
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 28 February 2023
Sec. Multiple Sclerosis and Neuroimmunology
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1137998
This article is part of the Research Topic MOGAD, Current Knowledge and Future Trends View all 13 articles
 Osman Corbali1,2
Osman Corbali1,2 Tanuja Chitnis1,2*
Tanuja Chitnis1,2*Myelin Oligodendrocyte Glycoprotein Antibody Disease (MOGAD) is a spectrum of diseases, including optic neuritis, transverse myelitis, acute disseminated encephalomyelitis, and cerebral cortical encephalitis. In addition to distinct clinical, radiological, and immunological features, the infectious prodrome is more commonly reported in MOGAD (37–70%) than NMOSD (15–35%). Interestingly, pediatric MOGAD is not more aggressive than adult-onset MOGAD, unlike in multiple sclerosis (MS), where annualized relapse rates are three times higher in pediatric-onset MS. MOGAD pathophysiology is driven by acute attacks during which T cells and MOG antibodies cross blood brain barrier (BBB). MOGAD lesions show a perivenous confluent pattern around the small veins, lacking the radiological central vein sign. Initial activation of T cells in the periphery is followed by reactivation in the subarachnoid/perivascular spaces by MOG-laden antigen-presenting cells and inflammatory CSF milieu, which enables T cells to infiltrate CNS parenchyma. CD4+ T cells, unlike CD8+ T cells in MS, are the dominant T cell type found in lesion histology. Granulocytes, macrophages/microglia, and activated complement are also found in the lesions, which could contribute to demyelination during acute relapses. MOG antibodies potentially contribute to pathology by opsonizing MOG, complement activation, and antibody-dependent cellular cytotoxicity. Stimulation of peripheral MOG-specific B cells through TLR stimulation or T follicular helper cells might help differentiate MOG antibody-producing plasma cells in the peripheral blood. Neuroinflammatory biomarkers (such as MBP, sNFL, GFAP, Tau) in MOGAD support that most axonal damage happens in the initial attack, whereas relapses are associated with increased myelin damage.
MOG is a transmembrane protein found on the outer surface of the central nervous system myelin and a marker of mature oligodendrocytes (1, 2). It constitutes only a small portion of the myelin (0.05%), and its possible roles include cell adhesion, microtubule stability, and receptor function (1, 3, 4).
High titers of autoantibodies targeting MOG are identified in various demyelinating diseases, including optic neuritis, transverse myelitis, acute disseminated encephalomyelitis (ADEM), and cerebral cortical encephalitis. These are now recognized as a spectrum of diseases associated with MOG antibodies, MOGAD (5). Despite heterogeneous presentation and clinical overlap between MOGAD, multiple sclerosis (MS), and neuromyelitis optica spectrum disease (AQP4-IgG+, NMOSD), distinctive radiologic, pathological, lab, and clinical features of MOGAD have been identified (Table 1), and most recently an international MOGAD diagnostical criteria has been proposed (63).
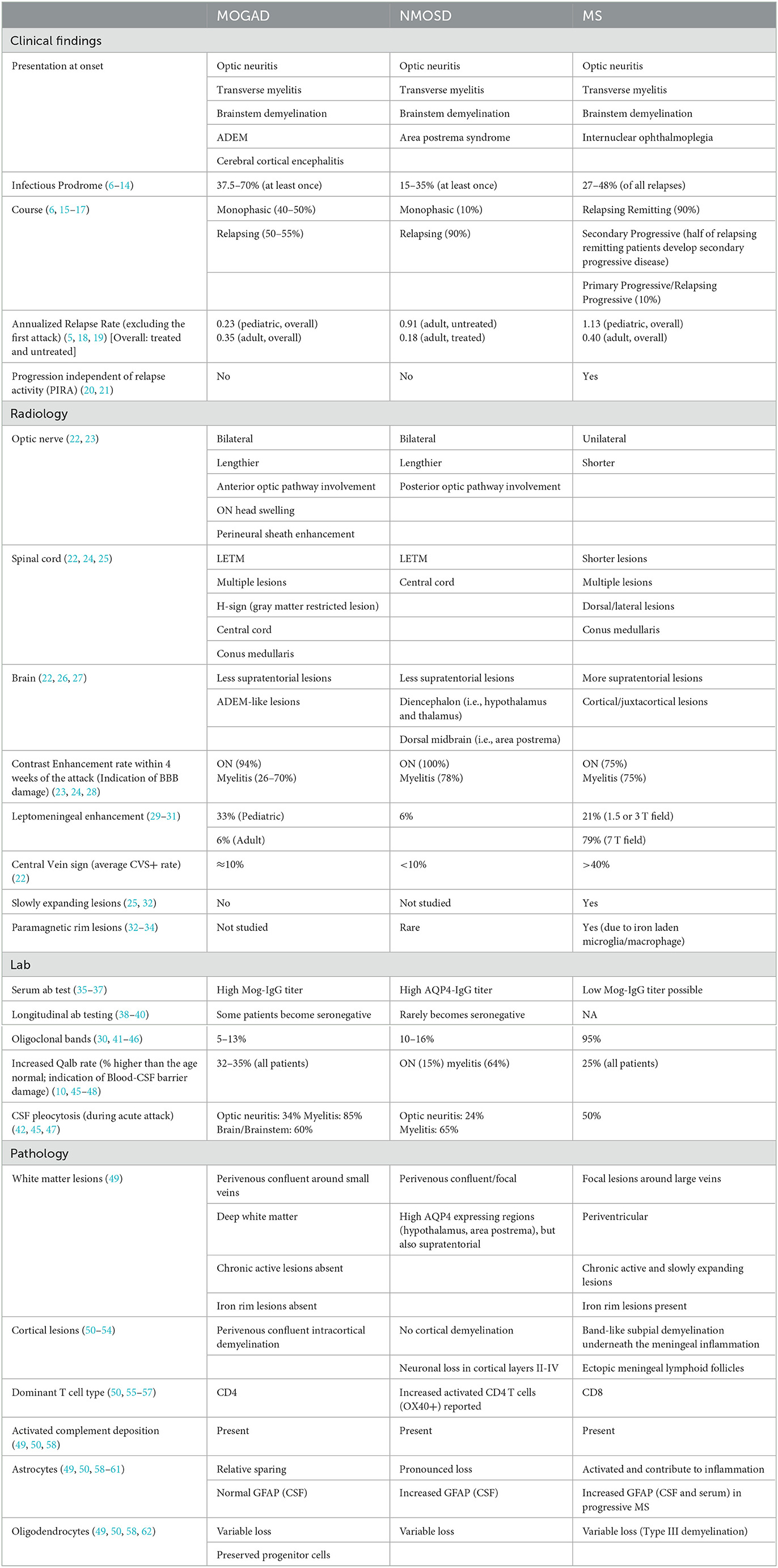
Table 1. Distinctive features of MOGAD.
Autoimmunity in MOGAD starts at the periphery by activation of T cells and production of autoantibodies and eventually transfer of these immune mediators into the CNS (5, 38). How humoral immunity and cellular immunity cooperate in MOGAD pathogenesis is an intriguing subject. This review will highlight major distinctive clinical, radiological, and histopathological features of MOGAD and summarize how different immune compartments contribute disease pathogenesis. For this purpose, we will use evidence from human and animal studies such as MOG-induced experimental autoimmune encephalomyelitis (EAE) models, as some of these models closely resemble MOGAD compared to MS due to relapsing and remitting course (vs. progressive disease) and MOG as the autoimmune trigger (64–66).
Distinctive clinical, radiological, and histopathological features and laboratory findings of MOGAD are summarized below (Table 1).
MOGAD most commonly presents as bilateral optic neuritis, transverse myelitis, ADEM, or, less commonly, cerebral cortical encephalitis. Brainstem demyelination could also occur in MOGAD; however, area postrema syndrome or internuclear ophthalmoplegia is associated with NMOSD or MS, respectively (5, 67).
The monophasic course is more common in MOGAD (40–50%), and the remaining half of the MOGAD cases experience a relapsing course, which is associated with persistent high titers of MOG-IgG (15, 16). In contrast, most NMOSD patients (90%) have a relapsing course (6). In MS, most patients first experience a relapsing-remitting phase (90%), half of which develop secondary progressive disease (17). Progression in MOGAD or NMOSD is relapse dependent; however, progression independent of relapse activity is well-established with MS (20, 21).
Pediatric MOGAD is not more aggressive than adult-onset MOGAD. The overall annualized relapse rate (ARR) in MOGAD, excluding the first attack, is 0.23 in pediatric and 0.35 in adult MOGAD patients (5). This is different from MS, where pediatric MS patients display a more inflammatory phenotype and therefore have higher ARR than adult-onset MS patients, with an ARR of 1.13 in pediatric-onset vs. 0.40 in adult-onset MS (18).
An infectious prodrome (at least once during the disease course) is commonly reported with MOGAD, varying from 37 to 70% (7–10). MOG-IgG+ optic neuritis patients had 37% and 67% preceding infection in two series (3/8 and 6/9) (7, 8). In MOG-IgG+ ADEM patients preceding infection was present in 70% (12/17, including three patients with vaccinations) (9). Jarius et al. reported an infectious prodrome in 40% (15/37) of MOGAD patients, which included mostly optic neuritis or myelitis patients but also some ADEM or cerebellitis cases (10). In NMOSD, the infectious prodrome is reported in 15–35% (6, 11, 12). Earlier studies with multiple sclerosis showed that 27–48% of all MS relapses were associated with infections if patients followed longitudinally (13, 14).
Optic neuritis is bilateral and lengthier (compared to MS), and the anterior optic pathway is more commonly involved (vs. posterior in NMOSD) in MOGAD (Figure 1) (22, 23, 68). Optic nerve head swelling and perineural sheath enhancement are other typical features of MOGAD, indicating increased blood—optic nerve barrier breakdown (22, 68).
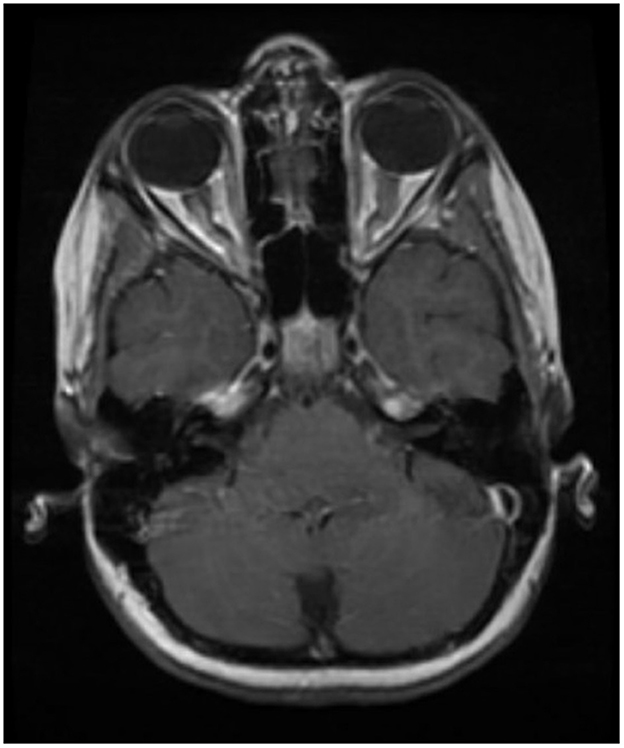
Figure 1. Typical optic nerve involvement in MOGAD. Bilateral and longer lesions involving anterior parts of the optic nerve.
Spinal cord involvement is seen as longitudinal extensive transverse myelitis (LETM) in MOGAD and NMOSD (22, 24, 25). Multiple spinal cord lesions are frequent in MOGAD and MS (both >60%) (24). Gray matter restricted (H sign) or central cord involvement is a typical appearance of MOGAD lesions on axial MRI (24, 25). On the other hand, shorter and dorsal/lateral spinal cord lesions are more typical of MS (25). Conus medullaris involvement is seen in MOGAD or MS (22).
Typical brain involvement in MOGAD is ADEM-like fluffy lesions (25). The number of supratentorial lesions is fewer in MOGAD and NMOSD than in MS (22). High AQP4 expressing regions such as diencephalon (hypothalamus and thalamus) or dorsal midbrain (area postrema) are commonly affected in NMOSD (26). In MS, more supratentorial lesions are typically found in cortical, juxtacortical, or periventricular areas (27).
Contrast (Gadolinium) enhancement is a measure of BBB breakdown commonly found in MOGAD lesions. Within 4 weeks of the symptom onset, ON has enhancing pattern in most MOGAD (94%), NMO (100%) and MS (75%) patients (23). Myelitis, on the other hand, has an enhancement rate of 27–70% of MOGAD patients and around 75% of NMO or MS patients within 4 weeks of the symptom onset (24, 28).
Leptomeningeal enhancement, an indicator of leptomeningeal inflammation, was less frequently reported in MOGAD than in MS (29–31, 69). Gadde et al. reported the presence of LME in 33% (7/21) of a pediatric cohort. In comparison, Cobo-Calvo et al. reported in 6% (3/49) of their adult cohort (29, 30). Further studies are needed to determine if LME prevalence is higher (using 7T MRI) and if LME presence is associated with relapse activity in MOGAD. In MS, LME presence is reported as 79% with 7T MRI, while with lower field (1.5 or 3T MRI), prevalence is 21% (31). In NMOSD, it is also infrequent (6%) (31).
Central vein sign, defined as lesions with central vein identifiable by MRI, is commonly seen in MS (>40%), while their frequency is much lower in MOGAD (≈10%) or NMOSD (< 10%) (22).
Slowly expanding lesions are not present in MOGAD, while present in MS (25, 32). Paramagnetic rim lesions (PRL) associated with iron-laden microglia and macrophages, are present in MS; however, they have not been investigated in MOGAD (32–34).
White matter lesions in MOGAD exhibit a confluent pattern around small veins, while in MS, focal lesions form around larger veins detectable by MRI (central vein sign) (50). Chronic active or slowly expanding lesions and iron rim lesions are absent in MOGAD while present in MS (50).
Cortical lesions in MOGAD also have perivenous confluent patterns and intracortical demyelination (50). In MS, subpial demyelination underneath the meningeal inflammation is common (51, 52). There are also ectopic lymphoid follicles found in MS, which are important aspect of chronic and progressive inflammation. In NMOSD, there is no cortical demyelination, while there is a neuronal loss in cortical layers II-IV (53, 54).
The dominant T cell type is CD4 in MOGAD, whereas CD8 is in MS (50, 55, 56). Astrocytes are spared in MOGAD, while pronouncedly decreased in NMOSD, and activated and proinflammatory in MS (49, 50, 58). Differential astrocyte involvement in these three diseases is also supported by GFAP levels in serum and CSF (59–61). GFAP-CSF levels are increased in the NMOSD patients but not in MOGAD compared to HC (60). In MS, progressive patients have higher GFAP both in serum and CSF compared to RRMS and HC (59). Conversely, oligodendrocytes are variably lost in all three diseases, while in MOGAD, progenitor cells are preserved as they do not yet express MOG (49, 50, 58, 62).
Oligoclonal band presence is low in MOGAD (5–13%), similar to NMOSD (10–16%) (30, 41–43, 45, 46). In MS, however, OCB positivity is found in the majority of the cases (95%) (44).
CSF pleocytosis in MOGAD is quite common during relapses and even higher in the spinal cord (85%) or brain/brainstem (60%) involvement compared to optic neuritis (34%) (42). In NMOSD, a similar trend is present, with CSF pleocytosis in 24% of optic neuritis and 65% of myelitis (45). In MS, 50% of the patients have pleocytosis during relapses (47).
Increased albumin CSF/serum ratio (QAlb), a measure of blood-CSF barrier dysfunction, is also a feature of MOGAD. In two separate studies, almost one-third of MOGAD patients (32 and 32.4%) had increased albumin CSF/serum ratio (QAlb) (10, 46). This ratio was even higher in patients with a history of a spinal cord, brain, or brainstem involvement (10/21; 47.6%) (10). Elevated QAlb values are reported at a similar frequency with MS (29.5%, n = 606) (70). On the other hand, QAlb levels are reported variably for NMOSD (increased in up to 50–80% of patients), which implies implying blood-CSF barrier dysfunction in MOGAD may not be as severe as it is in NMOSD (45, 46, 71).
CSF cytokine/chemokine profile of MOGAD shows increased proinflammatory cytokines (Figure 2, created with BioRender.com), including Th1 (TNF-α, IFNγ), Th2 (IL13), Th17 (IL6, IL8, G-CSF, GM-CSF), Treg (IL10) and B cell (CXCL12, APRIL, BAFF, CXCL13, CCL19) related and other (IL-1ra, MCP-1, MIP-1a) cytokines/chemokines (72, 73).
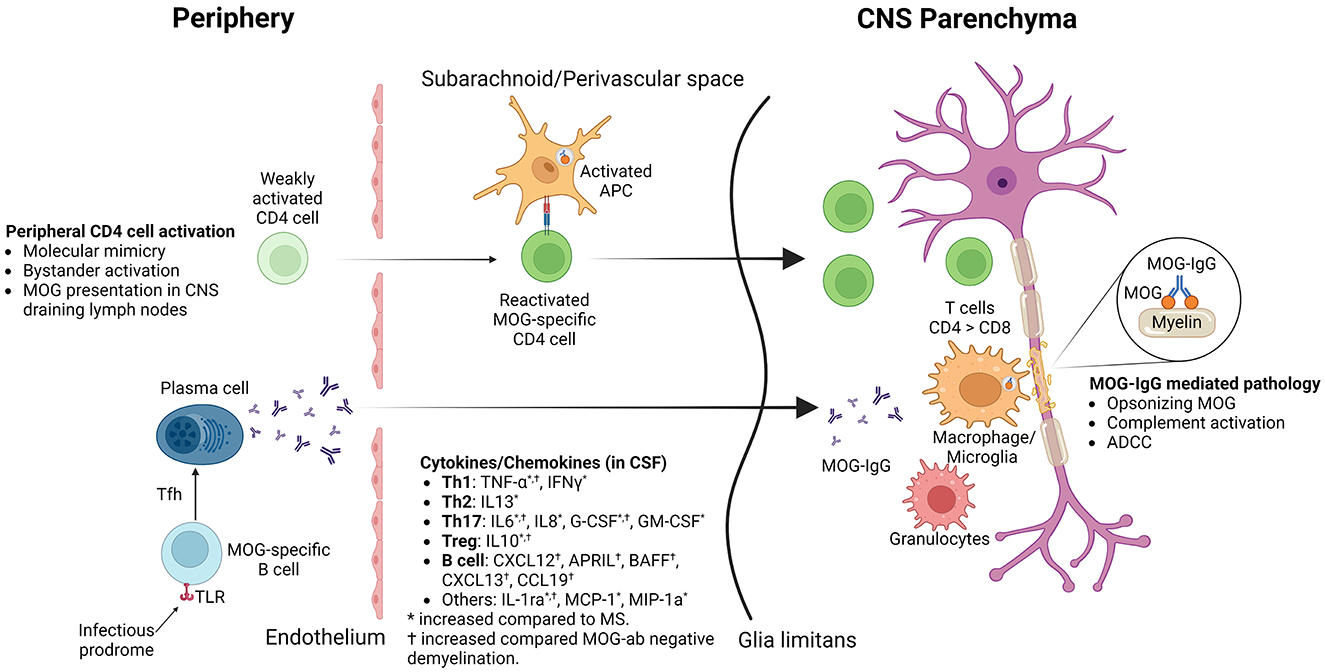
Figure 2. Pathophysiological actors in MOGAD.
The prevailing concept for autoimmunity in MOGAD is the outside-in model, where autoantibodies and activated immune cells in the peripheral blood cross the blood-brain barrier at the time of attack/relapse (Figure 2) (41, 74).
The central tolerance toward MOG may not be well developed in the thymus, preventing the elimination of MOG reactive T cells by negative selection (1, 75, 76). In the thymus, the expression of a self-antigen in the epithelial cells eliminates lymphocytes with a strong affinity to a self-antigen (central tolerance) and also allows some self-reactive T cells to develop into Tregs (peripheral tolerance) (77). MOG expression in the human thymus is variably reported. In two studies analyzing the thymus, MOG RNA was not detected, while in another study, MOG was detected in isolated medullary thymic epithelial cells (78–81). There is no protein-level study assessing MOG expression in human thymic tissue. Even if MOG expression in the thymus is low or present, central tolerance is not a perfect process, and peripheral tolerance mechanisms are needed to suppress self-reactive lymphocytes. Peripheral tolerance mechanisms include anergy or apoptosis of self-reactive T cells through the absence of costimulatory molecules or the presence of inhibitory molecules (such as PD1 or CTLA) and regulatory T cells (77). From MOG-induced EAE models and human MOGAD, we know that tolerance against MOG could be disrupted.
MOG reactive T cells, present due to a compromised tolerance against MOG, could be activated upon antigen-specific or non-specifically through mechanisms such as MOG peptide presentation, molecular mimicry, or bystander activation. MOG peptides could be present in the periphery, such as in cervical lymph nodes draining CNS, or rarely in a tumor expression MOG (82, 83). MOG antibodies, on the other hand, could facilitate recognition of trace amounts of MOG present in the periphery leading to T cell activation (84). Infections could cause bystander activation and molecular mimicry. Milk protein Butyrophilin and small Hepatitis B surface antigen are reported to have cross-immunoreactivity with MOG; however, pathophysiological consequences of these molecular mimicries have not been established (85, 86).
Genetic risk factors associated with the autoimmune etiology of MOGAD are not widely explored. HLA genotyping studies did not identify a significant allele in two Dutch and UK cohorts (87, 88). However, a study in the Chinese Han cohort observed that DQB1*05:02 and DRB1*16:02 alleles and DQB1*05:02-DRB1*16:02 haplotype were more frequent in pediatric-onset MOGAD patients and DQB1*05:02-DRB1*16:02 haplotype was associated with higher initial EDSS and relapse risk (89).
An infectious prodrome is commonly reported in MOGAD patients (37.5 to 70%) (7–10). An infectious prodrome could stimulate the underlying autoimmune processes by bystander activation, molecular mimicry, and epitope spreading. Infections could also disrupt peripheral tolerance by increasing costimulatory molecules and MHC-II expression on antigen-presenting cells (APCs) (90). This would lead to increased avidity of the interaction between APCs and self-reactive CD4+ T cells, which would otherwise go to anergy or apoptosis due to weak T cell stimulation.
Bystander activation of self-reactive B and T cells is one mechanism that could explain infectious prodrome commonly preceding attacks. In fact, pro-inflammatory cytokines that increase during infections, such as IL6 and TNFα, are also found to increase in CSF of MOGAD patients (38, 72, 73). In addition, Toll-Like receptor (TLR) activation caused by viruses could convert MOG-specific B cells into MOG-ab secreting plasmablasts (91). Separately, infections could directly or indirectly affect BBB as well (92, 93).
Since the COVID-19 breakout, multiple case studies reported COVID-19 infection preceding MOGAD onset (94, 95). The median time between COVID-19 to MOGAD diagnosis was 6 days (range: −7 to 45 days), while in a few cases, MOGAD diagnosis preceded or was concomitant with COVID-19 (94, 95). Further studies will show if the MOGAD incidence rate has increased after the pandemic.
The serum level of MOG IgG is an essential diagnostic and clinical biomarker. There is no consensus cut-off value for a diagnostic titer, which changes from center to center (35). Autoantibodies detected in MOGAD patients are of IgG1 type (96). MOG IgM levels do not correlate with anti-MOG IgG levels and could provide false positive results (96, 97). Persistent MOG-IgG positivity is associated with increased relapse risk (9). In monophasic MOGAD, MOG-IgG titers decrease over time, whereas in relapsing MOGAD, MOG-IgG titers tend to stay high (98).
Paired serum and CSF MOG-IgG positivity is found in more than half (56%) of the MOGAD patients (99). Some MOGAD patients are MOG-IgG seronegative and CSF positive, and CSF-restricted MOG-IgG may not always coexist with OCB positivity or elevated IgG index in the CSF (71). Therefore, in a strong clinical context (such as seronegative NMOSD or ADEM), CSF testing could help with MOGAD diagnosis (99, 100). Furthermore, CSF MOG-IgG positivity is associated with worse outcomes (99).
Detection of MOG-IgG antibodies in the serum by a live cell-based assay is the gold standard for diagnosis (35). Patient serum samples are incubated with live HEK293 cells expressing full-length MOG protein on their membrane, followed by secondary staining with anti-human IgG (H+L or Fc) or IgG1 (Fc) secondary antibodies. The analysis is done either by immune fluorescence microscopy or flow cytometry (35, 36).
Antibody response to MOG could be monoclonal or polyclonal. Mayer et al. tested the human MOG IgG binding pattern for seven different mutant human MOG proteins and mouse MOG proteins (101). They found that half of the patients showed decreased binding only to P42S (Proline to Serine) mutant, whereas about a third of the patients showed decreased binding to multiple mutants. These results indicated that an epitope containing P42 (proline at position 42) is the primary target of MOG IgG in half of the patients, and many patients have a polyclonal antibody response. Interestingly, immunoreactive epitopes are temporally stable, and there is no evidence of intramolecular epitope spreading (96, 101).
Pathogenicity of patient-derived purified MOG-IgGs was shown by Spadaro et al. by intrathecally injecting human MOG IgGs in an adoptive transfer EAE model (induced by MBP or MOG-specific T cells transferred to Lewis rats) (102). In this experiment MOG-antibodies were not pathogenic alone, and provided a second hit when interacted with T cells. When coupled with MBP-specific T cells, which are alone encephalitogenic and disrupt BBB, MOG abs mediated MS type II demyelination, characterized by complement (C9neo) and immunoglobulin deposition. On the other hand, when coupled with MOG-specific T cells, which do not induce clinical disease by themselves, MOG abs enhanced T cell recruitment and activation.
The source of the MOG abs in the CNS is mostly peripheral, although sometimes intrathecal production might be possible (41, 71, 74, 100). Through an impaired BBB (provided by activated T cells, infections, coexisting autoantibodies etc.), these MOG antibodies could enter the perivascular spaces and CNS, where they could contribute to disease pathology.
Suggested mechanisms for MOG antibody pathogenicity include opsonization of MOG, complement activation, antibody-dependent cellular cytotoxicity (ADCC), and anti-MOG ab-induced intracellular signaling cascade (3, 50, 84, 103, 104).
MOG-IgG opsonizes MOG and could activate myeloid antigen-presenting cells (APC) through Fc receptor binding (84, 104). These activated APCs can further stimulate MOG-specific T cells in the periphery or in perivascular spaces in the CNS (56).
The role of complement activation in MOGAD is still debated. IgG1 is a complement-fixing subtype, and there is evidence of C9neo deposition in the MOGAD histopathology (50). Furthermore, oligodendrocytes express relatively less surface complement regulatory proteins such as CR1, MCP, and HRF, making them more vulnerable to complement activation (106). In a multinational cohort study, serum-activated complement proteins (C3a, C5a, and Bb) were elevated in MOGAD patients compared to control groups (107). However, there was no correlation between activated complement protein levels in the serum and the clinical presentation (relapsing vs. monophasic or ADEM vs. ON vs. TM). On the other hand, complement activation in MOGAD is to a lesser extent, compared to AQP4 NMO (108). This might be because MOG-IgG has a bivalent binding pattern (both Fab subunits should bind to MOG), whereas AQP4-IgG has a monovalent binding pattern, which activates complement more efficiently (103, 108–111). This also indicates that anti-complement therapy may not be as successful as in NMO for MOGAD.
Another pathogenic mechanism is ADCC. Brilot et al. showed that MOG IgG binding to MOG induces Natural Killer cell-mediated killing of MOG-expressing cells in vitro (104).
MOG abs also have a direct downstream effect on oligodendrocytes (3). When Mog ab binds alone, it activates MAPK and AKT survival pathways and increases intracellular calcium levels, whereas cross-linking of the MOG abs leads to the activation of stress-related pathways and reduced cytoskeletal integrity.
B cells are part of the disease pathogenesis through the production of MOG antibodies; however, there is much to discover about their contribution to the MOGAD. For example, anti-CD20 therapy is relatively ineffective in most patients. In a large cohort, rituximab decreased the relapse rate by 37%, and only 33% of patients remained relapse-free after 2 years (105). Failure of B cell depleting therapy suggests alternative pathogenic mechanisms in these patients, which could be used for enhanced T cell activation and MOG ab production. Serum MOG ab levels and circulating MOG-specific B cells did not correlate in MOGAD patients, raising the possibility of different MOG ab sources (for example, CD20- plasma cells) (91).
CXCR4 expression is increased in CD19+ B cells in PBMCs from MOGAD patients (112). Interestingly, CXCL12 (also called stromal cell-derived factor 1), a ligand of CXCR4, is also found increased in the CSF (compared to MOG ab- demyelination) and serum (compared to MS) of MOGAD patients (73, 113). As, CXCL12/CXCR4 axis is related with chemotaxis, increased CXCL12 could contribute immune cell infiltration (such as T, B, and monocytes) in the MOGAD (114). B cells are present in the lesions of MOGAD, although fewer than T cells (56). However, the source of MOG-IgGs is presumably the periphery, except in some patients with intrathecal production or CSF-restricted positivity (41, 50, 56, 71, 74, 100). Therefore, it is not certain if B cells cross BBB during MOGAD relapses and contribute to lesion formation within the CNS.
Altered regulatory B cells in MOGAD are reported in a study (115). Decreased Breg/Bmem ratio and decreased IL10+ CD19+ cell frequency are accompanied by increased circulating follicular T helper cells in MOGAD (115).
T cells play a key role in MOGAD pathogenesis. First, MOG abs are IgG1 phenotype, so follicular T helper cells are essential for MOG-specific B cell class switching (96). Second, MOG abs are not pathogenic alone unless they are coupled with MOG-specific or encephalitogenic T cells (102). CD4+ T cells are the dominant inflammatory cell type in the lesions and have an essential role in disrupting BBB and creating a proinflammatory environment (50, 116).
MOG-specific T cells are first activated peripherally (90). Due to inadequate tolerance toward MOG, MOG-specific T cells could be present in the blood. Infections, molecular mimicry, and MOG peptide presentation could facilitate the activation of self-reactive T cells. Then, peripherally activated CD4+ T cells cross BBB and are reactivated by the MOG-laden APCs in the perivascular or subarachnoid spaces (56). This reactivation is followed by endothelial and microglial activation, allowing more T cells and autoantibodies to enter the perivascular space (90, 116).
The strength of T cell reactivation and chemokines in the CNS can facilitate parenchymal infiltration of the CD4+ T cells (90, 117, 118). Anti-MOG abs help APCs (such as macrophages) to present native MOG protein to T effector cells and enhance the reactivation of effector T cells in the CNS (84, 119). T cells could be reactive to different epitopes of MOG protein. Most immunogenic MOG epitopes were previously determined in EAE models and tested MS patients (120, 121). These include p35–55, p119–130, p181–195, and p186–200. The following study tested nine different MOG peptides (p1–20, p35–55, p64–80, p81–96, p99–107, p119–130, p181–195, p186–200, and p205–214) in MOGAD, AQP4+ NMO, MS, and HC, but didn't find a difference of MOG-specific T cells between any groups based on CFSE assay (122). This could be explained by the lack of central tolerance and, therefore, the presence of MOG reactive T cells even in HC. Another possibility is that native full MOG protein is required for an optimal T-cell response.
Th17 responses are higher in MOGAD patients. Our lab used different MOG peptides for in vitro stimulation and showed MOGAD patients had more IL17+ and IL17+IFNγ+ (double positive) central memory cells than healthy controls (123). When relapse and remission MOGAD samples were compared, there was an increased proportion of IL17+, IFNγ+, and IL17+IFNγ+ CMCs after stimulation with several individual MOG peptides in MOGAD patients at the time of relapse (123). Later, Horellou et al. stimulated PBMCs with recombinant full-length MOG protein (rhMOG) and observed increased IL17+ CD4 T cells in non-relapsing (monophasic) MOGAD patients upon rhMOG stimulation, but not in relapsing MOGAD or MS (124).
Tregs play an important role in peripheral immune tolerance. Horellou et al. reported an increased CD4+ Foxp3+ Treg population in the non-relapsing MOGAD group and decreased CD45RA-Foxp3+ Treg population in MOGAD relapsing group upon rhMOG stimulation (124). They hypothesized that this opposite response to MOG stimulation could contribute to the relapse mechanism. Besides, we don't know if Tregs are functional in MOGAD; however, inflammatory milieu (high IL6 and TNF) in MOGAD could render Tregs ineffective with suppressing (72, 113, 125).
Macrophage (CD68+) and granulocyte (hematoxylin-eosin) infiltration is reported in MOGAD lesions, especially in the perivascular demyelinating areas (50, 56). The presence of phagocytic macrophages indicates active demyelination, and MOG-laden macrophages could further activate T cells in the perivascular spaces (56).
IL-1β and IL-12p70, monocyte or dendritic cell-related cytokines, are increased in the serum of MOGAD patients (126). IL-1β levels were highest in the acute stage and lower in the chronic phase, indicating monocyte/macrophages play a role in the acute demyelinating stage (126). IL-1β also affects the permeability of endothelial cells in vitro human BBB model (127).
In a recent study of three patients (1 MOGAD, 1 RRMS, and 1 Healthy control), single-cell RNA sequencing of PBMCs revealed changes in monocyte signature in MOGAD compared to RRMS or HC, however, a larger, age and sex-matched cohort is needed to confirm these findings (112).
The increased neutrophil-to-lymphocyte ratio (NLR) in serum is also reported in MOGAD, with relapse samples having higher NLR values than the remission sample (128). This biomarker requires caution as many confounding factors, such as hospitalization and steroid treatment, could affect NLR.
Epitope spreading is not established in MOGAD, however, coexisting antibodies have been investigated. Kunchok et al. tested 17 neuronal IgGs in the CSF and serum of pediatric and adult MOGAD patients and found NMDA-R-IgG is the most frequent coexisting autoantibody (4% in children and 7% in adults) (129). On the other hand, anti-Aqp4 IgG and anti-MOG IgG rarely coexist (0.06%) (130).
Serum-IgG from demyelinating animal models or patients could also affect BBB permeability by affecting pericyte function and lymphocyte adhesion molecule (LAM) expression on endothelial cells (131, 132). Interestingly, anti-GRP78 antibodies were reported to be commonly present in acute MOGAD patients (10/15, 67%) and caused BBB dysfunction through increased LAM expression in endothelial cells and NF-κB activation (133).
In summary (Figure 2), MOGAD pathophysiology is driven by acute attacks during which T cells and MOG antibodies cross BBB. Initial activation of T cells in the periphery is followed by reactivation in the subarachnoid/perivascular spaces by MOG-laden APCs and inflammatory CSF milieu, which enables T cells to infiltrate CNS parenchyma. CD4 T cells, unlike CD8 in MS, are the dominant T cell type found in lesion histology. Granulocytes, macrophages/microglia, and activated complement are also found in the lesions, which could contribute to demyelination during acute relapses. MOG antibodies potentially contribute to pathology by opsonizing MOG, complement activation, and antibody-dependent cellular cytotoxicity. Stimulation of peripheral MOG-specific B cells through TLR stimulation or T follicular helper cells might help B cells differentiate into MOG antibody-producing plasma cells in the peripheral blood.
Different clinical presentations and lesion distribution seen in MOGAD is an intriguing topic. What is the difference between MOG-IgG-related optic neuritis, transverse myelitis, and tumefactive lesions in ADEM?
Higher antibody levels in serum can favor spinal cord involvement against the optic nerve. Jarius et al. reported that MOG antibody titers are higher during relapses compared to remission, and relapses involving myelitis have higher titers of MOG antibodies compared to isolated optic neuritis (48).
Expression levels of MOG protein across different parts of the CNS could contribute to differential lesion involvement. For example, Bettelli et al. reported that 2D2 TCR transgenic mice, which have MOG-specific T cell receptors, developed spontaneous autoimmune optic neuritis but not spinal cord lesions, which they associated with higher MOG expression in the optic nerve compared to the spinal cord (134). Comparative expression of human MOG protein in optic nerve and CNS has not been reported, although MOG expression in different parts of the human brain and spinal cord has been variably reported in the human protein atlas (80, 135). Consequently, we do not know if the most common lesion location (optic nerve in adults, brain in children) relates to MOG expression level in those tissues.
The Th17:Th1 ratio could affect lesion topography. PBMCs from MS patients were stimulated with MOG or MBP proteins, and higher Th17:Th1 upon MOG (recombinant human) stimulation was associated with spinal cord involvement (136). Interestingly, epitope-specific T cell functional avidity help determine Th17:Th1 ratio in MOG peptide-induced EAE model (C3HeB/Fej mice, p35–55, p79–90, p97–114 MOG peptides) (137). So, maybe also in MOGAD, different epitope/TCR reactivity in T helper cells brings differential Th17:Th1 balance, affecting the lesion topography.
The quantitative relationship between inflammatory T cells and autoimmune antibodies could explain the size and structure difference of lesions in MOGAD (50, 138). Lassmann et al. tested this hypothesis with encephalitogenic T cells (MBP-specific) and MOG antibodies in an EAE model. Higher numbers of T cells in the presence of MOG abs induced an ADEM-like phenotype with ubiquitous perivenous demyelination all over the brain stem and spinal cord and partly in the brain. In contrast, low T cell and high MOG ab titer induced focal demyelinating plaques like in MS.
Potential biomarkers investigated with MOGAD are listed below (Table 2). First, MOG-IgG is both a diagnostical and prognosis biomarker. High titers are needed for MOGAD diagnosis, and seronegative conversion is associated with decreased relapse risk (9). Furthermore, relapsing MOGAD patients have higher MOG-IgG titer at remission compared to monophasic MOGAD (139). Treatments such as IVIG, Plasmapheresis, or FcRn blockers target pathogenic antibodies in the blood, including MOG-IgG. IVIG has been used commonly for maintenance treatment in MOGAD, and potential therapeutic mechanisms include increasing clearance of pathogenic antibodies by saturating neonatal FcR (FcRN) and blockade of activating FcγRs (144). Similarly, anti-FcRn antibodies also increase the pathogenic ab clearance, and currently, rozanolixizumab, an anti-FcRn agent, is in phase 3 clinical trial for relapse prevention in MOGAD (NCT05063162).
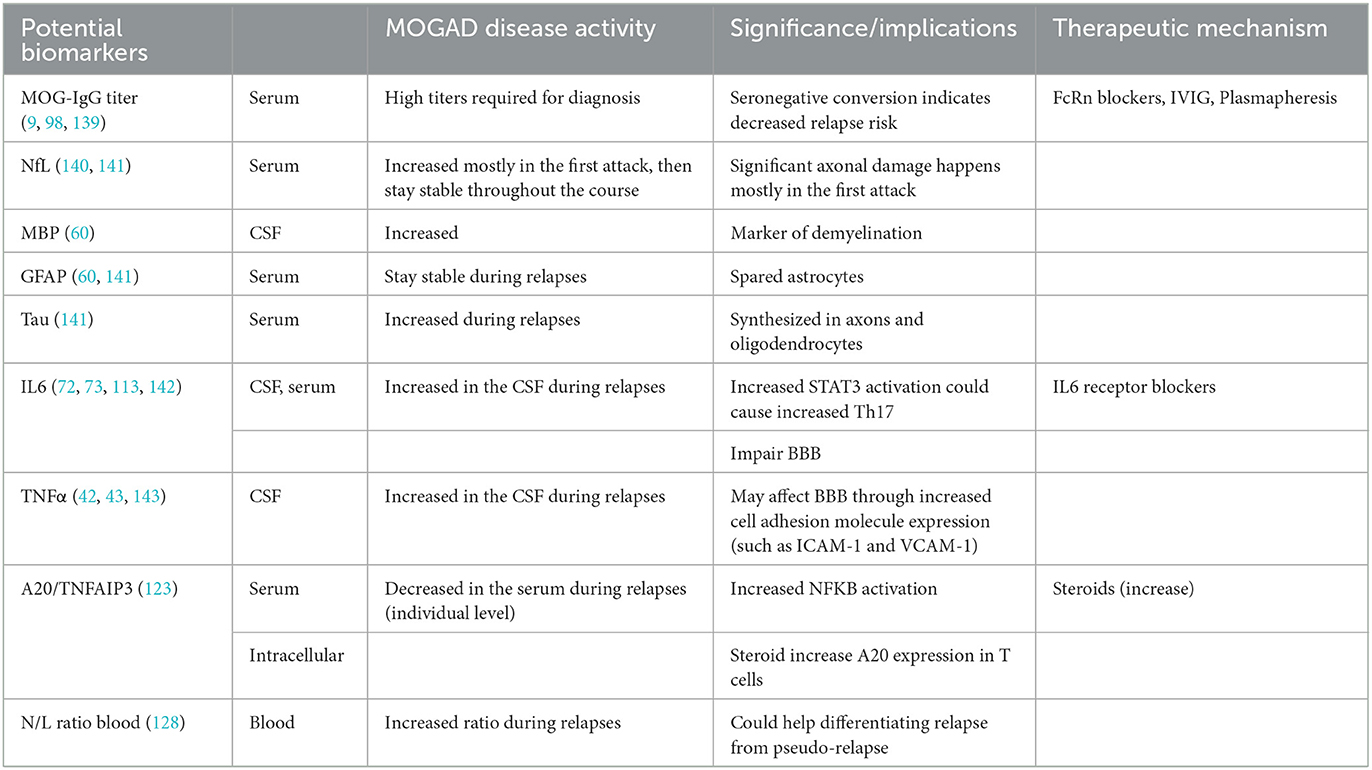
Table 2. Biomarkers investigated with MOGAD.
Serum NfL (sNfL) level is a biomarker for axonal damage and correlates with relapse activity in MS. A recent study evaluated longitudinal sNfL values from 18 MOGAD patients and found that median sNfL levels at the onset are higher compared to age-matched HC (140). Most follow-up sNfL values stayed stable or decreased over time, including relapse serum samples of 6 patients. In the following study, sNFL levels were not different between relapse and remission MOGAD samples (141). This observation supports that significant axonal damage happens in the first clinical attack (30, 140).
Increased MBP (CSF) and Tau levels (serum) in MOGAD suggest that there is myelin and oligodendrocyte damage (60, 141). Increased Tau levels in the relapse, but not sNFL, may support that the source of Tau could be damaged oligodendrocyte processes, not axons (141).
Serum GFAP levels are stable during relapses in MOGAD (141). This finding is compatible with the spared astrocytes seen in the biopsy. In NMOSD, astrocyte damage is pronounced and serum GFAP levels are increased during relapses (141). In MS, increased GFAP is seen with progressive disease (145).
IL6 and TNFα levels are increased during relapses in the CSF of MOGAD patients (43, 44). Recently, increased serum IL6 levels are also reported in MOGAD (113). Increased IL6 is associated with Th17 differentiation through IL6-STAT3 pathway and impaired BBB (142). Recently, a multinational study showed that IL6 receptor blockade with tocilizumab is therapeutically effective and safe in MOGAD and NMOSD (146). Increased TNFα, on the other hand, could also affect BBB permeability through leukocyte adhesion molecule expression such as ICAM-1 and VCAM-1 (143). Although, steroids may decrease TNFα synthesis, we do not have information about targeted therapies such as TNF receptor blocking, while few cases in the literature are diagnosed with MOGAD while on anti-TNF treatment due to preexisting other autoimmune diseases (147, 148).
We recently reported that A20, a negative regulator of the NF-κB pathway, is decreased in the serum during relapses on an individual level (123). Interestingly, steroids increase A20 expression in CD4 T cells. As well known, MOGAD patients are quite steroid-responsive, and it is difficult to taper steroids following relapses (149). In addition to inhibiting the NF-κB pathway through A20, steroids improve BBB and decrease activation of T cells, thereby minimizing the migration of lymphocytes into the brain (150–152).
NLR is increased in MOGAD patients during relapse, supporting high inflammatory activity in the periphery (128). However, this biomarker should be utilized carefully as factors such as hospitalization and steroid treatment affect NLR.
Micro RNAs (miRNAs) have not been fully explored in MOGAD. Only, a study measured miR-17, miR-18a, miR-20a, and miR-92a-1 in PBMCs (12 MOGAD, 12 HC) via qPCR and found them increased in MOGAD (153). In the EAE model, miRNA-17 increased Th17 and miRNA-20a decreased Treg fraction (153, 154). Further miRNA screening studies (serum or PBMCs) are needed to confirm these miRNAs and identify other miRNAs.
Developing a peripheral immune tolerance against MOG is a promising therapeutic method. For example, intradermal MOG vaccination increased MOG-specific Tregs and improved clinical outcomes in the macaque EAE model (rhMOG induced) (155). More recently, Ugur Sahin et al. used an mRNA vaccine to induce tolerance in a mouse EAE model (C57BL/6 mice, MOG p35-55 induced) (156). This mRNA vaccine provided a self-antigen presentation in a non-inflammatory context, expanded MOG-specific Tregs, and increased expression of inhibitory molecules such as PD-1 and CTLA4. Furthermore, when the mRNA vaccine was administered on days 7 and 10 after immunization with MOG p35–55, the development of EAE was prevented, and administration after the EAE onset alleviated the symptoms.
Frequent infectious prodrome and high IL6/IL17 associated cytokine-chemokine signature are important components of MOGAD (113). Within this inflammatory milieu in the peripheral blood, MOG-specific T cells, especially CD4+, are activated through bystander activation, molecular mimicry, or maybe MOG peptide presentation in CNS-draining lymph nodes. Peripherally activated T cells further open blood-brain barrier through reactivation in the perivascular spaces, allowing autoantibodies, complement, and more immune cells to enter to perivascular spaces and CNS (90, 116). In addition to CD4+ T cells, granulocytes, macrophages/microglia, and activated complement are also found in the lesions contributing to demyelination (50, 56).
In addition to being diagnostical and prognostic biomarker, MOG antibodies potentially contribute to pathology by opsonizing MOG, complement activation, and antibody-dependent cellular cytotoxicity. MOG antibodies enhance T cell-mediated inflammation in the CNS, however, MOG antibodies alone are not pathogenic (102). MOG-IgG is detected in the CSF of more than half of the MOGAD patients and associated with worse outcomes compared to CSF negative MOGAD patients (99). Since MOG antibodies require BBB dysfunction to enter CNS, a trigger involving T cell activation and BBB dysfunction is important for the onset.
BBB and blood-CSF barrier dysfunction in MOGAD is evidenced by contrast enhancing lesions and increased albumin quotient (32%) (10, 46). Increased pro-inflammatory cytokines (such as IL6, TNFα, IL-1β, MCP-1) could also contribute to BBB dysfunction (72, 73, 113). Available treatments such as plasmapheresis, IVIG (or FcRn blockers), steroids, and IL6 receptor blockers may affect BBB directly or indirectly through decreasing inflammatory cytokines and coexisting autoantibodies in the plasma.
Neuroinflammatory biomarkers (such as MBP, sNFL, GFAP, Tau) in MOGAD support that most axonal damage happens in the initial attack, as evidenced by increased sNFL (140, 141). Demyelination associated with myelin and oligodendrocyte damage is evidenced by increased MBP (CSF) and Tau levels (serum, during relapse) [51, 135].
Understanding the autoimmune etiology of MOGAD will help us to identify biomarkers, predict prognosis, and find targeted therapies. For example, therapeutic mechanisms targeting IL6, MOG-IgG (FcRn blocking or IVIG), or improving peripheral tolerance (Treg-inducing MOG-vaccine) are new avenues that will benefit our patients.
The first draft of the manuscript was written by OC. All authors commented on previous versions of the manuscript, contributed to the manuscript conception and design, read, and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Peschl P, Bradl M, Hoftberger R, Berger T, Reindl M. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. (2017) 8:529. doi: 10.3389/fimmu.2017.00529
2. Brunner C, Lassmann H, Waehneldt TV, Matthieu JM, Linington C. Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2',3'-cyclic nucleotide 3'-phosphodiesterase in the CNS of adult rats. J Neurochem. (1989) 52:296–304. doi: 10.1111/j.1471-4159.1989.tb10930.x
3. Marta CB, Montano MB, Taylor CM, Taylor AL, Bansal R, Pfeiffer SE. Signaling cascades activated upon antibody cross-linking of myelin oligodendrocyte glycoprotein: potential implications for multiple sclerosis. J Biol Chem. (2005) 280:8985–93. doi: 10.1074/jbc.M413174200
4. Johns TG, Bernard CC. The structure and function of myelin oligodendrocyte glycoprotein. J Neurochem. (1999) 72:1–9. doi: 10.1046/j.1471-4159.1999.0720001.x
5. Marignier R, Hacohen Y, Cobo-Calvo A. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol. (2021) 20:762–72. doi: 10.1016/S1474-4422(21)00218-0
6. Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflamm. (2012) 9:14. doi: 10.1186/1742-2094-9-14
7. Nakajima H, Motomura M, Tanaka K, Fujikawa A, Nakata R, Maeda Y, et al. Antibodies to myelin oligodendrocyte glycoprotein in idiopathic optic neuritis. BMJ Open. (2015) 5:e007766. doi: 10.1136/bmjopen-2015-007766
8. Ramanathan S, Reddel SW, Henderson A, Parratt JDE, Barnett M, Gatt PN, et al. Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm. (2014) 1:e40. doi: 10.1212/NXI.0000000000000040
9. López-Chiriboga AS, Majed M, Fryer J, Dubey D, McKeon A, Flanagan EP, et al. Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG-associated disorders. JAMA Neurol. (2018) 75:1355–63. doi: 10.1001/jamaneurol.2018.1814
10. Jarius S, Ruprecht K, Kleiter I. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation. (2016) 13:280. doi: 10.1186/s12974-016-0718-0
11. Jarius S, Wildemann B, Paul F. Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol. (2014) 176:149–64. doi: 10.1111/cei.12271
12. Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology. (1999) 53:1107–114. doi: 10.1212/WNL.53.5.1107
13. Sibley WA, Bamford CR, Clark K. Clinical viral infections and multiple sclerosis. Lancet. (1985) 1:1313–5. doi: 10.1016/S0140-6736(85)92801-6
14. Sibley WA, Foley JM. Infection and immunization in multiple sclerosis. Ann N Y Acad Sci. (1965) 122:457–66. doi: 10.1111/j.1749-6632.1965.tb20229.x
15. Cobo-Calvo A, Ruiz A, Rollot F, Arrambide G, Deschamps R, Maillart E, et al. Clinical features and risk of relapse in children and adults with myelin oligodendrocyte glycoprotein antibody-associated disease. Ann Neurol. (2021) 89:30–41. doi: 10.1002/ana.25909
16. Satukijchai C, Mariano R, Messina S, Sa M, Woodhall MR, Robertson NP, et al. Factors associated with relapse and treatment of myelin oligodendrocyte glycoprotein antibody-associated disease in the United Kingdom. JAMA Netw Open. (2022) 5:e2142780. doi: 10.1001/jamanetworkopen.2021.42780
17. Saguil A, Kane S, Farnell E. Multiple sclerosis: a primary care perspective. Am Fam Physician. (2014) 90:644–52.
18. Gorman MP, Healy BC, Polgar-Turcsanyi M, Chitnis T. Increased relapse rate in pediatric-onset compared with adult-onset multiple sclerosis. Arch Neurol. (2009) 66:54–9. doi: 10.1001/archneurol.2008.505
19. Poupart J, Giovannelli J, Deschamps R, Audoin B, Ciron J, Maillart E, et al. Evaluation of efficacy and tolerability of first-line therapies in NMOSD. Neurology. (2020) 94:e1645–56. doi: 10.1212/WNL.0000000000009245
20. Akaishi T, Misu T, Takahashi T, Takai Y, Nishiyama S, Fujimori J, et al. Progression pattern of neurological disability with respect to clinical attacks in anti-MOG antibody-associated disorders. J Neuroimmunol. (2021) 351:577467. doi: 10.1016/j.jneuroim.2020.577467
21. Akaishi T, Takahashi T, Misu T, Abe M, Ishii T, Fujimori J, et al. Progressive patterns of neurological disability in multiple sclerosis and neuromyelitis optica spectrum disorders. Sci Rep. (2020) 10:13890. doi: 10.1038/s41598-020-70919-w
22. Ciotti JR, Eby NS, Brier MR, Wu GF, Chahin S, Cross AH, et al. Central vein sign and other radiographic features distinguishing myelin oligodendrocyte glycoprotein antibody disease from multiple sclerosis and aquaporin-4 antibody-positive neuromyelitis optica. Mult Scler. (2022) 28:49–60. doi: 10.1177/13524585211007086
23. Ramanathan S, Prelog K, Barnes EH, Tantsis EM, Reddel SW, Henderson APD, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler. (2016) 22:470–82. doi: 10.1177/1352458515593406
24. Dubey D, Pittock SJ, Krecke KN. Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol. (2019) 76:301–9. doi: 10.1001/jamaneurol.2018.4053
25. Sechi E, Cacciaguerra L, Chen JJ, Mariotto S, Fadda G, Dinoto A, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD): a review of clinical and MRI features, diagnosis, and management. Front Neurol. (2022) 13:885218. doi: 10.3389/fneur.2022.885218
26. Wang KY, Chetta J, Bains P, Balzer A, Lincoln J, Uribe T, et al. Spectrum of MRI brain lesion patterns in neuromyelitis optica spectrum disorder: a pictorial review. Br J Radiol. (2018) 91:20170690. doi: 10.1259/bjr.20170690
27. Messina S, Mariano R, Roca-Fernandez A, Cavey A, Jurynczyk M, Leite MI, et al. Contrasting the brain imaging features of MOG-antibody disease, with AQP4-antibody NMOSD and multiple sclerosis. Mult Scler. (2022) 28:217–27. doi: 10.1177/13524585211018987
28. Mariano R, Messina S, Kumar K, Kuker W, Leite MI, Palace J. Comparison of clinical outcomes of transverse myelitis among adults with myelin oligodendrocyte glycoprotein antibody vs aquaporin-4 antibody disease. JAMA Netw Open. (2019) 2:e1912732. doi: 10.1001/jamanetworkopen.2019.12732
29. Gadde JA, Wolf DS, Keller S, Gombolay GY. Rate of leptomeningeal enhancement in pediatric myelin oligodendrocyte glycoprotein antibody-associated encephalomyelitis. J Child Neurol. (2021) 36:1042–6. doi: 10.1177/08830738211025867
30. Cobo-Calvo A, Ruiz A, Maillart E, Audoin B, Zephir H, Bourre B, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: the MOGADOR study. Neurology. (2018) 90:e1858–69. doi: 10.1212/WNL.0000000000005560
31. Ineichen BV, Tsagkas C, Absinta M, Reich DS. Leptomeningeal enhancement in multiple sclerosis and other neurological diseases: a systematic review and meta-Analysis. Neuroimage Clin. (2022) 33:102939. doi: 10.1016/j.nicl.2022.102939
32. Sinnecker T, Schumacher S, Mueller K, Pache F, Dusek P, Harms L, et al. MRI phase changes in multiple sclerosis vs neuromyelitis optica lesions at 7T. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e259. doi: 10.1212/NXI.0000000000000259
33. Maggi P, Sati P, Nair G, Cortese ICM, Jacobson S, Smith BR, et al. Paramagnetic rim lesions are specific to multiple sclerosis: an international multicenter 3T MRI study. Ann Neurol. (2020) 88:1034–42. doi: 10.1002/ana.25877
34. Martire MS, Moiola L, Rocca MA, Filippi M, Absinta M. What is the potential of paramagnetic rim lesions as diagnostic indicators in multiple sclerosis? Expert Rev Neurother. (2022) 8:1–9. doi: 10.1080/14737175.2022.2143265
35. Reindl M, Schanda K, Woodhall M. International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm. (2020) 7:674. doi: 10.1212/NXI.0000000000000674
36. Marchionatti A, Hansel G, Avila GU, Sato DK. Detection of MOG-IgG in clinical samples by live cell-based assays: performance of immunofluorescence microscopy and flow cytometry. Front Immunol. (2021) 12:642272. doi: 10.3389/fimmu.2021.642272
37. Levy M, Yeh EA, Hawkes CH, Lechner-Scott J, Giovannoni G. Implications of Low-Titer MOG Antibodies. Mult Scler Relat Disord. (2022) 59:103746. doi: 10.1016/j.msard.2022.103746
38. Mader S, Kumpfel T, Meinl E. Novel insights into pathophysiology and therapeutic possibilities reveal further differences between AQP4-IgG- and MOG-IgG-associated diseases. Curr Opin Neurol. (2020) 33:362–71. doi: 10.1097/WCO.0000000000000813
39. Di Pauli F, Berger T. Myelin oligodendrocyte glycoprotein antibody-associated disorders: toward a new spectrum of inflammatory demyelinating CNS disorders? Front Immunol. (2018) 9:2753. doi: 10.3389/fimmu.2018.02753
40. Waters P, Fadda G, Woodhall M, O'Mahony J, Brown RA, Castro DA, et al. Serial anti-myelin oligodendrocyte glycoprotein antibody analyses and outcomes in children with demyelinating syndromes. JAMA Neurol. (2020) 77:82–93. doi: 10.1001/jamaneurol.2019.2940
41. Höftberger R, Sepulveda M, Armangue T, Blanco Y, Rostásy K, Calvo AC, et al. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler. (2015) 21:866–74. doi: 10.1177/1352458514555785
42. Jarius S, Lechner C, Wendel EM, Baumann M, Breu M, Schimmel M, et al. Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 1: Results from 163 lumbar punctures in 100 adult patients. J Neuroinflammation. (2020) 17:261. doi: 10.1186/s12974-020-01824-2
43. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. (2017) 140:3128–38. doi: 10.1093/brain/awx276
44. Tumani H, Deisenhammer F, Giovannoni G, Gold R, Hartung HP, Hemmer B, et al. Revised McDonald criteria: the persisting importance of cerebrospinal fluid analysis. Ann Neurol. (2011) 70:520–1. doi: 10.1002/ana.22508
45. Jarius S, Paul F, Franciotta D, Ruprecht K, Ringelstein M, Bergamaschi R, et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci. (2011) 306:82–90. doi: 10.1016/j.jns.2011.03.038
46. Tanaka S, Hashimoto B, Izaki S, Oji S, Fukaura H, Nomura K. Clinical and immunological differences between MOG associated disease and anti AQP4 antibody-positive neuromyelitis optica spectrum disorders: blood-brain barrier breakdown and peripheral plasmablasts. Mult Scler Relat Disord. (2020) 41:102005. doi: 10.1016/j.msard.2020.102005
47. Diem L, Bürge M, Leichtle A, Hakim A, Chan A, Salmen A, et al. Pathological cerebrospinal fluid protein concentration and albumin quotient at relapse predicts short-term disability progression in multiple sclerosis: a retrospective single center observational study. Ther Adv Neurol Disord. (2020) 13:1756286420975909. doi: 10.1177/1756286420975909
48. Jarius S, Ruprecht K, Kleiter I, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflammation. (2016) 13:279. doi: 10.1186/s12974-016-0717-1
49. Lucchinetti CF, Guo Y, Popescu BF, Fujihara K, Itoyama Y, Misu T. The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol. (2014) 24:83–97. doi: 10.1111/bpa.12099
50. Höftberger R, Guo Y, Flanagan EP, Lopez-Chiriboga AS, Endmayr V, Hochmeister S, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. (2020) 139:875–92. doi: 10.1007/s00401-020-02132-y
51. Fischer MT, Wimmer I, Hoftberger R. Disease-specific molecular events in cortical multiple sclerosis lesions. Brain. (2013) 136:1799–815. doi: 10.1093/brain/awt110
52. Kawachi I, Lassmann H. Neurodegeneration in multiple sclerosis and neuromyelitis optica. J Neurol Neurosurg Psychiatry. (2017) 88:137–45. doi: 10.1136/jnnp-2016-313300
53. Popescu BFG, Parisi JE, Cabrera-Gómez JA, Newell K, Mandler RN, Pittock SJ, et al. Absence of cortical demyelination in neuromyelitis optica. Neurology. (2010) 75:2103–9. doi: 10.1212/WNL.0b013e318200d80c
54. Saji E, Arakawa M, Yanagawa K, Toyoshima Y, Yokoseki A, Okamoto K, et al. Cognitive impairment and cortical degeneration in neuromyelitis optica. Ann Neurol. (2013) 73:65–76. doi: 10.1002/ana.23721
55. Machado-Santos J, Saji E, Tröscher AR, Paunovic M, Liblau R, Gabriely G, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain. (2018) 141:2066–82. doi: 10.1093/brain/awy151
56. Takai Y, Misu T, Kaneko K, Chihara N, Narikawa K, Tsuchida S, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain. (2020) 143:1431–46. doi: 10.1093/brain/awaa102
57. Pohl M, Kawakami N, Kitic M. T cell-activation in neuromyelitis optica lesions plays a role in their formation. Acta Neuropathol Commun. (2013) 1:85. doi: 10.1186/2051-5960-1-85
58. Reich DS, Lucchinetti CF, Calabresi PA. Multiple Sclerosis. N Engl J Med. (2018) 378:169–80. doi: 10.1056/NEJMra1401483
59. Abdelhak A, Huss A, Kassubek J, Tumani H, Otto M. Serum GFAP as a biomarker for disease severity in multiple sclerosis. Sci Rep. (2018) 8:14798. doi: 10.1038/s41598-018-33158-8
60. Kaneko K, Sato DK, Nakashima I, Nishiyama S, Tanaka S, Marignier R, et al. Myelin injury without astrocytopathy in neuroinflammatory disorders with MOG antibodies. J Neurol Neurosurg Psychiatry. (2016) 87:1257–9. doi: 10.1136/jnnp-2015-312676
61. Schindler P, Grittner U, Oechtering J, Leppert D, Siebert N, Duchow AS, et al. Serum GFAP and NfL as disease severity and prognostic biomarkers in patients with aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder. J Neuroinflammation. (2021) 18:105. doi: 10.1186/s12974-021-02138-7
62. Marignier R, Nicolle A, Watrin C, Touret M, Cavagna S, Varrin-Doyer M, et al. Oligodendrocytes are damaged by neuromyelitis optica immunoglobulin G via astrocyte injury. Brain. (2010) 133:2578–91. doi: 10.1093/brain/awq177
63. Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: international MOGAD Panel proposed criteria. Lancet Neurol. (2023). doi: 10.1016/S1474-4422(22)00431-8
64. Pöllinger B, Krishnamoorthy G, Berer K, Lassmann H, Bösl MR, Dunn R, et al. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J Exp Med. (2009) 206:1303–16. doi: 10.1084/jem.20090299
65. Collongues N, Chanson JB, Blanc F, Steibel J, Lam CD, Shabbir A, et al. The Brown Norway opticospinal model of demyelination: does it mimic multiple sclerosis or neuromyelitis optica? Int J Dev Neurosci. (2012) 30:487–97. doi: 10.1016/j.ijdevneu.2012.05.004
66. Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. (2006) 129(Pt 8):1953–71. doi: 10.1093/brain/awl075
67. Hamza MM, Alas BF, Huang C, Quon JC, Amezcua L, Robers MV, et al. Internuclear ophthalmoplegia characterizes multiple sclerosis rather than neuromyelitis optica spectrum disease. J Neuroophthalmol. (2022) 42:239–45. doi: 10.1097/WNO.0000000000001534
68. Salama S, Khan M, Shanechi A, Levy M, Izbudak I, MRI. differences between MOG antibody disease and AQP4 NMOSD. Mult Scler. (2020) 26:1854–65. doi: 10.1177/1352458519893093
69. Absinta M, Vuolo L, Rao A. Gadolinium-based MRI characterization of leptomeningeal inflammation in multiple sclerosis. Neurology. (2015) 85:18–28. doi: 10.1212/WNL.0000000000001587
70. Gasperi C, Salmen A, Antony G, Bayas A, Heesen C, Kümpfel T, et al. Association of intrathecal immunoglobulin g synthesis with disability worsening in multiple sclerosis. JAMA Neurol. (2019) 76:841–9. doi: 10.1001/jamaneurol.2019.0905
71. Akaishi T, Takahashi T, Misu T, Kaneko K, Takai Y, Nishiyama S, et al. Difference in the source of anti-AQP4-IgG and anti-MOG-IgG antibodies in CSF in patients with neuromyelitis optica spectrum disorder. Neurology. (2021) 97:e1–e12. doi: 10.1212/WNL.0000000000012175
72. Kaneko K, Sato DK, Nakashima I, Ogawa R, Akaishi T, Takai Y, et al. CSF cytokine profile in MOG-IgG+ neurological disease is similar to AQP4-IgG+ NMOSD but distinct from MS: a cross-sectional study and potential therapeutic implications. J Neurol Neurosurg Psychiatry. (2018) 89:927–36. doi: 10.1136/jnnp-2018-317969
73. Kothur K, Wienholt L, Tantsis EM. B Cell, Th17, and Neutrophil related cerebrospinal fluid cytokine/chemokines are elevated in MOG antibody associated demyelination. PLoS ONE. (2016) 11:e0149411. doi: 10.1371/journal.pone.0149411
74. Sabatino JJ Jr, Probstel AK, Zamvil SS. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat Rev Neurosci. (2019) 20:728–45. doi: 10.1038/s41583-019-0233-2
75. Derbinski J, Schulte A, Kyewski B, Klein L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol. (2001) 2:1032–9. doi: 10.1038/ni723
76. Weber MS, Derfuss T, Metz I, Bruck W. Defining distinct features of anti-MOG antibody associated central nervous system demyelination. Ther Adv Neurol Disord. (2018) 11:1756286418762083. doi: 10.1177/1756286418762083
77. Abbas AL, Pillai A. Cellular and Molecular Immunology. Tenth ed. Philadelphia, PA: Elsevier (2021):337–363.
78. Gotter J, Brors B, Hergenhahn M, Kyewski B. Medullary epithelial cells of the human thymus express a highly diverse selection of tissue-specific genes colocalized in chromosomal clusters. J Exp Med. (2004) 199:155–66. doi: 10.1084/jem.20031677
79. Bruno R, Sabater L, Sospedra M, Ferrer-Francesch X, Escudero D, Martínez-Cáceres E, et al. Multiple sclerosis candidate autoantigens except myelin oligodendrocyte glycoprotein are transcribed in human thymus. Eur J Immunol. (2002) 32:2737–47. doi: 10.1002/1521-4141(2002010)32:10<2737::AID-IMMU2737>3.0.CO;2-0
80. Human, Protein Atlas proteinatlas.org.
81. Uhlen M, Fagerberg L, Hallstrom BM. Proteomics. Tissue-based map of the human proteome. Science. (2015) 347:1260419. doi: 10.1126/science.1260419
82. van Zwam M, Huizinga R, Melief MJ, Wierenga-Wolf AF, van Meurs M, Voerman JS, et al. Brain antigens in functionally distinct antigen-presenting cell populations in cervical lymph nodes in MS and EAE. J Mol Med. (2009) 87:273–86. doi: 10.1007/s00109-008-0421-4
83. Wildemann B, Jarius S, Franz J, Ruprecht K, Reindl M, Stadelmann C. MOG-expressing teratoma followed by MOG-IgG-positive optic neuritis. Acta Neuropathol. (2021) 141:127–31. doi: 10.1007/s00401-020-02236-5
84. Kinzel S, Lehmann-Horn K, Torke S. Myelin-reactive antibodies initiate T cell-mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol. (2016) 132:43–58. doi: 10.1007/s00401-016-1559-8
85. Bogdanos DP, Smith H, Ma Y, Baum H, Mieli-Vergani G, Vergani D, et al. study of molecular mimicry and immunological cross-reactivity between hepatitis B surface antigen and myelin mimics. Clin Dev Immunol. (2005) 12:217–24. doi: 10.1080/17402520500285247
86. Guggenmos J, Schubart AS, Ogg S, Andersson M, Olsson T, Mather IH, et al. Antibody cross-reactivity between myelin oligodendrocyte glycoprotein and the milk protein butyrophilin in multiple sclerosis. J Immunol. (2004) 172:661–8. doi: 10.4049/jimmunol.172.1.661
87. Bruijstens AL, Wong YYM, van Pelt DE, van der Linden PJE, Haasnoot GW, Hintzen RQ, et al. HLA association in MOG-IgG- and AQP4-IgG-related disorders of the CNS in the Dutch population. Neurol Neuroimmunol Neuroinflamm. (2020) 7:702. doi: 10.1212/NXI.0000000000000702
88. Grant-Peters M, Dos Passos GR, Yeung HY, Jacob A, Huda S, Leite MI, et al. No strong HLA association with MOG antibody disease in the UK population. Ann Clin Transl Neurol. (2021) 8:1502–7. doi: 10.1002/acn3.51378
89. Sun X, Qiu W, Wang J, Wang S, Wang Y, Zhong X, et al. Myelin oligodendrocyte glycoprotein-associated disorders are associated with HLA subtypes in a Chinese paediatric-onset cohort. J Neurol Neurosurg Psychiatry. (2020) 91:733–9. doi: 10.1136/jnnp-2019-322115
90. Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. (2009) 9:393–407. doi: 10.1038/nri2550
91. Winklmeier S, Schlüter M, Spadaro M, Thaler FS, Vural A, Gerhards R. Identification of circulating MOG-specific B cells in patients with MOG antibodies. Neurol Neuroimmunol Neuroinflamm. (2019) 6:625. doi: 10.1212/NXI.0000000000000625
92. In: Anaya JM, Shoenfeld Y, Rojas-Villarraga A, Levy RA, Cervera R eds. Autoimmunity: From Bench to Bedside. Colombia: Bogota (2013).
94. Johnsson M, Asztely F, Hejnebo S. SARS-CoV-2 a trigger of myelin oligodendrocyte glycoprotein-associated disorder. Ann Clin Transl Neurol. (2022). doi: 10.1002/acn3.51609
95. Lotan I, Nishiyama S, Manzano GS, Lydston M, Levy M. COVID-19 and the risk of CNS demyelinating diseases: A systematic review. Front Neurol. (2022) 13:970383. doi: 10.3389/fneur.2022.970383
96. Tea F, Lopez JA, Ramanathan S, Merheb V, Lee FXZ, Zou A, et al. Characterization of the human myelin oligodendrocyte glycoprotein antibody response in demyelination. Acta Neuropathol Commun. (2019) 7:145. doi: 10.1186/s40478-019-0786-3
97. Waters P, Woodhall M, O'Connor KC, Reindl M, Lang B, Sato DK, et al. MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e89. doi: 10.1212/NXI.0000000000000089
98. Wendel EM, Thonke HS, Bertolini A, Baumann M, Blaschek A, Merkenschlager A, et al. Temporal dynamics of MOG antibodies in children with acquired demyelinating syndrome. Neurol Neuroimmunol Neuroinflamm. (2022) 9:35. doi: 10.1212/NXI.0000000000200035
99. Carta S, Calvo ÁC, Armangué T, Saiz A, Lechner C, Rostásy K, et al. Significance of myelin oligodendrocyte glycoprotein antibodies in CSF: a retrospective multicenter study. Neurology. (2022) 3:10. doi: 10.1212/WNL.0000000000201662
100. Kwon YN, Kim B, Kim J-S, Mo H, Choi K, Oh S-I, et al. Myelin oligodendrocyte glycoprotein-immunoglobulin G in the CSF: clinical implication of testing and association with disability. Neurol Neuroimmunol Neuroinflamm. (2022) 9:1095. doi: 10.1212/NXI.0000000000001095
101. Mayer MC, Breithaupt C, Reindl M, Schanda K, Rostásy K, Berger T, et al. Distinction and temporal stability of conformational epitopes on myelin oligodendrocyte glycoprotein recognized by patients with different inflammatory central nervous system diseases. J Immunol. (2013) 191:3594–604. doi: 10.4049/jimmunol.1301296
102. Spadaro M, Winklmeier S, Beltrán E, Macrini C, Höftberger R, Schuh E, et al. Pathogenicity of human antibodies against myelin oligodendrocyte glycoprotein. Ann Neurol. (2018) 84:315–28. doi: 10.1002/ana.25291
103. Macrini C, Gerhards R, Winklmeier S, Bergmann L, Mader S, Spadaro M, et al. Features of MOG required for recognition by patients with MOG antibody-associated disorders. Brain. (2021) 144:2375–89. doi: 10.1093/brain/awab105
104. Brilot F, Dale RC, Selter RC, Grummel V, Kalluri SR, Aslam M, et al. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann Neurol. (2009) 66:833–42. doi: 10.1002/ana.21916
105. Whittam DH, Cobo-Calvo A, Lopez-Chiriboga AS, Pardo S, Gornall M, Cicconi S, et al. Treatment of MOG-IgG-associated disorder with rituximab: An international study of 121 patients. Mult Scler Relat Disord. (2020) 44:102251. doi: 10.1016/j.msard.2020.102251
106. Scolding NJ, Morgan BP, Compston DA. The expression of complement regulatory proteins by adult human oligodendrocytes. J Neuroimmunol. (1998) 84:69–75. doi: 10.1016/S0165-5728(97)00241-5
107. Keller CW, Lopez JA, Wendel E-M, Ramanathan S, Gross CC, Klotz L, et al. Complement activation is a prominent feature of MOGAD. Ann Neurol. (2021) 90:976–82. doi: 10.1002/ana.26226
108. Lerch M, Schanda K, Lafon E, Würzner R, Mariotto S, Dinoto A, et al. More efficient complement activation by anti-aquaporin-4 compared with anti-myelin oligodendrocyte glycoprotein antibodies. Neurol Neuroimmunol Neuroinflamm. (2023) 10:59. doi: 10.1212/NXI.0000000000200059
109. Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 M1/M23 isoforms and orthogonal arrays. J Biol Chem. (2011) 286:16516–24. doi: 10.1074/jbc.M111.227298
110. Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. (2014) 343:1260–3. doi: 10.1126/science.1248943
111. Soltys J, Liu Y, Ritchie A, Wemlinger S, Schaller K, Schumann H, et al. Membrane assembly of aquaporin-4 autoantibodies regulates classical complement activation in neuromyelitis optica. J Clin Invest. (2019) 129:2000–13. doi: 10.1172/JCI122942
112. Liu J, Yang X, Pan J. Single-cell transcriptome profiling unravels distinct peripheral blood immune cell signatures of RRMS and MOG antibody-associated disease. Front Neurol. (2021) 12:807646. doi: 10.3389/fneur.2021.807646
113. Bauer A, Rudzki D, Berek K, Dinoto A, Lechner C, Wendel EM, et al. Increased peripheral inflammatory responses in myelin oligodendrocyte glycoprotein associated disease and aquaporin-4 antibody positive neuromyelitis optica spectrum disorder. Front Immunol. (2022) 13:1037812. doi: 10.3389/fimmu.2022.1037812
114. Guyon A. CXCL12 chemokine and its receptors as major players in the interactions between immune and nervous systems. Front Cell Neurosci. (2014) 8:65. doi: 10.3389/fncel.2014.00065
115. Li X, Wang L, Zhou L, ZhangBao J, Miao MZ, Lu C, et al. The imbalance between regulatory and memory B cells accompanied by an increased number of circulating T-follicular helper cells in MOG-antibody-associated demyelination. Mult Scler Relat Disord. (2019) 36:101397. doi: 10.1016/j.msard.2019.101397
116. Bradl M, Reindl M, Lassmann H. Mechanisms for lesion localization in neuromyelitis optica spectrum disorders. Curr Opin Neurol. (2018) 31:325–33. doi: 10.1097/WCO.0000000000000551
117. Kawakami N, Lassmann S, Li Z, Odoardi F, Ritter T, Ziemssen T, et al. The activation status of neuroantigen-specific T cells in the target organ determines the clinical outcome of autoimmune encephalomyelitis. J Exp Med. (2004) 199:185–97. doi: 10.1084/jem.20031064
118. Gimenez MA, Sim J, Archambault AS, Klein RS, Russell JH, A. tumor necrosis factor receptor 1-dependent conversation between central nervous system-specific T cells and the central nervous system is required for inflammatory infiltration of the spinal cord. Am J Pathol. (2006) 168:1200–9. doi: 10.2353/ajpath.2006.050332
119. Flach AC, Litke T, Strauss J. Autoantibody-boosted T-cell reactivation in the target organ triggers manifestation of autoimmune CNS disease. Proc Natl Acad Sci U S A. (2016) 113:3323–8. doi: 10.1073/pnas.1519608113
120. Varrin-Doyer M, Shetty A, Spencer CM, Schulze-Topphoff U, Weber MS, Bernard CCA, et al. MOG transmembrane and cytoplasmic domains contain highly stimulatory T-cell epitopes in MS. Neurol Neuroimmunol Neuroinflamm. (2014) 1:e20. doi: 10.1212/NXI.0000000000000020
121. Shetty A, Gupta SG, Varrin-Doyer M. Immunodominant T-cell epitopes of MOG reside in its transmembrane and cytoplasmic domains in EAE. Neurol Neuroimmunol Neuroinflamm. (2014) 1:e22. doi: 10.1212/NXI.0000000000000022
122. Hofer LS, Ramberger M, Gredler V, Pescoller AS, Rostásy K, Sospedra M, et al. Comparative analysis of T-Cell responses to aquaporin-4 and myelin oligodendrocyte glycoprotein in inflammatory demyelinating central nervous system diseases. Front Immunol. (2020) 11:1188. doi: 10.3389/fimmu.2020.01188
123. Saxena S, Lokhande H, Gombolay G, Raheja R, Rooney T, Chitnis T. Identification of TNFAIP3 as relapse biomarker and potential therapeutic target for MOG antibody associated diseases. Sci Rep. (2020) 10:12405. doi: 10.1038/s41598-020-69182-w
124. Horellou P, de Chalus A, Giorgi L. Regulatory T Cells Increase After rh-MOG stimulation in non-relapsing but decrease in relapsing MOG antibody-associated disease at onset in children. Front Immunol. (2021) 12:679770. doi: 10.3389/fimmu.2021.679770
125. Korn T, Reddy J, Gao W. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. (2007) 13:423–31. doi: 10.1038/nm1564
126. Kwon YN, Kim B, Ahn S, Seo J, Kim SB, Yoon SS, et al. Serum level of IL-1beta in patients with inflammatory demyelinating disease: Marked upregulation in the early acute phase of MOG antibody associated disease (MOGAD). J Neuroimmunol. (2020) 348:577361. doi: 10.1016/j.jneuroim.2020.577361
127. Wong D, Dorovini-Zis K, Vincent SR. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp Neurol. (2004) 190:446–55. doi: 10.1016/j.expneurol.2004.08.008
128. Benetou C, Berti F, Hemingway C, Hacohen Y, Lim M. Neutrophil-to-lymphocyte ratio correlates with disease activity in myelin oligodendrocyte glycoprotein antibody associated disease (MOGAD) in children. Mult Scler Relat Disord. (2020) 45:102345. doi: 10.1016/j.msard.2020.102345
129. Kunchok A, Flanagan EP, Krecke KN. MOG-IgG1 and co-existence of neuronal autoantibodies. Mult Scler. (2021) 27:1175–86. doi: 10.1177/1352458520951046
130. Kunchok A, Chen JJ, McKeon A, Mills JR, Flanagan EP, Pittock SJ. Coexistence of myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies in adult and pediatric patients. JAMA Neurol. (2020) 77:257–9. doi: 10.1001/jamaneurol.2019.3656
131. Török O, Schreiner B, Schaffenrath J, Tsai H-C, Maheshwari U, Stifter SA, et al. Pericytes regulate vascular immune homeostasis in the CNS. Proc Natl Acad Sci U S A. (2021) 118:7118. doi: 10.1073/pnas.2016587118
132. Ulusoy C, Sekerdag E, Yilmaz V, Yilmaz AB, Atak D, Vural A, et al. Impact of autoimmune demyelinating brain disease sera on pericyte survival. Noro Psikiyatr Ars. (2021) 58:83–6. doi: 10.29399/npa.27350
133. Shimizu F, Ogawa R, Mizukami Y, Watanabe K, Hara K, Kadono C, et al. GRP78 antibodies are associated with blood-brain barrier breakdown in anti-myelin oligodendrocyte glycoprotein antibody-associated disorder. Neurol Neuroimmunol Neuroinflamm. (2022) 9(1). doi: 10.1212/NXI.0000000000001038
134. Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. (2003) 197:1073–81. doi: 10.1084/jem.20021603
135. Sjöstedt E, Zhong W, Fagerberg L, Karlsson M, Mitsios N, Adori C, et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science. (2020) 367:5947. doi: 10.1126/science.aay5947
136. Johnson MC, Pierson ER, Spieker AJ. Distinct T cell signatures define subsets of patients with multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e278. doi: 10.1212/NXI.0000000000000278
137. Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. (2008) 14:337–42. doi: 10.1038/nm1715
138. Lassmann H, Brunner C, Bradl M, Linington C. Experimental allergic encephalomyelitis: the balance between encephalitogenic T lymphocytes and demyelinating antibodies determines size and structure of demyelinated lesions. Acta Neuropathol. (1988) 75:566–76. doi: 10.1007/BF00686201
139. Gastaldi M, Foiadelli T, Greco G, Scaranzin S, Rigoni E, Masciocchi S, et al. Prognostic relevance of quantitative and longitudinal MOG antibody testing in patients with MOGAD: a multicentre retrospective study. J Neurol Neurosurg Psychiatry (. (2022). doi: 10.1136/jnnp-2022-330237
140. Mariotto S, Gastaldi M, Grazian L, Mancinelli C, Capra R, Marignier R, et al. NfL levels predominantly increase at disease onset in MOG-Abs-associated disorders. Mult Scler Relat Disord. (2021) 50:102833. doi: 10.1016/j.msard.2021.102833
141. Kim H, Lee E-J, Kim S, Choi L-K, Kim K, Kim HW, et al. Serum biomarkers in myelin oligodendrocyte glycoprotein antibody-associated disease. Neurol Neuroimmunol Neuroinflamm. (2020) 7:708. doi: 10.1212/NXI.0000000000000708
142. Takeshita Y, Fujikawa S, Serizawa K, Fujisawa M, Matsuo K, Nemoto J, et al. New BBB model reveals That IL-6 Blockade suppressed the BBB disorder, preventing onset of NMOSD. Neurol Neuroimmunol Neuroinflamm. (2021) 8:1076. doi: 10.1212/NXI.0000000000001076
143. O'Carroll SJ, Kho DT, Wiltshire R. Pro-inflammatory TNFalpha and IL-1beta differentially regulate the inflammatory phenotype of brain microvascular endothelial cells. J Neuroinflammation. (2015) 12:131. doi: 10.1186/s12974-015-0346-0
144. Nagelkerke SQ, Kuijpers TW. Immunomodulation by IVIg and the role of Fc-gamma receptors: classic mechanisms of action after all? Front Immunol. (2014) 5:674. doi: 10.3389/fimmu.2014.00674
145. Barro C, Healy BC, Liu Y, Saxena S, Paul A, Polgar-Turcsanyi M, et al. Serum GFAP and NfL levels differentiate subsequent progression and disease activity in patients with progressive multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2023) 10:52. doi: 10.1212/NXI.0000000000200052
146. Ringelstein M, Ayzenberg I, Lindenblatt G. Interleukin-6 receptor blockade in treatment-refractory MOG-IgG-associated disease and neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm. (2022) 9:1100. doi: 10.1212/NXI.0000000000001100
147. Okoye IS, Xu L, Walker J, Elahi S. The glucocorticoids prednisone and dexamethasone differentially modulate T cell function in response to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade. Cancer Immunol Immunother. (2020) 69:1423–36. doi: 10.1007/s00262-020-02555-2
148. Redenbaugh V, Flanagan EP, Floris V, Zara P, Bhatti MT, Sanchez F, et al. Exposure to TNF inhibitors is rare at MOGAD presentation. J Neurol Sci. (2022) 432:120044. doi: 10.1016/j.jns.2021.120044
149. Ramanathan S, Mohammad S, Tantsis E. Clinical course, therapeutic responses, and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. (2018) 89:127–37. doi: 10.1136/jnnp-2017-316880
150. Gelati M, Corsini E, Dufour A. High-dose methylprednisolone reduces cytokine-induced adhesion molecules on human brain endothelium. Can J Neurol Sci. (2000) 27:241–4. doi: 10.1017/S0317167100000883
151. Salvador E, Shityakov S, Forster C. Glucocorticoids and endothelial cell barrier function. Cell Tissue Res. (2014) 355:597–605. doi: 10.1007/s00441-013-1762-z
152. Van Laethem F, Baus E, Smyth LA, Andris F, Bex F, Urbain J, et al. Glucocorticoids attenuate T cell receptor signaling. J Exp Med. (2001) 193:803–14. doi: 10.1084/jem.193.7.803
153. Wang Y, Xie C, Song Y. miR-20a suppresses Treg differentiation by targeting Map3k9 in experimental autoimmune encephalomyelitis. J Transl Med. (2021) 19:223. doi: 10.1186/s12967-021-02893-4
154. Liu SQ, Jiang S, Li C, Zhang B, Li QJ. miR-17-92 cluster targets phosphatase and tensin homology and Ikaros family Zinc Finger 4 to promote TH17-mediated inflammation. J Biol Chem. (2014) 289:12446–56. doi: 10.1074/jbc.M114.550723
155. Fovet C-M, Stimmer L, Contreras V, Horellou P, Hubert A, Seddiki N, et al. Intradermal vaccination prevents anti-MOG autoimmune encephalomyelitis in macaques. EBioMedicine. (2019) 47:492–505. doi: 10.1016/j.ebiom.2019.08.052
Keywords: MOGAD, T cells, MOG (myelin oligodendrocyte glycoprotein), blood brain barrier (BBB), MOG-IgG, autoantibodies, pathophysiology-contemporary knowledge
Citation: Corbali O and Chitnis T (2023) Pathophysiology of myelin oligodendrocyte glycoprotein antibody disease. Front. Neurol. 14:1137998. doi: 10.3389/fneur.2023.1137998
Received: 05 January 2023; Accepted: 09 February 2023;
Published: 28 February 2023.
Edited by:
Sasitorn Siritho, Bumrungrad International Hospital, ThailandReviewed by:
Matteo Gastaldi, Neurological Institute Foundation Casimiro Mondino (IRCCS), ItalyCopyright © 2023 Corbali and Chitnis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tanuja Chitnis,  dGNoaXRuaXNAcmljcy5id2guaGFydmFyZC5lZHU=
dGNoaXRuaXNAcmljcy5id2guaGFydmFyZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.