 Clare I. Grady
Clare I. Grady Lisa M. Walsh
Lisa M. Walsh John D. Heiss
John D. Heiss
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 30 May 2023
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1154753
This article is part of the Research TopicEpigenetic approaches in targeting glioma development and progressionView all 5 articles
Epigenetic mechanisms allow cells to fine-tune gene expression in response to environmental stimuli. For decades, it has been known that mitochondria have genetic material. Still, only recently have studies shown that epigenetic factors regulate mitochondrial DNA (mtDNA) gene expression. Mitochondria regulate cellular proliferation, apoptosis, and energy metabolism, all critical areas of dysfunction in gliomas. Methylation of mtDNA, alterations in mtDNA packaging via mitochondrial transcription factor A (TFAM), and regulation of mtDNA transcription via the micro-RNAs (mir 23-b) and long noncoding RNAs [RNA mitochondrial RNA processing (RMRP)] have all been identified as contributing to glioma pathogenicity. Developing new interventions interfering with these pathways may improve glioma therapy.
Adult diffuse gliomas are Central nervous system (CNS) tumors arising from glial cells, most often astrocytes, oligodendrocytes, and ependymal cells. In 2016, the World Health Organization (WHO) published the fourth version of the CNS tumor classification system with the underlying concept of a multiple-input or layered diagnostic design based on histology, grading, and genomic markers (1). This new, layered classification of diffuse gliomas is better suited to clinical practice than earlier ones because it better predicts prognosis and the choice of therapies for biologically and genetically similar tumors. Glioblastoma multiforme (GBM) is the most prevalent and aggressive primary malignant brain tumor. It diffusely infiltrates the surrounding brain and is characterized by poor prognosis, with a five-year survival rate of 5.5%, despite multimodal therapy (2). Redundant signaling pathways and intratumoral heterogeneity contribute to the inability of conventional and targeted therapies to achieve remission (3–5). Whereas intratumoral heterogeneity traditionally was thought to arise from mutations accumulating and resulting in distinct genotypes, non-genetic heterogeneity from variations in regulatory mechanisms also plays a vital role. Of these regulatory mechanisms, the field of epigenetic modifications as they contribute to the development and progression of GBM has exploded, as shown by this Special Collection.
Epigenetics studies mitotically heritable and stable changes in gene expression resulting from DNA replication and transcription alterations rather than DNA sequence polymorphisms (mutations). Three epigenetic mechanisms have been identified: DNA methylation, histone modification, and noncoding RNA (ncRNA) associated gene silencing (6). DNA methylation involves adding a methyl group directly to a cytosine nucleotide followed by a guanine nucleotide. CpG is shorthand for 5’-C-phosphate-G-3′, where the cytosine and guanine are separated by only one phosphate group, distinguishing this single-stranded linear sequencing from the C-G base pairing for double-stranded sequences. CpGs are often surrounded by other CpGs forming a CpG Island (CGI). CpGs are commonly located in promoter regions because their methylation reduces the interaction between DNA and transcription factors (6). Histone modifications include acetylation, methylation, phosphorylation, and ubiquitylation of histone proteins. Histone modifications alter nucleosome DNA-histone interactions and can facilitate or prevent transcription (6). Noncoding RNAs (ncRNAs) have function despite not being translated into proteins. They participate in DNA methylation, histone modification, and direct gene silencing. Small ncRNAs consist of <200 nucleotides (nt) and include microRNAs (miRNA; 17–23 nt) and small interfering RNAs (siRNA; 20–30 nt). Long non-coding RNAs (lncRNAs) consist of >200 nt and include linear and circular RNAs (7).
Malignant tumors, including gliomas, favor abnormal energy production via aerobic glycolysis and show inherent resistance to apoptosis (8–10). Mitochondrial dysfunction could contribute to GBM pathophysiology by altering metabolic pathways and energy production, but these mechanisms are poorly understood. Mitochondria have a genome (mitogenome) to direct their function and communication with the nuclear genome (11). Recently, it has been shown that mitochondrial DNA (mtDNA), like nuclear DNA (nDNA), is regulated by epigenetic mechanisms through a process referred to as mitoepigenetics (Figure 1) (12).
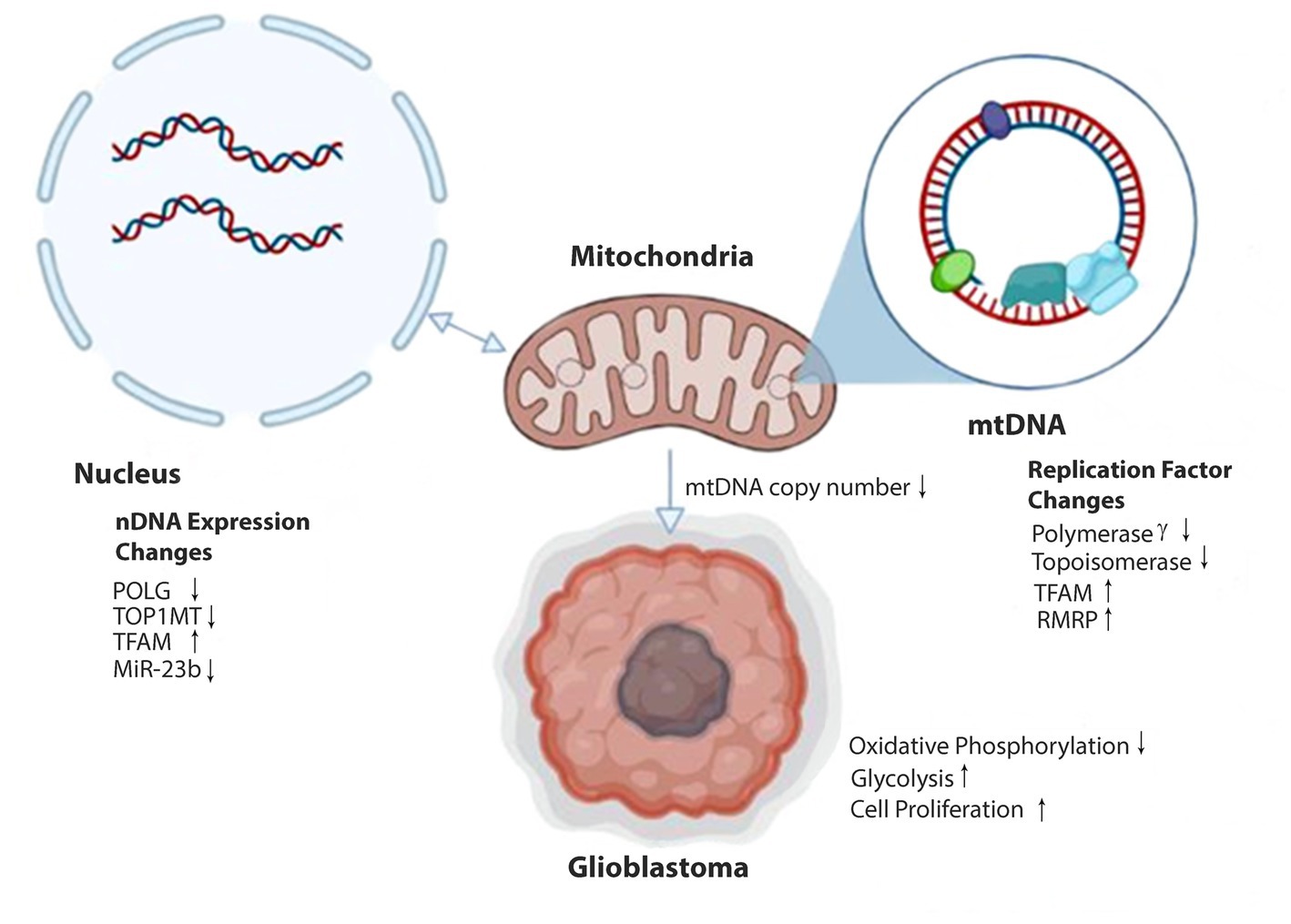
Figure 1. Epigenetic alterations affecting mitochondrial DNA expression in glioblastoma. Methylation of nDNA genes POLG and TOPIMT reduces production of Polymerase γ and Topoisomerase proteins and decreases transport and availability in the mitochondria (anterograde signaling). MiR-23b transcription is reduced in gliomas, increasing the expression of TFAM within the nucleus. Transport of IncRNA RMRP to the mitochondria is increased. Reduced Polymerase γ and Topoisomerase and elevated TFAM and RMRP alter mtDNA copy number and gene expression. Decreases in mtDNA copy number can signal epigenetic modifications of nDNA within the cell nucleus (retrograde signaling). Ultimately, these epigenetic modifications result in reduced oxidative phosphorylation capabilities in the mitochondria. In response, the stem cell adopts glycolytic metabolism and a proliferative glioblastoma phenotype.
Traditionally, mtDNA consists of 37 genes: 13 encoding polypeptide components of the respiratory chain, 22 tRNAs, and two rRNAs. mtDNA also contains a significant noncoding region termed the displacement loop (13). However, evidence is emerging that the mitochondrial transcriptome also includes mitochondrial-derived ncRNAs (14).
The mitochondrial proteome consists of approximately 1,500 proteins. nDNA encodes most respiratory chain components and proteins required for the synthesis, expression, and regulation of mitochondrial genes. The nuclear coding of most mitochondrial proteins explains why interest in GBM epigenetics has focused on nDNA rather than mtDNA (15). However, given the association between GBM pathogenesis and mitochondrial dysfunction, the epigenetic regulation of the small 16.6 kb mtDNA genome may be an untapped study area of incredible value. This review discusses the complexity of mitochondrial epigenetics, highlighting the roles mtDNA methylation, mtDNA packing, and ncRNA may play in GBM pathogenesis. We will describe epigenetic changes involving mitochondria that could enhance GBM pathogenicity. These include epigenetic changes to mtDNA directly altering mtDNA expression and epigenetic modifications of nDNA indirectly affecting mtDNA expression.
Mitochondrial epigenetics is a largely unexplored field, but the literature is rapidly growing as our understanding of mtDNA and the mitochondrial proteome deepens. The three most common types of nDNA epigenetic modification can be applied with some modification to mtDNA. Increasing evidence shows mtDNA methylation is associated with mtDNA transcriptional regulation and copy number (16, 17). Unlike nDNA, mtDNA lacks histones. mtDNA is packaged instead into nucleoid structures, in which mtDNA lies in the center surrounded by peripheral proteins (18). In 2011, scientists discovered ncRNAs inside mitochondria and created the first comprehensive map of the human mitochondrial transcriptome (14). We will review the three categories of epigenetic alterations in mtDNA (methylation, modification of nucleoid proteins, and ncRNA) in the context of glioma pathogenesis and potential therapeutic targets.
Methylation of the nuclear genome plays a critical role in mitochondrial function through its effect on mtDNA replication and copy number. Human embryonic and neural stem cells have extensive nDNA methylation in the early stages of development but become demethylated as cells differentiate and increase their mtDNA copy numbers (19, 20). An adequate copy number of mtDNA is essential to produce the machinery needed to engage in oxidative phosphorylation and meet ATP demands (21–23). The genome of glioblastoma cells stays heavily methylated. Many studies have shown that the mtDNA copy number is lower in glioma cells than in healthy cells (24–27). Low mtDNA copy number appears to lead glioma cells to rely on glycolysis to produce ATP instead of oxidative phosphorylation, promoting cell proliferation (28). Therefore, methylation of the nuclear and mitochondrial genomes of glioblastoma cells has been studied to understand how this epigenetic mechanism impacts mtDNA replication and mtDNA copy number (Table 1) (23, 28–30).
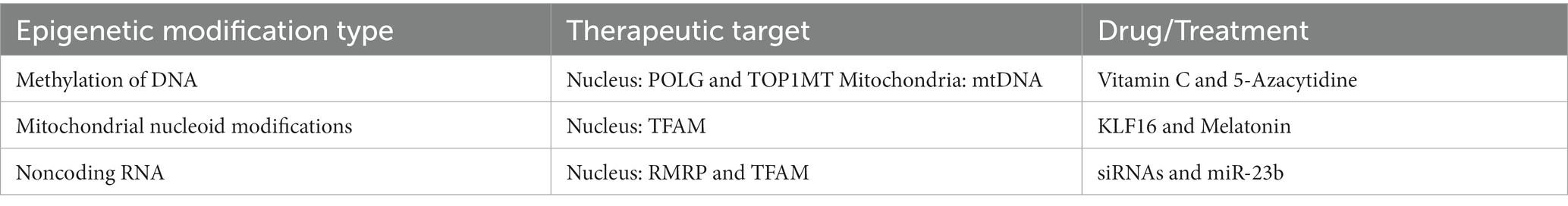
Table 1. Epigenetic modifications of DNA as therapeutic targets for potential drugs/treatments in gliomas.
MtDNA copy number is associated with the methylation of exon 2 of DNA polymerase gamma, POLG (28, 31). POLG, found on chromosome 15q25, encodes the mitochondrial-specific DNA polymerase, Pol γ, which is essential in mtDNA replication (32, 33). In the HSR-GBM1 model, exon 2 of POLG is highly methylated. However, using 5-azacytidine (5-Aza) resulted in significant mtDNA copy number increases in the HSR-GBM model (28). 5-Aza irreversibly binds the methylating enzyme DNA methyltransferase 1 (DMT1), reducing its ability to methylate DNA. Therefore, 5-Aza treatment reduced methylation of exon 2 of POLG, increased POLG expression, and increased mtDNA copy number (34). The use of demethylating agent 5-Aza also appeared to induce long-term differentiation of these cells (34).
The TOP1MT gene encodes for the mitochondrial-specific topoisomerase, which facilitates replication and transcription of mtDNA by relieving tension and supercoiling (35). Exon 8 and intron 9 of the TOP1MT gene were heavily methylated in the HSR-GBM1 model. However, TOP1MT expression and mtDNA copy number increased in response to the 5-Aza treatment (23). This further suggests that the demethylation of mtDNA replication factors leads to an increased mtDNA copy number.
Vitamin C enhances ten-eleven translocation methylcytosine dioxygenase (TET1) activity, which demethylates the 5th position of the pyrimidine ring of cytosine on POLG. Like 5-Aza treatments, VitC demethylation of POLG increased POLG expression and mtDNA copy number. HSR-GBM1 cells given VitC treatment did not differentiate fully (36). These studies demonstrated VitC and 5-Aza as potential treatments, although inhibition of DNMT with 5-Aza may be more effective than Vitamin C in decreasing methylation and upregulating mtDNA copy number. 5- Aza and Vitamin C are DNA demethylation agents that promote cell differentiation and may affect various cancers (34, 37–39). Few studies have examined these agents’ anti-glioma effects. One case study demonstrated intravenous vitamin C treatment benefiting a patient with GBM, suggesting the broader applicability of Vitamin C for glioma therapy (40). However, vitamin C enhanced glioblastoma invasiveness. Vitamin C deficiency reduced glioblastoma proliferation in vitro (41, 42). 5-Aza’s had better outcomes than VitC in the HSR-GBM1 model, supporting its testing in other models and potential in clinical trials.
Global demethylation of the HSR-GBM1 nuclear genome modified tumor-specific genes and critical mtDNA transcription and replication factors beyond POLG and TOP1MT (23). Demethylation downregulated a subunit of the SEC61 translocon complex (SEC61G) and upregulated the PR domain containing 16 (PRDM16) and serine/threonine-protein kinase (WNK2). DNA demethylating treatments affected cell death, growth, and differentiation pathways (43–45). Demethylation treatment upregulated epidermal growth factor 2 (EGFR2), B-Cell lymphoma 2 (BCL2), telomerase reverse transcriptase (TERT), and MYC. These tumor markers interact with mtDNA transcription and replication factors. Demethylation regulates cellular fate, cellular differentiation, and mtDNA replication (23, 46–49).
Evidence suggests that nuclear and mitochondrial genomic modifications contribute to glioma tumorigenesis (29). When mtDNA levels were decreased to different extents in HSR-GBM1 cells, cells experienced nDNA methylation changes to restore mtDNA copy numbers (29). This further exemplifies the importance of considering nDNA’s role in mtDNA replication when developing therapies targeting mtDNA copy number.
The methylation of mtDNA has not been studied extensively. Scientists debated for years on whether mtDNA could be methylated (50, 51). However, multiple studies have since confirmed mtDNA methylated sites (23, 52–54). Furthermore, mtDNA methylation contributed to mtDNA copy number and tumorigenesis in GBM. Sun et al. used naïve osteosarcoma cells with their original mtDNA (143B143B), and mtDNA depleted 143B cell lines repopulated with donor mtDNA from HSR-GBM1 cells (143BGBM) and human neural stem cells (143BNSC) (30). Each cell population was analyzed at an early and late growth stage. Cell lines with mtDNA derived from hNSCs (143BNSC) had higher levels of mtDNA methylation than cell lines with mtDNA derived from GBM or 143B (143BGBM or 143B143B) cell lines, suggesting decreased mtDNA methylation is initially necessary to enforce a tumor phenotype (30). The mtDNA copy number in 143BGBM and 143B143B cell lines also increased significantly from the early to late stages, suggesting that increased mtDNA copy number encouraged early tumorigenesis (30, 55). However, although mtDNA methylation decreases and mtDNA copy number increases as tumors first progress, after tumorigenesis is started by sufficient mtDNA copy number, methylation of mtDNA increases. This late event in tumorigenesis restricts mtDNA replication and maintains mtDNA copy numbers in GBM cell lines lower than in non-tumorigenic cell lines (30). This indicates that the mtDNA methylation changes that result in abnormal energy production in GBM occur in later stages of tumorigenesis.
These results give insight into mtDNA’s role in tumorigenesis. They suggest that mtDNA demethylation contributes to increases in mtDNA copy number, which could support demethylation therapy for GBM. The demethylating agents, 5Aza and VitC, significantly reduced mtDNA methylation and could prove helpful as therapies (23). Additional studies of demethylating agents will show if they can change nDNA and mtDNA epigenetics and tumor pathogenesis in glioma and glioblastoma.
Mitochondrial DNA (mtDNA) has no associated histone protein like nDNA The mitochondria’s nucleoid contains mtDNA centrally surrounded by core and peripheral proteins referred to as core and peripheral nucleoid factors (18). Core factors are crosslinked to mtDNA and include (1) transcription factor A, mitochondrial (TFAM), (2) mitochondrial single-strand binding protein (mtSSB), (3) DNA polymerase subunit gamma (POLG), (4) mtRNA polymerase (POLRMT), (5) Lon protease, and (6) DNA helicase Twinkle (56). Initially, the roles of these factors in transcription, translation, and cell-wide signaling were shown. More recently, it has become clear that several package proteins epigenetically modulate mtDNA, like histones modulate nDNA (57).
Mitochondrial transcription factor A (TFAM) is a 24-kDa protein encoded by a nuclear gene on chromosome 10. It was initially identified as a transcription factor for mtDNA (58) but is now thought dispensable for mtDNA transcription in vitro but crucial for packaging mtDNA within the mitochondria (59). TFAM has two high-mobility group (HMG) domains. These are DNA binding motifs that, upon binding, induce a U-shape confirmation in mtDNA (60, 61). U-turn bending recruits mitochondrial RNA polymerase to the mitochondrial light strand promoter (LSP) site. It is thought that the degree of bending may affect transcriptional activation efficacy (62). The mammalian mitochondria contain about 1 TFAM protein per 15–18 base pairs of mtDNA, making it abundant enough to coat the entire mitochondrial genome (63).
Lee et al. explored the association of TFAM with GBM and if TFAM antagonism could be an anti-GBM therapeutic strategy (64). In their study, a Western blot analysis with an anti-TFAM antibody showed markedly increased protein expression of TFAM in GBM cell lines, especially U343-MG and U373-MG cells. Quantitative real-time PCR (qRT-PCR) showed elevated mRNA levels of TFAM in the U251-MG, U343-MG, and U373-MG GBM cell lines. Human GBM tissues were also stained with anti-TFAM antibodies. Tumor tissue had considerably more TFAM staining than surrounding tissue. The differential expression of TFAM in the REMBRANDT cohort was analyzed and found to have significantly higher TFAM gene transcript levels in GBM, astrocytoma, and oligodendroglioma than normal controls (p < 0.0001) (64).
Correia et al. investigated transcript levels of TFAM as they related to GBM overall survival time (65). Using QT-PCR to quantify expression levels of TFAM, they compared TFAM expression of non-neoplastic brain tissue to two GBM subgroups: survival time under 12 months and survival time over 24 months. Although both GBM subgroups had significantly higher levels of TFAM than non-neoplastic brain tissue, the TFAM expression was higher in the 24-month survivors than the 12-month survivors (65).
Pediatric high-grade gliomas (pHGG) are the deadliest childhood CNS cancers. They are characterized by K27M mutations in histones H3.1 and H3.3 and G34R mutations in H3.3 (H3.3G34R) (66–68). H3.3G34R mutations are almost exclusive to hemispheric pHGG and occur in adolescents and young adults (69). Siddaway et al. unexpectedly discovered H3.3G34R localizes to the mitochondria at a higher rate than wild-type H3.3 (70). Furthermore, TFAM associated exclusively with the H3.3G34R mutated histone and not the wild-type histone. The authors hypothesized that H3.3G34R had a metabolic effect on the pHGG cells. They generated a metabolomic profile of H3.3 wild type versus H3.3G34R and showed enriched TCA cycle metabolites and a higher level of mitochondrial metabolism in these cells (70). H3.3G34R may be a useful epigenetic marker for TFAM in pHGG.
Although these studies elucidate an association between gliomas and TFAM, the mechanism underlying TFAM’s contribution to glioma pathogenesis is poorly understood. Levels of TFAM expression in GBM patient specimens did not correlate with GBM survival. Levels of TFAM and mtDNA transcription efficacy may have a complex, non-linear relationship: as TFAM increases, mtDNA assumes a more favorable mtDNA conformation for mtDNA transcription. However, a specific point may exist when TFAM coats mtDNA, reducing transcription efficacy. The best stoichiometry between TFAM and template DNA has not been established in GBM.
Nonetheless, TFAM may be a strong candidate target for glioma therapeutic interventions. Chen et al. investigated the relationship between Kruppel-like factor (KLF) 16 and TFAM in glioma cell proliferation (71). KLF members are zinc finger-containing transcription factors that regulate oncogenic or tumor-suppressive genes by binding GC-rich DNA sequences in gene promoter regions (72). The expression of KLF16 was found to be robustly reduced in six glioma cell lines and glioma tissues via Western blot studies and real-time PCR analysis. Furthermore, survival analysis showed glioma patients with low KLF16 had a poor prognosis (HR = 2.328, 95% CI = 1.387–4.017, p < 0.01). Given these results, KLF16 was postulated to have a tumor-suppressive role in glioma progression. Real-time PCR indicated TFAM expression was downregulated in KLF16-elevated cells and upregulated in KLF16-silenced cells. Using a chromatin immunoprecipitation (ChIP) assay, they showed a high binding affinity of endogenous KLF16 to the GC-rich basic transcriptional element in the TFAM nDNA promoter, indicating KLF16 directly repressed TFAM expression and could serve as a potential therapeutic target (71).
The effects of melatonin (N-acetyl-5-methoxytryptamine), a hormone synthesized from serotonin, on mitochondria have been widely explored, but little is known about how melatonin affects mtDNA and TFAM expression. Franco et al. investigated the relationship between TFAM and melatonin using the GBM cell line U87MG (73). When melatonin was incubated with these cells, the mRNA expression of TFAM and protein levels decreased. The reduction of TFAM resulted in reduced gene expression of mitochondrial NADH dehydrogenase 1, elevated reactive oxygen species (ROS) production, and decreased cell viability. Lastly, they showed that melatonin acts synergistically with temozolomide (TMZ). Cell viability was reduced by 34% by 3 mM melatonin and by 45% by TMZ. Combined treatment of melatonin and TMZ reduced viability by 87% (73).
Additionally, the exact epigenetic mechanism that results in upregulation of TFAM in GBM is unknown. Characterizing this mechanism may be beneficial as it could provide us with targets to reduce TFAM expression in GBM to restore a normal phenotype. One possible target could be nuclear respiratory factor 1, which has been found to suppress the TFAM promoter when methylated in vitro (74). However, future studies must look at this effect in GBM models before a therapy can be developed.
These studies demonstrate TFAM plays an integral role in the structure of mtDNA and transcriptional regulation and may serve as a novel epigenetic target for glioma therapy.
Noncoding RNAs (ncRNAs) have regulatory and structural functions, not protein template activity. They regulate gene expression by adjusting RNA processing and mRNA stability, modification, and translation (75). NcRNAs represent 70% of the nuclear genome in humans and the third category of epigenetic processes contributing to glioma development and progression (76). Advances in deep sequencing have revealed that the mitochondrial transcriptome results from a complex regulatory, expression, and processing network. ncRNAs participate in mitochondrial gene regulation (14). Two types of ncRNAs have been found inside mitochondria. The first is nuclear-encoded ncRNAs (nuclear-ncRNAs) involved in directional signaling from the nucleus to the mitochondria (anterograde signaling). The second type is the mitochondria-encoded ncRNAs (mt-ncRNAs). The study of mt-ncRNAs is groundbreaking because, until recently, the mitogenome was thought only to include genes encoding polypeptides, tRNAs, and rRNAs.
Several studies mapped many long noncoding RNAs and small noncoding RNAs to the mitochondrial genome. Mercer et al. identified 31 novel miRNAs expressed from 17 distinct loci. The majority (84%) were derived from tRNA genes (14). Rackham et al. identified three lncRNAs with sequences uniquely aligned to the mitochondrial genes encoding ND5, ND6, and Cyt b (77). Interestingly, ND6 is the least abundant mitochondrial encoded protein, perhaps because lncND6 downregulates its expression (77).
Conversely, nuclear-encoded RNAs have been found within the mitochondria. They are thought to regulate the mitochondrial genome by associating with Argonaute (AGO) proteins, forming the RNA-induced silencing complex (RISC) core, and exerting RNA interference (78). Thirteen nuclear miRNAs were found within the mitochondria associating with AGO and mitochondrial mRNA, implying RNAi may regulate mitochondrial biogenesis and function (78). Analysis of the mitochondrial transcriptome showed a nuclear-lncRNA part of the mitochondrial RNA processing endoribonuclease (14). Termed RMRP, this RNA part is important for mtDNA replication and RNA processing. RMRP helps endonuclease cleave mitochondrial RNA at a priming site of mtDNA replication (79). Given the growing cohort of ncRNAs and their epigenetic influence on mitochondrial expression, several studies have postulated this mechanism may contribute to glioma pathogenesis and can be targeted with new therapeutic agents.
The LncRNA RNA component of mitochondrial RNA processing (RMRP) was first found to promote carcinogenesis in gastric cancer. RMRP expression was recently investigated in low-grade (grade I-II) to high-grade (grade III-IV) glioma cell lines and tissues (80). Glioma tissues expressed significantly more RMRP than normal brain tissues in qRT-PCR experiments (80). Furthermore, lncRNA RMRP upregulation is significantly associated with advanced tumor grade and low Karnofsky Performance Score (KPS), indicating RMRP up-regulation may be involved in glioma progression. Knockdown of RMRP significantly decreased the proliferation of glioma cell lines in vitro. These findings suggest that reducing the expression of lncRNA RMRP impairs the transcription of mtDNA and inhibits malignant phenotypes of glioma cells (80).
Liu et al. performed experiments on lncRNAs, including RMRP, to decide if they were involved in regulating the TMZ resistance in gliomas (81). The top 100 upregulated lncRNAs in glioma tumor tissues were identified, including RMRP. RMRP expression levels were higher in tumors isolated from patients with relapsing glioma after TMZ treatment than tumors from TMZ-naïve patients. Liu et al. investigated the role of RMRP in TMZ resistance. Three siRNAs were synthesized for RMRP knockdown. RMRP knockdown increased the cell apoptosis rate 2.6-fold. RMRP depletion weakened the TMZ resistance of TMZ-resistant glioma cell lines and TMZ-treated glioma xenograft tumors (81). These studies demonstrate RMRP antagonism as a potential strategy for increasing the therapeutic efficacy of TMZ against glioblastoma.
As previously mentioned, mitochondrial transcription factor A (TFAM) is a core protein within the mitochondrial nucleoid structure responsible for creating favorable conformations of mtDNA and increasing transcription efficacy (62). TFAM can act alone as an epigenetic mechanism in glioma pathogenesis. Its susceptibility to miRNA regulation creates an added layer of mitoepigenetic control. MiR-23b is a miRNA highly expressed in several cancers and associated with tumorigenesis (82). Its role in gliomas was investigated using glioma cell lines and tissue specimens. MiR-23b expression levels measured by real-time RT-PCR were significantly lower in glioma than in normal brain tissue. Like other TFAM studies, this study showed that TFAM expression was significantly increased in glioma tissues and positively correlated to the malignancy grade. Cell lines overexpressing TFAM demonstrated increased proliferation and invasiveness. The 3′ untranslated region of TFAM was found to be a direct target of mIR-23b. Cell lines overexpressing miR-23b had decreased proliferation and invasiveness (80). These results suggest TFAM may be a direct target of epigenetic control via miR-23b (Table 1).
Mitochondrial deregulation is a GBM marker (83). Sixty percent of solid tumor patients have detectable mtDNA mutations within their body fluid cell-free DNA (84, 85). Epigenetic modifications of nDNA and mtDNA contribute to the malignant features and treatment resistance of glioma patients with nDNA and mtDNA mutations. Researchers are testing anti-glioma therapeutic strategies targeting epigenetic modifications in tumor cells and animal models. Treatments demethylating the POLG, TOP1MT, and TFAM genes attempt to increase tumor cell mtDNA copy number, differentiation, and apoptosis (83). Potential therapeutic agents targeting the POLG, TOP1MT, and TFAM genes include Vitamin C, 5-azacytidine, and melatonin. Another epigenetic therapeutic target is lncRNA RMRP expression. Reducing lncRNA RMRP expression levels in glioma restored the sensitivity of tumor cells to TMZ (Table 1). Inhibiting mtDNA transcription may also decrease glioma cell proliferation and invasion (80).
Glioblastoma and other malignant tumors manifest metabolic reprogramming called the Warburg effect, in which cellular glucose uptake is increased, and the glucose metabolite pyruvate is metabolized anaerobically to lactate. This effect occurs even in cancer cells with functional mitochondria under normoxic conditions (86). Glioblastoma also aerobically metabolizes glucose-derived pyruvate and fatty acids in the mitochondria in actively proliferating, high-oxygen-consuming tumor cells (87, 88). Damaged mitochondria accumulate in tumor cells due to impaired mitophagy and produce reactive oxygen species (ROS) that damage and mutate genomic and mtDNA and enhance genomic instability and oncogenesis (89). Glioblastoma combination therapies can be designed to include agents antagonizing the tumorigenic effects of the Warburg effect and mitochondrial genetic and epigenetic aberrations, like tumor proliferation, invasion, free radical production, impaired mitophagy, and reduced apoptosis (90).
Mitochondria participate in many biological processes, including metabolism, apoptosis, and cellular proliferation. It has been well-reported that the tumorigenicity of malignant tumors, including gliomas, is related to abnormal energy production and inherent resistance to apoptosis. Understanding mtDNA’s contribution to these processes goes beyond the proteins the mitochondrial genome expresses. We are beginning to appreciate how epigenetic mechanisms regulate mtDNA expression and contribute to tumor pathogenicity (Figure 1). Further understanding of mtDNA methylation, alterations in nucleoid packaging of mtDNA, and regulation of mtDNA by noncoding RNAs in glioma cell lines and tissue samples will uncover novel mechanisms underlying glioma progression that may be amenable to targeted therapies.
CG: conceptualization, writing original draft, review and editing, and visualization. LW: writing, review and editing, and visualization. JH: conceptualization, writing, review and editing, and supervision. All authors contributed to the article and approved the submitted version.
This research was funded by the Intramural Research Program of the National Institute of Neurological Diseases and Stroke at the National Institutes of Health, Project Number 1ZIA NS003052-15.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Louis, DN, Perry, A, Reifenberger, G, von Deimling, A, Figarella-Branger, D, Cavenee, WK, et al. The World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
2. Ostrom, QT, Gittleman, H, Liao, P, Vecchione-Koval, T, Wolinsky, Y, Kruchko, C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro-Oncology. (2017) 19:v1–v88. doi: 10.1093/neuonc/nox158
3. Stommel, JM, Kimmelman, AC, Ying, H, Nabioullin, R, Ponugoti, AH, Wiedemeyer, R, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. (2007) 318:287–90. doi: 10.1126/science.1142946
4. Nathanson, DA, Gini, B, Mottahedeh, J, Visnyei, K, Koga, T, Gomez, G, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science. (2014) 343:72–6. doi: 10.1126/science.1241328
5. Gilbert, MR, Dignam, JJ, Armstrong, TS, Wefel, JS, Blumenthal, DT, Vogelbaum, MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. (2014) 370:699–708. doi: 10.1056/NEJMoa1308573
6. Al Aboud, NM, Tupper, C, and Jialal, I. Genetics, Epigenetic Mechanism. Treasure Island (FL): Stat Pearls Publishing LLC (2022).
7. Han, Y, and He, X. Integrating Epigenomics into the Understanding of Biomedical Insight. Bioinform Biol Insights. (2016) 10:267–89. doi: 10.4137/BBI.S38427
8. Furnari, FB, Fenton, T, Bachoo, RM, Mukasa, A, Stommel, JM, Stegh, A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. (2007) 21:2683–710. doi: 10.1101/gad.1596707
9. Ziegler, DS, Kung, AL, and Kieran, MW. Anti-apoptosis mechanisms in malignant gliomas. J Clin Oncol. (2008) 26:493–500. doi: 10.1200/JCO.2007.13.9717
10. Seyfried, TN, and Mukherjee, P. Targeting energy metabolism in brain cancer: review and hypothesis. Nutr Metab (Lond). (2005) 2:30. doi: 10.1186/1743-7075-2-30
12. Manev, H, and Dzitoyeva, S. Progress in mitochondrial epigenetics. Biomol Concepts. (2013) 4:381–9. doi: 10.1515/bmc-2013-0005
13. Andrews, RM, Kubacka, I, Chinnery, PF, Lightowlers, RN, Turnbull, DM, and Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. (1999) 23:147. doi: 10.1038/13779
14. Mercer, TR, Neph, S, Dinger, ME, Crawford, J, Smith, MA, Shearwood, AM, et al. The human mitochondrial transcriptome. Cells. (2011) 146:645–58. doi: 10.1016/j.cell.2011.06.051
15. Calvo, S, Jain, M, Xie, X, Sheth, SA, Chang, B, Goldberger, OA, et al. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat Genet. (2006) 38:576–82. doi: 10.1038/ng1776
16. D'Aquila, P, Bellizzi, D, and Passarino, G. Mitochondria in health, aging and diseases: the epigenetic perspective. Biogerontology. (2015) 16:569–85. doi: 10.1007/s10522-015-9562-3
17. Iacobazzi, V, Castegna, A, Infantino, V, and Andria, G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol Genet Metab. (2013) 110:25–34. doi: 10.1016/j.ymgme.2013.07.012
18. Gilkerson, R, Bravo, L, Garcia, I, Gaytan, N, Herrera, A, Maldonado, A, et al. The mitochondrial nucleoid: integrating mitochondrial DNA into cellular homeostasis. Cold Spring Harb Perspect Biol. (2013) 5:a011080. doi: 10.1101/cshperspect.a011080
19. Lister, R, Pelizzola, M, Dowen, RH, Hawkins, RD, Hon, G, Tonti-Filippini, J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. (2009) 462:315–22. doi: 10.1038/nature08514
20. Ziller, MJ, Müller, F, Liao, J, Zhang, Y, Gu, H, Bock, C, et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. (2011) 7:e1002389. doi: 10.1371/journal.pgen.1002389
21. Facucho-Oliveira, JM, and St John, JC. The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev Rep. (2009) 5:140–58. doi: 10.1007/s12015-009-9058-0
22. Facucho-Oliveira, JM, Alderson, J, Spikings, EC, Egginton, S, and St John, JC. Mitochondrial DNA replication during differentiation of murine embryonic stem cells. J Cell Sci. (2007) 120:4025–34. doi: 10.1242/jcs.016972
23. Sun, X, Johnson, J, and St John, JC. Global DNA methylation synergistically regulates the nuclear and mitochondrial genomes in glioblastoma cells. Nucleic Acids Res. (2018) 46:5977–95. doi: 10.1093/nar/gky339
24. Liang, BC, and Hays, L. Mitochondrial DNA copy number changes in human gliomas. Cancer Lett. (1996) 105:167–73. doi: 10.1016/0304-3835(96)04276-0
25. Soltész, B, Pös, O, Wlachovska, Z, Budis, J, Hekel, R, Strieskova, L, et al. Mitochondrial DNA copy number changes, heteroplasmy, and mutations in plasma-derived exosomes and brain tissue of glioblastoma patients. Mol Cell Probes. (2022) 66:101875. doi: 10.1016/j.mcp.2022.101875
26. Dickinson, A, Yeung, KY, Donoghue, J, Baker, MJ, Kelly, RD, McKenzie, M, et al. The regulation of mitochondrial DNA copy number in glioblastoma cells. Cell Death Differ. (2013) 20:1644–53. doi: 10.1038/cdd.2013.115
27. Lai, RK, Chen, Y, Guan, X, Nousome, D, Sharma, C, Canoll, P, et al. Genome-wide methylation analyses in glioblastoma multiforme. PLoS One. (2014) 9:e89376. doi: 10.1371/journal.pone.0089376
28. Lee, W, Johnson, J, Gough, DJ, Donoghue, J, Cagnone, GL, Vaghjiani, V, et al. Mitochondrial DNA copy number is regulated by DNA methylation and demethylation of POLGA in stem and cancer cells and their differentiated progeny. Cell Death Dis. (2015) 6:e1664. doi: 10.1038/cddis.2015.34
29. Sun, X, and St John, JC. Modulation of mitochondrial DNA copy number in a model of glioblastoma induces changes to DNA methylation and gene expression of the nuclear genome in tumours. Epigenetics Chromatin. (2018) 11:53. doi: 10.1186/s13072-018-0223-z
30. Sun, X, Vaghjiani, V, Jayasekara, WSN, Cain, JE, and St John, JC. The degree of mitochondrial DNA methylation in tumor models of glioblastoma and osteosarcoma. Clin Epigenetics. (2018) 10:157. doi: 10.1186/s13148-018-0590-0
31. Kelly, RD, Mahmud, A, McKenzie, M, Trounce, IA, and St John, JC. Mitochondrial DNA copy number is regulated in a tissue specific manner by DNA methylation of the nuclear-encoded DNA polymerase gamma A. Nucleic Acids Res. (2012) 40:10124–38. doi: 10.1093/nar/gks770
32. Ropp, PA, and Copeland, WC. Cloning and characterization of the human mitochondrial DNA polymerase, DNA polymerase gamma. Genomics. (1996) 36:449–58. doi: 10.1006/geno.1996.0490
33. Kaguni, LS, and Olson, MW. Mismatch-specific 3′----5′ exonuclease associated with the mitochondrial DNA polymerase from Drosophila embryos. Proc Natl Acad Sci U S A. (1989) 86:6469–73. doi: 10.1073/pnas.86.17.6469
34. Christman, JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. (2002) 21:5483–95. doi: 10.1038/sj.onc.1205699
35. Zhang, H, Meng, LH, and Pommier, Y. Mitochondrial topoisomerases and alternative splicing of the human TOP1mt gene. Biochimie. (2007) 89:474–81. doi: 10.1016/j.biochi.2006.11.002
36. Blaschke, K, Ebata, KT, Karimi, MM, Zepeda-Martínez, JA, Goyal, P, Mahapatra, S, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. (2013) 500:222–6. doi: 10.1038/nature12362
37. Braiteh, F, Soriano, AO, Garcia-Manero, G, Hong, D, Johnson, MM, Silva Lde, P, et al. Phase I study of epigenetic modulation with 5-azacytidine and valproic acid in patients with advanced cancers. Clin Cancer Res. (2008) 14:6296–301. doi: 10.1158/1078-0432.CCR-08-1247
38. Sajadian, SO, Tripura, C, Samani, FS, Ruoss, M, Dooley, S, Baharvand, H, et al. Vitamin C enhances epigenetic modifications induced by 5-azacytidine and cell cycle arrest in the hepatocellular carcinoma cell lines HLE and Huh7. Clin Epigenetics. (2016) 8:46. doi: 10.1186/s13148-016-0213-6
39. Liu, M, Ohtani, H, Zhou, W, Ørskov, AD, Charlet, J, Zhang, YW, et al. Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc Natl Acad Sci U S A. (2016) 113:10238–44. doi: 10.1073/pnas.1612262113
40. Baillie, N, Carr, AC, and Peng, S. The Use of Intravenous Vitamin C as a Supportive Therapy for a Patient with Glioblastoma Multiforme. Antioxidants (Basel). (2018) 7:115. doi: 10.3390/antiox7090115
41. Ramírez, E, Jara, N, Ferrada, L, Salazar, K, Martínez, F, Oviedo, MJ, et al. Glioblastoma Invasiveness and Collagen Secretion Are Enhanced by Vitamin C. Antioxid Redox Signal. (2022) 37:538–59. doi: 10.1089/ars.2021.0089
42. Jara, N, Ramirez, E, Ferrada, L, Salazar, K, Espinoza, F, González-Chavarría, I, et al. Vitamin C deficient reduces proliferation in a human periventricular tumor stem cell-derived glioblastoma model. J Cell Physiol. (2021) 236:5801–17. doi: 10.1002/jcp.30264
43. Lu, Z, Zhou, L, Killela, P, Rasheed, AB, Di, C, Poe, WE, et al. Glioblastoma proto-oncogene SEC61gamma is required for tumor cell survival and response to endoplasmic reticulum stress. Cancer Res. (2009) 69:9105–11. doi: 10.1158/0008-5472.CAN-09-2775
44. Lei, Q, Liu, X, Fu, H, Sun, Y, Wang, L, Xu, G, et al. miR-101 reverses hypomethylation of the PRDM16 promoter to disrupt mitochondrial function in astrocytoma cells. Oncotarget. (2016) 7:5007–22. doi: 10.18632/oncotarget.6652
45. Moniz, S, Martinho, O, Pinto, F, Sousa, B, Loureiro, C, Oliveira, MJ, et al. Loss of WNK2 expression by promoter gene methylation occurs in adult gliomas and triggers Rac1-mediated tumour cell invasiveness. Hum Mol Genet. (2013) 22:84–95. doi: 10.1093/hmg/dds405
46. Morrish, F, and Hockenbery, D. MYC and mitochondrial biogenesis. Cold Spring Harb Perspect Med. (2014) 4:a014225. doi: 10.1101/cshperspect.a014225
47. Autret, A, and Martin, SJ. Emerging role for members of the Bcl-2 family in mitochondrial morphogenesis. Mol Cell. (2009) 36:355–63. doi: 10.1016/j.molcel.2009.10.011
48. Taylor, TE, Furnari, FB, and Cavenee, WK. Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance. Curr Cancer Drug Targets. (2012) 12:197–209. doi: 10.2174/156800912799277557
49. Sahin, E, Colla, S, Liesa, M, Moslehi, J, Müller, FL, Guo, M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. (2011) 470:359–65. doi: 10.1038/nature09787
50. Maekawa, M, Taniguchi, T, Higashi, H, Sugimura, H, Sugano, K, and Kanno, T. Methylation of mitochondrial DNA is not a useful marker for cancer detection. Clin Chem. (2004) 50:1480–1. doi: 10.1373/clinchem.2004.035139
51. Hong, EE, Okitsu, CY, Smith, AD, and Hsieh, CL. Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol Cell Biol. (2013) 33:2683–90. doi: 10.1128/MCB.00220-13
52. Bellizzi, D, D'Aquila, P, Scafone, T, Giordano, M, Riso, V, Riccio, A, et al. The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. (2013) 20:537–47. doi: 10.1093/dnares/dst029
53. Shock, LS, Thakkar, PV, Peterson, EJ, Moran, RG, and Taylor, SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A. (2011) 108:3630–5. doi: 10.1073/pnas.1012311108
54. Liu, B, Du, Q, Chen, L, Fu, G, Li, S, Fu, L, et al. CpG methylation patterns of human mitochondrial DNA. Sci Rep. (2016) 6:23421. doi: 10.1038/srep23421
55. Lee, WT, Cain, JE, Cuddihy, A, Johnson, J, Dickinson, A, Yeung, KY, et al. Mitochondrial DNA plasticity is an essential inducer of tumorigenesis. Cell Death Dis. (2016) 2:16016. doi: 10.1038/cddiscovery.2016.16
56. Bogenhagen, DF, Rousseau, D, and Burke, S. The layered structure of human mitochondrial DNA nucleoids. J Biol Chem. (2008) 283:3665–75. doi: 10.1074/jbc.M708444200
57. Alam, TI, Kanki, T, Muta, T, Ukaji, K, Abe, Y, Nakayama, H, et al. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. (2003) 31:1640–5. doi: 10.1093/nar/gkg251
58. Fisher, RP, and Clayton, DA. A transcription factor required for promoter recognition by human mitochondrial RNA polymerase. Accurate initiation at the heavy- and light-strand promoters dissected and reconstituted in vitro. J Biol Chem. (1985) 260:11330–8. doi: 10.1016/S0021-9258(17)39184-6
59. Shutt, TE, Bestwick, M, and Shadel, GS. The core human mitochondrial transcription initiation complex: It only takes two to tango. Transcription. (2011) 2:55–9. doi: 10.4161/trns.2.2.14296
60. Parisi, MA, and Clayton, DA. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science. (1991) 252:965–9. doi: 10.1126/science.2035027
61. Rubio-Cosials, A, Battistini, F, Gansen, A, Cuppari, A, Bernadó, P, Orozco, M, et al. Protein Flexibility and Synergy of HMG Domains Underlie U-Turn Bending of DNA by TFAM in Solution. Biophys J. (2018) 114:2386–96. doi: 10.1016/j.bpj.2017.11.3743
62. Morozov, YI, Parshin, AV, Agaronyan, K, Cheung, AC, Anikin, M, Cramer, P, et al. A model for transcription initiation in human mitochondria. Nucleic Acids Res. (2015) 43:3726–35. doi: 10.1093/nar/gkv235
63. Kukat, C, Wurm, CA, Spåhr, H, Falkenberg, M, Larsson, NG, and Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci U S A. (2011) 108:13534–9. doi: 10.1073/pnas.1109263108
64. Lee, H, Park, J, Tran, Q, Kim, D, Hong, Y, Cho, H, et al. Mitochondrial transcription factor A (TFAM) is upregulated in glioma. Mol Med Rep. (2017) 15:3781–6. doi: 10.3892/mmr.2017.6467
65. Correia, RL, Oba-Shinjo, SM, Uno, M, Huang, N, and Marie, SK. Mitochondrial DNA depletion and its correlation with TFAM, TFB1M, TFB2M and POLG in human diffusely infiltrating astrocytomas. Mitochondrion. (2011) 11:48–53. doi: 10.1016/j.mito.2010.07.001
66. Khuong-Quang, DA, Buczkowicz, P, Rakopoulos, P, Liu, XY, Fontebasso, AM, Bouffet, E, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. (2012) 124:439–47. doi: 10.1007/s00401-012-0998-0
67. Schwartzentruber, J, Korshunov, A, Liu, X-Y, Jones, DTW, Pfaff, E, Jacob, K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. (2012) 482:226–31. doi: 10.1038/nature10833
68. Wu, G, Broniscer, A, McEachron, TA, Lu, C, Paugh, BS, Becksfort, J, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. (2012) 44:251–3. doi: 10.1038/ng.1102
69. Mackay, A, Burford, A, Carvalho, D, Izquierdo, E, Fazal-Salom, J, Taylor, KR, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell. (2017) 32:520–537.e5. doi: 10.1016/j.ccell.2017.08.017
70. Siddaway, R, Canty, L, Pajovic, S, Milos, S, Coyaud, E, Sbergio, SG, et al. Oncohistone interactome profiling uncovers contrasting oncogenic mechanisms and identifies potential therapeutic targets in high grade glioma. Acta Neuropathol. (2022) 144:1027–48. doi: 10.1007/s00401-022-02489-2
71. Chen, X, Li, S, Ke, Y, Wu, S, Huang, T, Hu, W, et al. KLF16 suppresses human glioma cell proliferation and tumourigenicity by targeting TFAM. Artif Cells Nanomed Biotechnol. (2018) 46:608–15. doi: 10.1080/21691401.2018.1431654
72. Shields, JM, and Yang, VW. Identification of the DNA sequence that interacts with the gut-enriched Krüppel-like factor. Nucleic Acids Res. (1998) 26:796–802. doi: 10.1093/nar/26.3.796
73. Franco, DG, Moretti, IF, and Marie, SKN. Mitochondria transcription factor a: a putative target for the effect of melatonin on U87MG malignant glioma cell line. Molecules. (2018) 23:1129. doi: 10.3390/molecules23051129
74. Choi, YS, Kim, S, Lee, HK, Lee, K-U, and Pak, YK. In vitro methylation of nuclear respiratory factor-1 binding site suppresses the promoter activity of mitochondrial transcription factor A. Biochem Biophys Res Commun. (2004) 314:118–22. doi: 10.1016/j.bbrc.2003.12.065
75. Yao, RW, Wang, Y, and Chen, LL. Cellular functions of long noncoding RNAs. Nat Cell Biol. (2019) 21:542–51. doi: 10.1038/s41556-019-0311-8
76. Morris, KV, and Mattick, JS. The rise of regulatory RNA. Nat Rev Genet. (2014) 15:423–37. doi: 10.1038/nrg3722
77. Rackham, O, Shearwood, AM, Mercer, TR, Davies, SM, Mattick, JS, and Filipovska, A. Long noncoding RNAs are generated from the mitochondrial genome and regulated by nuclear-encoded proteins. RNA. (2011) 17:2085–93. doi: 10.1261/rna.029405.111
78. Bandiera, S, Rüberg, S, Girard, M, Cagnard, N, Hanein, S, Chrétien, D, et al. Nuclear outsourcing of RNA interference components to human mitochondria. PLoS One. (2011) 6:e20746. doi: 10.1371/journal.pone.0020746
79. Chang, DD, and Clayton, DA. Mouse RNAase MRP RNA is encoded by a nuclear gene and contains a decamer sequence complementary to a conserved region of mitochondrial RNA substrate. Cells. (1989) 56:131–9. doi: 10.1016/0092-8674(89)90991-4
80. Feng, W, Li, L, Xu, X, Jiao, Y, and Du, W. Up-regulation of the long non-coding RNA RMRP contributes to glioma progression and promotes glioma cell proliferation and invasion. Arch Med Sci. (2017) 13:1315–21. doi: 10.5114/aoms.2017.66747
81. Liu, T, Hu, J, Han, B, Tan, S, Jia, W, and Xin, Y. A positive feedback loop of lncRNA-RMRP/ZNRF3 axis and Wnt/β-catenin signaling regulates the progression and temozolomide resistance in glioma. Cell Death Dis. (2021) 12:952. doi: 10.1038/s41419-021-04245-y
82. Jin, L, Wessely, O, Marcusson, EG, Ivan, C, Calin, GA, and Alahari, SK. Prooncogenic factors miR-23b and miR-27b are regulated by Her2/Neu, EGF, and TNF-α in breast cancer. Cancer Res. (2013) 73:2884–96. doi: 10.1158/0008-5472.CAN-12-2162
83. Zhang, Y, Qu, Y, Gao, K, Yang, Q, Shi, B, Hou, P, et al. High copy number of mitochondrial DNA (mtDNA) predicts good prognosis in glioma patients. Am J Cancer Res. (2015) 5:1207–16.
84. Fliss, MS, Usadel, H, Caballero, OL, Wu, L, Buta, MR, Eleff, SM, et al. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science. (2000) 287:2017–9. doi: 10.1126/science.287.5460.2017
85. Leão Barros, MB, Pinheiro, DDR, and Borges, BDN. Mitochondrial DNA alterations in glioblastoma (GBM). Int J Mol Sci. (2021) 22:5855. doi: 10.3390/ijms22115855
86. Liberti, MV, and Locasale, JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. (2016) 41:211–8. doi: 10.1016/j.tibs.2015.12.001
87. Duraj, T, Garcia-Romero, N, Carrion-Navarro, J, Madurga, R, Mendivil, AO, Prat-Acin, R, et al. Beyond the Warburg effect: oxidative and glycolytic phenotypes coexist within the metabolic heterogeneity of glioblastoma. Cells. (2021) 10:202. doi: 10.3390/cells10020202
88. Klein, K, He, K, Younes, AI, Barsoumian, HB, Chen, D, Ozgen, T, et al. Role of mitochondria in cancer immune evasion and potential therapeutic approaches. Front Immunol. (2020) 11:573326. doi: 10.3389/fimmu.2020.573326
89. Porporato, PE, Filigheddu, N, Pedro, JMB, Kroemer, G, and Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. (2018) 28:265–80. doi: 10.1038/cr.2017.155
Keywords: mitoepigenetics, epigenetics, glioma, mitochondria, glioblastoma, mtDNA, methylation, noncoding RNA
Citation: Grady CI, Walsh LM and Heiss JD (2023) Mitoepigenetics and gliomas: epigenetic alterations to mitochondrial DNA and nuclear DNA alter mtDNA expression and contribute to glioma pathogenicity. Front. Neurol. 14:1154753. doi: 10.3389/fneur.2023.1154753
Edited by:
John Bianco, Princess Maxima Center for Pediatric Oncology, NetherlandsReviewed by:
Hernando Lopez-Bertoni, Johns Hopkins University, United StatesCopyright © 2023 Grady, Walsh and Heiss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clare I. Grady, Y2xhcmUuaS5ncmFkeUBtZWRzdGFyLm5ldA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.