
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 14 April 2023
Sec. Pediatric Neurology
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1131490
This article is part of the Research TopicCategorizing Neurodevelopmental Disorders: Themes Shared Across Genetic DiagnosesView all 7 articles
 Alshaimaa Alzahrani1Maha Alshalan1
Alshaimaa Alzahrani1Maha Alshalan1 Mohammed Alfurayh2
Mohammed Alfurayh2 Abdulaziz Bin Akrish2
Abdulaziz Bin Akrish2 Najlaa A. Alsubeeh1
Najlaa A. Alsubeeh1 Fuad Al Mutairi1,3*
Fuad Al Mutairi1,3*Background: Calcium ions are involved in several human cellular processes; nevertheless, the relationship between calcium channelopathies (CCs) and autism spectrum disorder (ASD) or intellectual disability (ID) has been previously investigated. We delineate the spectrum of clinical phenotypes and the symptoms associated with a syndrome caused by an inherited gain-of-function mutation in CACNA1D in a family with a history of neuropsychiatric disorders. We also review the clinical and molecular phenotype of previously reported variants of CACNA1D.
Case presentation: We report the case of a 9-year-old female patient, diagnosed with ASD, severe ID, hyperactivity, and aggressive impulsive behaviors. The father, who was a 65-year-old at the time of his death, had ID and developed major depressive disorder with catatonic features and nihilistic delusion, followed by rapidly progressive dementia. He died after experiencing prolonged seizures followed by post-cardiac arrest. The patient’s sister was a 30-year-old woman, known to have a severe ID with aggressive behaviors and sleep disorders. The sister has been diagnosed with bipolar disorder and psychosis. Through whole exome sequencing, a heterozygous previously identified and functionally characterized missense likely pathogenic variant was identified in the CACNA1D gene NM_001128840.3: c.2015C > T (p.Ser672Leu). These findings are consistent with the genetic diagnosis of autosomal dominant primary aldosteronism, seizures, and neurological abnormalities. This variant was found in the heterozygous status in the patient, her father, and her affected sister.
Conclusion: This case report will help to determine the key clinical features of this syndrome, which exhibits variable clinical presentations.
Autism spectrum disorder (ASD) is a group of conditions in which the person experiences significant difficulties in socialization, communication, and language (1). In addition, people with ASD exhibit unusual repetitive behaviors (2). Despite the lack of a clear mechanism, genetics have a significant role in the etiology of ASD; previous research studies conducted on families and twins have elucidated specific genetic causes for isolated cases, and a number of candidate genes and chromosomal regions have been indicated as relevant across multiple studies, including 2q, 7q, 15q, and 17q (3, 4). Genome-wide studies using genotyping microarrays, whole-exome sequencing, and whole-genome sequencing have identified a rapidly growing number of genes linked to ASD; furthermore, cumulative discoveries of de novo mutations have pointed the field in various directions, including the converging actions of ASD-associated mutations and progressive use of translational strategies (in animals and humans) to understand the outcomes of replicated ASD-associated mutations (5–7).
Calcium ions are involved in several human cellular processes including corticogenesis, transcription, and synaptogenesis and play a major role in the development of neurodevelopmental disorders (8). Since the calcium signaling pathway was recognized among the top three pathways involved in ASD, many discoveries of point mutations in the α1-subunit of L-type calcium channels (CaV1.2) and many other genes encoding voltage-gated calcium channel (VGCC) subunits, such as CACNA1B-F encoding pore-forming subunits, CACNB1 and CACNB2 encoding auxiliary β-subunits, and CACNA2D3 and CACNA2D4 for auxiliary α2-δ-subunits have been associated with ASD (9).
Voltage-gated calcium channels (VGCCs) are transmembrane proteins that are activated by depolarization, triggering Ca2+ influx into neurons and other excitable cells, which are composed of a pore-forming α1 subunit and auxiliary α2δ and β subunits (10). Until now, 10-mammalian VGCCs have been identified, and based on their activation threshold. CaVs are classified as high or low voltage activated (HVA and LVA) (11). One of the three main families of the LVA subgroup is the L-type Ca2+ channels (LTCCs; Cav1), which consists of four subtypes: Cav1.1, Cav1.2, Cav1.3, and Cav1.4 (12). The main functions of Cav1.1 include the excitation–contraction coupling in skeletal muscles and the regulation of transcription in these muscles. The main functions of Cav1.2 include excitation–contraction coupling in cardiac and smooth muscles, hormone secretion, and neurotransmitter release. The functions of Cav1.3 are similar to those of Cav1.2, with the addition of cardiac pace-making and auditory transduction. The Cav1.4 plays a role in visual transduction. Cav1.2 and Cav1.3 are expressed in the most electrically excitable cells, including various areas of the brain (13, 14) (Table 1). The variants in CACNA1C, CACNA1D, CACNA1F, and CACNB2 cause gain-of-function by preventing voltage-dependent inactivation of their subunits (Cav1.2, Cav1.3, Cav1.4, and Cavβ2), leading to an excessive influx of Ca2+, alteration in kinetics (activation and inactivation), and dominant-negative effects of truncated forms of alpha1 subunits (8). Variants located in CACNA1A and CACNA1H are the cause of the loss of function in encoded subunits (Cav2.1 and Cav3.2), resulting in decreased channel activity (16).
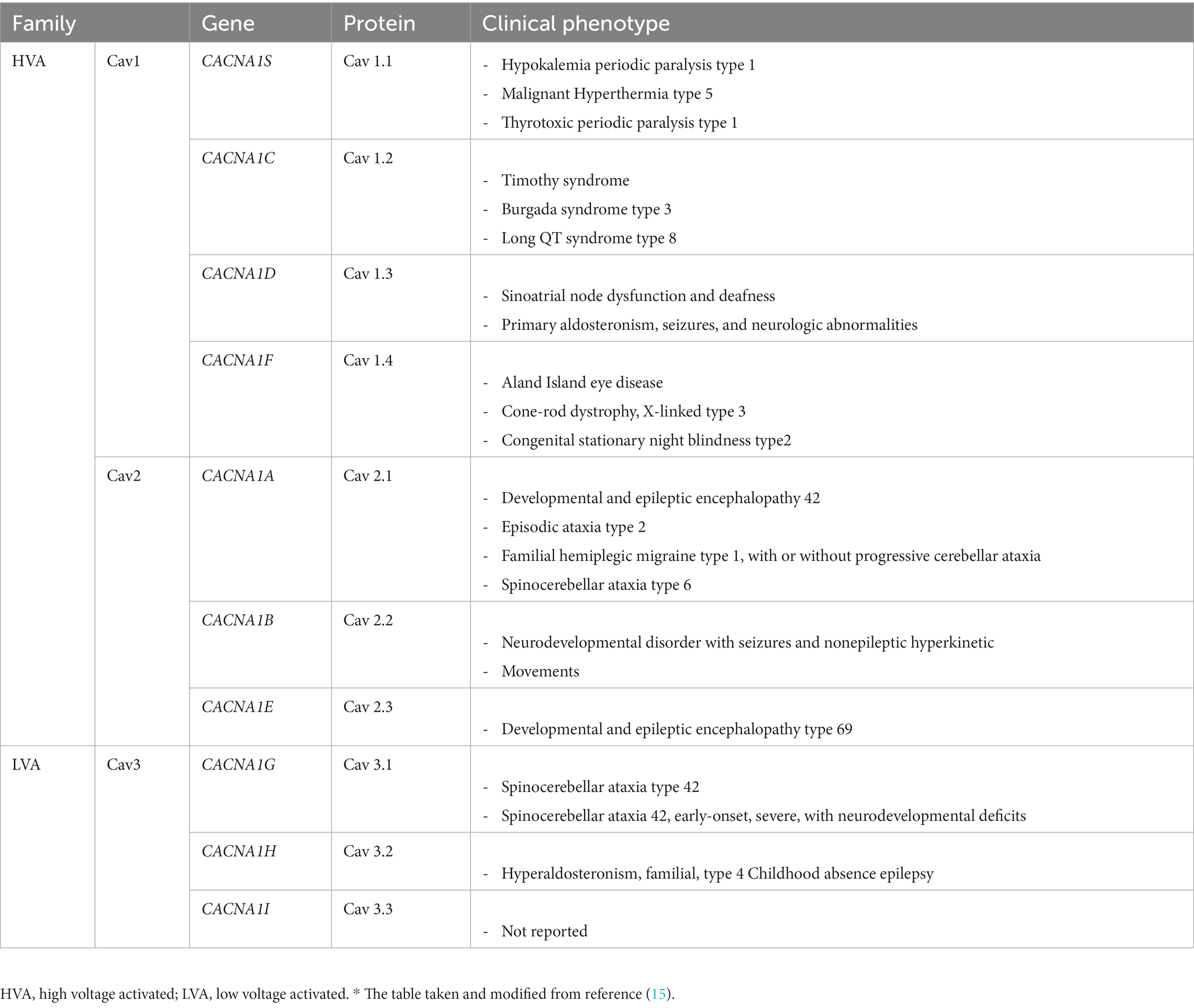
Table 1. Voltage-gated Ca2+ channels: classification and human genetic disease.*
In total, 10 genes encode the CaVα1-pore-forming subunit of CaVs (CACNA1). Variations in CACNA1 genes were found to be associated with a variable degree of ASD and other neurodevelopmental disorders. Most variants exhibited a gain-of-function effect. Generally, severe to profound ID/GDD was observed more for the cases with gain-of-function variants as compared to those with loss-of-function. Specifically, CACNA1E, CACNA1G, CACNA1F, CACNA2D2, and CACNA1A were associated with more severe phenotypes (8).
Based on their pharmacology and sequence similarity, CaVα1 subunits are subdivided into three subfamilies (CaV1, CaV2, and CaV3) (17). The CACNA1D gene encodes CaV1.3. This channel contributes to the rhythmic activity of the sinoatrial node and is thereby involved in the regulation of heart rate (18). Disease-causing variants in the CACNA1D gene have been associated with autosomal dominant primary aldosteronism, seizures, and neurologic abnormalities (OMIM 615474); and autosomal recessive sinoatrial node dysfunction and deafness (OMIM 614896). Similarly, individuals with variants in the CACNA1D gene have been revealed among individuals with ASD (19). Symptoms range from ASD with intellectual disability to seizures, muscle hypotonia, and global developmental delay in more severely affected patients. Some patients also exhibit additional endocrine symptoms (primary aldosteronism or hyperinsulinism) due to the expression of Cav1.3 in adrenal zona glomerulosa cells and pancreatic β-cells. The functional analysis of these reported variants has shown typical gain-of-function changes (20).
In this article, we present a previously reported variant of CACNA1D. We delineate the spectrum of clinical phenotypes and the symptoms associated with a syndrome caused by the inherited CANCA1D missense mutation in a family with a history of neuropsychiatric disorders.
The subject of the case report is a 9-year-old girl who was born at full term via normal spontaneous vaginal delivery to a healthy mother following an uneventful pregnancy. The neonatal history was unremarkable. Her mother started to seek medical advice at the age of 4 years as the child exhibited speech and language delay with challenging behaviors. These challenging behaviors took the form of aggressiveness, self-injurious behaviors, stereotyped and repetitive motor mannerisms with hand flapping and body rocking, lack of sleep, and attention-deficit hyperactivity-like disorders.
There were not any concerns regarding her gross motor skills; she sat at 7 months and walked at 14 months and continued to be appropriate for her age in all stages. Her fine motor skills were delayed, she could only hand grasp a pen for scribbling and was not able to dress with a significant stereotypic movement of her hands. Her cognitive and language skills were significantly delayed, she could count from 1 to 10, and she said repeated non-comprehensive limited words and sentences. In the development clinic, she was diagnosed with ASD and severe intellectual disability at the age of 6 years based on DSM-5 criteria. The assessment revealed deficits in social–emotional reciprocity, in nonverbal communicative behaviors used for social interaction, and in developing and maintaining relationships. The assessment also revealed that she had repetitive speech and motor movements, usually with her hands. On examination, her growth parameters were within normal percentiles, and she exhibited no gross dysmorphic features, normal neurological status, and no evidence of cardiovascular abnormalities. Her blood pressure was normal, but she was agitated and hyperactive and made no eye contact during the examination. At the age of 6 years, she was started on risperidone 0.5 mg daily and gradually increased to twice per day, which did not show significant improvement. Later at the age of 8 years, she was started additionally on methylphenidate 5 mg twice per day, which led to little improvement in sleep patterns and no change in hyperactivity or aggressive impulsive behaviors.
The parents were non-consanguineous. The mother is 50 years old, and she is not known to have any chronic medical illnesses. The father died at the age of 65 years. Although he was independent in activities of daily living, he was illiterate and was diagnosed to have ID in the form of impaired executive functions. He did not have any behavioral disorders and showed good interpersonal communication. He was diagnosed with hypertension and type 2 diabetes and developed stage 3 chronic kidney disease and coronary artery disease that required percutaneous coronary intervention. In the last 6 months of his life, he developed seizures that were controlled with levetiracetam 250 mg twice per day. All his neuroimaging was unremarkable, including magnetic resonance imaging (MRI) of the brain and computed tomography (CT) angiography of the brain. After 3 months, he started to have mood disturbances and became isolated, with a lack of sleep, loss of appetite, and suicidal ideation but no suicide attempt. An adult mental health team diagnosed him to have a major depressive disorder with catatonic features and nihilistic delusion, followed by rapidly progressive dementia, where he was started on sertraline 25 mg daily. He died after suffering prolonged seizures followed by post-cardiac arrest the next day.
The patient has six siblings, all of whom except one have no medical concerns. She is a 30-year-old female patient who was diagnosed to have severe ID with aggressive behaviors and sleep disorders. At the age of 14 years, she was diagnosed to have bipolar disorder and psychosis, as she has visual and auditory hallucinations, she was given citalopram 2 mg and quetiapine 200 mg daily with a poor response. Her symptoms have currently improved on aripiprazole 5 mg daily and olanzapine 5 mg once before sleep time.
Laboratory investigations for the patient, including basic and neurometabolic workups, chromosomal analysis, and array comparative genomic hybridization, were unremarkable. A hearing test returned normal results. WES was performed for molecular diagnosis using standard methods.
Informed consent for genetic analysis, clinical data, and images was obtained from the family, and it was secured at King Abdulaziz Medical City. Clinical investigations were conducted according to the declaration of Helsinki. Genetic analyses were performed in the context of the clinical genetic program by experienced clinical geneticists. Clinical data, pictures, DNA specimens, and other biological materials were collected, used, and stored after signed informed consent was obtained from the participating subjects/families.
Blood samples were collected from the two sisters and parents in EDTA tubes, and Genomic DNA was extracted from the stored EDTA peripheral blood using the QIAampDNA mini kit (Qiagen®) following the manufacturer’s instructions. The final genomic DNA quantity and integrity were performed using NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, United States) and Qubit® 3.0 Fluorometer (Thermo Fisher Scientific) following the manufacturer’s protocol. We then proceeded with whole exome sequencing (WES). Double-stranded DNA capture baits against approximately 36.5 Mb of the human coding exome (targeting >98% of the coding RefSeq from the human genome build GRCh37/hg19) were used to enrich target regions from fragmented genomic DNA with the Twist Human Core Exome Plus kit. The generated library was sequenced on an Illumina platform to obtain at least 20x coverage depth for >98% of the targeted bases. An in-house bioinformatics pipeline, including read alignment to GRCh37/hg19 genome assembly, variant calling, annotation, and comprehensive variant filtering, was applied. All variants with minor allele frequency (MAF) of less than 1% in the gnomAD database and disease-causing variants reported in HGMD® in ClinVar were considered. The investigation for relevant variants was focused on coding exons and flanking +/−20 intronic nucleotides of genes with clear gene-phenotype evidence. All potential modes of inheritance patterns were considered. In addition, provided family history and clinical information were used to evaluate identified variants with respect to their pathogenicity and causality. Variants were categorized into five classes (pathogenic, likely pathogenic, VUS, likely benign, and benign). All variants related to the phenotype of the patient were reported. The laboratory had established stringent quality criteria and validation processes for variants detected by NGS. Variants with low quality and/or unclear zygosity were confirmed by orthogonal methods. Consequently, a specificity of >99.9% for all reported variants was warranted.
To identify the genetic cause of the disorder in the patient and her affected family members, we performed quad whole-exome sequencing, including the index patient, the parents, and the affected sister using standard methods.
WES revealed a heterozygous previously identified and functionally characterized missense likely pathogenic variant in the CACNA1D gene NM_001128840.3:c.2015C > T (p.Ser672Leu). This result was consistent with the genetic diagnosis of autosomal dominant primary aldosteronism, seizures, and neurological abnormalities. This variant was also found in her father and in her affected sister in heterozygous status but was absent in her healthy mother. No other variants that could explain the patient’s phenotype were detected. The variant was classified as likely pathogenic according to American College of Medical Genetics (ACMG) guidelines.
This variant has previously been described by Hofer et al., who provided evidence that gain-of-function CACNA1D mutations, such as S672L, but not loss-of-function mutations, such as S672W, were associated with a high risk of neurodevelopmental disorders including autism (20). As the variant has been previously associated with the disease phenotype, this was likely the correct molecular diagnosis.
The identified variant (c.2015C > T; p.Ser672Leu) was Sanger sequenced in all members of the family using standard methods. The variant segregated perfectly with the disease phenotype within the family.
This substitution (c.215C > T; p.S672L) was predicted to be deleterious by several online computational prediction tools (Mutation Taster; Sorting Intolerant from Tolerant). Further segregation analysis of other siblings supported the pathogenicity of this variant. The identified variant was not observed in the ExAC or gnomAD database, thus confirming the pathogenic nature of the variant.
Previous studies have linked germline CACNA1D gene mutations in extended clinical phenotypes with certain neuropsychiatric and neurodevelopmental disorders, childhood epilepsies, and ASD (21). Heterozygous missense gain-of-function mutations are associated with neuropsychiatric disorders, developmental delay, and ASD. Gain-of-function variants in the CACNA1D gene have been identified as somatic mutations in aldosterone-producing adenoma (22). These findings support the idea that homozygous loss-of-function mutations, such as frameshift, premature stop codon, and splice-donor defect, result in a loss of channel function that causes deafness, bradycardia, and sinoatrial node arrhythmia (human sinoatrial node dysfunction and deafness syndrome, OMIM 614896) (15).
In this case report, we describe a case that is not related to the previous cases with the S672L variant in CACNA1D and with similar phenotypes of ASD (23); however, the patient in this case is reported to have inherited the variant from her father, whose clinical presentation mainly included ID, psychiatric illness, and seizures, which developed near the end of his life, but no autistic features. Moreover, her sister is known to have ID and a psychiatric disorder and exhibits aggressiveness and impulsive behaviors; however, she is not known to have autistic features either. None of the five cases with S672L has hypertension, hyperaldosteronism, or evidence of hyperinsulinemia (Table 2).
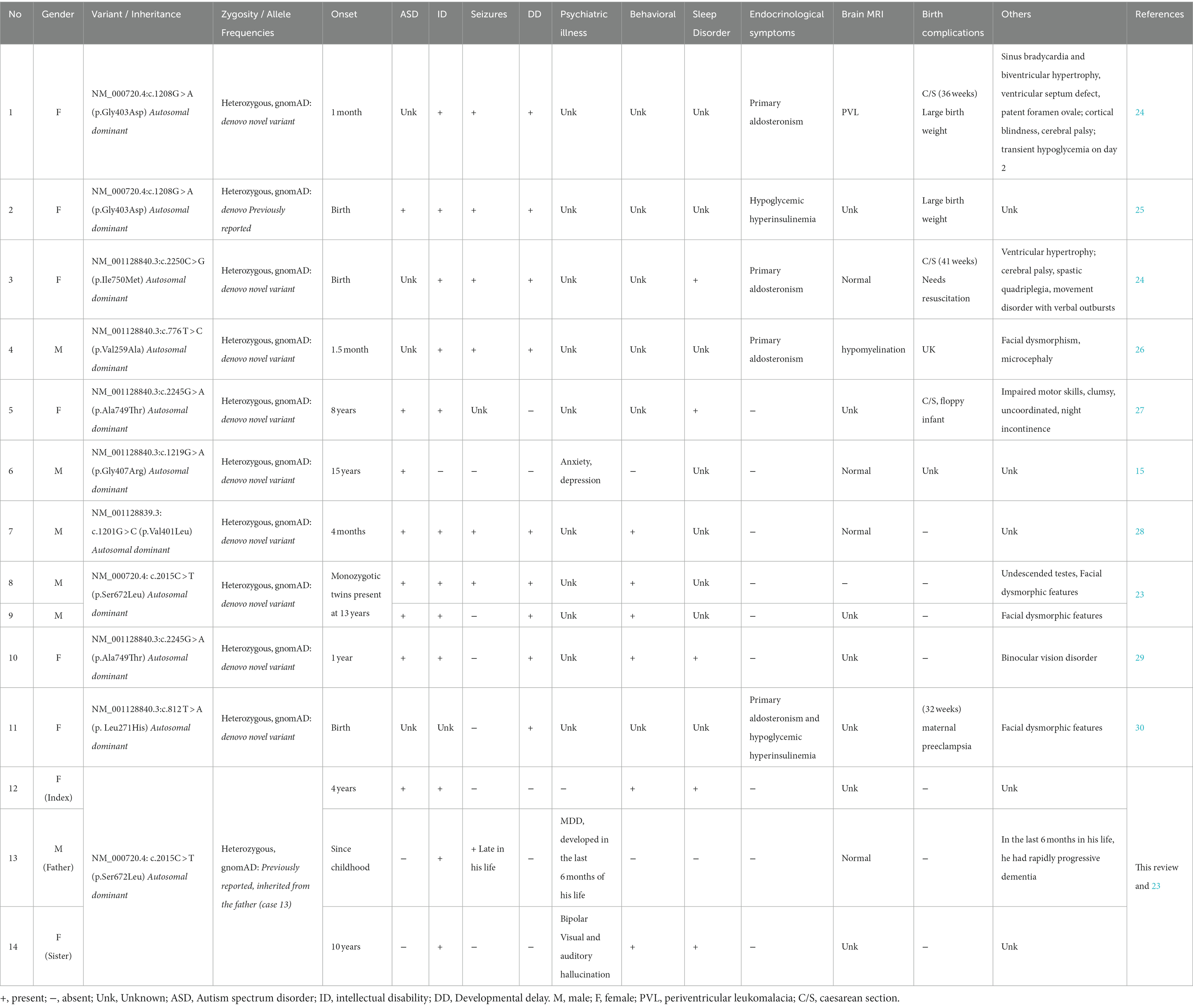
Table 2. Clinical phenotypes of reported cases with heterozygous missense gain of function mutation of CACNA1D.
We searched all the studies related to the CACNA1D gene that were published in PubMed between 2000 and 2022; detailed information from these articles is organized in Tables 2, 3. From the three cases described in this case study and previous clinical reports of 11 individuals, we attempted to delineate the spectrum of clinical phenotypes of missense gain-of-function mutations in the CACNA1D gene (15, 24–31).
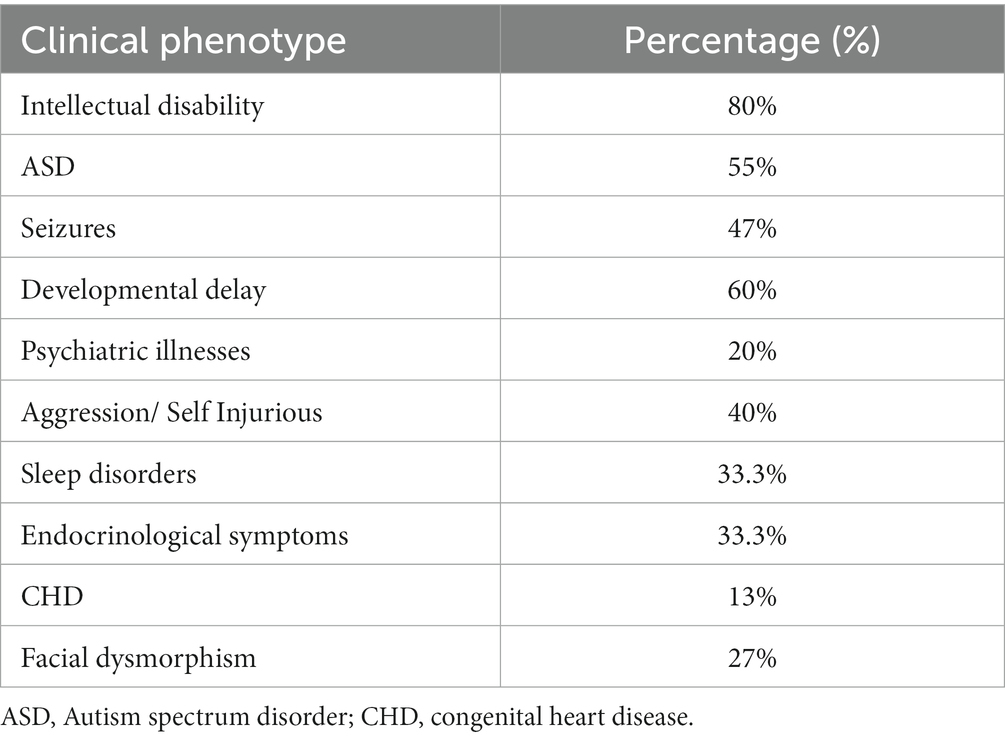
Table 3. Reported clinical phenotypes in patients with CACNA1D missense gain of function mutations.
Most (85%) patients presented with intellectual impairment or disability (only three cases did not report ID, two of which did not detail any clinical phenotype). Over half (55%) of the patients presented with ASD, and it was the presenting clinical phenotype of the subject of this case study (case 12); however, the subject’s father and sister (case 13 and case 14) were not clinically diagnosed to have ASD, which could be related to their late clinical presentation or misdiagnosis of psychiatric illnesses instead of ASD. Aggressiveness and self-injurious behaviors were detected in all cases with the S672L variant except one (case 14). In addition, these phenotypes were detected in two individuals with different variants (A749T and L271H; case 10 and case 12, respectively), and this clinical phenotype accounts for approximately 40% of the reported cases. Endocrine symptoms were not consistent and were only being observed in a third of the cases. They were the first clinical presentation that required immediate therapeutic intervention in patients with primary aldosteronism, seizures, and neurologic abnormalities and in the individuals affected by congenital hyperinsulinemic hypoglycemia (G403D) and (L271H) (24, 25, 30). Their absence does not rule out a CACNA1D channelopathy, and their presence cannot be predicted from the variant gating change. Furthermore, they should be monitored for symptoms of primary aldosteronism (case 11) and altered glucose homeostasis even if not present in the peri- and post-natal periods. No clear dysmorphic facial features were reported, and they are not consistent clinical features in the CACNA1D gene mutation as they are found in only 28% of the reported cases.
As discussed above, Cav1.3 L-type channels are also present in other tissues. Although the homozygous loss of Cav1.3 function results in congenital deafness and sinoatrial node dysfunction, it is currently unclear whether the heterozygous gain-of-function gating changes of the reported variants result in clinically relevant hearing-related symptoms or directly cause the cardiac abnormalities observed in some cases (15) (Table 2).
Hofer et al. functionally characterized the potential changes in the S652L variant. This mutation significantly shifted the voltage dependence of activation and steady-state inactivation to more negative potentials (~13–17 mV), further increasing window currents at subthreshold voltages. Moreover, it slowed tail currents and increased Ca2+ levels during action potential-like stimulations, which is characteristic of gain-of-function changes (20). An understanding of the effects of coding mutations on protein function and interaction can guide the treatment and management strategies. The heterozygous CACNA1D variants alter gating changes that increase Cav1.3 channel activity. Thus, the inhibition of Cav1.3 channel activity with drugs appears to be a potential treatment option in the affected individuals. The S672L mutation increased the sensitivity of Cav1.3 to inhibition by the dihydropyridine LTCC blocker isradipine by 3- to 4-fold (20). Although LTCC blockers may normalize the mutation-induced increase in channel function after diagnosis, the mutation may have already caused permanent developmental deficiencies that are resistant to drug treatment, and none of the reported cases showed a conclusive effect of this treatment on neurological or behavioral disorders (24). Therefore, the clinical effect of LTCC blockers needs to be tested in small clinical trials involving affected individuals.
This case report and all previous clinical reports shed light on the link between the CACNA1D gene mutation and neurodevelopmental disorders. A broad spectrum of clinical phenotypes was observed, ranging from ASD, with or without seizures and psychiatric disorders, to severe global developmental delay. The missense gain-of-function mutation in CACNA1D is inherited in autosomal dominant mode with complete penetrance, as shown in this case study. The minority of the reported cases have endocrinological disorders, such as hyperaldosteronism and hyperinsulinism, but they should be monitored as they might manifest later in life.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
AA participated in writing the background with significance, case presentations, discussion, conclusion, references, designating tables, and read and approved the final manuscript. MaA, MoA, AB, and NA helped draft the manuscript, read and agree on the final manuscript, and formatted the manuscript according to the journal’s guidelines. FA supervised the overall research, preparing the manuscript reading, and approving the final manuscript. All authors contributed to the article and approved the submitted version.
We would like to express our gratitude to the family of the patient for their cooperation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1.McPartland, J, and Volkmar, FR. Autism and related disorders. Handb Clin Neurol. (2012) 106:407–18. doi: 10.1016/B978-0-444-52002-9.00023-1
2.Volkmar, FR, Lord, C, Bailey, A, Schultz, RT, and Klin, A. Autism and pervasive developmental disorders. J Child Psychol Psychiatry. (2004) 45:135–70. doi: 10.1046/j.0021-9630.2003.00317.x
3.Colvert, E, Tick, B, McEwen, F, Stewart, C, Curran, SR, Woodhouse, E, et al. Heritability of autism spectrum disorder in a UK population-based twin sample. JAMA Psychiat. (2015) 72:415–23. doi: 10.1001/jamapsychiatry.2014.3028
4.Rosenberg, RE, Law, JK, Yenokyan, G, McGready, J, Kaufmann, WE, and Law, PA. Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch Pediatr Adolesc Med. (2009) 163:907–14. doi: 10.1001/archpediatrics.2009.98
5.O'Roak, BJ, Stessman, HA, Boyle, EA, Witherspoon, KT, Martin, B, Lee, C, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun. (2014) 5:5595. doi: 10.1038/ncomms6595
6.De Rubeis, S, He, X, Goldberg, AP, Poultney, CS, Samocha, K, Cicek, AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. (2014) 515:209–15. doi: 10.1038/nature13772
7.Willsey, AJ, Sanders, SJ, Li, M, Dong, S, Tebbenkamp, AT, Muhle, RA, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cells. (2013) 155:997–1007. doi: 10.1016/j.cell.2013.10.020
8.Kessi, M, Chen, B, Peng, J, Yan, F, Yang, L, and Yin, F. Calcium channelopathies and intellectual disability: a systematic review. Orphanet J Rare Dis. (2021) 16:219. doi: 10.1186/s13023-021-01850-0
9.Skafidas, E, Testa, R, Zantomio, D, Chana, G, Everall, IP, and Pantelis, C. Predicting the diagnosis of autism spectrum disorder using gene pathway analysis. Mol Psychiatry. (2014) 19:504–10. doi: 10.1038/mp.2012.126
10.Simms, BA, and Zamponi, GW. Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron. (2014) 82:24–45. doi: 10.1016/j.neuron.2014.03.016
11.Felix, R, Calderon-Rivera, A, and Andrade, A. Regulation of high-voltage-activated ca(2+) channel function, trafficking, and membrane stability by auxiliary subunits. Wiley Interdiscip Rev Membr Transp Signal. (2013) 2:207–20. doi: 10.1002/wmts.93
12.Striessnig, J, Bolz, HJ, and Koschak, A. Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels. Pflugers Arch. (2010) 460:361–74. doi: 10.1007/s00424-010-0800-x
13.Zamponi, GW, Striessnig, J, Koschak, A, and Dolphin, AC. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev. (2015) 67:821–70. doi: 10.1124/pr.114.009654
14.Nanou, E, and Catterall, WA. Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron. (2018) 98:466–81. doi: 10.1016/j.neuron.2018.03.017
15.Ortner, NJ, Kaserer, T, Copeland, JN, and Striessnig, J. Correction to: De novo CACNA1D Ca(2+) channelopathies: clinical phenotypes and molecular mechanism. Pflugers Arch. (2020) 472:1105. doi: 10.1007/s00424-020-02436-8
16.Liao, X, and Li, Y. Genetic associations between voltage-gated calcium channels and autism spectrum disorder: a systematic review. Mol Brain. (2020) 13:96. doi: 10.1186/s13041-020-00634-0
17.Andrade, A, Brennecke, A, Mallat, S, Brown, J, Gomez-Rivadeneira, J, Czepiel, N, et al. Genetic associations between voltage-gated calcium channels and psychiatric disorders. Int J Mol Sci. (2019) 20:3537. doi: 10.3390/ijms20143537
18.Striessnig, J, Pinggera, A, Kaur, G, Bock, G, and Tuluc, P. L-type Ca(2+) channels in heart and brain. Wiley Interdiscip Rev Membr Transp Signal. (2014) 3:15–38. doi: 10.1002/wmts.102
19.Pinggera, A, Lieb, A, Benedetti, B, Lampert, M, Monteleone, S, Liedl, KR, et al. CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type calcium channels. Biol Psychiatry. (2015) 77:816–22. doi: 10.1016/j.biopsych.2014.11.020
20.Hofer, NT, Tuluc, P, Ortner, NJ, Nikonishyna, YV, Fernandes-Quintero, ML, Liedl, KR, et al. Biophysical classification of a CACNA1D de novo mutation as a high-risk mutation for a severe neurodevelopmental disorder. Mol Autism. (2020) 11:4. doi: 10.1186/s13229-019-0310-4
21.Torok, F, Tezcan, K, Filippini, L, Fernandez-Quintero, ML, Zanetti, L, Liedl, KR, et al. Germline de novo variant F747S extends the phenotypic spectrum of CACNA1D Ca2+ channelopathies. Hum Mol Genet. (2023) 32:847–59. doi: 10.1093/hmg/ddac248
22.Heyes, S, Pratt, WS, Rees, E, Dahimene, S, Ferron, L, Owen, MJ, et al. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog Neurobiol. (2015) 134:36–54. doi: 10.1016/j.pneurobio.2015.09.002
23.Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature. (2015) 519:223–8. doi: 10.1038/nature14135
24.Scholl, UI, Goh, G, Stolting, G, de Oliveira, RC, Choi, M, Overton, JD, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet. (2013) 45:1050–4. doi: 10.1038/ng.2695
25.Flanagan, SE, Vairo, F, Johnson, MB, Caswell, R, Laver, TW, Lango Allen, H, et al. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr Diabetes. (2017) 18:320–3. doi: 10.1111/pedi.12512
26.Semenova, NA, Ryzhkova, OR, Strokova, TV, and Taran, NN. The third case report a patient with primary aldosteronism, seizures, and neurologic abnormalities (PASNA) syndrome de novo variant mutations in the CACNA1D gene. Zh Nevrol Psikhiatr Im S S Korsakova. (2018) 118:49–52. doi: 10.17116/jnevro201811812149
27.O'Roak, BJ, Vives, L, Girirajan, S, Karakoc, E, Krumm, N, Coe, BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. (2012) 485:246–50. doi: 10.1038/nature10989
28.Pinggera, A, Mackenroth, L, Rump, A, Schallner, J, Beleggia, F, Wollnik, B, et al. New gain-of-function mutation shows CACNA1D as recurrently mutated gene in autism spectrum disorders and epilepsy. Hum Mol Genet. (2017) 26:2923–32. doi: 10.1093/hmg/ddx175
29.Cannon, SC. Voltage-sensor mutations in channelopathies of skeletal muscle. J Physiol. (2010) 588:1887–95. doi: 10.1113/jphysiol.2010.186874
30.De Mingo Alemany, MC, Mifsud Grau, L, Moreno Macian, F, Ferrer Lorente, B, and Leon, CS. A de novo CACNA1D missense mutation in a patient with congenital hyperinsulinism, primary hyperaldosteronism and hypotonia. Channels. (2020) 14:175–80. doi: 10.1080/19336950.2020.1761171
Keywords: case report, CACNA1D gene, autism (ASC), pervasive development disorder, childhood neurologic disorders, seizure, intellectual disability
Citation: Alzahrani A, Alshalan M, Alfurayh M, Bin Akrish A, Alsubeeh NA and Al Mutairi F (2023) Case Report: Clinical delineation of CACNA1D mutation: New cases and literature review. Front. Neurol. 14:1131490. doi: 10.3389/fneur.2023.1131490
Edited by:
Madelyn Gillentine, Seattle Children’s Hospital, United StatesReviewed by:
Elena Bacchelli, University of Bologna, ItalyCopyright © 2023 Alzahrani, Alshalan, Alfurayh, Bin Akrish, Alsubeeh and Al Mutairi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fuad Al Mutairi, YWxtdXRhaXJpZnVAbW5naGEubWVkLnNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.