
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 20 March 2023
Sec. Dementia and Neurodegenerative Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1134225
This article is part of the Research TopicDementia and Neurodegenerative Diseases – Case Report Collection 2022View all 25 articles
 Ling Ling Rong1*
Ling Ling Rong1* Nicholas J. Lannen1Evan C. Tank1Jessica L. Feistel1Christopher J. Therasse2Anvita Potluri1Muhib Khan1Jiangyong Min1
Nicholas J. Lannen1Evan C. Tank1Jessica L. Feistel1Christopher J. Therasse2Anvita Potluri1Muhib Khan1Jiangyong Min1Background: Creutzfeldt–Jakob disease (CJD) is a rare, rapidly progressive, and uniformly fatal neurodegenerative disease. The reported incidence of CJD is 1 to 2 per million people worldwide annually, with fewer than 1,000 cases in the United States per year. In this study, we report a unique case series on temporo-spatial clusters of CJD cases in West Michigan.
Methods: A total of five CJD cases consisting of two temporal clusters were seen from July 2021 to June 2022 at Corewell Health West hospitals. All patients had brain MRI, EEG, and CSF tests. Four patients underwent autopsies.
Results: All patients' MRIs showed characteristic CJD patterns. Four patients had positive CJD panels in CSF. One patient had typical CJD EEG findings. Four patients were confirmed as sporadic CJD by autopsy. All patients died within 3 months after CJD was suspected.
Discussion: All patients lived within a 90-mile radius of Grand Rapids, MI, and two lived in the same county. West Michigan has a population of 1.6 million people, and the four counties where five patients lived have a combined population of 395,104, indicating CJD's new case rate of 3.1 and 12.5 per million people, respectively. Corewell Health is one of the three major healthcare systems in West Michigan. The actual incidence of CJD in West Michigan is likely even higher. This dense temporal and spatial cluster of CJD cases poses a serious public health challenge and warrants urgent investigation.
Creutzfeldt–Jakob disease (CJD) is a transmissible, rapidly progressive neurological disease, caused by misfolded prion protein in the brain (1). The reported prevalence of CJD is 1 to 2 per million people worldwide annually and less than 1,000 cases in the United States per year (2–5)1 CJD subtypes include sporadic, genetic, iatrogenic, and variant CJD with 85–90% of cases being sporadic (2, 6, 7). sCJD can further be divided into subtypes including MM/(MV)1, MM2, VV1, VV2, and MV2 based on disease-related prion protein features and prion protein genotype in the host at the methionine (M) and valine (V) polymorphic codon 129 (8–11)1. The clinical diagnostic criteria for probable sCJD are rapidly progressive dementia plus at least two of the following: myoclonus, visual or cerebellar signs, pyramidal/extrapyramidal signs, and akinetic mutism. Vertigo, headache, and neuropsychiatric symptoms can also present. Patients gradually lose mobility, speech, and progress into a comatose state (2, 6, 7, 12, 13). Despite extensive research since its initial description 100 years ago, CJD remains an incurable disease with a survival of 4–12 months from symptom onset in the vast majority of patients (2, 6, 7).
Over the past few decades, there have been increased reports on sCJD. Some studied regional or national geographical distribution or temporal occurrence, but their cases occurred during periods of 9 to 15 years (14–18). Few case series focused on cases with similar clinical presentation without patients' geographic information (19, 20), or on cases over 5 years in the same region (21).
Our case series includes two temporal clusters of CJD cases in one region. Within 1 year from July 2021 to June 2022, we observed five CJD cases at Corewell Health West Butterworth (BW) and Blodgett (BL) Hospitals in Grand Rapids, Michigan (MI). These two hospitals are 3 miles apart. All five cases had supportive brain magnetic resonance imaging (MRI), four of them had supportive cerebrospinal fluid (CSF) findings, and one case had a characteristic electroencephalogram (EEG) pattern. All patients died within 3 months after CJD was suspected. We report this dense temporo-spatial cluster of CJD cases to call for an urgent investigation by public health officials.
Patient 1: A 67-year-old white woman who worked as a clinic manager was admitted on 14 July 2021 to BW due to rapid neurological decline. Her initial symptom was severe insomnia starting in mid-January 2021. By February 2021, her symptoms progressed to vertigo, diplopia, and imbalance. By May 2021, she was not able to function at work due to cognitive impairment. Her family noticed intermittent “childlike” behavior. On admission, she was fully alert, awake, and oriented with normal cranial nerves. Montreal Cognitive Assessment (MoCA) testing revealed profound deficits with a corrected score of 17/30.
Patient 2: A 78-year-old white man who was a semi-retired funeral home director was admitted on 31 July 2021 to BW for rapidly progressive cognitive decline along with dysfunctional gait, abnormal speech, and intermittent body jerking. In early May 2021, he started to have intermittent hand weakness, paresthesia, and forgetfulness. Due to unsteadiness, he began using a cane in June 2021 but quickly progressed to a walker. Outpatient electromyography and nerve conduction velocity (EMG/NCV) studies were unrevealing. MRI brain reported a 3-mm subacute infarct in left caudate head and ventriculomegaly. In the same month, he developed expressive aphasia progressing to a paucity of speech. Over 2 weeks, he had a catastrophic decline with excessive daytime somnolence. He was unable to perform activities of daily living and developed alternating urinary incontinence and retention. On admission, he was somnolent but easily startled by auditory stimuli. He followed limited, one-step commands and moved all extremities but was oriented to self only.
Patient 3: A 77-year-old white man who was a semi-retired attorney was transferred to BL for continuous video EEG monitoring on 03 May 2022 from an outside hospital (OSH), which was 75 miles east of Grand Rapids. In mid-March 2022, he complained of “brain fog” after he started medication for his newly diagnosed hypertension. The dyscognia persisted despite discontinuation of the anti-hypertensive. He developed visual disturbance and reported seeing his own fingers abnormally elongated, and his legs were fat and bowed. He was able to provide a full history and had an intact neurological examination on admission at the OSH 6 days before transfer to our facility. Brain MRI at OSH reported subtle cortical restricted diffusion involving both posterior temporoparietal and occipital regions. Levetiracetam was initiated after a 1-h EEG captured intermittent delta slowing over the left frontal region without rhythmicity. CSF at OSH was unremarkable except pending the CJD panel. Upon transfer to BL, he was awake and alert but with limited orientation, verbal output, and impaired abstraction. His motor and sensory examinations were intact.
Patient 4: A 78-year-old white woman and homemaker presented to the local emergency department on 03 June 2022 for cognitive decline over several months, accelerating over a few weeks before presentation. Her brain MRI reported multiple infarcts in different territories concerning global hypoxic ischemia; the on-call tele-stroke physician requested transfer to BW for further evaluation as the MRI pattern suggested CJD rather than hypoxic ischemia. The patient's initial symptom was intermittent forgetfulness, which started in August 2021 and had worsened since late December 2021. In March 2022, she developed “pressure in head” and complained “I do not feel my brain work.” The patient was diagnosed with anxiety and treated with anxiolytics without benefit. In late April 2022, gait abnormality and word-finding difficulties arose, and in May 2022, she developed auditory hallucinations. By June 2022, jerks in her upper extremities were noticed. Upon admission, she was oriented to person only and had impaired attention, reasoning, and verbal expression but had preserved motor strength and sensation.
Patient 5: A 64-year-old white woman who worked as a nurse was admitted to BL on 17 June 2022 for rapidly progressive cognitive decline. Her initial symptoms were “brain fog,” dizziness, and fatigue, starting in January 2022. By late April 2022, she endorsed imbalance, diplopia, and multiple falls along with visual hallucinations, paranoia, and memory dysfunctions. On admission, she was awake, alert, oriented to self and could only recognize her close friends. Her cranial nerves were intact. She had mild proximal weakness in both bilateral upper and lower extremities. The vibratory sensation of bilateral lower extremities was impaired in a length-dependent distribution. She required stabilization on standing (Figure 1).
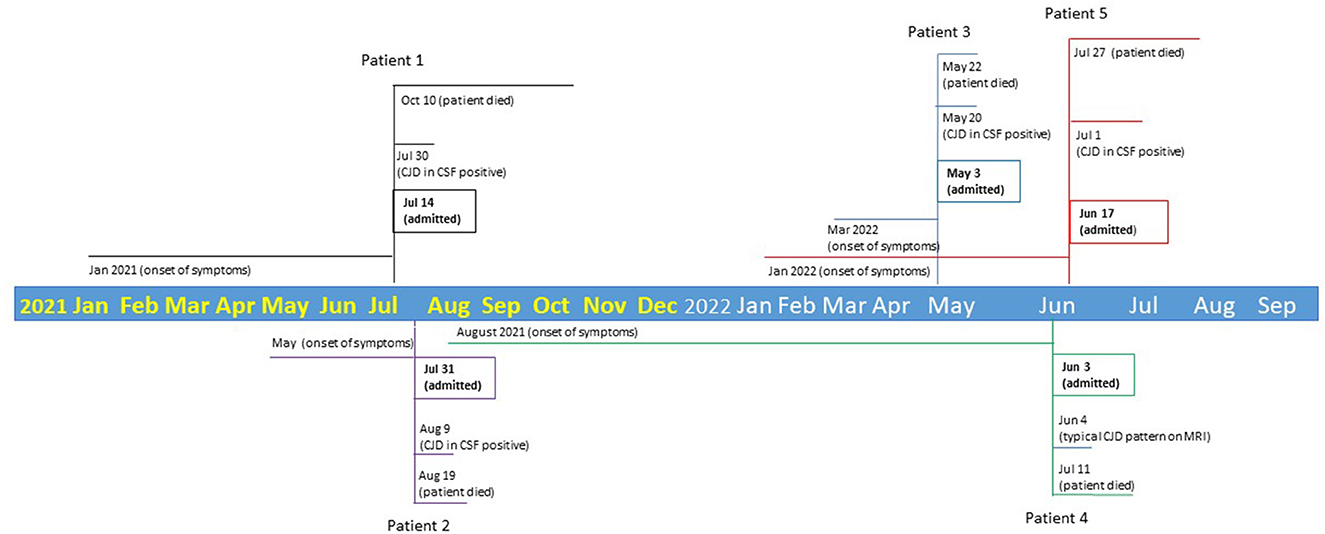
Figure 1. Timeline of two clusters of five CJD cases.
All five patients had initial or repeated 1.5 or 3 Tesla brain MRI with and without contrast (w/wo) at BW or BL, respectively, revealing restricted diffusion and corresponding hyperintense T2 FLAIR signal involving bilateral caudate nuclei and putamina in a symmetric pattern (patient 1); asymmetric diffusion restriction signals in the cerebral cortex of cingulum, left temporoparietal lobes, and caudate nucleus (patient 2); prominent multifocal diffusion restriction involving the bi-hemispheric cerebral cortex, more posteriorly and on the left compared to the right (patient 3); symmetric cortical diffusion restriction involving paramedian, lateral parietal cortices, temporal cortices, and to a lesser extent in the frontal lobe with involvement of left greater than the right (patient 4); restricted diffusion in the bilateral caudate nuclei (left > right) and the left mesial temporal lobe, including the amygdala, the hippocampus, and the forniceal column (patient 5) (Figure 2).
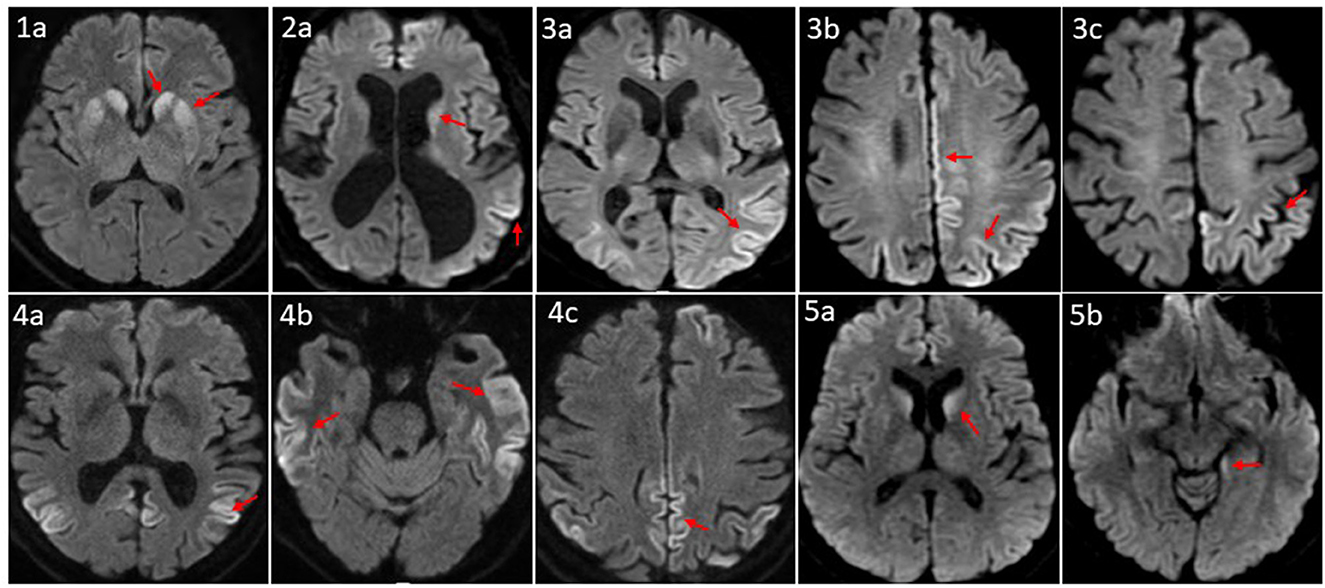
Figure 2. Axial MRI brain diffusion weighted images of five CID patients. (1a) Symmetric diffusion restriction in bilateral caudate nuclei and putamina. (2a) Subtle diffusion restriction involving the left caudate head, and left parletal cortex. (3a–c) Cortically based diffusion restriction involving the posterior left greater than right parietal lobes and paramedian left frontoparietal region. (4a–c) Intense cortically based restriction involving the paramedian and lateral parietal cortices, bilateral temporal cortices, and to a lesser extent the left greater that right frontal lobes. (5a, b) Relatively symmetric DWI within the caudate nuclei; subtle involvement of the posteromedial left hippocampus. Patient sequence was labeled from 1 to 5; red arrows point to sites of abnormal diffusion restriction.
EEG was performed on all patients. The EEG of patient 3 showed periodic sharp wave complexes (PSWC), a characteristic CJD pattern. Other patients' EEGs were unremarkable (patient 5), had non-specific rare generalized periodic discharges with triphasic morphology (patients 1 and 4); moderate bitemporal slowing with a right predominance (patient 1); generalized rhythmic delta activity (patient 2) (Figure 3). Blood, CSF basic tests, and Mayo Clinic autoimmune encephalopathy panel in serum (ENS2) were negative. The autoimmune encephalopathy panel and paraneoplastic panel in CSF were all negative. All patients' CSF CJD panels from the National Prion Disease Pathology Surveillance Center (NPDPSC) reported >98% likelihood of prion disease except patient 4 whose CJD likelihood and 14-3-3 proteins were inconclusive, RT-QuIC was negative, and T-tau level was high (19937 pg/ml) (Table 1).
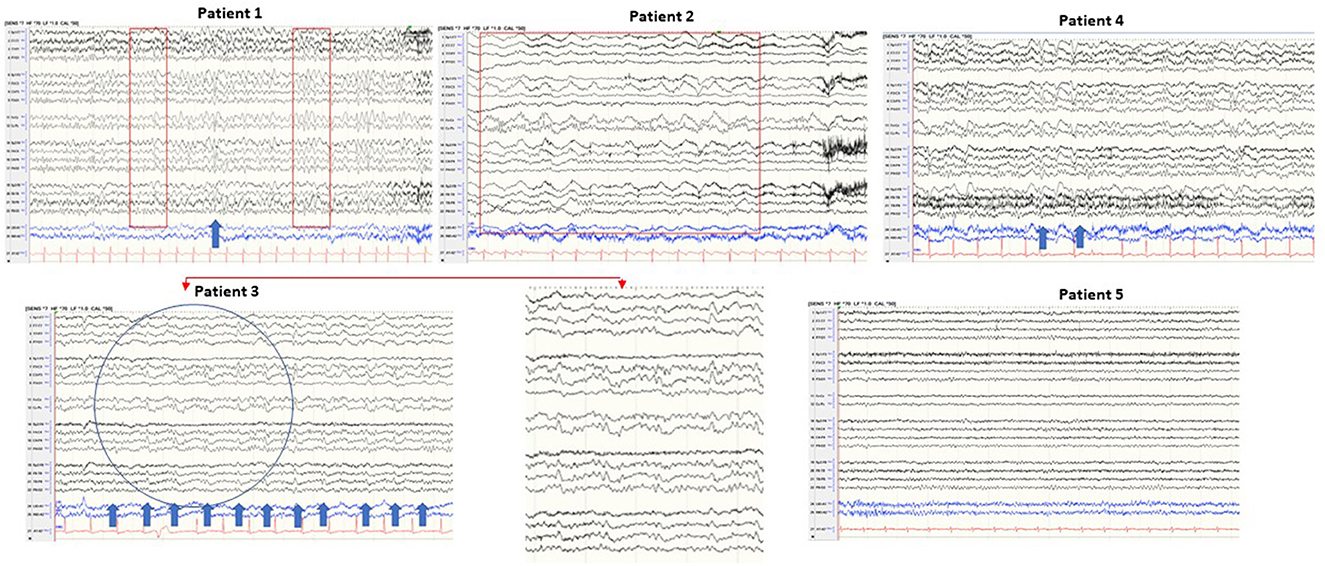
Figure 3. Typical samples of five patients in a standart anterior to posterior bipolar montage. Patient 1: background slowing and triphasic wave; patient 2: generalized rhythmic delta activity; patient 3: periodic sharp wave complexes (PSWC) at 1 Hz; patient 4: non-periodic triphasic wave; patient 5: normal EEG.
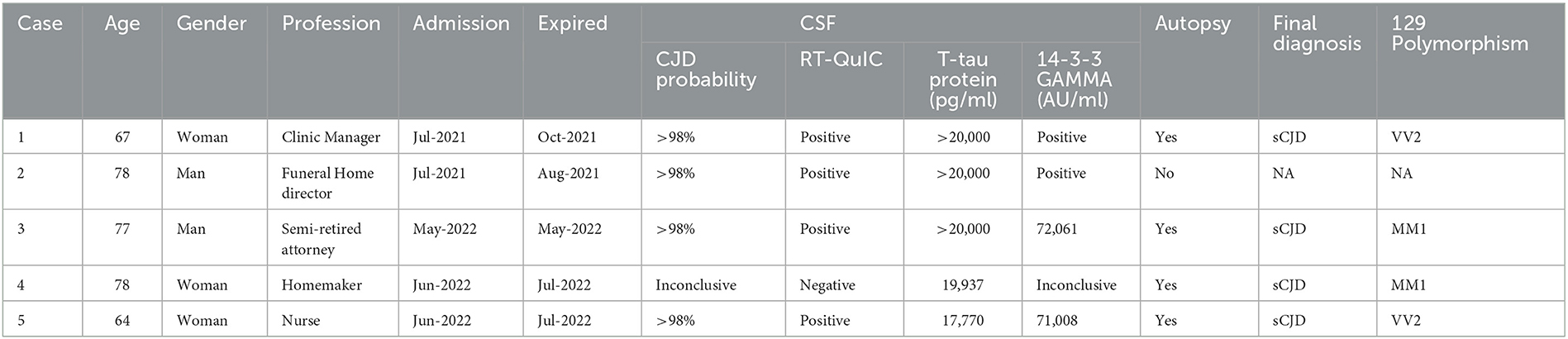
Table 1. Summary of patients' data, CSF CJD panel, and brain autopsy results.
All patients (except patient 4) received empirical treatment: five doses of 1,000 mg methylprednisolone intravenously daily, followed by five rounds of plasma exchange (patient 1); one dose of 400 mg/kg intravenous immunoglobulin (IVIg) (patient 2); five doses of 400 mg/kg IVIg alone daily (patient 3); or with five doses of IV methylprednisolone daily (patient 5). Patients did not have any benefits from the aforementioned treatment. Patients died in October 2021, August 2021, May 2022, July 2022, and July 2022, respectively. Four patients underwent autopsy and genetic analysis (the family of patient 2 declined autopsy). No gene mutation was detected in any patient. Except for codon 129 polymorphism, no other polymorphism was found in any patient. The final diagnosis was as follows: patients 1 and 5 were sCJD-VV2, and patients 3 and 4 were sCJD-MM1 (Table 1).
All patients in this case series had a rapid cognitive decline. Two patients also had visual disturbances (particularly patient 3 who presented with impaired visual perception at an early stage), and the illness progressed rapidly, possibly representing the Heidenhain variant of CJD (12, 22). On admission, the patients were 67, 78, 77, 78, and 64 years of age, and the durations from the time of symptoms onset to death were 8, 4, 3, 11, and 6 months, respectively. This is mostly consistent with sporadic CJD, which has reported a range of 55–75 years of peak onset age and a median survival duration of 4–12 months (2, 5, 7, 23). The durations from the time when positive CJD in CSF was reported (in patients 1, 2, 3, and 5) or from the time when brain MRI showed typical CJD patterns (patient 4) to death were 72, 10, 2, 26, and 43 days, respectively. All were shorter than the reported typical 4 to 6 months from diagnosis to death (5, 23).
Brain MRI, EEG, and advanced CSF studies are the most utilized diagnostic tests for CJD (7, 24–26). Brain MRI with diffusion-weighted imaging (DWI) has a sensitivity of 67–91% (27, 28) and a specificity of 97% for diagnosing sCJD (27). With increased awareness of sCJD, its diagnostic criteria, and improvement in MRI accessibility and scan quality, the sensitivity of MRI for CJD could reach 99% (28). Our five patients' brain MRI revealed typical sCJD patterns, that is hyperintensities in the cortical gray matter (cortical ribboning sign) and the deep nuclei (basal ganglia and thalamus). The cortical ribboning sign was proposed to be the biomarker in the prodromal phase of sCJD diagnosis (29, 30). The MRI of patient 1 demonstrated a symmetric pattern of bilateral DWI and T2 FLAIR correlated signal in caudate nuclei and putamina. MRIs of patients 2, 4, and 5 revealed asymmetric, cortically based DWI changes in the cingulate, the caudate nuclei, and the left temporoparietal cortex, and the MRI of patient 3 showed multifocal cortically based DWI pattern, more on the left. Park et al. found that being greater than 60 years of age and diffusion restriction in the caudate nucleus and putamen were independent prognostic factors of shorter survival duration in patients with sCJD (27) with median overall survival of 1.7 months compared to 14.2 months in the intermediate risk group. Radiographically, our five patients belong to the high-risk group.
RT-QuIC is a breakthrough technology for diagnosing CJD with specificity reaching 99–100% (31–33). Its sensitivity can increase from 77 to 96% after modified techniques (31). Patients' (1, 2, 3, and 5) CSF CJD panel reported a likelihood of CJD of more than 98%, positive RT-QuIC, high T-tau protein, and positive 14-3-3 protein (more than 71,000 Au/ml in patients 3 and 5, and no titer reported in patient 1 and 2). The CSF of patient 4 was inconclusive for CJD likelihood and 14-3-3 proteins. Her RT-QuIC was negative, but T-tau protein was 19,937 pg/ml. As per NPDPSC test report, the sensitivity of RT-QuIC is lower when specimens are discolored by blood. Shir et al. reported that elevated CSF 14-3-3 and T-tau proteins as well as clinical symptoms such as myoclonus and visual or cerebellar abnormalities are associated with shorter disease duration (7), which held true for patient 3.
EEG has a lower diagnostic value when compared to brain MRI and CJD panel in CSF. The reported sensitivity of EEG-specific abnormalities to diagnose probable sCJD ranged from 38.2 to 68.75% (34, 35). However, the characteristic EEG finding in CJD, periodic sharp wave complexes (PSWCs), has 86% specificity (36) and 95% positive predictive value (37). Our case series confirmed low sensitivity and high specificity of EEG for diagnosing CJD. Of the five patients, four patients showed EEG abnormality (80%) with 20% specific (patient 3 showed characteristic periodic sharp wave complexes PSWC at 1 Hz, bi-hemispheric with left predominance) and 60% non-specific abnormalities (patients 1, 2, and 5). Mundlamurri et al. reported that in the early stage of sCJD, patients' EEGs can be normal or non-specifically abnormal (35). In very early phases (1.67 months after onset and before the emergence of generalized PSWC) of sCJD, the predominant findings of EEG can be (1) lateralized periodic discharges (LPDs), (2) central sagittal sporadic epileptiform discharges (CSSEDs), and (3) focal epileptiform discharges (38). It is suggested that the early presence of the PSWC pattern has a prognostic value because these patients have significantly lower average survival time (39). The EEG of patient 3 captured PSWC on day 48 after the illness onset. He died 19 days after the EEG was done and 2 days after positive CJD in CSF was reported.
Brain biopsy or autopsy remains the gold standard for final diagnosis. Four patients underwent autopsy and genetic analysis. All four patients had sporadic CJD, of the two most common subtypes, sCJD-VV2 in Patients 1 and 5, and sCJD-MM1 in patients 3 and 4. Different subtypes have different clinical and neuropathological features, as well as survival times and test results (8, 40, 41). The sensitivity of RT-QuIC for detecting MM1 and VV2 is high (96.3%) but can be negative for MM2 and VV1 subtypes (33, 42). Younes et al. reported that MM1, MV1, and VV2 are related to short duration/fast progression, while MV2, VV1, and MM2 are associated with long duration/slow progression (43). Our case series revealed that patients 3 and 4 were sCJD-MM1 but with an 8-month difference in survival length; patients 1 and 5 were diagnosed with sCJD-VV2 with similar survival durations, 8 vs. 6 months. Patient 2 had a short survival duration; unfortunately, his family declined an autopsy.
Geographical clusters of sCJD have been reported, but most clusters contained cases distributed over many years (14, 17, 18, 44–48), and few were temporo-spatial clusters. A French cluster of three cases of CJD occurring in 1998 reported that two of the patients lived in the same village. Molecular and phenotypic analyses showed both patients were homozygous for methionine at the polymorphic codon 129 but one patient was MM1 while another had mixed features of MM1 and MM2 both clinically and histo-pathologically (48). RT-QuIC was not yet invented. A Japanese cluster of three CJD cases occurred between 1988 and 1989 near Fukuoka city; no hospitalization time was mentioned in the report, nor were CSF studies or codon 129 polymorphism analyses done on these patients (49). A cluster of four cases in Burlington, Ontario, Canada, between April 1989 and April 1990 with two additional cases on further inquiry, and a cluster of seven cases in Nassau County, New York, between mid-June 1999 and mid-June 2000 (50, 51) were reported without genetic studies. Some clustering was found later to be an aggregation of genetic CJD cases (52, 53).
Our five cases in two clusters were seen within 1 year in Grand Rapids, Michigan. Cluster one included patients 1 and 2, seen within 1 month from July to August of 2021; cluster two included patients 3, 4, and 5, observed within 1 month between May and June of 2022. All patients lived within a 90-mile radius of Grand Rapids. No interpersonal connections were identified among them. All patients were white with differing professions (Table 1). None of them had a family history of Creutzfeldt–Jakob disease, or personal history of corneal transplants, craniotomy, administration of human growth hormone derived from pools of pituitary glands, or surgical procedure at the same facility. However, families of patients 1, 2, and 4 reported consuming venison. More intriguingly, families and relatives of these three patients reported additional (at least four) possible or probable CJD cases occurring between 2007 and 2022 in their friends or communities (unpublished data). One of the patients was a 63-year-old white woman and mayor, who lived 35 miles from patient 2, and died of CJD in March 2022. Thus, such a wave of dense temporo-spatial clustering of CJD in West Michigan is very unusual and alarming.
Our case series does not support that CJD incidence has no geographical differences (4, 54). West Michigan has 1.6 million people, and the combined population of four counties where five patients lived is 395,104 in 2022, which makes the CJD new case rate 3.1 and 12.5 per million people in West Michigan and combined four counties, respectively, which is higher than reported 1 to 2 per million people worldwide and 350–710 cases in the United States annually (2–5)1. Adding the cases reported by our three patients' families, the new case occurrence would be even higher. Michigan disease surveillance system (MDSS) reported 19 CJD cases by 31 December 2022 and only 12 cases in 2018, and this reflects a 58% increase2 We do not have enough evidence to conclude that our two clusters are purely due to heightened awareness, more sensitive tests, and better ascertainment, nor could we be certain that they just simultaneously occurred (55). Our study has several limitations, including an observational study, a limited time period, not using the conventionally used solar year period, and a relatively small population and area in West Michigan. As such, this case series highlights only a possible trend. More research and evidence are certainly required to reach a conclusion. We have planned additional retrospective studies, which we expect will surmount these shortcomings. Epidemiological surveillance, research, development of new diagnostic technologies, and public health endeavors are critical (4, 56).
For five sCJD cases in two dense clusters within 1 year in Grand Rapids, MI is more than expected. Extensive screening in West Michigan may eventually arrive at a reliable incidence rate of CJD in this region. These two clusters along with additional cases reported by our patients' families warrant urgent investigation. Further research including epidemiological study regarding possible transmission events, common environmental factors that trigger CJD occurrence as well as continuous surveillance, and further improving diagnostic techniques are critical and necessary.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
LR: writing the original draft, selecting MRI images, and finalizing the manuscript. NL, JF, AP, MK, and JM: reviewing and editing. ET: selecting EEG pictures, draft reviewing, and editing. CT: selecting MRI images. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. ^https://case.edu/medicine/pathology/divisions/national-prion-disease-pathology-surveillance-center/human-prion-diseases
2. ^https://www.michigan.gov/mdhhs/-/media/Project/Websites/mdhhs/CDINFO/WSR/Current_WSR.pdf?rev=2219a79dac8f486fa870c4afcb339848&hash=F23C8EB5D0CCD5378F6A5AE8CF1CB954
2. Sitammagari K, Masood W. Creutzfeldt Jakob Disease. Treasure Island (FL): StatPearls Publishing. (2022).
3. Watson N, Brandel JP, Green A, Hermann P, Ladogana A, Lindsay T, et al. The importance of ongoing international surveillance for Creutzfeldt-Jakob disease. Nat Rev Neurol. (2021) 17:362–79. doi: 10.1038/s41582-021-00488-7
4. Kotkowski E, Cabot JH, Lacci JV, Payne DH, Cavazos JE, Romero RS, et al. Creutzfeldt-Jakob disease: In-hospital demographics report of national data in the United States from 2016 and review of a rapidly-progressive case. Clin Neurol Neurosurg. (2020) 197:106103. doi: 10.1016/j.clineuro.2020.106103
5. Uttley L, Carroll C, Wong R, Hilton DA, Stevenson M. Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect Dis. (2020) 20:e2–e10. doi: 10.1016/S1473-3099(19)30615-2
6. Centers for Disease Control and Prevention. Clinical and Pathologic Characteristics | Creutzfeldt-Jakob Disease, Classic (CJD) | Prion Disease | CDC. (2021).
7. Shir D, Lazar EB, Graff-Radford J, Aksamit A, Cutsforth-Gregory JK, Jones DT, et al. Analysis of clinical features, diagnostic tests, and biomarkers in patients with suspected Creutzfeldt-Jakob disease, 2014-2021. JAMA Netw Open. (2022) 5:e2225098. doi: 10.1001/jamanetworkopen.2022.25098
8. Bizzi A, Pascuzzo R, Blevins J, Moscatelli MEM, Grisoli M, Lodi R, et al. Subtype diagnosis of sporadic Creutzfeldt-Jakob disease with diffusion magnetic resonance imaging. Ann Neurol. (2021) 89:560–72. doi: 10.1002/ana.25983
9. Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. (1999) 46:224–233. doi: 10.1002/1531-8249(199908)46:2<224::AID-ANA12>3.0.CO;2-W
10. Gelpi E, Baiardi S, Nos C, Dellavalle S, Aldecoa I, Ruiz-Garcia R, et al. Sporadic Creutzfeldt-Jakob disease VM1: phenotypic and molecular characterization of a novel subtype of human prion disease. Acta Neuropathol Commun. (2022) 10:114. doi: 10.1186/s40478-022-01415-7
11. Parchi P, Strammiello R, Notari S, Giese A, Langeveld JPM, Ladogana A, et al. Incidence and Spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol. (2009) 118:659–71. doi: 10.1007/s00401-009-0585-1
12. Appleby BS, Appleby K, Crain BJ, Onyike CU, Wallin MT, Rabins PV. Characteristics of established and proposed sporadic Creutzfeldt-Jakob disease variants. Arch neurol. (2009) 66:208–15. doi: 10.1001/archneurol.2008.533
13. Krasnianski A, Kaune J, Jung K, Kretzschmar HA, Zerr I. First symptom and initial diagnosis in sporadic CJD patients in Germany. J Neurol. (2014) 261:1811–7. doi: 10.1007/s00415-014-7410-z
14. Xiao K, Pang MF, Zhao YQ, Gao LP, Wu YZ, Wang Y, et al. Difference of geographic distributions of the Chinese patients with prion diseases in the permanent resident places and referring places. Prion. (2022) 16:58–65. doi: 10.1080/19336896.2022.2080921
15. Puopolo M, Catelan D, Capellari S, Ladogana A, Sanguedolce A, Fedele A, et al. Spatial epidemiology of sporadic Creutzfeldt-Jakob disease in Apulia, Italy. Neuroepidemiology. (2020) 54:83–80. doi: 10.1159/000503234
16. Chamosa S, Tamayo I, Arteagoitia-Axpe JM, Juste RA, RodriguezMartinez AB, Zarraz-Imirizaldu JJ, et al. Geographical analysis of the sporadic Creutzfeldt-Jakob disease distribution in the autonomous community of the Basque Country for the period 1995-2008. Eur Neurol. (2014) 72:20–5. doi: 10.1159/000358298
17. Nakatani E, Nishimura T, Zhou B, Kaneda H, Teramukai S, Nagai Y, et al. Temporal and regional variations in sporadic Creutzfeldt-Jakob disease in Japan, 2001-2010. Epidemiol Infec. (2015) 143:1073–1078. doi: 10.1017/S0950268814001605
18. Klug GM, Wang H, Boyd A, Law M, Whyte S, Kaldor J, et al. Enhanced geographically restricted surveillance simulates sporadic Creutzfeldt-Jakob disease cluster. Brain. (2009) 132:493–501. doi: 10.1093/brain/awn303
19. Corriveau-Lecavalier N, Li W, Ramanan VK, Drubach D, Day GS, Jones DT. Three cases of Creutzfeldt-Jakob disease presenting with a predominant dysexecutive syndrome. J Neurol. (2022) 269:4222–8. doi: 10.1007/s00415-022-11045-7
20. Gomez-Mayordomo V, Kojovic M, Lopez-Valdes E, Alonso-Frech F, Horga A, Fernandez-Rodriguez R, et al. Functional neurological symptoms as initial presentation of Creutzfeldt-Jakob disease: case series. J Neurol. (2022) 270:1141–6. doi: 10.1007/s00415-022-11376-5
21. Bawa A, Zhang YH. Creutzfelft-Jakob Disease: A focused literature review and retrospective case series of five patients from a community hospital. J Neurol. Exper. Neurosci. (2022) 8:13–7. doi: 10.17756/jnen.2022-095
22. Kropp S, Schulz-Schaeffer WJ, Finkenstaedt M, Riedemann C, Windl O, Steinhoff BJ, et al. The Heidenhain variant of Creutzfeldt-Jakob disease. Arch Neurol. (1999) 56:55–61. doi: 10.1001/archneur.56.1.55
23. Staffaroni AM, Kramer AO, Casey M, Kang H, Rojas JC, Orru CD, et al. Association of blood and cerebrospinal fluid tau level and other biomarkers with survival time in sporadic Creutzfeldt-Jakob disease. JAMA Neurol. (2019) 76:969–77. doi: 10.1001/jamaneurol.2019.1071
24. Hermann P, Appleby B, Brandel JP, Caughey B, Collins S, Geschwind MD, et al. Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. Lancet Neurol. (2021) 20:235–46. doi: 10.1016/S1474-4422(20)30477-4
25. Zerr I. Laboratory diagnosis of Creutzfeldt-Jakob disease. N Engl J Med. (2022) 386:1345–1350. doi: 10.1056/NEJMra2119323
26. Schmitz M, Canaslan S, Espinosa JC, Fernandez-Borges N, Villar-Pique A, Zerr I. Validation of plasma and CSF neurofilament light chain as an early marker for sporadic Creutzfeldt-Jakob disease. Mol Neurobiol. (2022) 59:1–9. doi: 10.1007/s12035-022-02891-7
27. Park HY, Kim M, Suh CH, Kim SY, Shim WH, Kim SJ. Diagnostic value of diffusion -weighted brain magnetic resonance imaging in patients with sporadic Creutzfeldt-Jakob disease: a systematic review and meta-analysis. Eur Radiol. (2021) 31:9073–85. doi: 10.1007/s00330-021-08031-4
28. Jesuthasan A, Sequeira D, Hyare H, Odd H, Rudge P, Mok TH, et al. Assessing initial MRI reports for suspected CJD patients. J Neurol. (2022) 269:4452. doi: 10.1007/s00415-022-11087-x
29. Hamada Y, Deguchi K, Tachi K, Kita M, Nonaka W, Takata T, et al. Significance of cortical ribboning as a biomarker in the prodromal phase of sporadic Creutzfeldt-Jakob disease. Intern Med. (2022) 61:2667–2670. doi: 10.2169/internalmedicine.8354-21
30. Yasuda M, Sugiyama A, Hokkoku H, Suichi T, Ito K, Satoh K, et al. Propagation of diffusion-weighted MRI abnormalities in the preclinical stage of sporadic Creutzfeldt-Jakob disease. Neurology. (2022) 99:699–702. doi: 10.1212/WNL.0000000000201221
31. Orru CD, Groveman B, Hughson A, Zanusso G, Coulthart M, Caughey B. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. MBio. (2015) 6:e02451–14. doi: 10.1128/mBio.02451-14
32. Hermann P, Laux M, Glatzel M, Matschke J, Knipper T, Goebel S, et al. Validation and utilization of amended diagnostic criteria in Creutzfeldt-Jakob disease surveillance. Neurology. (2018) 91:e331–8. doi: 10.1212/WNL.0000000000005860
33. Rhoads DD, Wrona A, Foutz A, Blevins J, Glisic K, Person M, et al. Diagnosis of prion diseases by RT-QuIC results in improved surveillance. Neurology. (2020) 95:e1017–26. doi: 10.1212/WNL.0000000000010086
34. Qi C, Zhang JT, Zhao W, Xing XW, Yu SY. Sporadic Creutzfeldt-Jakob disease: A retrospective analysis of 104 cases. Eur Neurol. (2020) 83:65–72. doi: 10.1159/000507189
35. Mundlamurri RC, Shah R, Adiga MS, Chatterjee A, Gautham B, Raghavendra K, et al. EEG observations in probable sporadic CJD. Ann Indian Acad Neurol. (2020) 23:760–766. doi: 10.4103/aian.AIAN_672_20
36. Steinhoff BJ, Racker S, Herrendorf G, Poser S, Grosche S, Zerr I, et al. Accuracy and reliability of periodic sharp wave complexes in Creutzfeldt-Jakob disease. Arch Neurol. (1996) 53:162–6. doi: 10.1001/archneur.1996.00550020074017
37. Wieser HG, Schindler K, Zumsteg D. EEG in Creutzfeldt-Jakob disease. Clin Neurophysiol. (2006) 117:935–51. doi: 10.1016/j.clinph.2005.12.007
38. Matsubayashi T, Akaza M, Hayashi Y, Hamaguchi T, Satoh K, Kosami K, et al. Specific electroencephalogram features in the very early phases of sporadic Creutzfeldt-Jakob disease. J Neurol Sci. (2022) 437:120265. doi: 10.1016/j.jns.2022.120265
39. Castelli A, Placidi F, Bonomi CG, Giuliano FD, Martorana A, Pizzicannella G, et al. Periodic sharp wave complexes identify a distinctive phenotype in Creutzfeldt-Jakob disease. Clin Neurophysiol. (2022) 143:124–32. doi: 10.1016/j.clinph.2022.08.025
40. Tanev KS, Yilma M. An unusually presenting case of sCJD-the VV1 subtype. Clin Neurology and Neurosurg. (2009) 111:282–291. doi: 10.1016/j.clineuro.2008.09.017
41. Cali I, Puoti G, Smucny J, Curtiss PM, Cracco L, Kitamoto T, et al. Co-existence of PrPD types 1 and 2 in sporadic Creutzfeldt-Jakob disease of the VV subgroup: phenotypic and prion protein characteristics. Sci Rep. (2020) 10:1503. doi: 10.1038/s41598-020-58446-0
42. Carrasco A, Appleby BS, Cali I, Okhravi HR. Atypical case of VV1 Creutzfeldt-Jakob disease subtype: care report. Front Neurol. (2022) 13:875370. doi: 10.3389/fneur.2022.875370
43. Younes K, Rojas JC, Wolf A, Sheng-Yang GM, Paoletti M, Toller G, et al. Selective vulnerability to atrophy in sporadic Creutzfeldt-Jakob disease. Ann Clin Transl Neurol. (2021) 8:2283–1199. doi: 10.1002/acn3.51290
44. Linsell L, Cousens SN, Smith PG, Knight RSG, Zeidler M, Stewart G, et al. case-control study of sporadic Creutzfeldt-Jakob disease in the United Kingdom: analysis of clustering. Neurology. (2004) 63:2077–83. doi: 10.1212/01.WNL.0000145844.53251.BC
45. Collins S, Boyd A, Fletcher A, Kaldor J, Hill A, Farish S, et al. Creutzfeldt-Jakob disease cluster in an Australian rural city. Ann Neurol. (2002) 52:115–8. doi: 10.1002/ana.10224
46. D'Aignaux JH, Cousens SN, Delasnerie-Laupretre N, Brandel JP, Salomon D, Laplanche JL, et al. Analysis of the grographical distribution of sporadic Creutzfeldt-Jakob disease in France between 1992 and 1998. Int J Epidemiol. (2002) 31:490–5. doi: 10.1093/ije/31.2.490
47. Ruegger J, Stoeck K, Amsler L, Blaettler T, Zwahlen M, Aguzzi A, et al. case-control study of sporadic Creutzfeldt-Jakob disease in Switzerland: analysis of potential risk factors with regard to an increased CJD incidence in the years 2001-2004. BMC Public Health. (2009) 9:19. doi: 10.1186/1471-2458-9-18
48. Beaudry P, Parchi P, Peoc'h K, Desbordes P, Dartigues JF, Vital A, et al. A French cluster of Creutzfeldt-Jakob disease: 1 molecular analysis. Eur J Neurol. (2002) 9:457–62. doi: 10.1046/j.1468-1331.2002.00456.x
49. Arakawa K, Nagara H, Itoyama Y, Doh-ura K, Tomokane N, Tateishi J, et al. Clustering of three cases of Creutzfeldt-Jakob disease near Fukuoka city, Japan. Acta Neurol Scand. (1991) 84:445–7. doi: 10.1111/j.1600-0404.1991.tb04986.x
50. Nosal R, Kapoor A, Shanin R. Cluster of cases of Creutzfelft-Jakob disease—Ontario. Can Dis Wkly Rep. (1991) 17:12.
51. Adikari D, Farmer P. A cluster of Creutzfeldt-Jakob disease patients from Nassau county, New York, USA. Ann Clin Lab Sci. (2001) 31:211–2.
52. Golfdarb LG, Mitroya E, Brown P, Toh BH, Gaidsek DC. Mutation in codon 200 of scrpie amyloid protein gene in two clusters of Creutzfeldt-Jakob disease in Slovakia. Lancet. (1990) 336:514–5. doi: 10.1016/0140-6736(90)92073-Q
53. Miyakawa T, Iseki KIE, Kawanishi C, Sugiyama N, Onishi H, Yamada Y, et al. Japanese Creutzfeldt-Jakob disease patients exhibiting high incidence of the E200K PRNP mutation and located in the basin of a river. Neurol Res. (1998) 20:684–8. doi: 10.1080/01616412.1998.11740584
54. Shi Q, Gao C, Zhou W, Zhang BY, Chen JM, Tian C, et al. Surveillance for Creutzfeldt-Jakob disease in China from 2006 to 2007. BMC Public Health. (2008) 8:360. doi: 10.1186/1471-2458-8-360
55. Raubertas RF, Brown P, Cathala F, Brown I. The question of clustering of Creutzfeldt-Jakob disease. Am J Epidemiol. (1989) 129:146–54. doi: 10.1093/oxfordjournals.aje.a115103
Keywords: Creutzfeldt-Jacob disease, cluster, rapidly progressive dementia, prion, real-time quaking-induced conversion, West Michigan
Citation: Rong LL, Lannen NJ, Tank EC, Feistel JL, Therasse CJ, Potluri A, Khan M and Min J (2023) Case report: Two clusters of Creutzfeldt–Jakob disease cases within 1 year in West Michigan. Front. Neurol. 14:1134225. doi: 10.3389/fneur.2023.1134225
Received: 30 December 2022; Accepted: 23 February 2023;
Published: 20 March 2023.
Edited by:
Bruce Miller, University of California, San Francisco, United StatesReviewed by:
Ignazio Cali, Case Western Reserve University, United StatesCopyright © 2023 Rong, Lannen, Tank, Feistel, Therasse, Potluri, Khan and Min. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ling Ling Rong, bGluZ2xpbmcucm9uZ0Bjb3Jld2VsbGhlYWx0aC5jb20=; bGluZ2xpbmdyb25nQHlhaG9vLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.