 Xue Zhang1†
Xue Zhang1† Bin Liu
Bin Liu Xueli Fan
Xueli Fan- 1Department of Neurology, Binzhou Medical University Hospital, Binzhou, China
- 2Institute for Metabolic and Neuropsychiatric Disorders, Binzhou Medical University Hospital, Binzhou, China
Multiple sclerosis (MS) is an inflammatory and neurodegenerative disease that commonly results in nontraumatic disability in young adults. The characteristic pathological hallmark of MS is damage to myelin, oligodendrocytes, and axons. Microglia provide continuous surveillance in the CNS microenvironment and initiate defensive mechanisms to protect CNS tissue. Additionally, microglia participate in neurogenesis, synaptic refinement, and myelin pruning through the expression and release of different signaling factors. Continuous activation of microglia has been implicated in neurodegenerative disorders. We first review the lifetime of microglia, including the origin, differentiation, development, and function of microglia. We then discuss microglia participate in the whole processes of remyelination and demyelination, microglial phenotypes in MS, and the NF-κB/PI3K-AKT signaling pathway in microglia. The damage to regulatory signaling pathways may change the homeostasis of microglia, which would accelerate the progression of MS.
1. Microglia
1.1. The origin of microglia
Microglia were first discovered as a separate cell type by Pío del Río-Hortega in 1919. However, the exact origin of microglia remains unclear (1). Recently, an increasing number of studies have shown that microglia originate from yolk-sac progenitors during primitive hematopoiesis before the formation of bone marrow under homeostatic conditions (2) (Figure 1). Indeed, microglia were found in developing mouse embryos on embryonic day (E) 9.5 (3). On E9.5 of mouse embryonic development, immature A1 and A2 cells are localized on the surface of the developing brain. A1 cells began to express CD antigen 45 (CD45). During this period, myeloid markers are usually not expressed. Later, A1 cells further develop into A2 cells (4). Then, A2 cells migrate into the brain and begin expressing myeloid markers. A2 cells travel through the cerebral pial into the brain (5). It is presumed that many factors are involved in the process of microglial migration. No signaling molecule that can completely block microglia from entering the brain has been found (6).
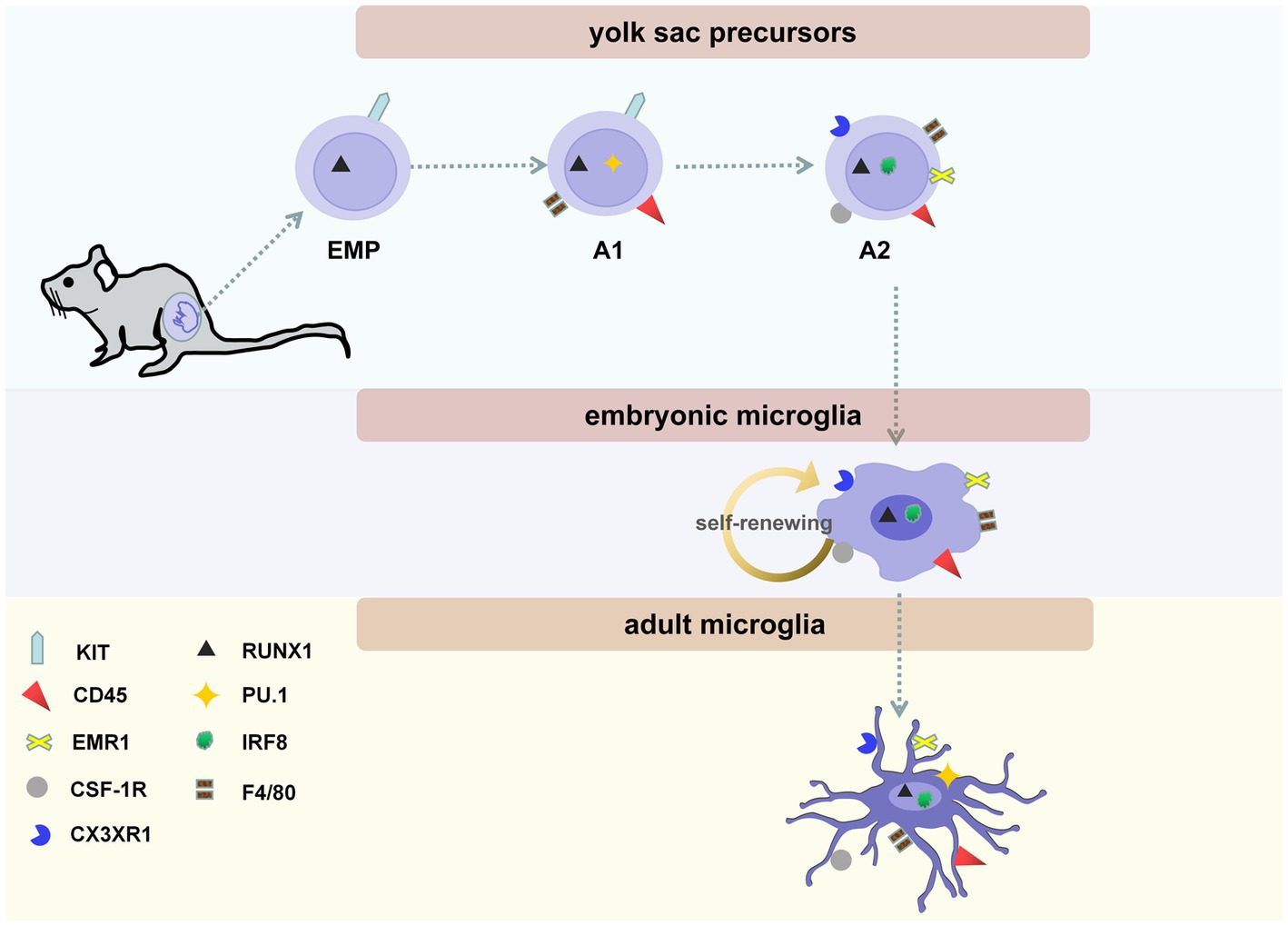
Figure 1. The three stages of microglial maturity. Microglial maturity at three stages: yolk sac precursors (including erythro-myeloid progenitors (EMPs), A1 and A2 cells), embryonic microglia, and adult microglia. On embryonic days (E)7.5–8.0, EMPs, which are KIT-positive, lineage marker-negative, can be found in the blood islands of the yolk sac. On E9.5, immature A1 and A2 cells are present on the surface of the developing brain. A1 cells begin to express CD antigen 45 (CD45); later, these cells develop into A2 cells. A2 cells begin expressing myeloid markers, including CX3C chemokine receptor 1 (CX3CR1, also called fractalkine receptor), colony-stimulating factor 1 receptor (CSF-1R, CD115), and emerin homolog 1 (EMR1, F4/80). In addition, the maturation of microglia is regulated by its endogenous transcription factors, including interferon regulatory factor 8 (IRF8), Runt-related transcription factor 1 (RUNX1), and PU.1. Embryonic microglia exhibit an amoeboid morphology without branching. Microglia are completely ramified on postnatal day (P)28 and maintain their ramified state until they become adult microglia.
1.2. The differentiation and development of microglia
In particular tissues, macrophages differentiate into different subtypes, such as Langerhans cells in the skin, alveolar macrophages, Kupffer cells, and microglia in the brain. Although these macrophages express many shared myeloid- and macrophage-specific markers, each tissue-resident population of macrophages have a distinct gene expression profile (7, 8). Microglia display specific transcriptomes and epigenomes, including transforming growth factor-beta (TGF-β), Spi-1 Proto-Oncogene (PU.1), interferon regulatory factor 8 (IRF8), and transmembrane protein 119 (Tmem119).
TGF-β1 signaling is indispensable for microglial survival. It is believed that the main downstream effectors of TGF-β signaling are small mothers against decapentaplegics (SMADs). In addition to controlling the activation state of microglia, TGF-β1 also maintains mature microglial homeostasis (9). For example, in TGF-β1-deficient mice, adult microglia fail to exhibit typical features, carry out their functions, and cannot survive (10). Other researchers have found that TGF-β1−/− mice not only exhibit neurological defects and marked microglial alterations but also do not show the microglia-enriched gene signature (11, 12).
PU.1 is the most abundantly expressed erythroblast transformation-specific (ETS) transcription factor in microglia in both mice and humans (13). PU.1 expression is correlated with microglial development and function. Stimulus-dependent transcription factors (SDTFs) can induce the activation of enhancer profiles, which mediate the response of microglia to injury (14). According to recent studies, the knockdown of PU.1 makes microglia more susceptible to death (15). In addition, microglial development is impaired in mice lacking PU.1 (16).
IRF8 is a heterodimeric partner of PU.1 that can also regulate the transcriptional programming governing microglial development (3). Studies have proven the existence of a positive feedback loop between PU.1 and IRF8. IRF8 and PU.1 directly target reciprocal gene transcription (17). PU.1 mediates IRF8 expression by acting directly on the IRF8 gene locus. Similarly, IRF8 regulates PU.1 expression through one of the upstream regulatory elements (URE) of the PU.1 locus. During microglial activation, the abovementioned positive feedback loops sustain high expression of PU.1 and IRF8.
Tmem119 is also a specific marker of microglia via analyzing microglial transcriptome datasets in the physiological CNS (18). Unlike other molecules, the function of Tmem119 is still unknown (19). Tmem119 was also found to be expressed in lymph nodes, skeletal muscle, and brown adipose tissue (20, 21). Beyond that, Tmem119 could not label all microglia (20). Microglia could attenuate the expression of Tmem119 under pathological conditions (19). Hence, there are some limitations to using Tmem 119.
1.3. The function of microglia in the healthy brain
Microglia have attracted increasing attention because of their diverse functions. Microglia in the developing CNS and early postnatal CNS, together with microglia in the adult brain, represent various functional entities. Microglia not only guide neurons and axons to form prenatal circuits but also prune synapses and regulate synaptic plasticity. Moreover, microglia can present antigens and participate in the process of myelination and myelin pruning. Hence, microglia play a crucial role in maintaining the homeostasis of the developing brain and adult brain.
1.3.1. Guidance and support
During prenatal development, microglia are located at the crossroads of significant neuronal migratory routes and axonal tract pathways, where they guide neurons and axons in forming prenatal circuits (22). Microglia participate in neurogenesis through the expression and release of signaling factors that impact the development and health of neurons, phagocytosing neural progenitor cells (NPCs) in the brain. Microglia also phagocytose growing axons during early brain development to regulate their growth, which helps shape new brain structures (6).
1.3.2. Synaptic pruning
The classical complement cascade, which involves C3, C1q, and CR3 reside, is known to play a key role in synaptic pruning in the brain. C1q activates the cascade, and then C3 coats the offenders and attracts receptor 3 (CR3; a heterodimer of CD11b) to tag “weaker” synapses. Depletion of any critical players in this cascade (C1q, C3, or CR3) increases the number of synapses (23). Synaptic pruning is also regulated by the CX3CL1-CX3CR1 signaling pathway. In CX3CR1-deficient mice, synaptic pruning is impaired to some extent (24).
1.3.3. Synaptic plasticity
Microglia mediate synaptic refinement in the postnatal brain (25). Microglia-induced synaptic plasticity is dependent on BDNF secretion, which increases the phosphorylation of neuronal tropomyosin kinase receptor type B (TrkB) (26). BDNF-TrkB signaling is involved in long-term potentiation (LTP) induction, which is the principal mechanism of synaptic plasticity (27). Changes in the number of synapses can also cause changes in synaptic plasticity, so synaptic pruning is also crucial for the regulation of synaptic plasticity (28).
1.3.4. Antigen presentation
It has been suggested that microglia are the primary antigen-presenting cells (APCs) in the CNS (29). Major histocompatibility complex (MHC) molecules are expressed at low levels in microglia (30). Under steady state conditions, MHC molecules are responsible for antigen presentation. Adult microglia also express genes associated with surveillance and the immune response, so they can constantly monitor the external environment. After stimulation, microglia can express de novo MHC and are able to develop into APCs (31).
1.3.5. Myelination and myelin pruning
In addition to these important functions, microglia secrete growth factors and support oligodendrocyte progenitor cells (OPC) and oligodendrocytes. Therefore, microglia can promote myelination under steady-state conditions. Under pathological conditions (for example, in MS), microglia also participate in the process of remyelination and demyelination. During the normal aging process, they also phagocytose excess myelin sheaths, which is called myelin pruning (23).
2. Microglia in multiple sclerosis
MS is the most common nontraumatic disabling disease among young adults (32). Additionally, MS is an inflammatory and neurodegenerative disease of the brain and spinal cord, and the characteristic pathological hallmark of MS is the loss of myelin, oligodendrocytes, and axons (33). Relapse remitting MS (RRMS), which accounts for 80–90% of MS cases, begins with episodes of neurological disorders and then partial or complete remission (34). Upon aging, patients with RRMS develop secondary progressive multiple sclerosis (SPMS) with fewer remissions and increasing clinical deterioration (35). Only 15% of MS patients have primary progressive MS (PPMS), which involves a steady and progressive loss of neurological function from the onset of the disease (36, 37). Progressive relapsing MS (PRMS) is the most uncommon form of MS, affecting approximately 5% of MS patients, and is characterized by a steady decline in health with unexpected spikes of deterioration and recovery (38). Although the clinical characteristics of MS are clear, the pathology of MS is a dynamic and continuous process (39).
Microglia provide continuous surveillance of the CNS microenvironment and initiate defense mechanisms to protect CNS tissue. Oligodendrocytes, the myelinating cells of the CNS (40), are often targets of autoimmune pathology during MS progression (41). There is emerging evidence that microglia actively contribute to inflammation that directly and indirectly contributes to neurodegeneration. Microglia are highly dynamic, so they can recognize changes in the cerebral parenchyma with their highly motile processes (2, 42). Microglia often keep a specific ramified morphology in healthy adult brain tissue. As soon as signs of injury, such as inflammation or tissue injury, are detected, microglia rapidly move toward the lesion site and transition into an activated state (43–45). Continued activation of microglia drives neuroinflammation and neurodegeneration (46).
2.1. Demyelination and myelin regeneration in MS: microglia and OPC
Myelin sheaths are generated by oligodendrocytes and wrap around axons (47, 48). As mentioned above, the characteristic pathological hallmark of MS is the loss of myelin, oligodendrocytes, and axons. Remyelination refers to the restoration of the myelin sheath around denuded axons (49) and is activated by the recruitment and differentiation of OPC (50). After migrating to the CNS, OPC adopt a premyelinating phenotype (preoligodendrocyes) and can wrap around axons. However, preoligodendrocytes are unable to form mature myelin. The expression of myelin basic protein (MBP) and myelin oligodendrocyte glycoprotein (MOG) indicates the development of mature oligodendrocytes (51). Mature oligodendrocytes contact and wrap neuronal axons with myelin while being connected to surviving myelin sheaths (52). While a substantial number of OPC are found in and around MS lesions, OPC recruitment and the differentiation of OPC into mature myelinating oligodendrocytes are impaired (53). Remyelination becomes less efficient as patients grow older and lesions become more chronic (50, 54). According to a number of studies, the development of OPC and oligodendrocytes is affected by protein products expressed by microglia (6, 55). For instance, microglia-conditioned medium increases the expression of platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and insulin-like growth factor 1 (IGF-1), which promote the survival and maturation of OPC.
Microglia are involved in both the de and remyelination phases of MS (Figure 2). From the onset of demyelination, microglia provide a favorable environment for myelin regeneration (56), recruiting new OPC, providing trophic support, repairing damaged tissue, and clearing debris (57). In the surrounding MS lesions, microglia express Semaphorin-3F (SEMA3F), which can attract OPC to damaged areas (58). Apart from that, studies have also found that microglia express 6,200 genes, including the upregulated genes Lrp1, Calr, CXCL10, CXCL13, Pdgfa, Pdgfb, Vegfa, Vegfb, TGF-β1, MMP12, and MMP14, in the cuprizone-induced demyelination model. These upregulated genes are involved in phagocytosing apoptotic cells and debris, recruiting OPC, and supporting oligodendrocyte remyelination, differentiation, and tissue remodeling (59). Microglia have been proven to be fundamental for remyelination (60). To evaluate the effect of microglia on myelination, researchers examined the myelination of oligodendrocytes in larvae lacking microglia. IRF8 and CSF-1R are necessary for microglial development (3, 61). To eliminate microglia in the early stages of development, researchers used an antisense morpholino oligonucleotide designed to block the translation of IRF8 messenger RNA and a CSF-1R inhibitor that was used previously in larval zebrafish (62). Under these conditions, oligodendrocytes exhibited more sheaths (23). Myelin debris inhibits OPC differentiation, which markedly affects remyelination (63). In CX3CR1 gene knockout mice, phagocytosis of microglia is dramatically reduced after treatment with cuprizone, resulting in the persistence of myelin debris and inhibition of proper remyelination due to impaired OPC recruitment (60, 64, 65). Phagocytosis of myelin debris by microglia is essential for the initiation of lesion repair (66). Triggering receptor expressed on myeloid cells (TREM2), which is expressed on the cell surface and binds polyanions, thus activating downstream signaling cascades through the adapter DAP12 (also called TYRO protein tyrosine kinase binding protein) (67), has been found to be related to phagocytosis (68). In Trem2−/− mice, clearance of myelin debris and axonal support are impaired, the number of oligodendrocytes is increased, and inflammatory mediators are expressed after long-term cuprizone treatment, resulting in persistent demyelination (69). Moreover, myelin interacts with microglial MER proto-oncogene tyrosine kinase (MERTK). MERTK is indispensable for myelin phagocytosis (65). In vitro studies have shown that microglia stimulated with TGF-β express increased MERTK receptors, which have a good ability to clear myelin debris (70). A series of studies revealed the multiple mechanisms by which microglia exert beneficial effects on OPC survival and maturation (71).
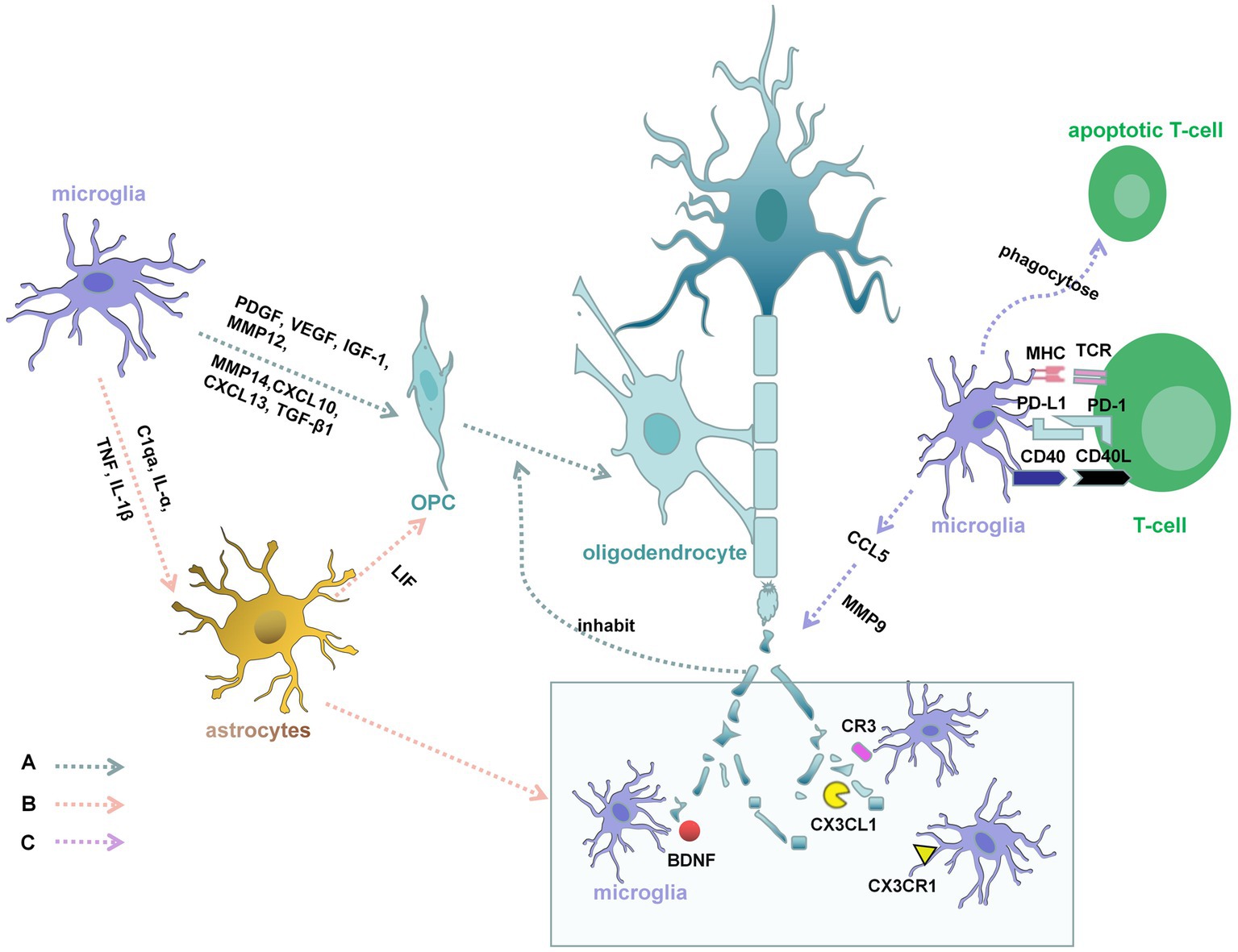
Figure 2. Demyelination and myelin regeneration in MS. A: Microglia can express platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), insulin-like growth factor 1 (IGF-1), CXCL10, CXCL3, TGF-β1, MMP12, and MMP14, which promote the survival and maturation of OPC and supporting oligodendrocyte remyelination. Myelin debris inhibits OPC differentiation, which markedly affects remyelination. From the onset of demyelination, microglia provide a favorable environment for clearing debris. The brain-derived neurotrophic factor (BDNF), CX3CR1, CX3CL1, and C3R are involved in this process. B: C1qa, interleukin-ɑ (IL-ɑ), and Tumor necrosis factor (TNF) secreted by microglia, which could increase the reactivity of astrocytes. Interleukin-1β (IL-1β) secreted by microglia, can activate astrocytes and allow the astrocytes to produce leukemia inhibitory factor (LIF). LIF can promote the differentiation of OPC into mature myelinating oligodendrocytes and relieve demyelination. Microglia are also recruited by astrocytes to the demyelinating lesions, which ensures the smooth process of phagocytosis. C: Encephalitogenic T cells might produce cytokines that directly activate microglia. Activated microglia are the principal source of CC and CXC-chemokines, such as CCL5. CCL5 promotes the activation and secretion of metalloproteinase-9 (MMP-9) in human microglia, which is in connection with the degradation of myelin proteins. Microglia also have a great ability to phagocytose apoptotic encephalitogenic T cells, which can influence the chemoattractive milieu.
2.2. Demyelination and myelin regeneration in MS: microglia and astrocytes
Classically, active MS lesions are highly inflammatory, showing infiltration of many lymphocytes, including CD8+ T cells, CD20+ B cells, macrophages, and CD4+ T cells (33, 72). In addition, resident cells of the CNS, including activated microglia and reactive astrocytes, are present in MS lesions (73). These infiltrating cells produce a large number of cytokines and other inflammatory factors that contribute to myelin destruction and axonal degradation. The degree of myelin regeneration depends on the severity of the disease. Microglia have an effect on the function of astrocytes (Figure 2). Researchers have discovered that microglia are indispensable for the activation of astrocytes by using cuprizone (CPZ)-induced mouse models of demyelination and remyelination (74). In mice that knock out cytokines secreted by microglia, including C1qa, interleukin-ɑ (IL-ɑ), and Tumor necrosis factor (TNF), the reactivity of astrocytes was reduced (75). Interleukin-1β (IL-1β) secreted by microglia, can activate astrocytes and allow the astrocytes to produce leukemia inhibitory factor (LIF) (76). LIF can promote the differentiation of OPC into mature myelinating oligodendrocytes and relieve demyelination in an animal model of multiple sclerosis (77). In further experiments, researchers have found astrocytes are also involved in the process of myelin debris removal (75). Microglia are recruited by astrocytes to the demyelinating lesions, which ensures the smooth process of phagocytosis (78). When the process is broken, OPC do not mature properly, resulting in the failure of remyelination. In MS, microglia participate in the whole processes of remyelination and demyelination (Figure 2).
2.3. Microglia and T cells interactions in MS
T lymphocytes are the basic organizers of most autoimmune responses, including MS (Figure 2). In MS, the encephalitogenic T cells might produce cytokines that directly activate microglia or through inducing the cerebral tissue damage indirectly activate microglia (79). Eugene D. Ponomarev first discovered that microglial activation in the CNS takes precedence over the appearance of neurological impairment symptoms in EAE and the peripheral macrophage cells flow over into the CNS, which further explains the activation of encephalitogenic T cells are necessary to the EAE onset (80). Activated microglia are the principal source of CC and CXC-chemokines, such as CCL2, CCL3, CCL4, CCL5, CCL12, and CX3CL1 (81). All of chemokines’ biological effects are performed by binding ligands to receptors. Chemokines play the main role in immunity and inflammation in MS. They attract the migration of pathogenic cells, interfering immune regulation of T-lymphocytes (82). Moreover, CCL5 promotes the activation and secretion of metalloproteinase-9 (MMP-9) in human microglia (83). MMP-9 is not only involved in the process of leukocyte extravasation but also in connection with the degradation of myelin proteins (84). Microglia also have a great ability to phagocytose apoptotic cells in the CNS, including encephalitogenic T cells, which can influence the chemoattractive milieu (85). Therefore, microglia may further disturb encephalitogenic T cells recruitment.
2.4. Paramagnetic rim lesions in MS
In MS, the white matter lesions are classified into active, inactive, and remyelinated lesions according to the distribution of inflammatory cells and the severity of demyelination (86). Slowly expanding chronic active lesions (CALs) mainly happen to those who occur 10 years after onset and CALs were seen more often in progressive MS than in relapsing disease (87). The paramagnetic rim lesions (PRLs) have been considered diagnostic MS biomarkers, which identify the CALs (88). Histological analysis has shown that PRLs correspond with iron-laden active microglia at the edges of CALs and indicate compartmentalized inflammation (89–91). When oligodendrocytes and myelin were damaged, microglia could phagocytose the iron released in the cells and the iron-laden microglia occurred at the MS lesions (92). Neuroimaging studies have revealed that the patients with progressive disease, compared to the patients with relapsing disease, have more rate of PRLs (93, 94). The clinical correlation about PRLs and more serious neurological impairments, is a more complex course. PRLs tend to the extent of involved lesions expanding 2 percent annually, according to a recent study on a new study on PRLs at 3-T MRI (93).
2.5. Microglial phenotypes in multiple sclerosis
As noted above, microglia have different functions in the embryonic, early postnatal, and adult stages and are multifunctional cells. Functional plasticity allows activated microglia to be polarized to either the M1 (pro-inflammatory) phenotype or the M2 (anti-inflammatory) phenotype (95). M1 microglia secrete pro-inflammatory mediators and oxidative compounds, including IL-1, IL-12, IL-23, Iba-1, CD68, IL-1β, TNF-α, and nitric oxide (NO). Additionally, MHC class II, costimulatory molecules, Fc receptors, and integrins are expressed in M1 microglia. Eventually, M1 microglia become cytotoxic and promote inflammation (67, 96, 97). In contrast, M2 microglia can promote tissue repair by releasing anti-inflammatory mediators such as IL-4, IL-10, and IL-13 and upregulating the expression of the M2 markers CD206 and arginase 1 (Arg1) (98). M2 microglia also secrete growth factors and neurotrophic factors, such as IGF-I, CSF1, nerve growth factor (NGF), BDNF, neurotrophin (NT)4/5, and glial cell-derived neurotrophic factor (GDNF).
Microglia are able to switch between the pro-inflammatory and anti-inflammatory states to maintain tissue homeostasis in response to the environment (99). In the experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis, both M1 and M2 microglia play vital roles in remyelination (51). In early demyelination, the M1 phenotype predominates, while M2 microglia are more abundant in later stages. It has been discovered that M2 microglia drive oligodendrocytes differentiation and remyelination. When remyelination starts, M1 microglia obviously transform into anti-inflammatory microglia. When remyelination is efficient, the M2 number is increased. Furthermore, M2 microglia-conditioned medium promotes the differentiation of oligodendrocytes, while the M1-conditioned medium inhibits this process (100). Despite this, microglial activation is not a simple dichotomy but is part of a spectrum of functional states (3). Emerging evidence suggests that the concept of M1/M2 microglia polarization may be outdated. In fact, transcriptome studies have shown that microglial activation is varied and context dependent (22).
Furthermore, according to their morphological characteristics, microglia can be divided into four types. These include what are known as ramified (micro somata but long ramifications), amoeboid (ramifications shorter, the cell somata larger), and phagocytic (no ramifications, just a rounded cell) (101). And dystrophic microglia are another cellular phenotype of microglia, which was considered to be one form of aging in microglia (102). Dystrophic microglia are rich among old people, and more and more recent studies discovered it also has been relationship to neurodegenerative diseases (103), such as MS. The morphology of dystrophic microglia is very different from hypertrophic microglia. They are characterized by fragmentation of cytoplasm, serious deformities of mitochondria, and distal branches becoming thinner (104). Research has suggested while M2 microglia play a protective role for CNS, finally they can convert to a dystrophic state (105). Dystrophic microglia produce more inflammatory mediators and loss the function of neuroprotection, which cause the disease to become worse (106).
As alluded to above, microglial activation is not a simple dichotomy. The whole-genome transcriptomics and unbiased proteomics studies have clarified problems associated with the microglial polarization response. The mixed phenotypes (intermediate phenotypes) have M1 and M2 markers at the same time, which occurs mainly in aging and various pathologic conditions, meaning M1 and M2 represent part of a spectrum of various activation phenotypes (107, 108). Even though an increasing number of people have realized that such a simplistic dichotomy (M1/M2) could not completely represent the complex phenotypes of microglia in vivo, this classification is important to the development of PET tracer (109). Under the circumstances, we interpret the M1 (pro-inflammatory) in connection with neurotoxic functions and the M2 (anti-inflammatory) involved in recovery and reduce harmful effects.
2.6. The PI3K-AKT pathway in microglia may be involved in MS
In MS, signal transduction pathway abnormalities in microglia may be the main causes of microglial activation and exacerbation of neuroimmune responses. Studies have demonstrated that the phosphatidylinositol 3-kinase (PI3K) and protein kinase B (AKT, also commonly known as protein kinase B (PKB)) signaling pathways play a crucial role in the modulating microglial activity and inflammatory responses (110). The PI3K-AKT pathway regulates cellular activities such as neuronal cell proliferation, migration, and plasticity. PI3K-AKT signaling in the brain seems to be closely related to microglial activity and activation. An increasing number of studies have shown that aberrant PI3K-AKT signaling participates in neurodegeneration and neuroinflammatory diseases (111).
Microglia express many key regulatory receptors that activate the PI3K-Akt pathway (112). As one of the first lines of immune defense, microglia express a number of different toll-like receptors (TLRs), including TLR2, TLR3, TLR4, TLR5, TLR7, and TLR9, on their surface (41). Among TLRs, TLR4 is an important receptor that mediates the inflammatory response and can interact with various immune stimulants. TLR4, which interacts with lipopolysaccharide (LPS), delivers downstream signals through myeloid differentiation primary response 88 (MyD88). Then, MyD88 phosphorylates tyrosine residues. Several studies have provided evidence that PI3K-AKT signaling is required for LPS-TLR4-dependent activation of microglia (113). Increased expression of TLR4 or constant TLR4 stimulation can lead to the subsequent activation of PI3K-AKT. PI3K is recruited via its p85 domain, which leads to downstream signal activation (114, 115). In this setting, microglia are constantly activated, and neuroinflammation is perpetuated. Increased TLR4 expression on microglia suppresses their polarization toward the anti-inflammatory phenotype and simultaneously prolongs the microglia-mediated proinflammatory response (116). In the brain, CSF-1R is primarily expressed by microglia (117). Contact of the CSF-1R receptor with microglia leads to oligomerization and the phosphorylation of its tyrosine residues, which are indispensable for PI3K recruitment (118). When expressed at high levels, CSF-1R and CSF-1 are considered mediators of demyelination in progressive MS, which exacerbates neuroinflammation due to the survival and constant proliferation of microglia (119). Furthermore, CX3CR1 activates the PI3K-AKT signaling pathway in a dose-dependent manner (51). CX3CR1 is a gene that is highly expressed in microglia. In summary, abnormal expression of TLR-4, CSF-1R, and CX3CR1 leads to aberrant PI3K-AKT signaling and impairs brain development, thus promoting the onset of neurodegenerative diseases such as MS.
Currently, the role of PI3K-AKT signaling in neuroimmunology is still incompletely understood. The PI3K-AKT pathway regulates microglia in response to various extracellular stimuli. It plays an important role in activating microglia to produce proinflammatory mediators after stimulation (110). Researchers have found the lipophilic amino alcohol 4b can attenuate the pathogenesis of EAE by inhibiting the PI3K-AKT pathway (120). The emodin also has therapeutic effect on EAE mice, which can down-regulate the expression of phosphorylated (p)-PI3K, p-Akt and further inhibit microglia activation and inflammation (121). However, the PI3K-AKT pathway may also play a neuroprotective role in different diseases. Researchers have also confirmed that PI3K-AKT pathway activation is important for oligodendrocytes survival and axonal myelination in the EAE model (122).
2.7. The NF-κB signaling pathway in microglia may be involved in MS
Another signaling pathway associated with MS is the nuclear factor-κB (NF-κB) signaling pathway. The NF-κB, expressed by many cells such as microglia, neurons, and astrocytes, plays a strong part in inflammation and immunity (123). The NF-κB signaling system is controlled by three interacting parts: NF-κB dimers, an inhibitor of NF-κB (IκB) regulators, and IkappaB kinase (IKK) complexes (124).
The activation of NF-κB is regulated by the canonical and non-canonical pathways. The canonical pathway can be activated by all kinds of stimuli, including cytokines, a number of pathogens, and different types of stress, which regulate proinflammatory gene expression (125). The non-canonical pathway was mainly activated by the TNF receptor superfamily members (126). This pathway contributed to maintaining immune homeostasis by participating in the development of the lymphoid tissues and various immune cells under healthy physiological conditions (126). In microglia, the pathway can be activated Fas ligand after the combination with TNF-α (126).
In MS, the activation of NF-κB in microglia is a reaction to injury to the CNS. Activated NF-κB leads to a cascade of signaling events, including the production of IL-1, which promotes microglia generating more proinflammatory cytokines, nitric oxide (NO), and neurotoxic reactive oxygen species (127). These cause a prominent toxic effect on the nervous system and exacerbate neuronal degeneration. Under the stimulation of pathology, the largest group of activated NF-κB form in the canonical pathway is the RelA: p50 (p65: p50) heterodimer, which plays a significant role in chronic inflammatory disease and neurodegenerative pathologies such as MS (125). Some compounds widespread in nature and some kinds of suppressors of cytokine signaling, perform a beneficial role in the treatment of EAE by downregulating NF-κB p65 signaling. Belinostat, the inhibitor of histone deacetylase, can meliorate symptoms of EAE through the downregulation of NF-κB p65 protein expression (128). Matrine (MAT), naturally present in Sophora flavescens, also alleviates the condition of EAE. MAT downregulates NF-κB p65 Phosphorylation, which also happened during the treatment courses of EAE (129). In LPS-induced microglia, the high-density lipoprotein (HDL) could significantly reduce the expression of TLR4 and NF-κB p65 (130). And in EAE, HDL could alleviate the soakage of inflammation cells in the spinal cord and brain, reducing the ratio of M1 microglia (130). Microglia also influence the development of EAE by regulating the noncanonical NF-κB pathway. Research showed that the noncanonical NF-kB pathway in microglia can interact with T cell-derived cytokine, which can accelerate the process of MS (131). Therefore, it has been a long-awaited goal to treat MS by directly targeting NF-κB pathway activation.
3. Conclusion
It is well known that remyelination failure is a significant challenge in the treatment of MS. Microglia, which are involved in myelin regeneration and play versatile roles in the pathogenesis of neuroinflammation and neurodegeneration, may be key therapeutic targets for diseases. Further studies are needed to better elucidate the precise roles of microglia in diseases and identify new therapeutic options that not only prevent new damage but also restore lost function.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (grant nos. 81701192 and 81901380), Shandong Provincial Natural Science Foundation, China (grant nos. ZR2017BH078 and ZR2017BC047), and Scientific Research Foundation of Binzhou Medical University (grant nos. BY2017KYQD15 and BY2016KYQD21).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tremblay, ME, Lecours, C, Samson, L, Sánchez-Zafra, V, and Sierra, A. From the Cajal alumni Achucarro and Rio-Hortega to the rediscovery of never-resting microglia. Front Neuroanat. (2015) 9:45. doi: 10.3389/fnana.2015.00045
2. Li, Q, and Barres, BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. (2018) 18:225–42. doi: 10.1038/nri.2017.125
3. Prinz, M, and Priller, J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. (2014) 15:300–12. doi: 10.1038/nrn3722
4. Gomez Perdiguero, E, Klapproth, K, Schulz, C, Busch, K, Azzoni, E, Crozet, L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
5. Perea, JR, García, E, Vallés-Saiz, L, Cuadros, R, Hernández, F, Bolós, M, et al. p38 activation occurs mainly in microglia in the P301S tauopathy mouse model. Sci Rep. (2022) 12:2130. doi: 10.1038/s41598-022-05980-8
6. Hammond, TR, Robinton, D, and Stevens, B. Microglia and the brain: complementary partners in development and disease. Annu Rev Cell Dev Biol. (2018) 34:523–44. doi: 10.1146/annurev-cellbio-100616-060509
7. Gosselin, D, Link, VM, Romanoski, CE, Fonseca, GJ, Eichenfield, DZ, Spann, NJ, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cells. (2014) 159:1327–40. doi: 10.1016/j.cell.2014.11.023
8. Lavin, Y, Winter, D, Blecher-Gonen, R, David, E, Keren-Shaul, H, Merad, M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cells. (2014) 159:1312–26. doi: 10.1016/j.cell.2014.11.018
9. Taylor, RA, Chang, CF, Goods, BA, Hammond, MD, Mac Grory, B, Ai, Y, et al. TGF-beta1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J Clin Invest. (2017) 127:280–92. doi: 10.1172/JCI88647
10. Wong, K, Noubade, R, Manzanillo, P, Ota, N, Foreman, O, Hackney, JA, et al. Mice deficient in NRROS show abnormal microglial development and neurological disorders. Nat Immunol. (2017) 18:633–41. doi: 10.1038/ni.3743
11. Spittau, B, Dokalis, N, and Prinz, M. The role of TGFbeta signaling in microglia maturation and activation. Trends Immunol. (2020) 41:836–48. doi: 10.1016/j.it.2020.07.003
12. Qin, Y, Garrison, BS, Ma, W, Wang, R, Jiang, A, Li, J, et al. A milieu molecule for TGF-beta required for microglia function in the nervous system. Cells. (2018) 174:156–171.e16. doi: 10.1016/j.cell.2018.05.027
13. Buttgereit, A, Lelios, I, Yu, X, Vrohlings, M, Krakoski, NR, Gautier, EL, et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol. (2016) 17:1397–406. doi: 10.1038/ni.3585
14. Holtman, IR, Skola, D, and Glass, CK. Transcriptional control of microglia phenotypes in health and disease. J Clin Invest. (2017) 127:3220–9. doi: 10.1172/JCI90604
15. Yeh, H, and Ikezu, T. Transcriptional and epigenetic regulation of microglia in health and disease. Trends Mol Med. (2019) 25:96–111. doi: 10.1016/j.molmed.2018.11.004
16. Gosselin, D, Skola, D, Coufal, NG, Holtman, IR, Schlachetzki, JCM, Sajti, E, et al. An environment-dependent transcriptional network specifies human microglia identity. Science. (2017) 356:eaal3222. doi: 10.1126/science.aal3222
17. Zhou, N, Liu, K, Sun, Y, Cao, Y, and Yang, J. Transcriptional mechanism of IRF8 and PU.1 governs microglial activation in neurodegenerative condition. Protein Cells. (2019) 10:87–103. doi: 10.1007/s13238-018-0599-3
18. Satoh, J, Kino, Y, Asahina, N, Takitani, M, Miyoshi, J, Ishida, T, et al. TMEM119 marks a subset of microglia in the human brain. Neuropathology. (2016) 36:39–49. doi: 10.1111/neup.12235
19. Bennett, ML, Bennett, FC, Liddelow, SA, Ajami, B, Zamanian, JL, Fernhoff, NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. (2016) 113:E1738–46. doi: 10.1073/pnas.1525528113
20. Vankriekelsvenne, E, Chrzanowski, U, Manzhula, K, Greiner, T, Wree, A, Hawlitschka, A, et al. Transmembrane protein 119 is neither a specific nor a reliable marker for microglia. Glia. (2022) 70:1170–90. doi: 10.1002/glia.24164
21. Tagliaferri, C, Wittrant, Y, Davicco, MJ, Walrand, S, and Coxam, V. Muscle and bone, two interconnected tissues. Ageing Res Rev. (2015) 21:55–70. doi: 10.1016/j.arr.2015.03.002
22. Colonna, M, and Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. (2017) 35:441–68. doi: 10.1146/annurev-immunol-051116-052358
23. Hughes, AN, and Appel, B. Microglia phagocytose myelin sheaths to modify developmental myelination. Nat Neurosci. (2020) 23:1055–66. doi: 10.1038/s41593-020-0654-2
24. Paolicelli, RC, Bolasco, G, Pagani, F, Maggi, L, Scianni, M, Panzanelli, P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. (2011) 333:1456–8. doi: 10.1126/science.1202529
25. Thion, MS, and Garel, S. On place and time: microglia in embryonic and perinatal brain development. Curr Opin Neurobiol. (2017) 47:121–30. doi: 10.1016/j.conb.2017.10.004
26. Bar, E, and Barak, B. Microglia roles in synaptic plasticity and myelination in homeostatic conditions and neurodevelopmental disorders. Glia. (2019) 67:2125–41. doi: 10.1002/glia.23637
27. Colucci-D'Amato, L, Speranza, L, Volpicelli, F, and Neurotrophic Factor, BDNF. Physiological functions and therapeutic potential in depression, neurodegeneration and brain cancer. Int J Mol Sci. (2020) 21:7777. doi: 10.3390/ijms21207777
28. Cornell, J, Salinas, S, Huang, HY, and Zhou, M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen Res. (2022) 17:705–16. doi: 10.4103/1673-5374.322423
29. Sanchez, J, DePaula-Silva, AB, Doty, DJ, Hanak, TJ, Truong, A, Libbey, JE, et al. The CSF1R-microglia axis has protective host-specific roles during neurotropic picornavirus infection. Front Immunol. (2021) 12:621090. doi: 10.3389/fimmu.2021.621090
30. Voet, S, Prinz, M, and van Loo, G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol Med. (2019) 25:112–23. doi: 10.1016/j.molmed.2018.11.005
31. Perry, VH. A revised view of the central nervous system microenvironment and major histocompatibility complex class II antigen presentation. J Neuroimmunol. (1998) 90:113–21. doi: 10.1016/S0165-5728(98)00145-3
32. Kobelt, G, Thompson, A, Berg, J, Gannedahl, M, Eriksson, J, the MSCOI Study Group, et al. New insights into the burden and costs of multiple sclerosis in Europe. Mult Scler. (2017) 23:1123–36. doi: 10.1177/1352458517694432
33. Psenicka, MW, Smith, BC, Tinkey, RA, and Williams, JL. Connecting neuroinflammation and neurodegeneration in multiple sclerosis: are oligodendrocyte precursor cells a nexus of disease? Front Cell Neurosci. (2021) 15:654284. doi: 10.3389/fncel.2021.654284
34. Dobson, R, and Giovannoni, G. Multiple sclerosis - a review. Eur J Neurol. (2019) 26:27–40. doi: 10.1111/ene.13819
35. Choi, S, Guo, L, and Cordeiro, MF. Retinal and brain microglia in multiple sclerosis and neurodegeneration. Cells. (2021) 10:1507. doi: 10.3390/cells10061507
36. McCarthy, MK, and Weinberg, JB. The immunoproteasome and viral infection: a complex regulator of inflammation. Front Microbiol. (2015) 6:21. doi: 10.3389/fmicb.2015.00021
37. Hurwitz, BJ. The diagnosis of multiple sclerosis and the clinical subtypes. Ann Indian Acad Neurol. (2009) 12:226–30. doi: 10.4103/0972-2327.58276
38. Jacques, FH. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. (2015) 84:963. doi: 10.1212/01.wnl.0000462309.76486.c5
39. Jia, X, Madireddy, L, Caillier, S, Santaniello, A, Esposito, F, Comi, G, et al. Genome sequencing uncovers phenocopies in primary progressive multiple sclerosis. Ann Neurol. (2018) 84:51–63. doi: 10.1002/ana.25263
40. Kuhn, S, Gritti, L, Crooks, D, and Dombrowski, Y. Oligodendrocytes in development, myelin generation and beyond. Cells. (2019) 8:1424. doi: 10.3390/cells8111424
41. O’Loughlin, E, Madore, C, Lassmann, H, and Butovsky, O. Microglial phenotypes and functions in multiple sclerosis. Cold Spring Harb Perspect Med. (2018) 8:a028993. doi: 10.1101/cshperspect.a028993
42. Madry, C, Kyrargyri, V, Arancibia-Cárcamo, IL, Jolivet, R, Kohsaka, S, Bryan, RM, et al. Microglial ramification, surveillance, and interleukin-1beta release are regulated by the two-pore domain K(+) channel THIK-1. Neuron. (2018) 97:299–312.e6. doi: 10.1016/j.neuron.2017.12.002
43. Sieger, D, Moritz, C, Ziegenhals, T, Prykhozhij, S, and Peri, F. Long-range Ca2+ waves transmit brain-damage signals to microglia. Dev Cell. (2012) 22:1138–48. doi: 10.1016/j.devcel.2012.04.012
44. Paladini, MS, Feng, X, Krukowski, K, and Rosi, S. Microglia depletion and cognitive functions after brain injury: from trauma to galactic cosmic ray. Neurosci Lett. (2021) 741:135462. doi: 10.1016/j.neulet.2020.135462
45. Hu, X, Leak, RK, Shi, Y, Suenaga, J, Gao, Y, Zheng, P, et al. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. (2015) 11:56–64. doi: 10.1038/nrneurol.2014.207
46. Hagan, N, Kane, JL, Grover, D, Woodworth, L, Madore, C, Saleh, J, et al. CSF1R signaling is a regulator of pathogenesis in progressive MS. Cell Death Dis. (2020) 11:904. doi: 10.1038/s41419-020-03084-7
47. Lloyd, AF, and Miron, VE. The pro-remyelination properties of microglia in the central nervous system. Nat Rev Neurol. (2019) 15:447–58. doi: 10.1038/s41582-019-0184-2
48. Yeung, MS, Zdunek, S, Bergmann, O, Bernard, S, Salehpour, M, Alkass, K, et al. Dynamics of oligodendrocyte generation and myelination in the human brain. Cells. (2014) 159:766–74. doi: 10.1016/j.cell.2014.10.011
49. Franklin, RJ, and Ffrench-Constant, C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. (2008) 9:839–55. doi: 10.1038/nrn2480
50. Moyon, S, Dubessy, AL, Aigrot, MS, Trotter, M, Huang, JK, Dauphinot, L, et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J Neurosci. (2015) 35:4–20. doi: 10.1523/JNEUROSCI.0849-14.2015
51. Gacem, N, and Nait-Oumesmar, B. Oligodendrocyte development and regenerative therapeutics in multiple sclerosis. Life. (2021) 11:327. doi: 10.3390/life11040327
52. Kury, P, Kremer, D, and Gottle, P. Drug repurposing for neuroregeneration in multiple sclerosis. Neural Regen Res. (2018) 13:1366–7. doi: 10.4103/1673-5374.235242
53. Boyd, A, Zhang, H, and Williams, A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. (2013) 125:841–59. doi: 10.1007/s00401-013-1112-y
54. McMurran, CE, Jones, CA, Fitzgerald, DC, and Franklin, RJM. CNS remyelination and the innate immune system. Front Cell Dev Biol. (2016) 4:38. doi: 10.3389/fcell.2016.00038
55. Kalafatakis, I, and Karagogeos, D. Oligodendrocytes and microglia: key players in myelin development, damage and repair. Biomol Ther. (2021) 11:1058. doi: 10.3390/biom11071058
56. Pons, V, and Rivest, S. Beneficial roles of microglia and growth factors in MS, a brief review. Front Cell Neurosci. (2020) 14:284. doi: 10.3389/fncel.2020.00284
57. Rawji, KS, Mishra, MK, and Yong, VW. Regenerative capacity of macrophages for remyelination. Front Cell Dev Biol. (2016) 4:47. doi: 10.3389/fcell.2016.00047
58. Williams, A, Piaton, G, Aigrot, MS, Belhadi, A, Theaudin, M, Petermann, F, et al. Semaphorin 3A and 3F: key players in myelin repair in multiple sclerosis? Brain. (2007) 130:2554–65. doi: 10.1093/brain/awm202
59. Davies, CL, and Miron, VE. Distinct origins, gene expression and function of microglia and monocyte-derived macrophages in CNS myelin injury and regeneration. Clin Immunol. (2018) 189:57–62. doi: 10.1016/j.clim.2016.06.016
60. Lampron, A, Larochelle, A, Laflamme, N, Préfontaine, P, Plante, MM, Sánchez, MG, et al. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med. (2015) 212:481–95. doi: 10.1084/jem.20141656
61. Shiau, CE, Kaufman, Z, Meireles, AM, and Talbot, WS. Differential requirement for irf8 in formation of embryonic and adult macrophages in zebrafish. PLoS One. (2015) 10:e0117513. doi: 10.1371/journal.pone.0117513
62. Green, LA, Nebiolo, JC, and Smith, CJ. Microglia exit the CNS in spinal root avulsion. PLoS Biol. (2019) 17:e3000159. doi: 10.1371/journal.pbio.3000159
63. Li, J, Zhang, L, Chu, Y, Namaka, M, Deng, B, Kong, J, et al. Astrocytes in oligodendrocyte lineage development and white matter pathology. Front Cell Neurosci. (2016) 10:119. doi: 10.3389/fncel.2016.00119
64. Healy, LM, Perron, G, Won, SY, Michell-Robinson, MA, Rezk, A, Ludwin, SK, et al. MerTK is a functional regulator of myelin phagocytosis by human myeloid cells. J Immunol. (2016) 196:3375–84. doi: 10.4049/jimmunol.1502562
65. Healy, LM, Jang, JH, Won, SY, Lin, YH, Touil, H, Aljarallah, S, et al. MerTK-mediated regulation of myelin phagocytosis by macrophages generated from patients with MS. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e402. doi: 10.1212/NXI.0000000000000402
66. Gruchot, J, Weyers, V, Göttle, P, Förster, M, Hartung, HP, Küry, P, et al. The molecular basis for remyelination failure in multiple sclerosis. Cells. (2019) 8:825. doi: 10.3390/cells8080825
67. Geladaris, A, Hausler, D, and Weber, MS. Microglia: the missing link to decipher and therapeutically control MS progression? Int J Mol Sci. (2021) 22:3461. doi: 10.3390/ijms22073461
68. Piccio, L, Buonsanti, C, Mariani, M, Cella, M, Gilfillan, S, Cross, A H, et al. Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. (2007) 37:1290–301. doi: 10.1002/eji.200636837
69. Poliani, PL, Wang, Y, Fontana, E, Robinette, ML, Yamanishi, Y, Gilfillan, S, et al. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest. (2015) 125:2161–70. doi: 10.1172/JCI77983
70. Raine, CS. Multiple sclerosis: the resolving lesion revealed. J Neuroimmunol. (2017) 304:2–6. doi: 10.1016/j.jneuroim.2016.05.021
71. Enrich-Bengoa, J, Manich, G, Dégano, IR, and Perálvarez-Marín, A. Deciphering the genetic crosstalk between microglia and oligodendrocyte precursor cells during demyelination and remyelination using transcriptomic data. Int J Mol Sci. (2022) 23:14868. doi: 10.3390/ijms232314868
72. Machado-Santos, J, Saji, E, Tröscher, AR, Paunovic, M, Liblau, R, Gabriely, G, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain. (2018) 141:2066–82. doi: 10.1093/brain/awy151
73. Frischer, JM, Weigand, SD, Guo, Y, Kale, N, Parisi, JE, Pirko, I, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol. (2015) 78:710–21. doi: 10.1002/ana.24497
74. Marzan, DE, Brügger-Verdon, V, West, BL, Liddelow, S, Samanta, J, and Salzer, JL. Activated microglia drive demyelination via CSF1R signaling. Glia. (2021) 69:1583–604. doi: 10.1002/glia.23980
75. Clarke, LE, Liddelow, SA, Chakraborty, C, Münch, AE, Heiman, M, and Barres, BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A. (2018) 115:E1896–905. doi: 10.1073/pnas.1800165115
76. Wies, MV, Di Pietro, AA, and Pasquini, LA. Microglia depletion as a therapeutic strategy: friend or foe in multiple sclerosis models? Neural Regen Res. (2023) 18:267–72. doi: 10.4103/1673-5374.346538
77. Steelman, AJ, Zhou, Y, Koito, H, Kim, SJ, Payne, HR, Lu, QR, et al. Activation of oligodendroglial Stat3 is required for efficient remyelination. Neurobiol Dis. (2016) 91:336–46. doi: 10.1016/j.nbd.2016.03.023
78. Matejuk, A, and Ransohoff, RM. Crosstalk between astrocytes and microglia: an overview. Front Immunol. (2020) 11:1416. doi: 10.3389/fimmu.2020.01416
79. Ioannides, ZA, Csurhes, PA, Swayne, A, Foubert, P, Aftab, BT, and Pender, MP. Correlations between macrophage/microglial activation marker sTREM-2 and measures of T-cell activation, neuroaxonal damage and disease severity in multiple sclerosis. Mult Scler J Exp Transl Clin. (2021) 7:205521732110197. doi: 10.1177/20552173211019772
80. Ponomarev, ED, Shriver, LP, Maresz, K, Pedras-Vasconcelos, J, Verthelyi, D, and Dittel, BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. (2007) 178:39–48. doi: 10.4049/jimmunol.178.1.39
81. Tsai, CF, Chen, GW, Chen, YC, Shen, CK, Lu, DY, Yang, LY, et al. Regulatory effects of quercetin on M1/M2 macrophage polarization and oxidative/antioxidative balance. Nutrients. (2021) 14:67. doi: 10.3390/nu14010067
82. Cui, LY, Chu, SF, and Chen, NH. The role of chemokines and chemokine receptors in multiple sclerosis. Int Immunopharmacol. (2020) 83:106314. doi: 10.1016/j.intimp.2020.106314
83. Long, H, Xie, R, Xiang, T, Zhao, Z, Lin, S, Liang, Z, et al. Autocrine CCL5 signaling promotes invasion and migration of CD133+ ovarian cancer stem-like cells via NF-kappaB-mediated MMP-9 upregulation. Stem Cells. (2012) 30:2309–19. doi: 10.1002/stem.1194
84. Seo, JE, Hasan, M, Han, JS, Kang, MJ, Jung, BH, Kwok, SK, et al. Experimental autoimmune encephalomyelitis and age-related correlations of NADPH oxidase, MMP-9, and cell adhesion molecules: the increased disease severity and blood-brain barrier permeability in middle-aged mice. J Neuroimmunol. (2015) 287:43–53. doi: 10.1016/j.jneuroim.2015.08.005
85. Chan, A, Hummel, V, Weilbach, FX, Kieseier, BC, and Gold, R. Phagocytosis of apoptotic inflammatory cells downregulates microglial chemoattractive function and migration of encephalitogenic T cells. J Neurosci Res. (2006) 84:1217–24. doi: 10.1002/jnr.21029
86. Filippi, M, Brück, W, Chard, D, Fazekas, F, Geurts, JJG, Enzinger, C, et al. Association between pathological and MRI findings in multiple sclerosis. Lancet Neurol. (2019) 18:198–210. doi: 10.1016/S1474-4422(18)30451-4
87. Hemond, CC, Reich, DS, and Dundamadappa, SK. Paramagnetic rim lesions in multiple sclerosis: comparison of visualization at 1.5-T and 3-T MRI. AJR Am J Roentgenol. (2022) 219:120–31. doi: 10.2214/AJR.21.26777
88. Hemond, CC, Baek, J, Ionete, C, and Reich, DS. Paramagnetic rim lesions are associated with pathogenic CSF profiles and worse clinical status in multiple sclerosis: a retrospective cross-sectional study. Mult Scler. (2022) 28:2046–56. doi: 10.1177/13524585221102921
89. Dal-Bianco, A, Grabner, G, Kronnerwetter, C, Weber, M, Kornek, B, Kasprian, G, et al. Long-term evolution of multiple sclerosis iron rim lesions in 7 T MRI. Brain. (2021) 144:833–47. doi: 10.1093/brain/awaa436
90. Absinta, M, Sati, P, Schindler, M, Leibovitch, EC, Ohayon, J, Wu, T, et al. Persistent 7-tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J Clin Invest. (2016) 126:2597–609. doi: 10.1172/JCI86198
91. Gillen, KM, Mubarak, M, Park, C, Ponath, G, Zhang, S, Dimov, A, et al. QSM is an imaging biomarker for chronic glial activation in multiple sclerosis lesions. Ann Clin Transl Neurol. (2021) 8:877–86. doi: 10.1002/acn3.51338
92. Yong, H, and Yong, VW. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nat Rev Neurol. (2022) 18:40–55. doi: 10.1038/s41582-021-00581-x
93. Absinta, M, Sati, P, Masuzzo, F, Nair, G, Sethi, V, Kolb, H, et al. Association of chronic active multiple sclerosis lesions with disability in vivo. JAMA Neurol. (2019) 76:1474–83. doi: 10.1001/jamaneurol.2019.2399
94. Maggi, P, Sati, P, Nair, G, Cortese, ICM, Jacobson, S, Smith, BR, et al. Paramagnetic rim lesions are specific to multiple sclerosis: an international multicenter 3T MRI study. Ann Neurol. (2020) 88:1034–42. doi: 10.1002/ana.25877
95. Ramirez, AI, de Hoz, R, Salobrar-Garcia, E, Salazar, JJ, Rojas, B, Ajoy, D, et al. The role of microglia in retinal neurodegeneration: Alzheimer's disease, Parkinson, and Glaucoma. Front Aging Neurosci. (2017) 9:214. doi: 10.3389/fnagi.2017.00214
96. Liu, R, Xu, NG, Yi, W, and Ji, C. Electroacupuncture attenuates inflammation after ischemic stroke by inhibiting NF-kappaB-mediated activation of microglia. Evid Based Complement Alternat Med. (2020) 2020:8163052. doi: 10.1155/2020/8163052
97. Wang, J, Wang, J, Wang, J, Yang, B, Weng, Q, and He, Q. Targeting microglia and macrophages: a potential treatment strategy for multiple sclerosis. Front Pharmacol. (2019) 10:286. doi: 10.3389/fphar.2019.00286
98. Colton, CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. (2009) 4:399–418. doi: 10.1007/s11481-009-9164-4
99. Orihuela, R, McPherson, CA, and Harry, GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. (2016) 173:649–65. doi: 10.1111/bph.13139
100. Miron, VE, Boyd, A, Zhao, JW, Yuen, TJ, Ruckh, JM, Shadrach, JL, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. (2013) 16:1211–8. doi: 10.1038/nn.3469
101. Savage, JC, Carrier, M, and Tremblay, ME. Morphology of microglia across contexts of health and disease. Methods Mol Biol. (2019) 2034:13–26. doi: 10.1007/978-1-4939-9658-2_2
102. Streit, WJ, Sammons, NW, Kuhns, AJ, and Sparks, DL. Dystrophic microglia in the aging human brain. Glia. (2004) 45:208–12. doi: 10.1002/glia.10319
103. Shahidehpour, RK, Higdon, RE, Crawford, NG, Neltner, JH, Ighodaro, ET, Patel, E, et al. Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol Aging. (2021) 99:19–27. doi: 10.1016/j.neurobiolaging.2020.12.003
104. Xue, QS, and Streit, WJ. Microglial pathology in down syndrome. Acta Neuropathol. (2011) 122:455–66. doi: 10.1007/s00401-011-0864-5
105. Rodriguez-Callejas, JD, Fuchs, E, and Perez-Cruz, C. Increased oxidative stress, hyperphosphorylation of tau, and dystrophic microglia in the hippocampus of aged Tupaia belangeri. Glia. (2020) 68:1775–93. doi: 10.1002/glia.23804
106. Tischer, J, Krueger, M, Mueller, W, Staszewski, O, Prinz, M, Streit, WJ, et al. Inhomogeneous distribution of Iba-1 characterizes microglial pathology in Alzheimer’s disease. Glia. (2016) 64:1562–72. doi: 10.1002/glia.23024
107. Grabert, K, Michoel, T, Karavolos, MH, Clohisey, S, Baillie, JK, Stevens, MP, et al. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci. (2016) 19:504–16. doi: 10.1038/nn.4222
108. Guo, S, Wang, H, and Yin, Y. Microglia polarization from M1 to M2 in neurodegenerative diseases. Front Aging Neurosci. (2022) 14:815347. doi: 10.3389/fnagi.2022.815347
109. Beaino, W, Janssen, B, Vugts, DJ, de Vries, HE, and Windhorst, AD. Towards PET imaging of the dynamic phenotypes of microglia. Clin Exp Immunol. (2021) 206:282–300. doi: 10.1111/cei.13649
110. Chu, E, Mychasiuk, R, Hibbs, ML, and Semple, BD. Dysregulated phosphoinositide 3-kinase signaling in microglia: shaping chronic neuroinflammation. J Neuroinflammation. (2021) 18:276. doi: 10.1186/s12974-021-02325-6
111. Xu, F, Na, L, Li, Y, and Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. (2020) 10:54. doi: 10.1186/s13578-020-00416-0
112. Cianciulli, A, Porro, C, Calvello, R, Trotta, T, Lofrumento, DD, and Panaro, MA. Microglia mediated neuroinflammation: focus on PI3K modulation. Biomol Ther. (2020) 10:137. doi: 10.3390/biom10010137
113. Zhou, LT, Wang, KJ, Li, L, Li, H, and Geng, M. Pinocembrin inhibits lipopolysaccharide-induced inflammatory mediators production in BV2 microglial cells through suppression of PI3K/Akt/NF-kappaB pathway. Eur J Pharmacol. (2015) 761:211–6. doi: 10.1016/j.ejphar.2015.06.003
114. Zhou, L, Liu, Z, Wang, Z, Yu, S, Long, T, Zhou, X, et al. Astragalus polysaccharides exerts immunomodulatory effects via TLR4-mediated MyD88-dependent signaling pathway in vitro and in vivo. Sci Rep. (2017) 7:44822. doi: 10.1038/srep44822
115. Zhu, H, Pan, L, Dai, Y, Zheng, D, and Cai, S. Role of TLR4/MyD88 signaling pathway in the occurrence and development of uremia-induced myocardial hypertrophy and possible mechanism. Evid Based Complement Alternat Med. (2021) 2021:7883643:1–9. doi: 10.1155/2021/7883643
116. Calvo-Rodriguez, M, García-Rodríguez, C, Villalobos, C, and Núñez, L. Role of toll like receptor 4 in Alzheimer’s disease. Front Immunol. (2020) 11:1588. doi: 10.3389/fimmu.2020.01588
117. Spangenberg, E, Severson, PL, Hohsfield, LA, Crapser, J, Zhang, J, Burton, EA, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun. (2019) 10:3758. doi: 10.1038/s41467-019-11674-z
118. Hosking, MP, Tirotta, E, Ransohoff, RM, and Lane, TE. CXCR2 signaling protects oligodendrocytes and restricts demyelination in a mouse model of viral-induced demyelination. PLoS One. (2010) 5:e11340. doi: 10.1371/journal.pone.0011340
119. NBB-PsyBöttcher, C, Schlickeiser, S, Sneeboer, MAM, Kunkel, D, Knop, A, et al. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat Neurosci. (2019) 22:78–90. doi: 10.1038/s41593-018-0290-2
120. da Silva, LC, Lima, IVA, da Silva, MCM, Corrêa, TA, de Souza, VP, de Almeida, MV, et al. A new lipophilic amino alcohol, chemically similar to compound FTY720, attenuates the pathogenesis of experimental autoimmune encephalomyelitis by PI3K/Akt pathway inhibition. Int Immunopharmacol. (2020) 88:106919. doi: 10.1016/j.intimp.2020.106919
121. Zheng, K, Lv, B, Wu, L, Wang, C, Xu, H, Li, X, et al. Protecting effect of emodin in experimental autoimmune encephalomyelitis mice by inhibiting microglia activation and inflammation via Myd88/PI3K/Akt/NF-kappaB signalling pathway. Bioengineered. (2022) 13:9322–44. doi: 10.1080/21655979.2022.2052671
122. Giacoppo, S, Pollastro, F, Grassi, G, Bramanti, P, and Mazzon, E. Target regulation of PI3K/Akt/mTOR pathway by cannabidiol in treatment of experimental multiple sclerosis. Fitoterapia. (2017) 116:77–84. doi: 10.1016/j.fitote.2016.11.010
123. Singh, SS, Rai, SN, Birla, H, Zahra, W, Rathore, AS, and Singh, SP. NF-kappaB-mediated neuroinflammation in Parkinson’s disease and potential therapeutic effect of polyphenols. Neurotox Res. (2020) 37:491–507. doi: 10.1007/s12640-019-00147-2
124. Mitchell, S, Vargas, J, and Hoffmann, A. Signaling via the NFkappaB system. Wiley Interdiscip Rev Syst Biol Med. (2016) 8:227–41. doi: 10.1002/wsbm.1331
125. Giridharan, S, and Srinivasan, M. Mechanisms of NF-kappaB p65 and strategies for therapeutic manipulation. J Inflamm Res. (2018) 11:407–19. doi: 10.2147/JIR.S140188
126. Yu, H, Lin, L, Zhang, Z, Zhang, H, and Hu, H. Targeting NF-kappaB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. (2020) 5:209. doi: 10.1038/s41392-020-00312-6
127. Wei, X, Cho, KS, Thee, EF, Jager, MJ, and Chen, DF. Neuroinflammation and microglia in glaucoma: time for a paradigm shift. J Neurosci Res. (2019) 97:70–6. doi: 10.1002/jnr.24256
128. Shen, Y, Yang, R, Zhao, J, Chen, M, Chen, S, Ji, B, et al. The histone deacetylase inhibitor belinostat ameliorates experimental autoimmune encephalomyelitis in mice by inhibiting TLR2/MyD88 and HDAC3/NF-kappaB p65-mediated neuroinflammation. Pharmacol Res. (2022) 176:105969. doi: 10.1016/j.phrs.2021.105969
129. Chu, Y, Jing, Y, Zhao, X, Wang, M, Zhang, M, Ma, R, et al. Modulation of the HMGB1/TLR4/NF-kappaB signaling pathway in the CNS by matrine in experimental autoimmune encephalomyelitis. J Neuroimmunol. (2021) 352:577480. doi: 10.1016/j.jneuroim.2021.577480
130. Hong, S, Niu, M, Meng, D, Li, A, Dong, Q, Zhang, J, et al. High-density lipoprotein reduces microglia activation and protects against experimental autoimmune encephalomyelitis in mice. Int Immunopharmacol. (2022) 105:108566. doi: 10.1016/j.intimp.2022.108566
Keywords: multiple sclerosis, microglia, remyelination, demyelination, signal transduction
Citation: Zhang X, Chen F, Sun M, Wu N, Liu B, Yi X, Ge R and Fan X (2023) Microglia in the context of multiple sclerosis. Front. Neurol. 14:1157287. doi: 10.3389/fneur.2023.1157287
Edited by:
Wassim Elyaman, Columbia University, United StatesReviewed by:
Hamza Coban, University of Connecticut, United StatesMarta Olah, Columbia University, United States
Copyright © 2023 Zhang, Chen, Sun, Wu, Liu, Yi, Ge and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangming Yi, YmlsbGFubkAxNjMuY29t; Ruli Ge, Z2VybGlAMTYzLmNvbQ==; Xueli Fan, eHVlbGlmYW5AeWVhaC5uZXQ=
†These authors have contributed equally to this work