 Ze-hua Lai1,2
Ze-hua Lai1,2 Hai-yan Zhou
Hai-yan Zhou Li-li Zeng
Li-li Zeng- 1Department of Neurology and Institute of Neurology, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Department of Neurology, Yangpu Hospital, Tongji University School of Medicine, Shanghai, China
Hereditary spastic paraplegia (HSP) is a group of neurodegenerative diseases with genetic and clinical heterogeneity characterized by spasticity and weakness of the lower limbs. It includes four genetic inheritance forms: autosomal dominant inheritance (AD), autosomal recessive inheritance (AR), X-linked inheritance, and mitochondrial inheritance. To date, more than 82 gene loci have been found to cause HSP, and SPG15 (ZFYVE26) is one of the most common autosomal recessive hereditary spastic paraplegias (ARHSPs) with a thin corpus callosum (TCC), presents with early cognitive impairment and slowly progressive leg weakness. Here, we reported a homozygous pathogenic variant in ZFYVE26. A 19-year-old Chinese girl was admitted to our hospital presenting with a 2-year progressive bilateral leg spasticity and weakness; early cognitive impairment; corpus callosum dysplasia; chronic neurogenic injury of the medulla oblongata supplied muscles; and bilateral upper and lower limbs on electromyogram (EMG). Based on these clinical and electrophysiological features, HSP was suspected. Exome sequencing of the family was performed by high-throughput sequencing, and an analysis of the patient showed a ZFYVE26 NM_015346: c.7111dupA p.(M2371Nfs*51) homozygous mutation. This case reported a new ZFYVE26 pathogenic variant, which was different from the SPG15 gene mutation reported earlier.
Introduction
Hereditary spastic paraplegia (HSP), also called spastic paraplegia (SPG), is a group of neurodegenerative diseases with genetic and clinical heterogeneity characterized by spasticity and weakness of the lower limbs (1), and the prevalence is ~1.8/100,000 (2). It includes four genetic inheritance forms, namely: autosomal dominant inheritance (AD), autosomal recessive inheritance (AR), X-linked inheritance, and mitochondrial inheritance (2). Until now, over 82 gene loci have been found to cause HSP (3–5). HSP patients may have either pure or complicated HSP, differing based on symptoms. Patients with pure HSP simply develop spasticity and weakness of the lower limbs (6), while patients with complicated HSP are often accompanied by other symptoms, such as early cognitive impairment, ataxia, visual disturbance, macular degeneration, dysarthria, and callosal agenesis (7).
SPG15 (Spastic Paraplegia type of 15, ZFYVE26) is one of the most common ARHSPs with thin corpus callosum (TCC) (8), and it presents with early cognitive impairment and slowly progressive leg weakness (9). ZFYVE26 gene is localized at 14p24.1, and it encodes a zinc finger protein with an FYVE domain called “spastizin” (10). It forms a protein complex with Spatacsin (SPG11) and KIAA0415 (SPG48), and participates in various cellular events such as membrane trafficking and signal transduction (10).
Here, we reported a homozygous mutation in ZFYVE26. A 19-year-old Chinese girl was admitted to our hospital presenting with a 2-year progressive bilateral leg spasticity and weakness, and early cognitive impairment; corpus callosum dysplasia and chronic neurogenic injury of the medulla oblongata supplied muscles; and bilateral upper and lower limbs on electromyogram (EMG). Based on these clinical features and the electrophysiological findings, HSP was suspected. Exome sequencing of the family was performed by high-throughput sequencing, and an analysis of the patient showed a c.7111dupA p.(M2371Nfs*51) (Exon 38) homozygous novel mutation in ZFYVE26 gene. This case reported a new ZFYVE26 pathogenic variant.
Case presentation
A 19-year-old girl was admitted to our hospital presenting with a 2-year progressive bilateral leg spasticity and weakness. Her medical history was not remarkable, and her family history was negative for genetic disease. Vital signs were in the normal ranges: body temperature, 36.7°C; respiratory rate, 22 breaths/min; pulse rate, 72 bpm; and blood pressure, 126/68 mm Hg. Neurological examinations revealed that the lower limbs' muscle strengths were of grades 3–5; hypermyotonia in her lower limbs, hyperreflexia in the knee and ankle reflexes, and bilateral Babinski signs (+), Chaddock signs (+), Gordon signs (+), and Oppenheim signs (+). Cranial nerve, the upper limb, and ataxia examination showed no abnormalities. She also had mild mental deficiency with a MoCA score of 22. Lab examinations, including metabolic, tumor marker, and immunity parameters, were all within normal ranges. Brain and cervical MRI revealed corpus callosum dysplasia and cervical disk herniation in C3/4 and C5/6. EMG reported that F wave latency for the ulnar nerve and the tibial nerve was normal; but EMG of the anterior tibial muscle showed fibrillations and positive sharp waves; and there were widened MUP time limit, increased wave amplitude, and increased polyphase wave during the light contraction in most muscles, and decreased recruitment phase during recontraction. These findings indicated that there was extensive chronic neurogenic electromyographic impairment; and anterior horn involvement was considered first (ball, cervix, and lumbar). The diagnosis of HSP was suspected. Biopsy of the right biceps muscle showed no obvious hyperplasia of connective tissue, no abnormalities in muscle bundle of the small vessel wall, and no inflammatory cells infiltration or abnormal deposits around blood vessels; and there was no muscle fiber atrophy, hypertrophy, necrosis, swirl or spiral change, no nuclei aggregation, and no vacuolar formation in muscle fiber. Therefore, neurogenic skeletal muscle injury was suspected. Second-generation gene sequencing test revealed that the patient carried a ZFYVE26 NM_015346: c.7111dupA p.(M2371Nfs*51) homozygous mutation, which came from her heterozygous parents (Figure 1). Combined with clinical symptoms, a diagnosis of SPG15 was made. The patient was given 5 mg of baclofen twice a day. However, there was no significant improvement in her symptoms on discharge, but her symptoms were alleviated gradually in 3 months of follow-up.
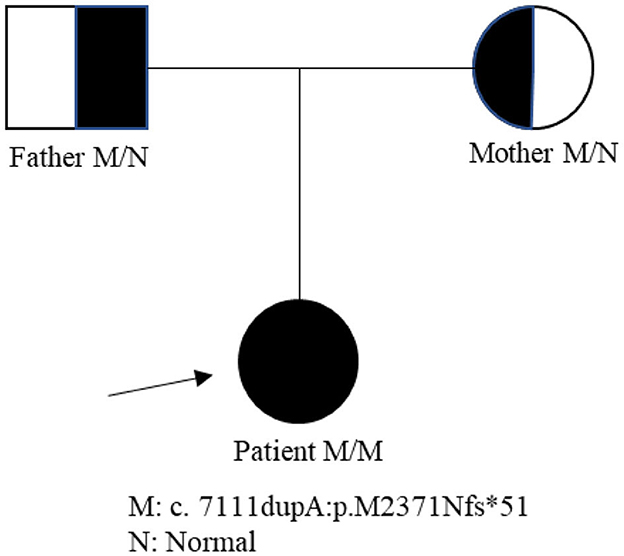
Figure 1. The patient's family tree of the novel mutation ZFYVE26.
Discussion
HSP, a kind of genetic neurodegenerative disease and clinical heterogeneity characterized by spasticity and weakness of the lower limbs, can be classified into two types, namely, the pure type (clinical manifestations include typical muscle spasms, hyperreflexia, clonus, gait disorder, and bladder dysfunction) (6) and the complicated type (besides the abovementioned symptoms, clinical manifestation includes early cognitive impairment, ataxia, visual disturbance, macular degeneration, dysarthria, and callosal agenesis) (11, 12).
The pathogenesis of HSP remains unknown, and the axonal degeneration caused by the various types of HSP has different molecular pathogenesis. There are four modes of mutation, namely, AD, AR, X-linked inheritance, and mitochondrial inheritance (13). To date, over 82 gene loci have been found to cause HSP (3). These genes are involved in many cellular events such as membrane trafficking, signal transduction, the morphology of the endoplasmic reticulum, microtubule dynamics and transport, mitochondrial function, lipid metabolism, and endosome/lysosome functions. The main pathological change of HSP is axonal degeneration, which may also be accompanied by other changes such as demyelination and loss of neurons. Axonal degeneration of the corticospinal tract (most obvious in the thoracic spinal cord) and fasciculus gracilis fibrosis (most obvious in the cervical spinal cord) have also been detected in autopsy.
SPG15 (ZFYVE26) is a kind of early-onset complex ARHSP, which is characterized by typical atrophy of the corpus callosum, and its main clinical symptoms include urinary urgency and incontinence, visual impairment, retinal and macular degeneration, nystagmus, mood fluctuation, mental impairment, ataxia, spastic paraplegia, dysarthria, arcus plantaris, lower limb spasm, callosal agenesis, clonicity, fecal incontinence, bladder sphincter dysfunction, peripheral axon neurodegeneration, distal muscle atrophy, and lower limb muscle weakness (9). Most patients with SPG15 had the first symptoms in their adolescence, some born to consanguineous family even had language delay in infancy and gait disorder at the age of 11 years (5). Our patient presented with classical complicated type of HSP symptoms, including the paralysis of motor neurons in both lower limbs, mild mental impairment, and callosal agenesis. She had progressive bilateral leg spasticity and weakness at the age of 17 years but had no visual or hearing impairment. However, 19 disease-related genes caused HSP manifesting progressive spasticity of the lower limbs and TCC, including SPG1, SPG11, SPG15, SPG21, SPG30, SPG32, SPG35, SPG44, SPG44(65), SPG46, SPG47, SPG48, SPG49, SPG50, SPG52, SPG54, SPG56, SPG63, and SPG71. Furthermore, the onset of these SPGs occurs mainly in children and adolescents and is often accompanied by intellectual disability. While it is hard to distinguish them from clinical symptoms, gene testing is good at detecting specific genetic mutations. Second-generation gene sequencing test proved that this patient had a ZFYVE26 NM_015346: c.7111dupA p.(M2371Nfs*51) homozygous mutation, and both her parents carried a ZFYVE26 NM_015346: c.7111dupA p.(M2371Nfs*51) heterozygous variant. However, this gene loci mutation had not been reported earlier. Therefore, the patient was diagnosed with SPG15, which was composed of AR.
ZFYVE26/Spastizin mutations increase immature autophagosomes and lead to autophagy defects. A complex that is composed of ZFYVE26/Spastizin, SPG11/Spatacsin and AP5 (adaptor-related protein complex 5) is important in autophagic lysosomal reformation (ALR) (10). Although both ZFYVE26 and SPG11 interact with RAB5A and RAB11, the two proteins regulating endosome trafficking and maturation, only ZFYVE26 mutations affect RAB protein interactions and activation and make the fusion between autophagosomes and endosomes defective (10). The ZFYVE26 c.7111dupA p.M2371Nfs*51 mutation identified in our patient is a novel mutation. This frameshift mutation possibly leads to the loss of some amino acid residues in the final protein product and causes loss of function. To date, there have been 62 mutations reported for SPG15 (Table 1) (5, 14–18, 21, 25, 26, 29, 31–38). There are 62.90% (39/62) missense mutations, 29.03% (18/62) deletion mutations, 3.23% (2/62) duplication mutations, and 8.06% (5/62) insertion mutations (Table 1). We searched published literature in PubMed and Web of Science databases and analyzed the clinical features of these 62 kinds of ZFYVE26 mutations in 84 patients (Table 1). Most patients first had walking gait disorder at young age (13.75 ± 7.85 years), and some had language delay in infancy. The main clinical characteristics of the patients with these mutations include low limbs weakness (98.81%), spasticity (96.43%), and amyotrophy (30.95%); cognitive impairment (39.29%), mental abnormality (50%), dysarthria (46.43%), TCC (47.62%), and white matter hyperintensities (WMH) (39.29%) on MRI; axonal peripheral polyneuropathy (35.71%), Babinski sign positive (27.39%), urinary problems (21.43%), and sphincter (5.95%); and ataxia (17.86%), retinal degeneration (19.05%), nystagmus (14.29%), clawfoot (13.10%), strephenopodia (1.19%), sensory abnormalities, action tremor (9), epilepsy (10.71%), hearing impairment (5.95%), upper limbs weakness (7.14%), spasticity (10.71%), and amyotrophy (5.95%).
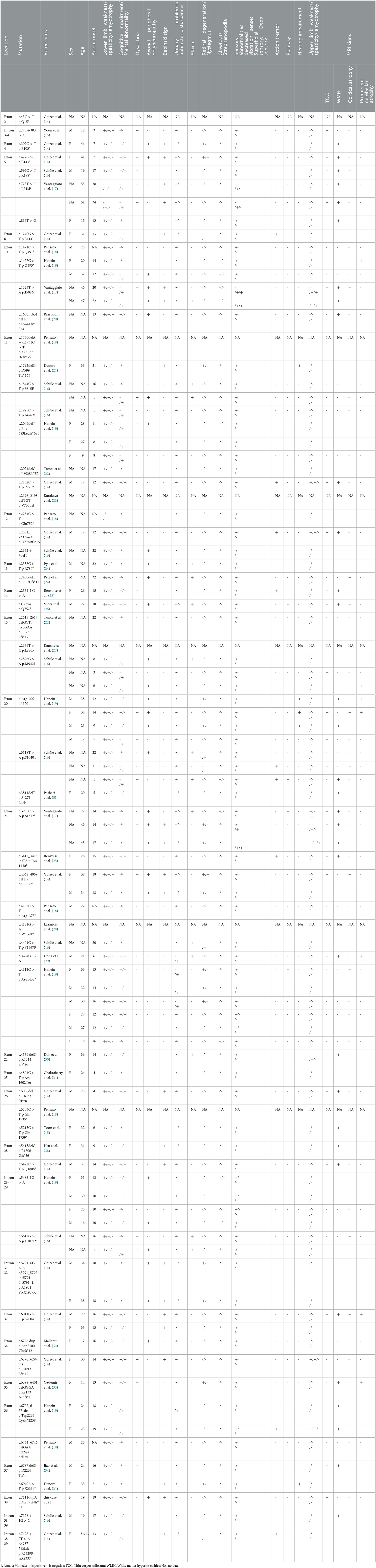
Table 1. SPG15 gene loci mutation and clinical features.
Options available for the treatment of spastic paraplegia are much less than its clinical and genetic types. Rehabilitation therapy and physical therapy are necessary for the maintenance of muscular strength and coordinated movement, and medications such as oral baclofen, intramuscular injections of botulinum toxin, or intrathecal injections of baclofen can relieve spasms. Although HSP has no impact on the lifespan of patients, it can cause serious disability. Genetic diagnosis and symptoms management are important. Early diagnosis and clinical intervention are also helpful to slow disease progression.
Conclusion
Here, we reported a new homozygous mutation of the ZFYVE26 gene, c.7111dupA p.(M2371Nfs*51) (Exon 38). There have been no previous reports on this genetic locus mutation with HSP. Gene testing plays an important role in the diagnosis of HSP, and family genetic lineage reveals the source of the pathogenic gene. With the rapid improvement of gene testing technology, the number of known HSP disease-causing gene is increasing, which brings a challenge to the early diagnosis and clinical evolution.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
Z-hL wrote this manuscript. X-yL and Y-yS collected clinical data and references. H-yZ and L-lZ guided the writing and revision of this manuscript. All authors contributed to the article and approved the submitted version.
Funding
The grant numbers and funding information: NSFC (82271313 to L-lZ), Shanghai Health Commission General Project (202240026 to L-lZ), and Clinical Research Plan of SHDC (SHDC2020CR2027B to L-lZ).
Acknowledgments
We thank Ruijin Hospital for providing a clinical platform, and the clinical work of all involved doctors and nurses. We also thank our patient and her family for their permission and support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1160110/full#supplementary-material
Abbreviations
HSP, Hereditary spastic paraplegia; AD, autosomal dominant inheritance; AR, autosomal recessive inheritance; TCC, thin corpus callosum; WMH, white matter hyperintensities; EMG, electromyogram; AP5, adaptor-related protein complex 5; ALR, autophagic lysosomal reformation.
References
1. Berciano J, Gazulla J, Infante J. History of ataxias and paraplegias with an annotation on the first description of striatonigral degeneration. Cerebellum. (2021) 21:531–44. doi: 10.1007/s12311-021-01328-6
2. Murala S, Nagarajan E, Bollu PC. Hereditary spastic paraplegia. Neurol Sci. (2021) 42:883–94. doi: 10.1007/s10072-020-04981-7
3. Mackay-Sim A. Hereditary spastic paraplegia: from genes, cells and networks to novel pathways for drug discovery. Brain Sci. (2021) 11:403. doi: 10.3390/brainsci11030403
4. Parodi L, Fenu S, Stevanin G, Durr A. Hereditary spastic paraplegia: More than an upper motor neuron disease. Rev Neurol. (2017) 173:352–60. doi: 10.1016/j.neurol.2017.03.034
5. Pashaei M, Davarzani A, Hajati R, Zamani B, Nafissi S, Larti F, et al. Description of clinical features and genetic analysis of one ultra-rare (SPG64) and two common forms (SPG5A and SPG15) of hereditary spastic paraplegia families. J Neurogenet. (2021) 35:84–94. doi: 10.1080/01677063.2021.1895146
6. Harding AE. Hereditary “pure” spastic paraplegia: a clinical and genetic study of 22 families. J Neurol Neurosurg Psychiatry. (1981) 44:871–83. doi: 10.1136/jnnp.44.10.871
7. Nan H, Shiraku H, Mizuno T, Takiyama Y. A p.Arg499His mutation in SPAST is associated with infantile-onset complicated spastic paraplegia: a case report and review of the literature. BMC Neurol. (2021) 21:439. doi: 10.1186/s12883-021-02478-0
8. Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology. (2014) 42:174–83. doi: 10.1159/000358801
9. Ebrahimi-Fakhari D, Alecu JE, Blackstone C. Spastic paraplegia 15. 2021 may 27. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al., editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (1993–2021).
10. Vantaggiato C, Panzeri E, Castelli M, Citterio A, Arnoldi A, Santorelli FM, et al. ZFYVE26/SPASTIZIN and SPG11/SPATACSIN mutations in hereditary spastic paraplegia types AR-SPG15 and AR-SPG11 have different effects on autophagy and endocytosis. Autophagy. (2019) 15:34–57. doi: 10.1080/15548627.2018.1507438
11. Lo Giudice T, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A. Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Expl Neurol. (2014) 261:518–39. doi: 10.1016/j.expneurol.2014.06.011
12. Finsterer J, Löscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci. (2012) 318:1–18. doi: 10.1016/j.jns.2012.03.025
13. Peng F, Sun YM, Quan C, Wang J, Wu JJ. Two novel homozygous mutations of CAPN1 in Chinese patients with hereditary spastic paraplegia and literatures review. Orphanet J Rare Dis. (2019) 14:83. doi: 10.1186/s13023-019-1053-1
14. Goizet C, Boukhris A, Maltete D, Guyant-Maréchal L, Truchetto J, Mundwiller E, et al. SPG15 is the second most common cause of hereditary spastic paraplegia with thin corpus callosum. Neurology. (2009) 73:1111–9. doi: 10.1212/WNL.0b013e3181bacf59
15. Yoon G, Baskin B, Tarnopolsky M, Boycott KM, Geraghty MT, Sell E, et al. Autosomal recessive hereditary spastic paraplegia-clinical and genetic characteristics of a well-defined cohort. Neurogenetics. (2013) 14:181–8. doi: 10.1007/s10048-013-0366-9
16. Schüle R, Schlipf N, Synofzik M, Klebe S, Klimpe S, Hehr U, et al. Frequency and phenotype of SPG11 and SPG15 in complicated hereditary spastic paraplegia. J Neurol Neurosurg Psychiatry. (2009) 80:1402–4. doi: 10.1136/jnnp.2008.167528
17. Vantaggiato C, Crimella C, Airoldi G, Polishchuk R, Bonato S, Brighina E, et al. Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain. (2013) 136:3119–39. doi: 10.1093/brain/awt227
18. Pensato V, Castellotti B, Gellera C, Pareyson D, Ciano C, Nanetti L, et al. Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain. (2014) 137:1907–20. doi: 10.1093/brain/awu121
19. Hanein S, Martin E, Boukhris A, Byrne P, Goizet C, Hamri A, et al. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet. (2008) 82:992–1002. doi: 10.1016/j.ajhg.2008.03.004
20. Riazuddin S, Hussain M, Razzaq A, Iqbal Z, Shahzad M, Polla DL, et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol Psychiatry. (2017) 22:1604–14. doi: 10.1038/mp.2016.109
21. Denora PS, Muglia M, Casali C, Truchetto J, Silvestri G, Messina D, et al. Spastic paraplegia with thinning of the corpus callosum and white matter abnormalities: further mutations and relative frequency in ZFYVE26/SPG15 in the Italian population. J Neurol Sci. (2009) 277:22–5. doi: 10.1016/j.jns.2008.09.039
22. Tunca C, Seker T, Akçimen F, Coskun C, Bayraktar E, Palvadeau R, et al. Revisiting the complex architecture of ALS in Turkey: Expanding genotypes, shared phenotypes, molecular networks, and a public variant database. Hum Mutat. (2020) 41:e7–e45. doi: 10.1002/humu.24055
23. Karakaya M, Storbeck M, Strathmann EA, Vedove AD, Hölker I, Altmueller J, et al. Targeted sequencing with expanded gene profile enables high diagnostic yield in non-5q-spinal muscular atrophies. Hum Mutat. (2018) 39:1284–1298. doi: 10.1002/humu.23560
24. Pyle A, Smertenko T, Bargiela D, Griffin H, Duff J, Appleton M, et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain. (2015) 138(Pt 2):276–83. doi: 10.1093/brain/awu348
25. Renvoisé B, Chang J, Singh R, Yonekawa S, FitzGibbon EJ, Mankodi A, et al. Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann Clin Transl Neurol. (2014) 1:379–89. doi: 10.1002/acn3.64
26. Vinci M, Fchera M, Antonino Musumeci S, Cali F, Aurelio Vitello G. Novel c.C2254T (p.Q752*) mutation in ZFYVE26 (SPG15) gene in a patient with hereditary spastic paraparesis. J Genet. (2018) 97:1469–72. doi: 10.1007/s12041-018-1038-1
27. Kancheva D, Atkinson D, De Rijk P, Zimon M, Chamova T, Mitev V, et al. Novel mutations in genes causing hereditary spastic paraplegia and Charcot-Marie-Tooth neuropathy identified by an optimized protocol for homozygosity mapping based on whole-exome sequencing. Genet Med. (2016) 18:600–7. doi: 10.1038/gim.2015.139
28. Lazaridis KN, Schahl KA, Cousin MA, Babovic-Vuksanovic D, Riegert-Johnson DL, Gavrilova RH, et al. Outcome of whole exome sequencing for diagnostic odyssey cases of an individualized medicine clinic: The mayo clinic experience. Mayo Clin Proc. (2016) 91:297–307. doi: 10.1016/j.mayocp.2015.12.018
29. Dong Y, Li XY, Wang XL, Xu F, Wang ZJ, Song Y, et al. Genetic, clinical and neuroimaging profiles of sporadic and autosomal recessive hereditary spastic paraplegia cases in Chinese. Neurosci Lett. (2021) 761:136108. doi: 10.1016/j.neulet.2021.136108
30. Koh K, Tsuchiya M, Nagasaka T, Shindo K, Takiyama Y. Decreasing 123I-ioflupane SPECT accumulation and 123I-MIBG myocardial scintigraphy uptake in a patient with a novel homozygous mutation in the ZFYVE26 gene. Neurol Sci. (2019) 40:429–41. doi: 10.1007/s10072-018-3603-z
31. Chakrabarty S, Vijayakumar N, Radhakrishnan K, Satyamoorthy K. Spastizin mutation in hereditary spastic paraplegia with thin corpus callosum. J Neurol. (2016) 263:2130–2. doi: 10.1007/s00415-016-8258-1
32. Mallaret M, Lagha-Boukbiza O, Biskup S, Namer IJ, Rudolf G, Anheim M, et al. SPG15: a cause of juvenile atypical levodopa responsive parkinsonism. J Neurol. (2014) 261:435–7. doi: 10.1007/s00415-013-7216-4
33. Özdemir TR, Gençpinar P, Arican P, Öztekin Ö, Dündar NO, Özyilmaz B, et al. A case of spastic paraplegia-15 with a novel pathogenic variant in ZFYVE26 gene. Int J Neurosci. (2019) 129:1198–202. doi: 10.1080/00207454.2019.1653293
34. Jiao B, Zhou Z, Hu Z, Du J, Liao X, Luo Y, et al. Homozygosity mapping and next generation sequencing for the genetic diagnosis of hereditary ataxia and spastic paraplegia in consanguineous families. Parkinsonism Relat Disord. (2020) 80:65–72. doi: 10.1016/j.parkreldis.2020.09.013
35. Pascual B, de Bot ST, Daniels MR, França MC Jr, Toro C, Riverol M, et al. “Ears of the Lynx” MRI Sign Is Associated with SPG11 and SPG15 Hereditary Spastic Paraplegia. AJNR Am J Neuroradiol. (2019) 40:199–203. doi: 10.3174/ajnr.A5935
36. Bibi F, Efthymiou S, Bourinaris T, Tariq A, Zafar F, Rana N, et al. Rare novel CYP2U1 and ZFYVE26 variants identified in two Pakistani families with spastic paraplegia. J Neurol Sci. (2020) 411:116669. doi: 10.1016/j.jns.2020.116669
37. Schicks J, Synofzik M, Pétursson H, Huttenlocher J, Reimold M, Schöls L, et al. Atypical juvenile parkinsonism in a consanguineous SPG15 family. Mov Disord. (2011) 26:564–6. doi: 10.1002/mds.23472
Keywords: hereditary spastic paraplegia, ZFYVE26, case report, novel homozygous mutation, SPG15
Citation: Lai Z-h, Liu X-y, Song Y-y, Zhou H-y and Zeng L-l (2023) Case report: Hereditary spastic paraplegia with a novel homozygous mutation in ZFYVE26. Front. Neurol. 14:1160110. doi: 10.3389/fneur.2023.1160110
Received: 06 February 2023; Accepted: 16 June 2023;
Published: 23 August 2023.
Edited by:
Bruce Miller, University of California, San Francisco, United StatesReviewed by:
Linda Gailite, Riga Stradinš University, LatviaLuke William Bonham, University of California, San Francisco, United States
Copyright © 2023 Lai, Liu, Song, Zhou and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-li Zeng, bGx6ZW5nQDEyNi5jb20=; Hai-yan Zhou, emhhaXlhbi5jb21AMTYzLmNvbQ==