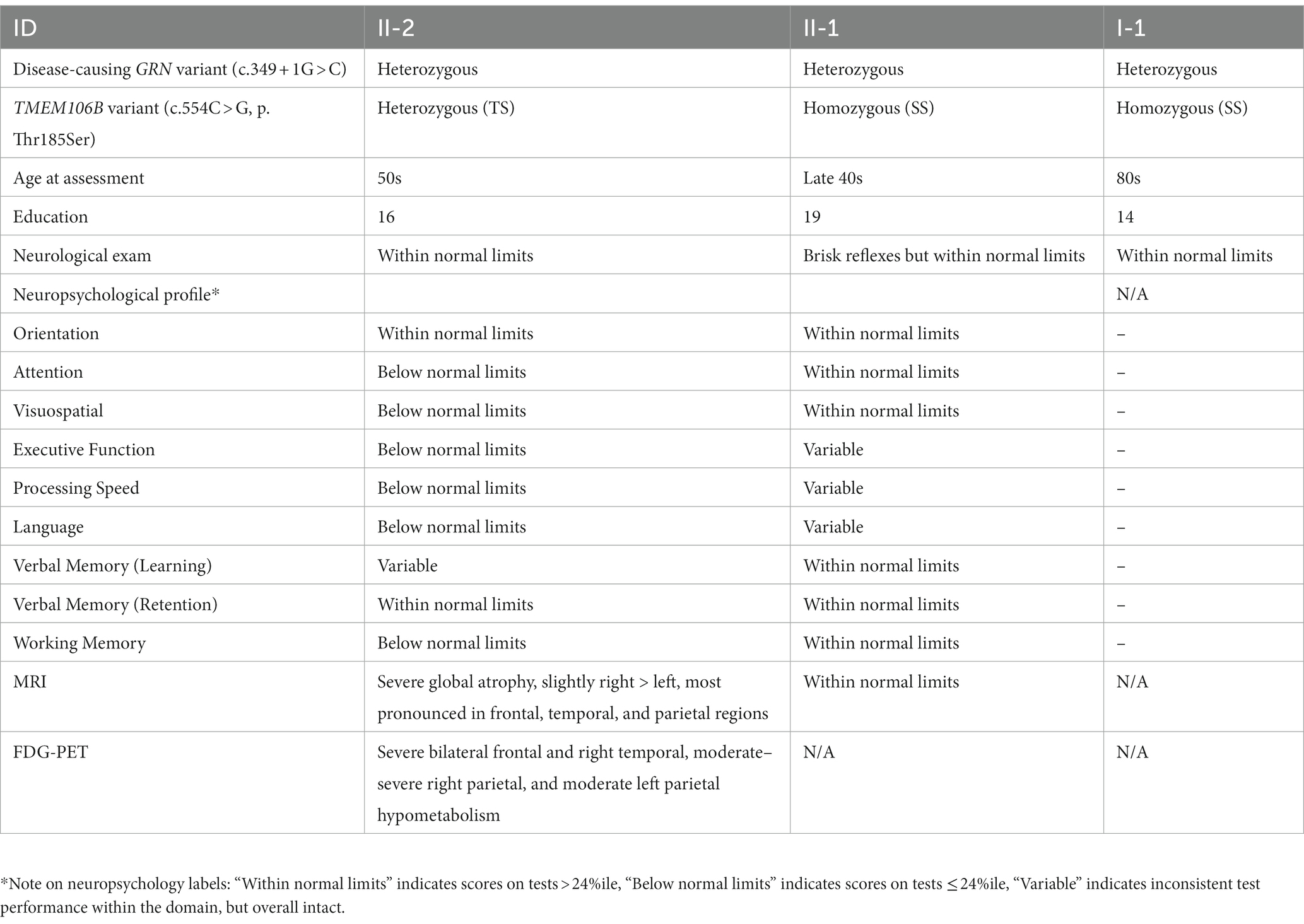
Table 1. Family member information and results of neurological assessment.

Figure 2. MRI images of the proband at evaluation. Portrayed are representative images from a T1-weighted sequence indicating severe global atrophy, slightly right > left, most pronounced in frontal, temporal, and parietal regions, and deemed consistent with behavioral variant frontotemporal dementia.
The patient’s family history was unremarkable (Figure 1C). The parents are still living (in their 80s) without any neurological or psychological symptoms. The patient has a healthy sibling (in their late 40s) and three healthy adult children. Much of the family history was lost due to grandparents and many other family members dying during the World War II era. Despite a lack of family history, we performed a full dementia-ALS genetic testing panel as well as expansion testing for the hexanucleotide repeat in C9orf72 in patient II-2. The only disease-causing variant identified was a previously reported variant, c.349 + 1G > C (Splice donor site), in GRN (19) This variant is predicted to lead to skipping of exon 3, introducing a premature termination codon, nonsense-mediated decay, and loss of progranulin protein.
In order to understand the origin of this mutation, the patient’s parents and sibling were also tested which revealed that both the parent (I-1) and sibling (II-1) also carried the mutation. We subsequently evaluated the patient’s parent (I-1) which revealed no current neuropsychiatric or behavioral symptoms. The psychiatric history was also unremarkable. The parent had no reported difficulties performing ADLs and IADLs and brief cognitive testing on the Mini Mental State Exam revealed intact cognition. On video examination, the parent was socially appropriate and there were no motor abnormalities or Parkinsonism. The patient’s sibling (II-1) was also evaluated, which revealed no significant cognitive changes, socially inappropriate behavior, or other symptoms consistent with bvFTD. The sibling did endorse some longstanding navigational problems as well as more recent subjective memory complaints, low mood, and anxiety. These recent changes were judged to be likely related to their concern about the patient’s condition and the recent familial genetic findings. Neuropsychological testing was largely within normal limits (Table 1). Neurological examination was normal, with the exception of brisk reflexes.
The family wished to understand the reason why the parent was asymptomatic despite carrying the same mutation. Since we previously identified TMEM106B as the only genome-wide significant modifier of disease risk in GRN mutation carriers using a population of symptomatic individuals, TMEM106B genetic testing was performed in all GRN mutation carriers from the family. This analysis revealed that the asymptomatic parent and sibling both carry two copies of the protective TMEM106B haplotype (rs3173615; SS at position 185) while the patient was found to be heterozygous (rs3173615, ST at position 185). Based on our previous study (4) the family was counseled that the parent and sibling have a theoretical 50% reduced risk to develop symptoms as compared to those with no protective haplotypes.
We present a GRN family with a proband presenting with classical young-onset FTD whose parent is an asymptomatic carrier in their 80s, possibly protected from developing disease symptoms because of the modifying effect of TMEM106B. This report is in line with our previously reported TMEM106B genetic study in unrelated GRN patients, in which very few of the symptomatic GRN carriers were homozygous for the TMEM106B protective haplotype (4), and suggests that the presence of at least one TMEM106B risk haplotype is required - as a permissive haplotype - to develop FTLD-GRN. This family report highlights the importance of genotyping FTD patients and relatives for their respective TMEM106B haplotypes, especially in at-risk GRN mutation carriers.
Given the diverse and repeatedly demonstrated modifying effect of TMEM106B on the development and presentation of neurodegenerative brain diseases, it is surprising that TMEM106B genotyping is usually not reported in scientific publications and is also not routinely implemented in diagnostic profiling. Even though the specific functional variant(s) responsible for the disease-modifying effect is not known, candidate functional variants are in strong LD (rs1990622 and rs3173615; r2 = 0.976 in European populations), and genotyping either one of the variants will provide the necessary information. We argue that TMEM106B genetic testing for the ‘risk haplotype’ could be established by sequencing for the rs3173615 variant as it is the only coding variant differentiating the permissive (T) from the protective (S) haplotype and therefore present in exome sequencing data, which is the predominant sequencing method used for genetic testing.
The lack of systematic genotyping of TMEM106B in GRN families has hampered identification of GRN carriers who are also homozygous for the TMEM106B ‘protective’ haplotype (and thus often remain without symptoms) simply because they do not show up in the clinic to participate in research studies. Moreover, the few GRN carriers who were reported to develop symptoms without carrying a TMEM106B risk haplotype may have developed disease due to additional risk factors or advanced aging or have in fact a different disease etiology. Detailed investigation of these individuals will be necessary as it is possible that the molecular mechanism underlying disease in GRN carriers that do not carry the permissive risk haplotype is mechanistically distinct; information that will be vital, especially in light of the development of gene-based therapies for GRN mutation carriers.
In sum, diagnostic testing for the TMEM106B haplotype does not only hold potential to improve genetic counseling in GRN families but will also facilitate further studies by enabling the collection of patient data and material (such as biofluids) crucial for research. A deeper understanding of the risk- and disease-modifying effect of TMEM106B will be essential and could hold the key towards new insights and therapeutic avenues for FTD and related neurodegenerative diseases.
Data from the proband, the parent, and sibling were obtained through research overseen and approved by the Institutional Review Boards at the Johns Hopkins School of Medicine and Columbia University Irving Medical Center; (JHM IRB00227492, CUIMC IRB-AAAS8862, CUIMC IRB-AAAP1303). The patient and family provided approval to share information in this case report and the family pedigree was anonymized. The changes do not affect the current description and conclusion of this report.
Clinical evaluations of the proband, the parent, and sibling were completed at Columbia University Irving Medical Center as part of their involvement in the following NIA-sponsored research studies: ARTFL-LEFFTDS Longitudinal Frontotemporal Lobar Degeneration (ALLFTD) and Neuroanatomical associations with the factor structure underlying neuropsychiatric symptoms in Alzheimer’s disease (NAPS).
Saliva samples from the proband, the parents, and sibling were collected and sent to Invitae Laboratory, San Francisco, California for genetic testing. A 33-gene Hereditary Amyotrophic Lateral Sclerosis, Frontotemporal Dementia, and Alzheimer’s Disease Panel was analyzed by next-generation sequencing followed by analysis of GRN (c.349 + 1G > C) and TMEM106B (c.554C > G).
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Institutional Review Boards at the Johns Hopkins School of Medicine and Columbia University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained for the publication of this case report.
EH and JG neurologically evaluated and counseled the proband and family. MM completed research testing with the proband and family. EH, JG, and MM helped to critically review and revise the manuscript. RR and JP provided insight into the interaction between progranulin and TMEM106B and drafted the manuscript. All authors made substantial contributions to the discussion of the content, reviewed, and edited the article.
The work was supported by the University of Antwerp Research Funds (BOF) and Vlaams Instituut voor Biotechnologie (VIB), as well as the National Institutes of Health (NIH) grants AG062268, UG3NS103870 and the ARTFL LEFFTDS Longitudinal Frontotemporal Lobar Degeneration grant (ALLFTD; U19AG063911). PJ is supported by a fellowship from Research Foundation—Flanders (FWO, application 11M1622N).
RR is a member of the Scientific Advisory Board of Arkuda Therapeutics and receives invention royalties from a patent related to progranulin.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Goldman, JS, and van Deerlin, VM. Alzheimer’s disease and frontotemporal dementia: the current state of genetics and genetic testing since the advent of next-generation sequencing. Mol Diagn Ther. (2018) 22:505–13. doi: 10.1007/s40291-018-0347-7
2. Olney, NT, Spina, S, and Miller, BL. Frontotemporal dementia. Neurol Clin. (2017) 35:339–74. doi: 10.1016/J.NCL.2017.01.008
3. Rademakers, R, Neumann, M, and MacKenzie, IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. (2012) 8:423–34. doi: 10.1038/nrneurol.2012.117
4. Pottier, C, Zhou, X, Perkerson, RB, Baker, M, Jenkins, GD, Serie, DJ, et al. Potential genetic modifiers of disease risk and age at onset in patients with frontotemporal dementia and GRN mutations: a genome-wide association study. Lancet Neurol. (2018) 17:548–58. doi: 10.1016/S1474-4422(18)30126-1
5. Nicholson, AM, and Rademakers, R. What we know about TMEM106B in neurodegeneration. Acta Neuropathol. (2016) 132:639–51. doi: 10.1007/S00401-016-1610-9
6. Feng, T, Lacrampe, A, and Hu, F. Physiological and pathological functions of TMEM106B: a gene associated with brain aging and multiple brain disorders. Acta Neuropathol. (2021) 141:327–39. doi: 10.1007/S00401-020-02246-3
7. van Deerlin, VM, Sleiman, PMA, Martinez-Lage, M, Chen-Plotkin, A, Wang, LS, Graff-Radford, NR, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. (2010) 42:234–9. doi: 10.1038/NG.536
8. Gallagher, MD, Posavi, M, Huang, P, Unger, TL, Berlyand, Y, Gruenewald, AL, et al. A dementia-associated risk variant near TMEM106B alters chromatin architecture and gene expression. Am J Hum Genet. (2017) 101:643–63. doi: 10.1016/j.ajhg.2017.09.004
9. Nicholson, AM, Finch, NA, Wojtas, A, Baker, MC, Perkerson, RB, Castanedes-Casey, M, et al. TMEM106B p.T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem. (2013) 126:781–91. doi: 10.1111/jnc.12329
10. Chen-Plotkin, AS, Unger, TL, Gallagher, MD, Bill, E, Kwong, LK, Volpicelli-Daley, L, et al. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci. (2012) 32:11213–27. doi: 10.1523/JNEUROSCI.0521-12.2012
11. Busch, JI, Martinez-Lage, M, Ashbridge, E, Grossman, M, van Deerlin, VM, Hu, F, et al. Expression of TMEM106B, the frontotemporal lobar degeneration-associated protein, in normal and diseased human brain. Acta Neuropathol Commun. (2014) 1:36. doi: 10.1186/2051-5960-1-36
12. Cruchaga, C, Graff, C, Chiang, HH, Wang, J, Hinrichs, AL, Spiegel, N, et al. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol. (2011) 68:581–6. doi: 10.1001/ARCHNEUROL.2010.350
13. Finch, N, Carrasquillo, MM, Baker, M, Rutherford, NJ, Coppola, G, Dejesus-Hernandez, M, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. (2011) 76:467–74. doi: 10.1212/WNL.0b013e31820a0e3b
14. Adams, HHH, Verhaaren, BFJ, Vrooman, HA, Uitterlinden, AG, Hofman, A, van Duijn, CM, et al. TMEM106B influences volume of left-sided temporal lobe and interhemispheric structures in the general population. Biol Psychiatry. (2014) 76:503–8. doi: 10.1016/j.biopsych.2014.03.006
15. Premi, E, Formenti, A, Gazzina, S, Archetti, S, Gasparotti, R, Padovani, A, et al. Effect of TMEM106B polymorphism on functional network connectivity in asymptomatic GRN mutation carriers. JAMA Neurol. (2014) 71:216–21. doi: 10.1001/jamaneurol.2013.4835
16. Li, Z, Farias, FHG, Dube, U, Del-Aguila, JL, Mihindukulasuriya, KA, Fernandez, MV, et al. The TMEM106B FTLD-protective variant, rs 1990621, is also associated with increased neuronal proportion. Acta Neuropathol. (2020) 139:45–61. doi: 10.1007/s00401-019-02066-0
17. Tropea, TF, Mak, J, Guo, MH, Xie, SX, Suh, E, Rick, J, et al. TMEM106B effect on cognition in Parkinson disease and frontotemporal dementia. Ann Neurol. (2019) 85:801–11. doi: 10.1002/ana.25486
18. Rhinn, H, and Abeliovich, A. Differential aging analysis in human cerebral cortex identifies variants in TMEM106B and GRN that regulate aging phenotypes. Cell Syst. (2017) 4:404–415.e5. doi: 10.1016/j.cels.2017.02.009
Keywords: frontotemporal dementia, progranulin, TMEM106B, disease penetrance, genetic counseling
Citation: Perneel J, Manoochehri M, Huey ED, Rademakers R and Goldman J (2023) Case report: TMEM106B haplotype alters penetrance of GRN mutation in frontotemporal dementia family. Front. Neurol. 14:1160248. doi: 10.3389/fneur.2023.1160248
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Victoria Fernandez, Washington University in St. Louis, United StatesCopyright © 2023 Perneel, Manoochehri, Huey, Rademakers and Goldman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosa Rademakers, cm9zYS5yYWRlbWFrZXJzQHVhbnR3ZXJwZW4udmliLmJl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.