 Xinyi Sun
Xinyi Sun Yehui Lv
Yehui Lv Jian Lin
Jian Lin- 1School of Basic Medical Sciences, Shanghai University of Medicine and Health Sciences, Shanghai, China
- 2Institute of Wound Prevention and Treatment, Shanghai University of Medicine and Health Sciences, Shanghai, China
- 3Chongming Hospital Affiliated to Shanghai University of Medicine and Health Sciences, Shanghai, China
Sudden unexpected death in epilepsy (SUDEP) is defined as a sudden, unexpected, non-traumatic, non-drowning death in a person with epilepsy. SUDEP is generally considered to result from seizure-related cardiac dysfunction, respiratory depression, autonomic nervous dysfunction, or brain dysfunction. Frequency of generalized tonic clonic seizures (GTCS), prone posture, and refractory epilepsy are considered risk factors. SUDEP has also been associated with inherited cardiac ion channel disease and severe obstructive sleep apnea. Most previous studies of SUDEP mechanisms have focused on cardiac and respiratory dysfunction and imbalance of the neural regulatory system. Cardiac-related mechanisms include reduction in heart rate variability and prolongation of QT interval, which can lead to arrhythmias. Laryngospasm and amygdala activation may cause obstructive and central apnea, respectively. Neural mechanisms include impairment of 5-HT and adenosine neuromodulation. The research to date regarding molecular mechanisms of SUDEP is relatively limited. Most studies have focused on p-glycoprotein, catecholamines, potassium channels, and the renin-angiotensin system, all of which affect cardiac and respiratory function.
Introduction
Sudden unexpected death in epilepsy (SUDEP) is defined as a sudden, unexpected, non-traumatic, non-drowning death in a person with epilepsy, witnessed or unwitnessed, in which an autopsy does not reveal an anatomical or toxicological cause of death (1). In a large Chinese community cohort of 1,562 epileptic patients, 15 experienced suspected SUDEP during the 5-year follow-up period; SUDEP incidence was 2.34 per 1,000 person-years (2). Sudden epileptic death is believed to be related to cardiac dysfunction, respiratory depression, autonomic nervous dysfunction, and brain dysfunction during seizure; however, the exact mechanism is unclear (3–5). The purpose of this review is to summarize the current knowledge regarding mechanisms of SUDEP.
SUDEP risk factors
The frequency of generalized tonic-clonic seizures (GTCS) is considered the most important clinical risk factor for SUDEP (3, 6–8): the higher the GTCS frequency, the higher the risk of SUDEP. Data from a pooled analysis of SUDEP risk factors indicate that patients who have one to two GTCS per year are nearly three times as likely to experience SUDEP than patients who do not have GTCS; patients who have more than 50 GTCS per year are more than 14 times as likely to experience SUDEP (8). Prone positioning during seizures is an important risk factor for accidental death. Most instances of SUDEP occur after a generalized seizure; patients are usually found in the prone position (9, 10). In one study, 73.3% of SUDEP patients died in the prone position and prone position was significantly associated with SUDEP (11). Refractory epilepsy is also a risk factor for SUDEP (12). SUDEP accounts for 5–30% of deaths in all epileptic patients and up to 50% of deaths in patients with refractory epilepsy (13). Furthermore, risk of SUDEP is also higher in males, patients who have had epilepsy for many years, patients with ion channel or arrhythmia-related gene mutations, patients with neurological comorbidities, and patients taking multiple antiepileptic agents (8, 14, 15, 17). The occurrence of SUDEP is also associated with genes related to cardiac arrythmia and ion channels, especially the mutation related to long QT syndrome (LQTS), which may increase the risk of sudden death when combined with epilepsy (16–18). Severe obstructive sleep apnea has also been associated with increased risk of SUDEP (19). Seizure incidence is significantly lower in obstructive sleep apnea patients who receive positive airway pressure therapy than patients who are untreated (20). Structural brain damage may also be a SUDEP risk factor. Changes in brain structures and networks involved in central autonomic nerve and respiratory control have been observed in SUDEP patients and those at high-risk for SUDEP (21). These changes are mainly changes in gray matter volume in the hippocampus, amygdala, and thalamus (13, 22–24).
SUDEP mechanisms
Most studies which have examined the mechanisms underlying SUDEP have focused on cardiac and respiratory dysfunction and imbalance within the neural regulation system.
Cardiac dysfunction
Epilepsy may induce various transient cardiac effects, including heart rate changes, heart rate variability (HRV), arrhythmia, cardiac arrest and other electrocardiographic abnormalities (25). Acute and adaptive changes in heart rhythm in epileptic patients is one potential pathogenic SUDEP mechanism (18). To some extent, HRV reflects the balance of the sympathetic and parasympathetic autonomic nervous system divisions. An increase in HRV indicates increased parasympathetic activity while a decrease in HRV indicates a relative increase in sympathetic activity (26). In addition, HRV in epileptic patients decreases in the interictal period, especially in patients with temporal lobe epilepsy and drug resistant epilepsy (27). Moreover, reduction in HRV is associated with higher risk of SUDEP (28). Prolongation of the QT interval may be an important cause of ventricular arrhythmias in epileptic patients (29). Chahal et al. (17) reported that a prolonged QT interval in epileptic patients was associated with increased mortality. In particular, long QT syndrome, an inherited cardiac ion channel disease, is characterized by prolonged ventricular repolarization and ventricular arrhythmia, which may cause syncope or sudden cardiac death (30). In a dog model of long QT syndrome, anticonvulsant drugs can trigger torsade de pointes (31), which can progress to ventricular fibrillation and sudden death (32). Repeated seizures may also cause structural changes in the heart, which is another potential SUDEP mechanism. Pansani et al. (33) reported that repeated seizures in rats with epilepsy may damage the function and structure of the heart through regulation of microRNA that leads to myocardial cell hypertrophy and myocardial fibrosis. Similar pathological changes have also been reported in autopsy studies of SUDEP patients (34).
Respiratory dysfunction
Central and obstructive apnea and respiratory arrest have also been suspected as mechanisms underlying SUDEP. Hypoxemia caused by obstructive laryngospasm and subsequent respiratory arrest may be a mechanism of accidental sudden death in epileptic patients (35, 36). Tavee et al. (37) reported severe laryngospasm, continuous inspiratory wheezing, and cyanosis during a GTCS in a patient with refractory epilepsy. In animal studies, laryngospasm has been associated with seizure-associated reflux of gastric acid into the throat (35, 38) as well as seizure-associated increased recurrent laryngeal nerve discharge (36). Reflux is probably the cause of epilepsy-associated laryngeal spasm. In a rat epilepsy model, ST segment elevation on electrocardiography, intermittent apnea, and electroencephalography narrowing due to hypoxia were observed after gastric reflux entered the throat (38). Another rat study reported that blocking reflux into the esophagus could eliminate sudden epileptic death (35).
Amygdala activation may cause central apnea and sudden death in epileptic patients. Spread of seizure activity to the amygdala induces central apnea and decreased oxygen saturation (39–41). An area in the human amygdala that inhibits respiration and elicits apnea has been identified in children with epilepsy (42). Many patients with epilepsy are completely unaware of their apnea and do not report dyspnea (41). In addition, in a mouse model of SUDEP, electrolytic damage of the amygdala significantly reduced the incidence of seizure-induced respiratory arrest (S-IRA) and death (43). However, other studies have reported that seizures involving the amygdala are not accompanied by apnea/hypoventilation or that apnea/hypoventilation precedes the seizure. These findings indicate that amygdala involvement may not be important for induction of apnea/hypoventilation in all seizures (44).
Pulmonary edema/congestion is the most common pathological lung finding in SUDEP patients (34). In a forensic analysis of nine SUDEP cases, all exhibited pulmonary edema, pulmonary congestion, alveolar hemorrhage, and pulmonary small bronchiole wall contraction (45). GTCS are associated with neurogenic pulmonary edema (NPE). In post-ictal pulmonary edema, GTCS are the most frequently reported type (46). In one post-ictal neurogenic pulmonary edema study, five of 47 patients had symptoms of pulmonary edema and all five had GTCS (47). Moreover, the presence of an abnormality on chest radiography is significantly associated with the duration of the preceding GTCS (48). In animal models of epilepsy, pulmonary vascular pressure increases in proportion to the duration of seizure. This induced hypertension discharges fluid from the vascular compartment into the pulmonary parenchyma, causing pulmonary edema (49). This may be the mechanism of neurogenic pulmonary edema caused by epilepsy.
Neurotransmitter dysfunction
5-hydroxytryptamine (5-HT) and adenosine may participate in the pathophysiological mechanism of SUDEP (50). 5-HT plays a neuroregulatory role in respiratory control. It provides tension and excitability drive for multiple components of the respiratory network, detects changes in tissue pH/CO2, and regulates ventilation by affecting neurotransmitter release (51). In DBA/1 mice, S-IRA is related to a defect in 5-HT neurotransmission in the dorsal raphe nucleus (52). Light stimulation of 5-HT neurons in the dorsal raphe nucleus and use of selective serotonin reuptake inhibitors and the antiepileptic drug fenfluramine can enhance the effect of 5-HT and reduce S-IRA incidence (52–54). 5-HT3 and 5-HT4 receptors may be involved in the above mechanism (53, 54). In a rat epilepsy model, seizures induced by pilocarpine can cause depletion of 5-HT in the hippocampus and significantly damage serotonergic neurons in the raphe nucleus (55). Adenosine signaling has a variety of beneficial and harmful effects in the context of epilepsy. Inhibition of adenosine, which leads to respiratory dysfunction during seizure, may be an important SUDEP mechanism (56). In DBA/2 epileptic mice, blockade of adenosine metabolism was significantly associated with increased incidence of S-IRA, while adenosine A2 receptor antagonists were significantly associated with lower incidence (57). A1 receptor activation with specific agonists can inhibit drug-resistant epileptic events in human temporal cortex slices from drug-resistant patients (58). In patients with temporal lobe epilepsy and hippocampal sclerosis, density of cortical A2A receptors was significantly lower in those with a higher risk of SUDEP, which suggests impaired neuroglial dysfunction and adenosine regulation in these patients. In addition, amygdala A1 receptor density was increased in the high-risk patients, which may contribute to peri-ictal amygdala dysfunction in SUDEP (59). Adenosine is closely associated with SUDEP and adenosine receptors may play an important role.
Molecular mechanism
P-glycoprotein
P-glycoprotein (P-gp) may be involved in SUDEP, which is the main cause of death in patients with refractory epilepsy (60). Multidrug resistance in patients with refractory epilepsy is primarily related to overexpression of ABC transporters such as P-gp (61, 62). Regardless of metabolic biotransformation, the biological distribution of antiseizure medications and their metabolites depends on functional expression of ABC transporters in the blood–brain barrier, intestine, liver, and kidney (61). However, high-frequency uncontrolled seizures can induce expression of ABC transporters such as P-gp in excretory organs and cells which normally do not express them such as neurons and cardiomyocytes; this increases the risk of refractory epilepsy (61). The expression of P-gp in neurons and myocardial cells can significantly reduce the resting membrane potential (−60 to −10 mV) and affect function in a manner that predisposes to epilepsy, malignant arrhythmia, and sudden accidental death (61, 62). Auzmendi et al. (60) reported that repeated induction of seizures in Wistar rats causes P-gp expression, electrocardiography changes, and increased mortality; these findings may be related to depolarization caused by myocardial cell P-gp expression.
Catecholamines
Status epilepticus causes release of a large amount of catecholamines (63), which can cause myocardial ischemia, calcium ion overload, oxidative stress, and mitochondrial dysfunction that lead to cardiac damage (64). In animal studies, repeated induction of S-IRA in DBA/1 mice can cause ventricular calcification necrosis; the incidence and lesion size depend on the total number of S-IRA episodes (65). Verrier et al. (66) introduced the concept of “epileptic heart,” which is “a heart and coronary vasculature damaged by chronic epilepsy as a result of repeated surges in catecholamines and hypoxemia leading to electrical and mechanical dysfunction.”
K+ channel
Kv1.1 belongs to the Shaker subfamily of voltage-gated potassium channels and is widely expressed throughout the nervous system, serving as a key regulator of neuronal excitability (67, 68). In wild-type mice, Kv1.1 can be detected in brain nuclei associated with heart and lung function including the basolateral amygdala nucleus, dorsal respiratory group nuclei, dorsal motor nucleus of the vagus nerve, nucleus ambiguus, ventral respiratory column nuclei, and the pontine respiratory group nuclei. It is also found in the posterior trapezoidal nucleus and central area nucleus, which are crucial for chemical sensing (69). Neurons in the posterior trapezium nucleus directly regulate respiration in response to CO2/hydrogen ion changes in tissues and control respiration by integrating information from several respiratory centers, including the raphe medulla (70). Kv1.1 subunits control spontaneous excitatory synaptic activity of pyramidal neurons in the basolateral amygdala (71). Kcna knockout mice lack the Kv1.1 subunit and are used as a genetic model of SUDEP (72). In these mice, the inhibitory control of interneurons in the central lateral amygdala nucleus is reduced and abnormal parasympathetic transmission leads to impaired neural control of cardiac rhythm (69, 71, 73). With Kv1.1 deficiency, seizures may cause proliferation of glial cells in the nuclei of the heart and lung centers, which may cause abnormal breathing (69). Kv1.1 deficiency also reduces the inhibitory control of interneurons in the central lateral amygdala and the overexcitation related to seizure inhibition (71). In Kv1.1-deficient mice, epileptic seizures cause abnormal parasympathetic nerve transmission, which leads to impaired neural control of heart rhythm and malignant arrhythmias (73).
In animals with chronic epilepsy, levels of the Kv4.2 myocardial voltage-gated potassium channel are decreased (74). The Kv4.2 subunit contributes to the pore-forming region of channels that express a transient A-type potassium ion current in hippocampal CA1 pyramidal cell dendrites. It is the main medium of hyperpolarized A-type current in the brain, plays an important role in signal processing and synaptic integration, and is a key regulator of neuronal excitability (75, 76). Compared with wild-type mice, the latency to seizure and status epilepticus onset is lower in Kv4.2 knockout mice (76). Silencing of Kv4.2 is mediated by miR-324-5p (75, 77). In epileptic mice, increased Kv4.2 mRNA silencing causes decreased Kv4.2 protein level and production of type A current; its role in regulating neuronal excitability is also limited (77). Inhibition of miR-324-5p can reduce the frequency of spontaneous seizures and epileptic spikes between seizures and produce neuroprotective and antiepileptic effects (75, 77).
Renin-angiotensin system
The renin-angiotensin system has also been implicated in SUDEP (78). Angiotensin II is the main peptide of the system. Various signal pathways in the central nervous system are stimulated by angiotensin II receptor-1 (ATR1) and angiotensin II receptor-2 (ATR2) (79). Activation of ATR1 is pro-inflammatory and pro-epileptogenic (80). Angiotensin converting enzyme and ATR1 are upregulated in the brain of rats with repetitive seizures (81). We speculate that repeated seizures will lead to upregulation of ATR1 in the brain, causing pro-inflammatory and pro-epileptogenic effects that may lead to SUDEP. Other renin-angiotensin system pathways in the nervous system include angiotensin-(1–7) binding to the receptor Mas (82, 83). Angiotensin-(1–7) participates in the learning and memory process that takes place in the central marginal region of the brain. In chronically stimulated epileptic rats, levels of thimet oligopeptidase [the main enzyme involved in generation of angiotensin-(1–7)], angiotensin-(1–7), and receptor Mas transcripts are elevated (82). However, the effect of this on risk of SUDEP is unknown.
Other mechanisms
Oxygen-conserving reflexes (OCR), amygdala rapid kindling (ARK), and central nervous system damage owing to repeated GTCS may also be potential mechanisms of SUDEP. Biggs et al. (84) reported that in epileptic rats, induction of OCR causes fluctuations in heart rate and respiratory rate similar to human SUDEP. The ability of the carotid body to stimulate respiratory restart appears to be impaired during seizures (85). Totola et al. (86) reported that in epileptic rats, the number of Fos-immunoreactive neurons in the posterior trapezoidal nucleus, raphe magnus nucleus, and nucleus tractus solitarius decreased after ARK; in addition, the ventilatory volume decreased significantly. ARK damages respiratory neurons in the brain stem, resulting in respiratory dysfunction. After a single GTCS, the blood–brain barrier exhibits signs of inflammation, neuronal damage, and transitory destruction (87). Therefore, repeated GCS attacks may cause central nervous system damage and SUDEP.
To sum up, catecholamine, P-gp and ATR1 may participate in SUDEP as important molecular biomarkers. The gene mutation related to potassium channel may also involve in its occurrence (Figure 1).
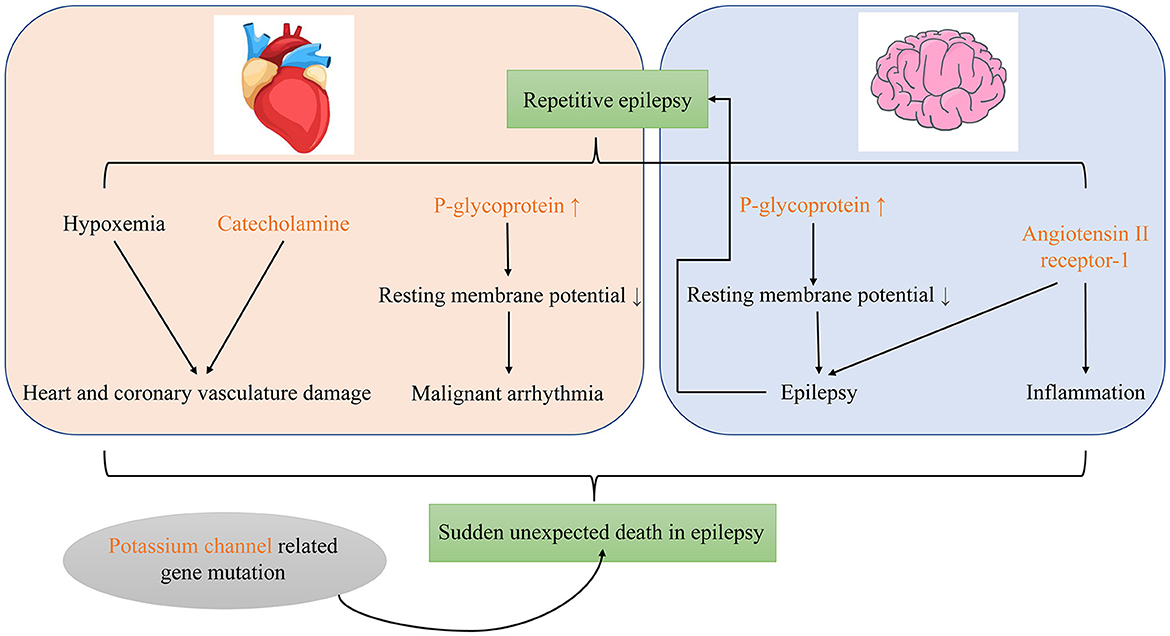
Figure 1. Possible molecular mechanisms of sudden unexpected death in epilepsy.
Discussion
Among the various SUDEP risk factors, frequency of GTCS appears to be the most important. Prone position, male sex, chronic epilepsy, ion channel or arrhythmia-related gene mutations, neurological comorbidities, polytherapy, long QT syndrome, obstructive sleep apnea, and structural brain damage are other potential factors.
Previous studies of SUDEP mechanisms have focused on malignant arrhythmias, myocardial cell hypertrophy, myocardial fibrosis, central and obstructive apnea, pulmonary edema, and abnormal regulation of the neurotransmitters 5-HT and adenosine. Research of underlying molecular mechanisms has been limited; therefore, SUDEP is often difficult to distinguish from other causes of sudden death (88).
There are many possible molecular mechanisms of SUDEP. First, epileptic seizures induce the expression of P-gp in neurons and myocardial cells, which reduces resting membrane potential and predisposes to development of epilepsy, malignant arrhythmias, and sudden death. Second, after status epilepticus, a large amount of catecholamines are released, which can cause myocardial ischemia, calcium overload, oxidative stress, and mitochondrial dysfunction. In turn, these may cause myocardial damage. Third, abnormal potassium channels may increase the risk of cardiopulmonary dysfunction during seizures. Fourth, repeated seizures lead to upregulation of ATR1 in the brain, causing pro-inflammatory and pro-epileptogenic effects. Finally, OCR, ARK, and central nervous system damage caused by repeated GTCS may be involved as well.
The mechanism of SUDEP is complex and most previous studies have focused on cardiac and respiratory dysfunction and imbalance of the neural regulatory system. Through a systematic literature review, we speculate on the mechanism of certain SUDEP cases as follows: (1) Repeated seizures cause chronic structural damage to the brain, especially the respiratory and cardiac centers. This cumulative damage causes cumulative increased risk of sudden death. (2) During seizures, especially GTCS, sudden neurological disorder and cardiac respiratory dysfunction may cause sudden death. (3) Deletions in ion channel genes deletion increase the risk of cardiopulmonary dysfunction during seizures. Overall, this review summarizes the existing mechanisms and molecular mechanisms of SUDEP, hoping to update the research progress and provide useful reference for forensic scholars in routine cases.
Author contributions
XS and YL designed the framework of the review and drafted the manuscript. YL and JL provided supervision and contributed to manuscript writing and editing. All authors have read and approved the latest version of the manuscript.
Funding
This work was funded by the Shanghai Sailing Plan (21YF1418800), the National Innovative Foundation Project for Students (202210262058) and the Shanghai Key Laboratory of Forensic Medicine, Academy of Forensic Science (KF1902).
Acknowledgments
We thank Edanz for editing the language of a draft of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Shankar R, Donner EJ, McLean B, Nashef L, Tomson T. Sudden unexpected death in epilepsy (SUDEP): what every neurologist should know. Epileptic Disord. (2017) 19:1–9. doi: 10.1684/epd.2017.0891
2. Ge Y, Ding D, Zhang Q, Yang B, Wang T, Li B, et al. Incidence of sudden unexpected death in epilepsy in community-based cohort in China. Epilepsy Behav. (2017) 76:76–83. doi: 10.1016/j.yebeh.2017.08.024
3. Zhao H, Long L, Xiao B. Advances in sudden unexpected death in epilepsy. Acta Neurol Scand. (2022) 146:716–22. doi: 10.1111/ane.13715
4. Garg D, Sharma S. Sudden unexpected death in epilepsy (SUDEP): what pediatricians need to know. Indian Pediatr. (2020) 57:890–94. doi: 10.1007/s13312-020-1986-4
5. Bernardi J, Aromolaran KA, Aromolaran AS. Neurological disorders and risk of arrhythmia. Int J Mol Sci. (2020) 22:188. doi: 10.3390/ijms22010188
6. Ryvlin P, Rheims S, Lhatoo SD. Risks and predictive biomarkers of sudden unexpected death in epilepsy patient. Curr Opin Neurol. (2019) 32:205–12. doi: 10.1097/WCO.0000000000000668
7. O'Neal TB, Shrestha S, Singh H, Osagie I, Ben-Okafor K, Cornett EM, et al. Sudden unexpected death in epilepsy. Neurol Int. (2022) 14:600–13. doi: 10.3390/neurolint14030048
8. Shorvon S, Tomson T. Sudden unexpected death in epilepsy. Lancet. (2011) 378:2028–38. doi: 10.1016/S0140-6736(11)60176-1
9. Ali A, Wu S, Issa NP, Rose S, Towle VL, Warnke P, et al. Association of sleep with sudden unexpected death in epilepsy. Epilepsy Behav. (2017) 76:1–6. doi: 10.1016/j.yebeh.2017.08.021
10. Oguz Akarsu E, Sahin E, Ozel Yildiz S, Bebek N, Gurses C, Baykan B. Peri-ictal prone position is associated with independent risk factors for sudden unexpected death in epilepsy: a controlled video-EEG monitoring Unit Study. Clin EEG Neurosci. (2018) 49:197–205. doi: 10.1177/1550059417733385
11. Liebenthal JA, Wu S, Rose S, Ebersole JS, Tao JX. Association of prone position with sudden unexpected death in epilepsy. Neurology. (2015) 84:703–9. doi: 10.1212/WNL.0000000000001260
12. Barot N, Nei M. Autonomic aspects of sudden unexpected death in epilepsy (SUDEP). Clin Auton Res. (2019) 29:151–60. doi: 10.1007/s10286-018-0576-1
13. Ellis SP, Szabo CA. Sudden unexpected death in epilepsy: incidence, risk factors, and proposed mechanisms. Am J Forensic Med Pathol. (2018) 39:98–102. doi: 10.1097/PAF.0000000000000394
14. Mastrangelo M, Esposito D. Paediatric sudden unexpected death in epilepsy: from pathophysiology to prevention. Seizure. (2022) 101:83–95. doi: 10.1016/j.seizure.2022.07.020
15. Asadi-Pooya AA, Sperling MR. Clinical features of sudden unexpected death in epilepsy. J Clin Neurophysiol. (2009) 26:297–301. doi: 10.1097/WNP.0b013e3181b7f129
16. Manolis TA, Manolis AA, Melita H, Manolis AS. Sudden unexpected death in epilepsy: the neuro-cardio-respiratory connection. Seizure. (2019) 64:65–73. doi: 10.1016/j.seizure.2018.12.007
17. Chahal CAA, Salloum MN, Alahdab F, Gottwald JA, Tester DJ, Anwer LA, et al. Systematic review of the genetics of sudden unexpected death in epilepsy: potential overlap with sudden cardiac death and arrhythmia-related genes. J Am Heart Assoc. (2020) 9:e012264. doi: 10.1161/JAHA.119.012264
18. Bleakley LE, Soh MS, Bagnall RD, Sadleir LG, Gooley S, Semsarian C, et al. Are variants causing cardiac arrhythmia risk factors in sudden unexpected death in epilepsy? Front Neurol. (2020) 11:925. doi: 10.3389/fneur.2020.00925
19. Cheng JY. Risk of sudden unexpected death in people with epilepsy and obstructive sleep apnea. Epilepsy Res. (2021) 176:106729. doi: 10.1016/j.eplepsyres.2021.106729
20. Somboon T, Grigg-Damberger MM, Foldvary-Schaefer N. Epilepsy and sleep-related breathing disturbances. Chest. (2019) 156:172–81. doi: 10.1016/j.chest.2019.01.016
21. Allen LA, Harper RM, Lhatoo S, Lemieux L, Diehl B. Neuroimaging of sudden unexpected death in epilepsy (SUDEP): insights from structural and resting-state functional MRI studies. Front Neurol. (2019) 10:185. doi: 10.3389/fneur.2019.00185
22. Patodia S, Paradiso B, Ellis M, Somani A, Sisodiya SM, Devinsky O, et al. Characterisation of medullary astrocytic populations in respiratory nuclei and alterations in sudden unexpected death in epilepsy. Epilepsy Res. (2019) 157:106213. doi: 10.1016/j.eplepsyres.2019.106213
23. Allen LA, Vos SB, Kumar R, Ogren JA, Harper RK, Winston GP, et al. Cerebellar, limbic, and midbrain volume alterations in sudden unexpected death in epilepsy. Epilepsia. (2019) 60:718–29. doi: 10.1111/epi.14689
24. Allen LA, Harper RM, Guye M, Kumar R, Ogren JA, Vos SB, et al. Altered brain connectivity in sudden unexpected death in epilepsy (SUDEP) revealed using resting-state fMRI. Neuroimage Clin. (2019) 24:102060. doi: 10.1016/j.nicl.2019.102060
25. Costagliola G, Orsini A, Coll M, Brugada R, Parisi P, Striano P. The brain-heart interaction in epilepsy: implications for diagnosis, therapy, and SUDEP prevention. Ann Clin Transl Neurol. (2021) 8:1557–68. doi: 10.1002/acn3.51382
26. Goldberger JJ, Challapalli S, Tung R, Parker MA, Kadish AH. Relationship of heart rate variability to parasympathetic effect. Circulation. (2001) 103:1977–83. doi: 10.1161/01.CIR.103.15.1977
27. Myers KA, Sivathamboo S, Perucca P. Heart rate variability measurement in epilepsy: how can we move from research to clinical practice? Epilepsia. (2018) 59:2169–78. doi: 10.1111/epi.14587
28. DeGiorgio CM, Miller P, Meymandi S, Chin A, Epps J, Gordon S, et al. RMSSD, a measure of vagus-mediated heart rate variability, is associated with risk factors for SUDEP: the SUDEP-7 inventory. Epilepsy Behav. (2010) 19:78–81. doi: 10.1016/j.yebeh.2010.06.011
29. Schurr JW, Grewal PK, Fan R, Rashba E. QT interval measurement in ventricular pacing: implications for assessment of drug effects and pro-arrhythmia risk. J Electrocardiol. (2022) 70:13–8. doi: 10.1016/j.jelectrocard.2021.11.029
30. Lankaputhra M, Voskoboinik A. Congenital long QT syndrome: a clinician's guide. Intern Med J. (2021) 51:1999–2011. doi: 10.1111/imj.15437
31. van der Linde H, Kreir M, Teisman A, Gallacher DJ. Seizure-induced Torsades de pointes: in a canine drug-induced long-QT1 model. J Pharmacol Toxicol Methods. (2021) 111:107086. doi: 10.1016/j.vascn.2021.107086
32. Haddad PM, Anderson IM. Antipsychotic-related QTc prolongation, torsade de pointes and sudden death. Drugs. (2002) 62:1649–71. doi: 10.2165/00003495-200262110-00006
33. Pansani AP, Ghazale PP, Dos Santos EG, Dos Santos Borges K, Gomes KP, Lacerda IS, et al. The number and periodicity of seizures induce cardiac remodeling and changes in micro-RNA expression in rats submitted to electric amygdala kindling model of epilepsy. Epilepsy Behav. (2021) 116:107784. doi: 10.1016/j.yebeh.2021.107784
34. Nascimento FA, Tseng ZH, Palmiere C, Maleszewski JJ, Shiomi T, McCrillis A, et al. Pulmonary and cardiac pathology in sudden unexpected death in epilepsy (SUDEP). Epilepsy Behav. (2017) 73:119–25. doi: 10.1016/j.yebeh.2017.05.013
35. Budde RB, Arafat MA, Pederson DJ, Lovick TA, Jefferys JGR, Irazoqui PP. Acid reflux induced laryngospasm as a potential mechanism of sudden death in epilepsy. Epilepsy Res. (2018) 148:23–31. doi: 10.1016/j.eplepsyres.2018.10.003
36. Nakase K, Kollmar R, Lazar J, Arjomandi H, Sundaram K, Silverman J, et al. Laryngospasm, central and obstructive apnea during seizures: defining pathophysiology for sudden death in a rat model. Epilepsy Res. (2016) 128:126–39. doi: 10.1016/j.eplepsyres.2016.08.004
37. Tavee J, Morris H. Severe postictal laryngospasm as a potential mechanism for sudden unexpected death in epilepsy: a near-miss in an EMU. Epilepsia. (2008) 49:2113–7. doi: 10.1111/j.1528-1167.2008.01781.x
38. Mandal R, Budde R, Lawlor GL, Irazoqui P. Utilizing multimodal imaging to visualize potential mechanism for sudden death in epilepsy. Epilepsy Behav. (2021) 122:108124. doi: 10.1016/j.yebeh.2021.108124
39. Nobis WP, González Otárula KA, Templer JW, Gerard EE, VanHaerents S, Lane G, et al. The effect of seizure spread to the amygdala on respiration and onset of ictal central apnea. J Neurosurg. (2019) 132:1313–23. doi: 10.3171/2019.1.JNS183157
40. Nobis WP, Schuele S, Templer JW, Zhou G, Lane G, Rosenow JM, et al. Amygdala-stimulation-induced apnea is attention and nasal-breathing dependent. Ann Neurol. (2018) 83:460–71. doi: 10.1002/ana.25178
41. Dlouhy BJ, Gehlbach BK, Kreple CJ, Kawasaki H, Oya H, Buzza C, et al. Breathing inhibited when seizures spread to the amygdala and upon amygdala stimulation. J Neurosci. (2015) 35:10281–9. doi: 10.1523/JNEUROSCI.0888-15.2015
42. Rhone AE, Kovach CK, Harmata GI, Sullivan AW, Tranel D, Ciliberto MA, et al. A human amygdala site that inhibits respiration and elicits apnea in pediatric epilepsy. JCI Insight. (2020) 5:852. doi: 10.1172/jci.insight.134852
43. Marincovich A, Bravo E, Dlouhy B, Richerson GB. Amygdala lesions reduce seizure-induced respiratory arrest in DBA/1 mice. Epilepsy Behav. (2021) 121:106440. doi: 10.1016/j.yebeh.2019.07.041
44. Park K, Kanth K, Bajwa S, Girgis F, Shahlaie K, Seyal M. Seizure-related apneas have an inconsistent linkage to amygdala seizure spread. Epilepsia. (2020) 61:1253–60. doi: 10.1111/epi.16518
45. Du Y, He GY, Yao L, Ren P, Pang L, Wang WD. Forensic analysis of 9 cases of sudden unexpected death in epilepsy. Fa Yi Xue Za Zhi. (2022) 38:490–94. doi: 10.12116/j.issn.1004-5619.2020.400616
46. Romero-Osorio OM, Abaunza-Camacho JF, Sandoval-Briceno D. Postictal pulmonary oedema: a review of the literature. Rev Neurol. (2019) 68:339–45. doi: 10.33588/rn.6808.2018356
47. Mahdavi Y, Surges R, Nikoubashman O, Olaciregui Dague K, Brokmann JC, Willmes K, et al. Neurogenic pulmonary edema following seizures: a retrospective computed tomography study. Epilepsy Behav. (2019) 94:112–17. doi: 10.1016/j.yebeh.2019.02.006
48. Kennedy JD, Hardin KA, Parikh P, Li CS, Seyal M. Pulmonary edema following generalized tonic clonic seizures is directly associated with seizure duration. Seizure. (2015) 27:19–24. doi: 10.1016/j.seizure.2015.02.023
49. Simon RP. Epileptic sudden death: animal models. Epilepsia. (1997) 38:S35–7. doi: 10.1111/j.1528-1157.1997.tb06124.x
50. Ruthirago D, Julayanont P, Karukote A, Shehabeldin M, Nugent K. Sudden unexpected death in epilepsy: ongoing challenges in finding mechanisms and prevention. Int J Neurosci. (2018) 128:1052–60. doi: 10.1080/00207454.2018.1466780
51. Hodges MR, Richerson GB. The role of medullary serotonin (5-HT) neurons in respiratory control: contributions to eupneic ventilation, CO2 chemoreception, and thermoregulation. J Appl Physiol. (2010) 108:1425–32. doi: 10.1152/japplphysiol.01270.2009
52. Zhang H, Zhao H, Zeng C, Van Dort C, Faingold CL, Taylor NE, et al. Optogenetic activation of 5-HT neurons in the dorsal raphe suppresses seizure-induced respiratory arrest and produces anticonvulsant effect in the DBA/1 mouse SUDEP model. Neurobiol Dis. (2018) 110:47–58. doi: 10.1016/j.nbd.2017.11.003
53. Faingold CL, Randall M, Zeng C, Peng S, Long X, Feng HJ. Serotonergic agents act on 5-HT(3) receptors in the brain to block seizure-induced respiratory arrest in the DBA/1 mouse model of SUDEP. Epilepsy Behav. (2016) 64:166–70. doi: 10.1016/j.yebeh.2016.09.034
54. Tupal S, Faingold CL. Serotonin 5-HT(4) receptors play a critical role in the action of fenfluramine to block seizure-induced sudden death in a mouse model of SUDEP. Epilepsy Res. (2021) 177:106777. doi: 10.1016/j.eplepsyres.2021.106777
55. Lin WH, Huang HP, Lin MX, Chen SG, Lv XC, Che CH, et al. Seizure-induced 5-HT release and chronic impairment of serotonergic function in rats. Neurosci Lett. (2013) 534:1–6. doi: 10.1016/j.neulet.2012.12.007
56. Purnell B, Murugan M, Jani R, Boison D. The good, the bad, and the deadly: adenosinergic mechanisms underlying sudden unexpected death in epilepsy. Front Neurosci. (2021) 15:708304. doi: 10.3389/fnins.2021.708304
57. Faingold CL, Randall M, Kommajosyula SP. Susceptibility to seizure-induced sudden death in DBA/2 mice is altered by adenosine. Epilepsy Res. (2016) 124:49–54. doi: 10.1016/j.eplepsyres.2016.05.007
58. Klaft ZJ, Hollnagel JO, Salar S, Calişkan G, Schulz SB, Schneider UC, et al. Adenosine A1 receptor-mediated suppression of carbamazepine-resistant seizure-like events in human neocortical slices. Epilepsia. (2016) 57:746–56. doi: 10.1111/epi.13360
59. Patodia S, Paradiso B, Garcia M, Ellis M, Diehl B, Thom M, et al. Adenosine kinase and adenosine receptors A(1) R and A(2A) R in temporal lobe epilepsy and hippocampal sclerosis and association with risk factors for SUDEP. Epilepsia. (2020) 61:787–97. doi: 10.1111/epi.16487
60. Auzmendi J, Buchholz B, Salguero J, Canellas C, Kelly J, Men P, et al. Pilocarpine-induced status epilepticus is associated with p-glycoprotein induction in cardiomyocytes, electrocardiographic changes, and sudden death. Pharmaceuticals. (2018) 11:21. doi: 10.3390/ph11010021
61. Czornyj L, Auzmendi J, Lazarowski A. Transporter hypothesis in pharmacoresistant epilepsies. Is it at the central or peripheral level? Epilepsia Open. (2022) 7:S34–46. doi: 10.1002/epi4.12537
62. Auzmendi J, Akyuz E, Lazarowski A. The role of P-glycoprotein (P-gp) and inwardly rectifying potassium (Kir) channels in sudden unexpected death in epilepsy (SUDEP). Epilepsy Behav. (2021) 121:106590. doi: 10.1016/j.yebeh.2019.106590
63. Hawkes MA, Hocker SE. Systemic complications following status epilepticus. Curr Neurol Neurosci Rep. (2018) 18:7. doi: 10.1007/s11910-018-0815-9
64. Du Y, Demillard LJ, Ren J. Catecholamine-induced cardiotoxicity: a critical element in the pathophysiology of stroke-induced heart injury. Life Sci. (2021) 287:120106. doi: 10.1016/j.lfs.2021.120106
65. Zhao H, Zhang H, Schoen FJ, Schachter SC, Feng HJ. Repeated generalized seizures can produce calcified cardiac lesions in DBA/1 mice. Epilepsy Behav. (2019) 95:169–74. doi: 10.1016/j.yebeh.2019.04.010
66. Verrier RL, Pang TD, Nearing BD, Schachter SC. The epileptic heart: concept and clinical evidence. Epilepsy Behav. (2020) 105:106946. doi: 10.1016/j.yebeh.2020.106946
67. D'Adamo MC, Liantonio A, Rolland JF, Pessia M, Imbrici P. Kv11 channelopathies: pathophysiological mechanisms and therapeutic approaches. Int J Mol Sci. (2020) 21:935. doi: 10.3390/ijms21082935
68. Glasscock E. Kv1.1 channel subunits in the control of neurocardiac function. Channels. (2019) 13:299–307. doi: 10.1080/19336950.2019.1635864
69. Dhaibar HA, Hamilton KA, Glasscock E. Kv11 subunits localize to cardiorespiratory brain networks in mice where their absence induces astrogliosis and microgliosis. Mol Cell Neurosci. (2021) 113:103615. doi: 10.1016/j.mcn.2021.103615
70. Mulkey DK, Hawkins VE, Hawryluk JM, Takakura AC, Moreira TS, Tzingounis AV. Molecular underpinnings of ventral surface chemoreceptor function: focus on KCNQ channels. J Physiol. (2015) 593:1075–81. doi: 10.1113/jphysiol.2014.286500
71. Thouta S, Zhang Y, Garcia E, Snutch TP. K(v)11 channels mediate network excitability and feed-forward inhibition in local amygdala circuits. Sci Rep. (2021) 11:15180. doi: 10.1038/s41598-021-94633-3
72. Dhaibar H, Gautier NM, Chernyshev OY, Dominic P, Glasscock E. Cardiorespiratory profiling reveals primary breathing dysfunction in Kcna1-null mice: implications for sudden unexpected death in epilepsy. Neurobiol Dis. (2019) 127:502–11. doi: 10.1016/j.nbd.2019.04.006
73. Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL. Kv11 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. (2010) 30:5167–75. doi: 10.1523/JNEUROSCI.5591-09.2010
74. Lai YC Li N, Lawrence W, Wang S, Levine A, Burchhardt DM, et al. Myocardial remodeling and susceptibility to ventricular tachycardia in a model of chronic epilepsy. Epilepsia Open. (2018) 3:213–23. doi: 10.1002/epi4.12107
75. Gross C, Yao X, Engel T, Tiwari D, Xing L, Rowley S, et al. MicroRNA-mediated downregulation of the potassium channel Kv42 contributes to seizure onset. Cell Rep. (2016) 17:37–45. doi: 10.1016/j.celrep.2016.08.074
76. Barnwell LF, Lugo JN, Lee WL, Willis SE, Gertz SJ, Hrachovy RA, et al. Kv42 knockout mice demonstrate increased susceptibility to convulsant stimulation. Epilepsia. (2009) 50:1741–51. doi: 10.1111/j.1528-1167.2009.02086.x
77. Tiwari D, Brager DH, Rymer JK, Bunk AT, White AR, Elsayed NA, et al. MicroRNA inhibition upregulates hippocampal A-type potassium current and reduces seizure frequency in a mouse model of epilepsy. Neurobiol Dis. (2019) 130:104508. doi: 10.1016/j.nbd.2019.104508
78. Szczurkowska PJ, Polonis K, Becari C, Hoffmann M, Narkiewicz K, Chrostowska M. Epilepsy and hypertension: the possible link for sudden unexpected death in epilepsy? Cardiol J. (2021) 28:330–35. doi: 10.5603/CJ.a2019.0095
79. Kusmirowska K, Kowalski A, Rebas E. Angiotensins as neuromodulators. Postepy Biochem. (2012) 58:478–84.
80. Ramos AJ. Brain angiotensin system: a new promise in the management of epilepsy? Clin Sci. (2021) 135:725–30. doi: 10.1042/CS20201296
81. Pereira MG, Becari C, Oliveira JA, Salgado MC, Garcia-Cairasco N, Costa-Neto CM. Inhibition of the renin-angiotensin system prevents seizures in a rat model of epilepsy. Clin Sci. (2010) 119:477–82. doi: 10.1042/CS20100053
82. Pereira MG, Souza LL, Becari C, Duarte DA, Camacho FR, Oliveira JA, et al. Angiotensin II-independent angiotensin-(1-7) formation in rat hippocampus: involvement of thimet oligopeptidase. Hypertension. (2013) 62:879–85. doi: 10.1161/HYPERTENSIONAHA.113.01613
83. Gouveia TL, Frangiotti MI, de Brito JM, de Castro Neto EF, Sakata MM, Febba AC, et al. The levels of renin-angiotensin related components are modified in the hippocampus of rats submitted to pilocarpine model of epilepsy. Neurochem Int. (2012) 61:54–62. doi: 10.1016/j.neuint.2012.04.012
84. Biggs EN, Budde R, Jefferys JGR, Irazoqui PP. Ictal activation of oxygen-conserving reflexes as a mechanism for sudden death in epilepsy. Epilepsia. (2021) 62:752–64. doi: 10.1111/epi.16831
85. Biggs EN, Budde RB, Jefferys JGR, Irazoqui PP. Carotid body stimulation as a potential intervention in sudden death in epilepsy. Epilepsy Behav. (2022) 136:108918. doi: 10.1016/j.yebeh.2022.108918
86. Totola LT, Malheiros-Lima MR, Delfino-Pereira P, Del Vecchio F, Souza FC, Takakura AC, et al. Amygdala rapid kindling impairs breathing in response to chemoreflex activation. Brain Res. (2019) 1718:159–68. doi: 10.1016/j.brainres.2019.05.015
87. Nass RD, Wagner M, Surges R, Holdenrieder S. Time courses of HMGB1 and other inflammatory markers after generalized convulsive seizures. Epilepsy Res. (2020) 162:106301. doi: 10.1016/j.eplepsyres.2020.106301
Keywords: epilepsy, mortality, sudden unexpected death in epilepsy, mechanism, P-glycoprotein, catecholamines, K+ channel, renin-angiotensin system
Citation: Sun X, Lv Y and Lin J (2023) The mechanism of sudden unexpected death in epilepsy: A mini review. Front. Neurol. 14:1137182. doi: 10.3389/fneur.2023.1137182
Received: 04 January 2023; Accepted: 20 January 2023;
Published: 06 February 2023.
Edited by:
Beixu Li, Shanghai University of Political Science and Law, ChinaReviewed by:
Hui Li, Shanghai Criminal Science and Technology Institution, ChinaHeng Zhang, Anhui Medical University, China
Copyright © 2023 Sun, Lv and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yehui Lv,  bHZ5aF8xNUBzdW1ocy5lZHUuY24=
bHZ5aF8xNUBzdW1ocy5lZHUuY24=