
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 24 May 2023
Sec. Neurological Biomarkers
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1124145
This article is part of the Research Topic Biomarkers in Neurodegenerative Diseases for Early Detection, Diagnosis, Prognosis, and Prediction View all 6 articles
 Sonia Monge-García1
Sonia Monge-García1 María-Salud García-Ayllón2,3,4*José Sánchez-Payá1,5Ruth Gasparini-Berenguer1María-Ángeles Cortés-Gómez2,3,4
María-Salud García-Ayllón2,3,4*José Sánchez-Payá1,5Ruth Gasparini-Berenguer1María-Ángeles Cortés-Gómez2,3,4 Javier Sáez-Valero1,3,4
Javier Sáez-Valero1,3,4 José-Antonio Monge-Argilés1,6*
José-Antonio Monge-Argilés1,6*Background: Alzheimer's disease (AD) accompanied by psychotic symptoms (PS) has a poor prognosis and may be associated with imbalances in key neural proteins such as alpha-synuclein (AS).
Aim: The aim of the study was to evaluate the diagnostic validity of AS levels in the cerebrospinal fluid (CSF) as a predictor of the emergence of PS in patients with prodromal AD.
Materials and methods: Patients with mild cognitive impairment were recruited between 2010 and 2018. Core AD biomarkers and AS levels were measured in CSF obtained during the prodromal phase of the illness. All patients who met the NIA-AA 2018 criteria for AD biomarkers received treatment with anticholinesterasic drugs. Follow-up evaluations were conducted to assess patients for the presence of psychosis using current criteria; the use of neuroleptic drugs was required for inclusion in the psychosis group. Several comparisons were made, taking into account the timing of the emergence of PS.
Results: A total of 130 patients with prodromal AD were included in this study. Of these, 50 (38.4%) met the criteria for PS within an 8-year follow-up period. AS was found to be a valuable CSF biomarker to differentiate between the psychotic and non-psychotic groups in every comparison made, depending on the onset of PS. Using an AS level of 1,257 pg/mL as the cutoff, this predictor achieved at least 80% sensitivity.
Conclusion: To our knowledge, this study represents the first time that a CSF biomarker has shown diagnostic validity for prediction of the emergence of PS in patients with prodromal AD.
The diagnosis of Alzheimer's disease (AD) must be restricted to people who exhibit positive biomarkers along with specific disease phenotypes (1). Behavioral and psychological symptoms are close to universally present in AD patients, and may include psychotic symptoms (PS) (2).
Psychotic symptoms are defined as the presence of delusions and/or hallucinations (2, 3), these being the most widely used diagnostic criteria for psychosis, as proposed by Jeste and Finkel (4) and revised by Cummings et al. (3). Over half of people with AD experience PS during their illness (5, 6). In particular, systematic reviews indicate a cross-sectional prevalence of approximately 40%, although it is recognized that lower rates occur in community populations and higher rates in clinical settings (2).
Psychotic symptoms have been found to be associated with rapid cognitive decline in AD (7, 8), and their emergence may represent a distinct phenotype (9). However, the association between AD psychosis and cognitive decline does not appear to be attributable to factors such as age at onset of AD, disease duration, sex, race, education, or family psychiatric history (10). PS are also associated with more rapid progression of functional impairment, hospital admission, earlier admission to institutional care, and increased mortality (11–15). Some studies have suggested that a sharper trajectory of decline occurs among people who develop PS, even before the onset of PS (8, 12, 16, 17). The use of antipsychotic drugs to treat PS in dementia is associated with greater mortality (18) and with adverse events (19).
This evidence suggests that a different underlying biological and/or genetic predisposition may be present in these individuals, and that the occurrence of PS represents a more severe AD phenotype (20); currently, however, there are no methods for predicting the occurrence of psychosis in AD patients.
The postulated neural mechanisms of AD psychosis include disturbances in cholinergic muscarinic receptors and altered concentrations of serotonin, tau protein, kalirin, and dopamine receptors (2). AD may be associated with selective alterations in dopamine receptor density (21, 22). Overexpression of wild-type alpha-synuclein (AS) decreases dopamine neurotransmission (23), and dopamine modifies aggregation of AS in the nervous system, resulting in greater abundance of AS oligomers (24). Lewy bodies, which are insoluble aggregates composed mainly of phosphorylated AS, are found in approximately 30%−50% of people with AD (25, 26); their presence contributes to the risk of psychosis and excess cognitive burden (21).
Various authors have published reports of alterations in CSF levels of AS in patients with AD, including in the prodromal phase of the illness (27–38). Increased CSF levels of AS have been found in patients with prodromal AD compared with controls and in patients with MCI attributable to Lewy body disease. Notably, however, a small subgroup of patients with MCI-AD and PS have been found to display low levels of CSF AS (37).
This study aimed to extend the sample size with a large cohort and to estimate the diagnostic validity of CSF levels of AS as a predictor of the emergence of PS in patients with prodromal AD.
This single-center retrospective cohort study included patients with mild cognitive impairment (MCI) in accordance with the Petersen criteria (39). These patients were recruited from the outpatient dementia consultation clinic at the Neurology Service of the Dr. Balmis General University Hospital (Alicante, Spain) between 2010 and 2018.
Patients were included in the study if they were aged over 55 years; had concordant clinical and neuropsychological diagnoses; exhibited a positive profile for AD biomarkers in their CSF (40); had an MMSE score ≥ 22, IQCODE score < 80, Barthel index ≥ 90, Neuropsychiatric Inventory (NPI) score < 8 (41), and Lawton–Brody scale (IADL) score ≥ 4; underwent clinical follow-up for > 2 years; and were treated with an anticholinesterasic drug following the diagnosis of prodromal AD (42). Informed consent was obtained from patients before their participation in this study and before lumbar puncture (LP) was performed. Patients who had dementia or other neurological, psychiatric, or medical diseases that could contribute to cognitive deterioration or psychosis according to the criteria established by Cummings et al. (3) at the time of inclusion; patients were also excluded if they had an UPDRS III score > 4 at inclusion, had an MRI Fazekas scale score >2 (43), were receiving anticoagulant therapy, did not provide informed consent, had a Yesavage score >5 for depression, or had a Pittsburgh sleep quality index > 7 (44).
All patients underwent physical and neurological examination, neuropsychological studies, cerebral magnetic resonance imaging, blood tests, and LP.
The NIA-AA criteria were used to evaluate conversion of MCI to AD (42). A control group was included, consisting of patients with acute or chronic headache (n =12) or pain syndrome (n =7) who did not undergo cognitive decline during the follow-up period.
APOE genotype was available for only 81 of the patients, because this was not technically possible to obtain during the first 5 years of recruitment.
Enrolled patients were evaluated every 6–12 months to check for the development of clinical dementia criteria (42). All the patients with AD met the dementia criteria within 2 years after LP and had been receiving anticholinesterasic treatment since the diagnosis of prodromal AD.
Psychotic symptoms were considered to be present when the patient scored >9 on the hallucinations + delusions (F x S) items of the NPI test (41), after exclusion of acute illness, delirium, or recent changes in treatment (3). No pharmacologic media had been used previously in any cases. With the agreement of the caregivers, neuroleptic treatment was initiated at a minimally effective dose.
All CSF samples were obtained between 10:00 and 14:00. LP was performed by a neurologist using a 20 × 3.5-gauge needle. CSF was collected in standard polypropylene tubes, centrifuged for 10 min at 1,500 g, and then aliquoted in propylene tubes. Samples were stored at −80°C. Only samples with < 50 red blood cells/μL were included (28).
Aβ42, total tau (T-tau), and phosphorylated tau 181 (p-tau181) were measured via commercial ELISA (Innotest, Innogenetic/Fujirebio, Ghent, Belgium) following the manufacturer's instructions. Assays were tested blind with respect to clinical diagnosis 6 months after LP.
Aβ42 > 800 pg/mL, T-tau < 350 pg/mL, and p-tau181 < 56,5 pg/mL were considered normal values. Patients were considered to have an AD CSF profile when at least Aβ42 and p-tau181 were abnormal, as per the 2018 NIA-AA criteria (40).
AS levels in the CSF were measured using the LEGEND MAX human AS ELISA kit with a precoated plate (BioLegend, San Diego, CA, USA) according to the manufacturer's instructions. Assays were performed in May 2021 in triplicate and blinded with respect to clinical diagnosis. This assay has previously been validated in a Europe-wide inter-laboratory study (45). Since a higher inter-assay CV was observed when the analysis was performed with different kit batches, all samples were analyzed with plates from the same batch, and one CSF sample was measured with each of the plates for standardization of AS levels between plates. CSF samples from each of the different groups were included on each plate. Luminescence detection was carried out using a BMG Labtech LUMIstar Optima.
The Kolmogorov–Smirnov test was used to analyze the distribution of each quantitative variable. The Student's t-test (for parametric variables) and the Mann–Whitney U test (for non-parametric variables) were used to compare groups and subgroups. The chi-squared test or Fisher's exact test was used for qualitative variables.
Receiver operating characteristic (ROC) curve analysis was performed to determine the optimal cutoff for prediction of AS and the associated area under the curve (AUC). The optimal cutoff value was defined accounting for the highest sensitivity and specificity. Following this, the sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) for AS with the determined cutoff point were all calculated. In all hypothesis tests, a p ≤ 0.05 was determined to represent statistical significance. Correlations between motor/cognitive scores and AS levels and other CSF biomarkers in the diagnostic groups and subgroups were examined using Spearman's rho. The statistical package SPSS 21.0 was used for statistical analyses.
The study was approved by the Ethics Committees of the Dr. Balmis General University Hospital (ref. number: PI2020/250) and the Universidad Miguel Hernández (ref. number: PRL.IN.JSV.01.21).
Figure 1 shows the distribution of patients across each group. A total of 50 patients with prodromal AD (38.4%) had developed PS (AD-PS group) 8 years after their initial diagnosis, with 37 of these (28%) having done so within the first 4 years after diagnosis.
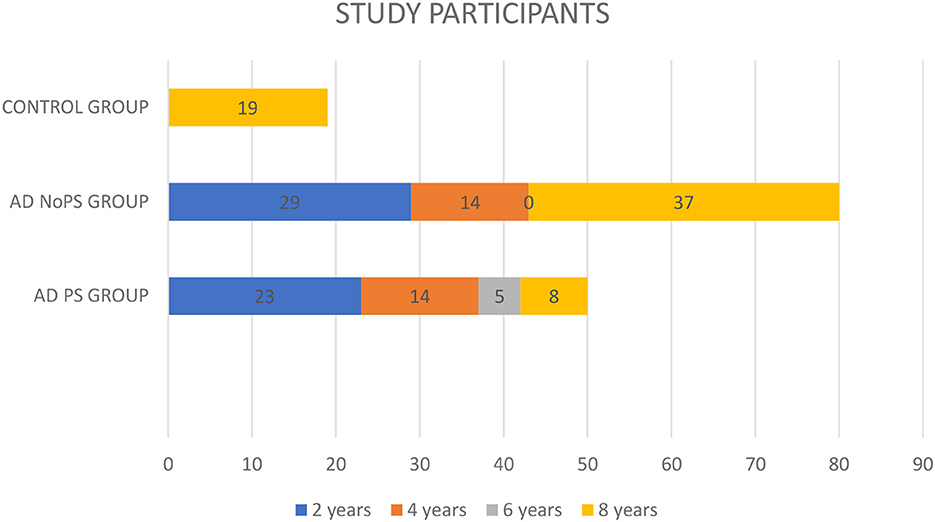
Figure 1. Follow-up of the study participants.
The study also included 80 patients without PS (AD-No PS group): 37 (28%) remained free of PS over 8 years of follow-up. However, the follow-up period was < 4 years for 43 patients (33%) in this group. The minimum follow-up period was 2 years.
The AD-PS group exhibited lower values for AS (p < 0.0001), the ratio of AS/p-tau181 (p < 0.006), p-tau181 levels (p < 0.01), and Aβ42 levels (p < 0.02) (Table 1, Supplementary Table 1). Clinically, the AD-PS group had lower NPI scores (p < 0.01) and UPDRS III scores (p < 0.007). No other differences were identified across the remaining variables. Correlation coefficients representing the associations between motor/cognitive scores and AS levels and other CSF biomarkers in the diagnostic groups are presented in Table 2. In both groups, CSF level of AS was positively correlated with T-tau (AD-PS group: ρ = 0.55, P < 0.0001; AD-No PS group: ρ = 0.41, P < 0.01), p-tau181 (AD-PS group: ρ = 0.56, P < 0.0001; AD-No PS group: ρ = 0.57, P < 0.001), NPI score (AD-PS group: ρ = 0.52, P < 0.01; AD-No PS group: ρ = 0.5, p < 0.05), and UPDRS (III) score (AD-PS group: ρ = 0.51, P < 0.01; AD-No PS group: ρ = 0.48, P < 0.05).
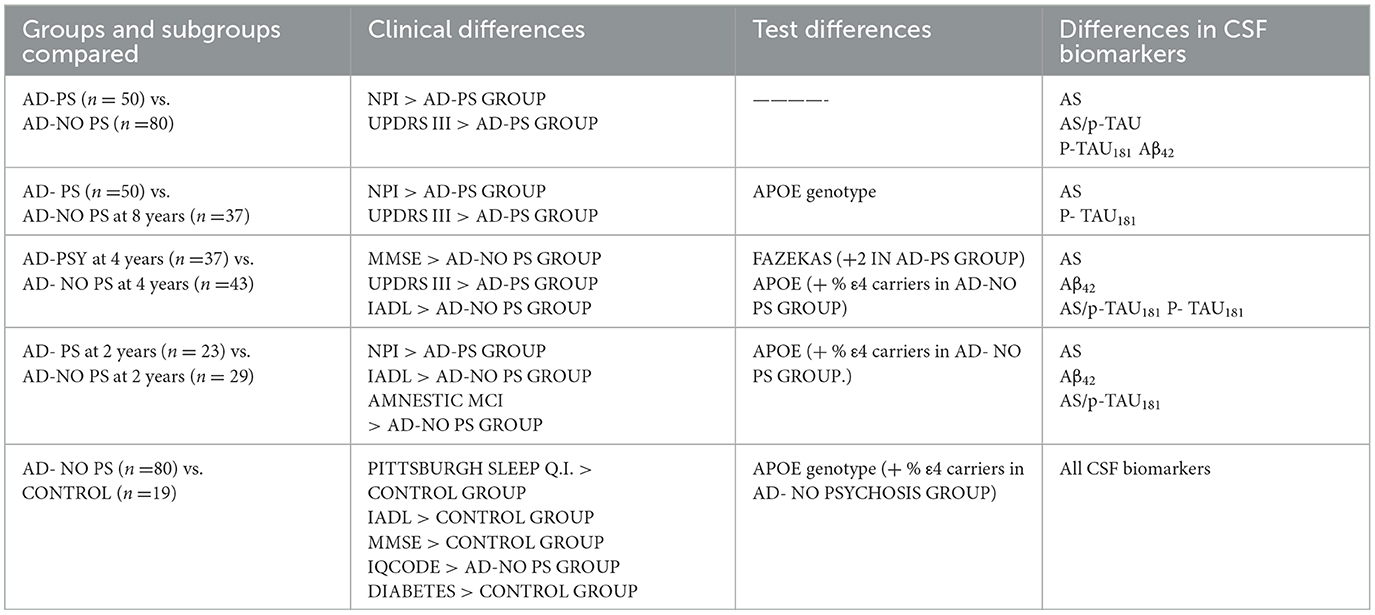
Table 1. Significant differences between groups and subgroups.
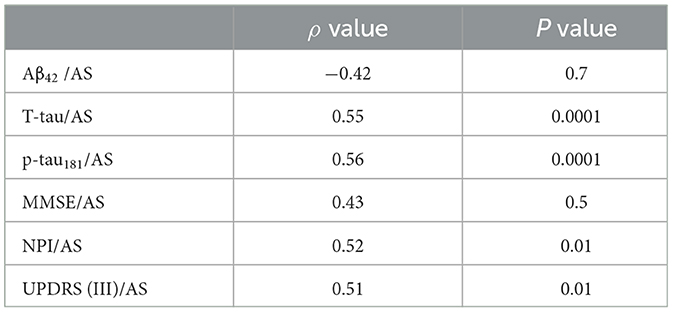
Table 2A. Spearman correlations between motor/cognitive scores and AS levels and other CSF biomarkers in the diagnostic groups and subgroups. Psychosis group (AD-PS).
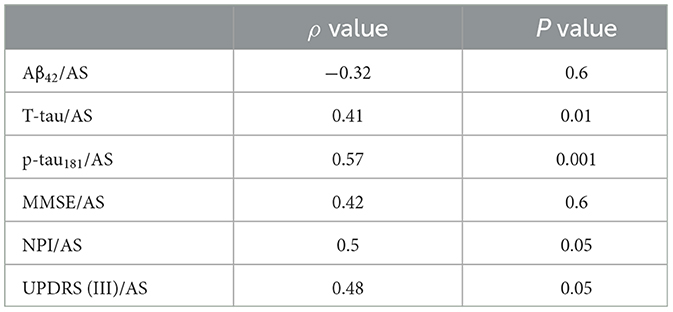
Table 2B. No-psychosis group (AD-No PS).
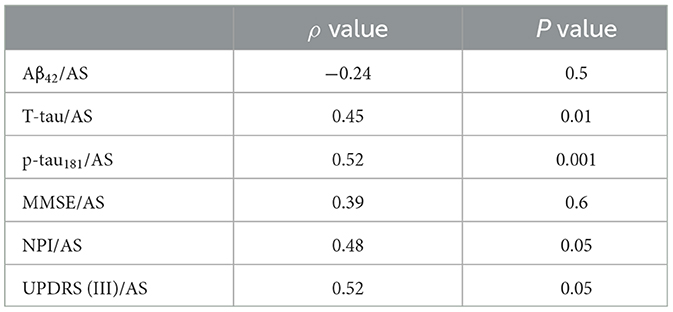
Table 2C. AD no-psychosis group at 8-year follow-up.
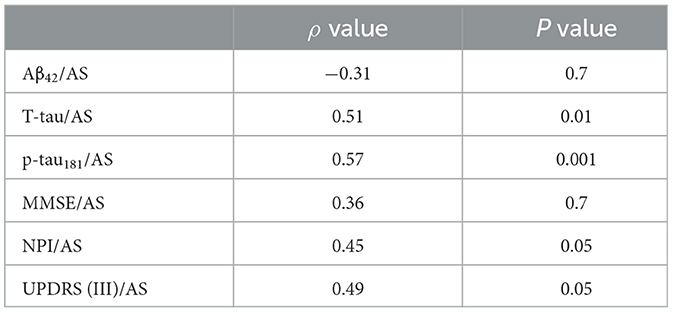
Table 2D. AD no-psychosis group at 4-year follow-up.
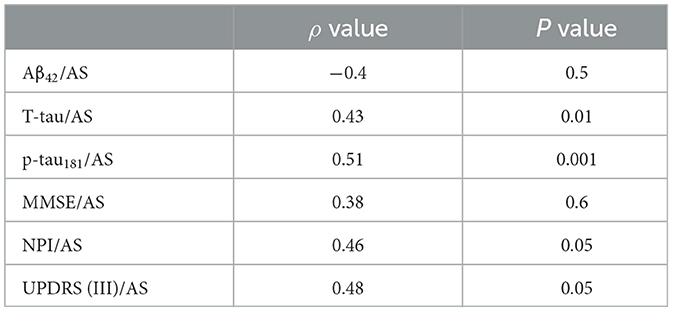
Table 2E. AD no-psychosis group at 2-year follow-up.
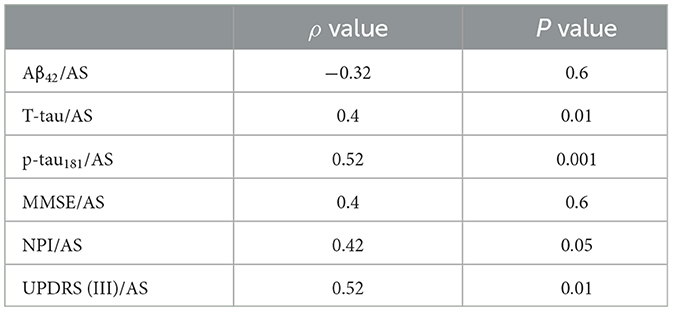
Table 2F. AD psychosis group at 4-year follow-up.
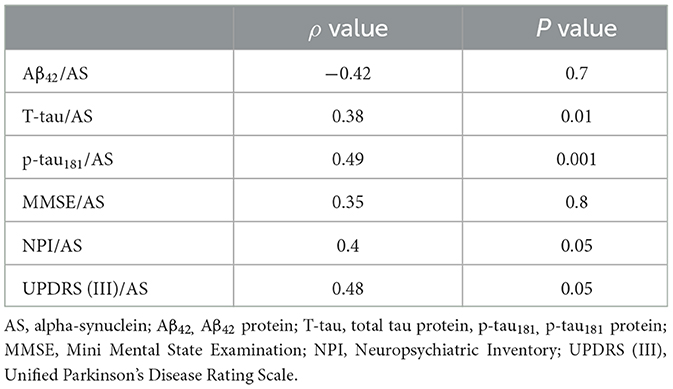
Table 2G. AD psychosis group at 2-year follow-up.
Comparisons between subgroups were carried out to assess the predictive value of the biomarkers and clinical parameters for PS during follow-up.
As compared with the AD-no PS subgroup 8 years after diagnosis (n = 37), the AD-PS group (n = 50) still exhibited lower AS levels (p < 0.006) and p-tau181 (p < 0.01), and higher scores on the NPI (p < 0.001) and the UPDRS III (p < 0.002). No other differences were found in the remaining variables, except in relation to APOE genotype (p < 0.001); however, these data were only available for a subset of patients (only 19 patients from the AD-PS group and two from the AD-No PS subgroup 8 years after diagnosis) (Table 1, Supplementary Table 2). Correlation coefficients representing the associations between motor/cognitive scores and AS levels and other CSF biomarkers in the diagnostic groups and subgroups are presented in Table 2. In the AD-No PS subgroup with 8 years of follow-up, CSF level of AS CSF was positively correlated with T-tau (ρ = 0.45, p < 0.01), p-tau181 (ρ = 0.52, p < 0.001), NPI score (ρ = 0.48, p < 0.05), and UPDRS (III) score (ρ = 0.52, p < 0.05) (Tables 2A–C).
In a comparison of the subgroups limited to those patients with at least 4 years of follow-up, the AD-PS subgroup (n = 37) exhibited lower levels of AS (p < 0.001), Aβ42 (p < 0.004), and p-tau181 (p < 0.01) and a lower ratio of AS/p-tau181 (p < 0.02) as compared with the AD-No PS subgroup (n = 43). Clinically, the AD-PS subgroup had lower MMSE (p < 0.01) and IADL scores (p < 0.02) and higher UPDRS III scores (p < 0.04). Finally, the proportion of patients in this subgroup with the APOE- ε4 genotype was lower (p < 0.05; analysis limited to 61 of 80 patients), and patients in this subgroup had higher Fazekas MRI scores for white matter pathology (p < 0.02). No other differences were found in the remaining variables (Table 1, Supplementary Table 3). Correlation coefficients representing the associations between motor/cognitive scores and AS levels and other CSF biomarkers in the diagnostic subgroups are presented in Tables 2D, F. In both subgroups, CSF level of AS was positively correlated with T-tau (AD-PS subgroup: ρ = 0.4, p < 0.01; AD-No PS subgroup: ρ = 0.51, p < 0.01), p-tau181 (AD-PS subgroup: ρ = 0.52, p < 0.001; AD-No PS subgroup: ρ = 0.57, p < 0.001), NPI score (AD-PS subgroup: ρ = 0.42, p < 0.05; AD-No PS subgroup: ρ = 0.45, p < 0.05), and UPDRS (III) score (AD-PS subgroup: ρ = 0.52, p < 0.01; AD-No PS subgroup: ρ = 0.49, p < 0.05).
Since we were investigating the prognostic value of the biomarkers, we also compared subgroups at the shortest period of follow-up: that is, patients with only a 2-year follow-up period. This AD-PS subgroup (n =23) exhibited lower levels of AS (p < 0.002) and Aβ42 (p < 0.02) and a lower ratio of AS/p-tau181 (p < 0.05) as compared with the AD-No PS subgroup (n =29). The AD-PS subgroup also had a lower rate of amnestic MCI (p < 0.02), a lower proportion of patients with the APOE- ε4 genotype (p < 0.05, data limited to 42 of 52 patients), and lower IADL scores (p < 0.02), but they had higher NPI scores (p < 0.02). No other differences were found in the remaining variables (Supplementary Table 4). Correlation coefficients representing the associations between motor/cognitive scores and AS levels and other CSF biomarkers in the diagnostic subgroups are presented in Table 2. In both subgroups, CSF level of AS was positively correlated with T-tau (AD-PS subgroup: ρ = 0.38, p < 0.01; AD-No PS subgroup: ρ = 0.43, p < 0.01), p-tau181 (AD-PS subgroup: ρ = 0.49, p < 0.001; AD-No PS subgroup: ρ = 0.51, p < 0.001), NPI score (AD-PS subgroup: ρ = 0.40, p < 0.05; AD-No PS subgroup: ρ = 0.46, p < 0.05), and UPDRS (III) score (AD-PS subgroup: ρ = 0.48, p < 0.01; AD-No PS subgroup: ρ = 0.48, p < 0.05) (Tables 2E, G).
In comparison to the control group, the AD-No PS group exhibited higher levels of AS (p < 0.04), T-tau (p < 0.0001), and p-tau181 (p < 0.0001); higher ratios of T-tau/Aβ42 (p < 0.0001) and p-tau181/Aβ42 (p < 0.0001); lower levels of Aβ42 (p < 0.0001); and a lower AS/p-tau181 ratio (p < 0.001). Clinically, they had higher IQCODE scores (p < 0.0001) and a higher incidence of the APOE- ε4 genotype (p < 0.01; data limited to 61 of 99 patients), but had lower scores on the Pittsburgh sleep quality index (p < 0.02), MMSE (p < 0.0001), and IADL (p < 0.001). No other differences were found (Table 1, Supplementary Table 5).
ROC curves were defined in order to assess the diagnostic value of AS. Taking an AS level of 1,257 as the cutoff, the use of this threshold showed sensitivity equal to or >80% in differentiating between the groups and subgroups analyzed (Table 3). The NPV reached 80% in differentiating between the AD-PS and AD-No PS groups (Table 3).
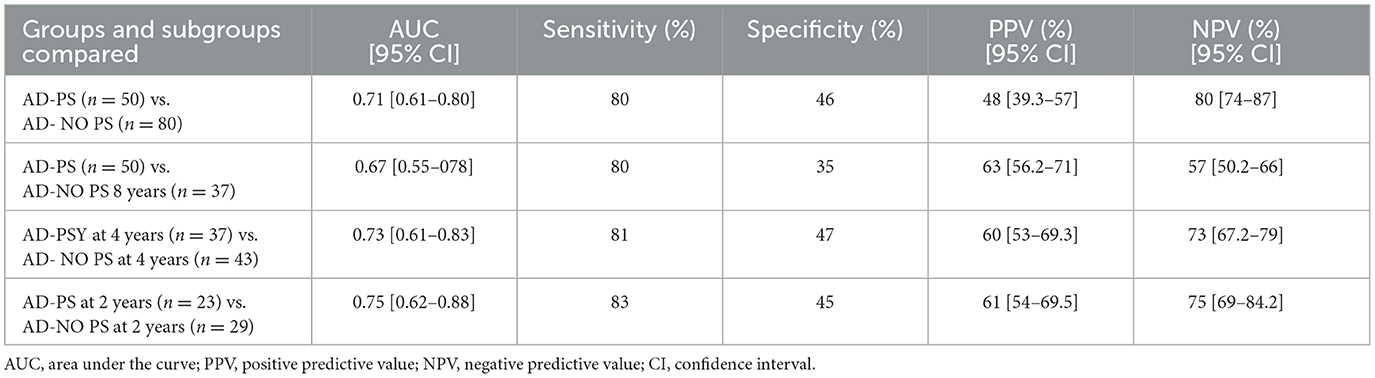
Table 3. ROC curves for AS level at a cutoff of 1,257 pg/ml as a differentiator between AD groups and subgroups.
This study indicated that AS is a valuable CSF biomarker for prediction of PS in a prodromal AD patient cohort. Among the other CSF biomarkers, p-tau181, AS/p-tau181 ratio, and Aβ42 also reached the threshold, but displayed lower diagnostic validity for this purpose.
The presence of psychotic symptoms in AD is reportedly associated with AS and/or tau cerebral pathologies (8). The pathogenic relationship between AS and PS is recognized because of their common implication of cerebral dopamine levels (23, 24). Increased AS affects dopamine neurotransmission at multiple levels, particularly in decreasing dopamine synthesis (23). Moreover, dopamine influences the aggregation of AS in the nervous system, which results in AS oligomers and unique dopamine-induced oligomeric conformations (24). Selective alterations in dopamine receptor density have been found postmortem and in vivo in patients with AD and PS; these alterations may be associated with distinct clinical profiles (21, 22).
In the present study, lower CSF levels of AS were found in the AD-PS group than in the AD-No PS group. Such a decrease may be associated with the cerebral deposition of AS, forming Lewy bodies in alpha-synucleinopathies (46, 47). A number of studies have reported AS pathology in approximately 50% of autopsied patients with AD (25, 36). Patients with AD and AS tend to exhibit amplified deterioration, typically enduring more severe symptoms and shorter duration of survival (47), which may contribute to a distinctive clinical profile within AD (9, 22). Evidence from other studies suggests that AS might be involved in the development of AD from the very early stages of Aβ pathology formation (48), as well as tau hyperphosphorylation (26). These data suggest that AS is involved in the pathophysiology of AD (26). Early identification of this condition in patients with AD, including quantification of CSF AS, should enable provision of a better treatment plan and improvements in prognosis (37).
The AD-No PS group exhibited higher levels of AS than the control group, particularly during the first 4 years after diagnosis. These results were in line with those of previous studies, with short follow-up periods and without follow-up, than have taken PS into account in the clinical description or evolution (27–29, 32–36). The increase in AS observed in AD patients is based on evidence on elevated AS in the brain tissue of patients with AD (49) and/or the neuronal damage related to AD (33). This increase in CSF levels of AS in AD patients is associated with the accumulation of amyloid plaques (26) and tau proteins (36, 50, 51). These results are in accordance with the lower levels of Aβ42, higher levels of p-tau181, and higher AS/p-tau181 ratio observed in the AD-No PS group in the present study. In a previous study, PS was found to be associated with tau phosphorylation abnormalities (8). Nevertheless, although that association should be female-specific, the incidence of PS in AD should be more likely among men because of the nature of AS pathology (8). To our knowledge, no gender differences have been described in the relationship of PS with Aβ42 protein and the ratio of AS/p-tau181.
In this study, the decrease in the aforementioned ratio among the AD-PS group was more attributable to lower levels of AS than to a clear decrease in the levels of p-tau181. Another independent report, on a longitudinal study of AD, has found that lower values for this ratio predict faster cognitive decline (35).
The sensitivity of AS in differentiating between the AD-PS and AD-No PS groups is notable, considering the ability to predict the occurrence of PS, which requires specific pharmacologic intervention. The emergence of PS is associated with endogenous and exogenous factors, including underlying biological and/or genetic predispositions in the individual (2). Regarding endogenous factors, of the dopaminergic factors mentioned above, certain alterations to the cholinergic (2, 52–56) and serotoninergic systems (57, 58) are related to the occurrence of PS in AD. In terms of exogenous factors, sleep quality, familial relationships with caregivers, medical antecedents, schooling, and capacity to engage in activities of daily living should influence the emergence of PS in AD (2). No significant differences at the time of inclusion were found between the AD groups on any of these variables, including demographic and radiological data, indicating the high level of homogeneity between the groups included in this study.
Clinical NPI and UPDRS (III) scores increased among the PS group over the period following inclusion. These data may be concordant with abnormal dopaminergic status, which is probably related to the amount of cortical and subcortical Lewy bodies (59). To date, the concept of mixed AD + dementia with Lewy bodies is accepted in neuropathological settings as a difficult clinical diagnosis, with a lack of biomarkers to assist identification (60). Quantification of CSF level of AS may facilitate this objective (61). Positive correlations were observed between AS levels, such scores, and tau protein levels in both AD groups and in the subgroups. These findings in the initial stages of AD support the involvement of AS in the pathogenesis of AD (27, 32, 52).
Alzheimer's disease is considered to be a clinical–biological entity (1). Our data and data presented in previous publications (27, 28, 32–37) support the added value of measurement of CSF levels of AS in further characterization of the CSF AD biomarker profile. The contradictory results published in some previous reports regarding the potential use of CSF level of AS as a diagnostic biomarker for AD may be attributable to various factors, including reduced sample size, uncertain diagnosis and/or uncontrolled follow-up, enrolment of patients at different stages of the disease, differences in age, and lack of control for blood contamination, among others (29, 62–65). The current AT (N) classification for a biological definition of AD has been defined as flexible, meaning that new biomarkers can be added when they become available (40), and there is a need for more biomarkers, such as AS, indicating other aspects of the mechanisms of the disease (66–69).
Beta-synuclein is another member of the synuclein family and is emerging as a reliable synaptic marker in CSF and blood for AD and prion disease (70, 71). In the study by Barba et al., increased levels of CSF AS were observed in pre-AD patients, but not in MCI-AD or dementia-AD. Beta-synuclein was elevated in all AD continuum subgroups. Elevated CSF levels of both beta-synuclein and AS in pre-AD may reflect the earliest synaptic dysfunction occurring in AD. Decreased AS levels may indicate the presence of α-synucleinopathy, whereas beta-synuclein concentrations are not influenced by the presence of synucleinopathy or by blood contamination, which instead affects AS measurements.
In this study, the handling of the samples was performed as per recommended operating procedures (72). CSF samples with < 50 red blood cells/μL were included (28), and the reagents used had been validated in a Europe-wide inter-laboratory study (45). These points of analytical and methodological validity are critical for the value of these results. Nevertheless, in relation to protein-misfolding cyclic amplification (PMCA) and real-time quaking-induced conversion (RT-QuIC), ultrasensitive protein amplification assays for the detection of misfolded protein aggregates could also offer diagnostic reliability for AS levels in CSF. In particular, RT-QuIC assay has demonstrated high specificity and sensitivity for detection of CSF AS aggregation in patients with synucleinopathies when compared to AD patients and controls (73–75). The possibility of analyzing the cholinergic, glutamatergic, and serotoninergic pathways with these techniques should be of great interest for completion of the study of PS in AD patients.
The main limitation of this study was the lack of neuropathological confirmation. Nevertheless, to our knowledge, the long clinical follow-up period of the patients was among the longest across all published studies. The clinical and biological data were very conclusive. However, the ability of caregivers to control behavioral disturbances was not tested, and a portion of the AD-No PS group had an incomplete period of follow-up in which the emergence of PS could be excluded. Finally, the lack of data on APOE genotype in patients recruited during the first 5 years of the study was another limitation.
In conclusion, the quantification of CSF AS in patients with prodromal AD allows for prediction of the emergence of PS during the subsequent 8 years with high sensitivity. In this regard, early identification will enable the provision of better treatment plans and improvements in prognosis. Future biomarker panels, including the biological study of cholinergic and serotoninergic pathways, will probably enable more complete prediction of PS in AD.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by Ethical Committee of University General Hospital Dr. Balmis. Alicante, Ref number: PI2020/250. The patients/participants provided their written informed consent to participate in this study.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This study was partially funded by an ISABIAL grant (2020-286) to J-AM-A and by the Direcció General de Ciència I Investigació, Generalitat Valenciana to JS-V (AICO/2021/308). M-ÁC-G was supported by a BEFPI fellowship from the Generalitat Valenciana.
The authors are thankful to the outpatient unit and the Biobank at the Dr. Balmis General University Hospital. We also acknowledge support for acquisition of some of the reagents from Novartis Spain, Danone Spain, KRKA Spain, and Neuraxpharm Spain.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1124145/full#supplementary-material
AD, Alzheimer's disease; AS, alpha-synuclein; PS, psychotic symptoms; CSF, cerebrospinal fluid; LP, lumbar puncture; MCI, mild cognitive impairment; IADL, instrumental activities of daily living; MMSE, Mini Mental State Examination; IQCODE, Informant Questionnaire on Cognitive Decline in the Elderly; NPI, Neuropsychiatric Inventory; PSQI, Pittsburgh Sleep Quality Index; UPDRS III, Unified Parkinson Disease Rating Scale; A Aβ1−42 protein; T-tau, total tau protein; P-tau181, phosphorylated tau 181.
1. Dubois B, Villain N, Frisoni GB, Rabinovici GD, Sabbagh M, Cappa S, et al. Clinical diagnosis of Alzheimer's disease: recommendations of the International Working Group. Lancet Neurol. (2021) 20:484–96. doi: 10.1016/S1474-4422(21)00066-1
2. Ballard C, Kales HC, Lyketsos C, Aarsland D, Creese B, Mills R, et al. Psychosis in Alzheimer's disease. Curr Neurol Neurosci Rep. (2020) 20:57. doi: 10.1007/s11910-020-01074-y
3. Cummings J, Cortez-Pinto L, Cruz M, Fischer CE, Gerritsen DL, Grossberg GT, et al. Criteria for psychosis in major and mild neurocognitive disorders: International Psychogeriatric Association. Consensus clinical and research definition. Am J Geriatr Psychiatry. (2020) 28:1256–69. doi: 10.1016/j.jagp.2020.09.002
4. Jeste DV, Finkel SI. Psychosis of Alzheimer's disease and related dementias: diagnostic criteria for a distinct syndrome. Am J Geriatr Psychiatry. (2000) 8:29–34. doi: 10.1097/00019442-200002000-00004
5. Haupt M, Kurz A, Janner M. A 2-year follow-up of behavioural and psychological symptoms in Alzheimer's disease. Dement Geriatr Cogn Disord. (2000) 11:147–52. doi: 10.1159/000017228
6. Ballard C, Gray A, Ayre G. Psychotic symptoms, aggression and restlessness in dementia. Rev Neurol (Paris). (1999) 155:S44–52.
7. Stern RG, Mohs RC, Davidson M, Schmeidler J, Silverman J, Kramer-Ginsberg E, et al. A longitudinal study of Alzheimer's disease: measurement, rate and predictors of cognitive deterioration. Am J Psychiatry. (1994) 151:390–6. doi: 10.1176/ajp.151.3.390
8. Koppel J, Acker C, Davies P, Lopez OL, Jimenez H, Azose M, et al. Psychotic Alzheimer's disease is associated with gender-specific tau phosphorylation abnormalities. Neurobiol Aging. (2014) 35:2021–8. doi: 10.1016/j.neurobiolaging.2014.03.003
9. Sweet RA, Nimgoankar VL, Devlin B, Jeste DV. Psychotic symptoms in Alzheimer's disease: evidence for a distinct phenotype. Mol Psychiatry. (2003) 8:383–92. doi: 10.1038/sj.mp.4001262
10. Ropacki SA, Jeste DV. Epidemiology of and risk factors for psychosis of Alzheimer's disease: a review of 55 studies published from 1990 to 2003. Am J Psychiatr. (2005) 162:2022–30. doi: 10.1176/appi.ajp.162.11.2022
11. Scarmeas N, Brandt J, Albert M, Hadjigeorgiou G, Papadimitriou A, Dubois B, et al. Delusions and hallucinations are associated with worse outcome in Alzheimer disease. Arch Neurol. (2005) 62:1601–8. doi: 10.1001/archneur.62.10.1601
12. Peters ME, Schwartz S, Han D, Rabins PV, Steinberg M, Tschanz JT, et al. Neuropsychiatric symptoms as predictors of progression to severe Alzheimer's dementia and death: the Cache County dementia progression study. Am J Psychiatr. (2015) 172:460–5. doi: 10.1176/appi.ajp.2014.14040480
13. Wadsworth LP, Lorius N, Donovan NJ, Locascio JJ, Rentz DM, Johnson KA, et al. Neuropsychiatric symptoms and global functional impairment along the Alzheimer's continuum. Dement Geriatr Cogn Disord. (2012) 34:96–111. doi: 10.1159/000342119
14. Zahodne LB, Ornstein K, Cosentino S, Denavand DP, Stern Y. Longitudinal relationships between Alzheimer disease progression and psychosis, depressed mood and agitation/aggression. Am J Geriatr Psychiatry. (2015) 23:130–40. doi: 10.1016/j.jagp.2013.03.014
15. Beydoun MA, Beydoun HA, Gamaldo AA, Rostant OS, Dore GA, Zonderman AB, et al. Nationwide inpatient prevalence, predictors, and outcomes of Alzheimer's disease among older adults in the United States, 2002-2012. J Alzheimers Dis. (2015) 48:361–75. doi: 10.3233/JAD-150228
16. Paulsen JS, Ready RE, Stout JC, Salmon DP, Thal LJ, Grant I, et al. Neurobehaviors and psychotic symptoms in Alzheimer's disease. J Int Neuropsychol Soc. (2000) 6:815–20. doi: 10.1017/S1355617700677081
17. Weamer EA, Emanuel JE, Varon D, Miyahara S, Wilkosz PA, Lopez OL, et al. The relationship of excess cognitive impairment in MCI and early Alzheimer's disease to the subsequent emergence of psychosis. Int Psychogeriatr. (2009) 21:78–85. doi: 10.1017/S1041610208007734
18. Kales HC, Kim HM, Zivin K, Valenstein M, Scyfried LS, Chiang C, et al. Risk of mortality among individual antipsychotics in patients with dementia. Am J Psychiatry. (2012) 169:71–9. doi: 10.1176/appi.ajp.2011.11030347
19. Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry. (2006) 14:191–210. doi: 10.1097/01.JGP.0000200589.01396.6d
20. Murray PS, Kumar S, DeMichele-Sweet MAA, Sweet RA. Psychosis in Alzheimer's disease. Biol Psychiatry. (2014) 75:542–52. doi: 10.1016/j.biopsych.2013.08.020
21. Sweet RA, Hamilton RL, Healy MT, Wisniewski SR, Henteleff R, Pollock BG, et al. Alterations of striatal dopamine receptor binding in Alzheimer disease are associated with Lewy body pathology and antemortem psychosis. Arch Neurol. (2001) 58:466–72. doi: 10.1001/archneur.58.3.466
22. Reeves, S, McLachlan E., Bertrand J., D'Antonio F., Brownings S., Nair A., et al. (2017). Therapeutics window of dopamine D2/3 receptors occupancy to treat psychosis in Alzheimer's disease. Brain. 140, 1117–1127. doi: 10.1093/brain/aww359
23. Butler B, Sambo D, Khoshbouei H. Alpha-synuclein modulates dopamine neurotransmission. J Chem Neuroanat 83-84. (2017) 41–9. doi: 10.1016/j.jchemneu.2016.06.001
24. Mor DE, Daniels MJ, Ischiropoulos H. The usual suspects, dopamine and alpha-synuclein, conspire to cause neurodegeneration. Mov Disord. (2019) 34:167–79. doi: 10.1002/mds.27607
25. Holtzer R, Irizarry MC, Sanders J, Hyman BT, Wegesin DJ, Riba A, et al. Relation of quantitative indexes of concurrent α-synuclein abnormalities to clinical outcome in autopsy-proven Alzheimer disease. Arch Neurol. (2006) 63:226–30. doi: 10.1001/archneur.63.2.226
26. Twohig D, Nielsen HM. Alpha-synuclein in the pathophysiology of Alzheimer's disease. Mol Neurodegener. (2019) 14:23. doi: 10.1186/s13024-019-0320-x
27. Toledo JB, Korff A, Shaw LM, Trojanowski JQ, Zhang J. Alpha-synuclein improves diagnostic and prognostic performance of tau an Aβ in Alzheimer's disease. Acta Neuropathol. (2013) 126:683–97. doi: 10.1007/s00401-013-1148-z
28. Korff A, Liu C, Ginghina C, Shi M, Zhang J. Alpha-synuclein in cerebrospinal fluid of Alzheimer's disease and mild cognitive impairment. J Alzheimers Dis. (2013) 36:679–88. doi: 10.3233/JAD-130458
29. Berge G, Sando SB, Albrektsen G, Lauridsen C, Moller I, Grontvedt GR, et al. Alpha-synuclein measured in cerebrospinal fluid from patients with Alzheimer's disease, mild cognitive impairment, or healthy controls: a two years follow-up study. BMC Neurol. (2016) 16:180. doi: 10.1186/s12883-016-0706-0
30. Hansson, O, Hall S., Öhrfelt A., Zetterberg H, Blennow K., Minthon L., Nägga K., et al. (2014). Levels of CSF alpha-synuclein oligomers are increased in Parkinson's disease with dementia and dementia with Lewy bodies compared to Alzheimer'disease. Alzheimers Res Ther 6, 25. doi: 10.1186/alzrt255
31. Mackin RS, Insel P, Zhang J, Mohlenhoff B, Galasko D, Weiner M, et al. CSF alpha-synuclein and Lewy body-like symptoms in normal controls, mild cognitive impairment and Alzheimer'disease. J Alzheimers Dis. (2014) 43:1007–16. doi: 10.3233/JAD-141287
32. Slaets S, Vanmechelen E, Le Bastard N, Decraemer H, Vandijck M, Martin JJ, et al. Increased CSF Alpha-synuclein levels in Alzheimer's disease: correlation with tau levels. Alzheimers Dement. (2014) 10:S290–8. doi: 10.1016/j.jalz.2013.10.004
33. Majbour NK, Chiasserini D, Vaikath NN, Eusebi P, Tokuda T, van de Berg W, et al. Increased levels of total but not oligomeric or phosphorylated forms of alpha-synuclein in patients diagnosed with probable Alzheimer's disease. Sci Rep. (2017) 7:40263. doi: 10.1038/srep40263
34. Shi M, Tang L, Toledo JB, Ginghina C, Wang H, Aro P, et al. CSF alpha-synuclein contributes to the differential diagnosis of Alzheimer's disease. Alzheimers Dement. (2018) 14:1052–62. doi: 10.1016/j.jalz.2018.02.015
35. Wang H, Stewart T, Toledo JB, Ginghina C, Tang L, Atik A, et al. A longitudinal study of total and phosphorylated alpha-synuclein with other biomarkers in CSF of Alzheimer's disease and mild cognitive impairment. J Alzheimers Dis. (2018) 61:1541–53. doi: 10.3233/JAD-171013
36. Twohig D, Rodríguez-Vieitez E, Sando SB, Berge G, Lauridsen C, Moller I, et al. The relevance of CSF alpha-synuclein levels to sporadic and familial Alzheimer's disease. Acta Neuropathol Commun. (2018) 6:130. doi: 10.1186/s40478-018-0624-z
37. García-Ayllón, M. S., Monge-Argilés J. A., Monge-García V., Navarrete F., Cortés-Gómez M. A., Sánchez-Payá J., et al. (2019). Measurement of CSF alpha-synuclein improves early differential diagnosis of mild cognitive impairment due to Alzheimer's disease. J Neurochem 150, 218–230. doi: 10.1111/jnc.14719
38. Bousiges O, Philippi N, Lavaux T, Perret-Liaudet A, Lachmann I, Schaeffer-Agalède C, et al. Differential diagnostic value of total alpha-synuclein assay in the CSF between Alzheimer's disease and dementia with Lewy bodies from the prodromal stage. Alzheimers Res Ther. (2020) 12:120. doi: 10.1186/s13195-020-00684-5
39. Artero S, Petersen R, Touchon J, Ritchie K. Revised criteria for mild cognitive impairment: validation within a longitudinal population study. Dement Geriatr Cogn Disord. (2006) 22:465–70. doi: 10.1159/000096287
40. Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. (2018) 14:535–62. doi: 10.1016/j.jalz.2018.02.018
41. Cummings J. The Neuropsychiatric Inventory: assessing psychopathology in dementia patients. Neurology. (1997) 48:S10–6. doi: 10.1212/WNL.48.5_Suppl_6.10S
42. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CRJr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the NIA-AA workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. (2011) 7:263–9. doi: 10.1016/j.jalz.2011.03.005
43. Fazekas F, Kleiner R, Offenbacher H, Payer F, Schmidt R, Kleinert G, et al. The morphologic correlate of incidental punctate white matter hyperintensities on MR images. Am J Neuroradiol. (1991) 12:915–21.
44. Hita-Contreras F, Martínez-López E, Latorre-Román PA, Garrido F, Santos MA, Martínez-Amat A. Reliability and validity of the Spanish version of the Pittsburgh sleep quality index (PSQI) in patients with fibromyalgia. Rheumatol Int. (2014) 34:929–36. doi: 10.1007/s00296-014-2960-z
45. Kruse N, Mollenhauer B. Validation of a commercially available enzyme-linked immunoabsorbent assay for the quantification of human alpha-synuclein in CSF. J Immunol Methods. (2015) 426:70–5. doi: 10.1016/j.jim.2015.08.003
46. Tokuda T, Salem SA, Allsop D, Mizuno T, Nakagawa M, Qureshi MM, et al. Decreased alpha-synuclein in CSF of aged individuals and subjects with Parkinson's disease. Biochem Biophys Res Commun. (2006) 349:162–6. doi: 10.1016/j.bbrc.2006.08.024
47. Wennstrom M, Surova Y, Hall S, Nilsson C, Minthon L, Bostrom F, et al. Low CSF levels of both alpha-synuclein and alpha-synuclein cleaving enzyme neurosin in patients with synucleinopathy. PLoS ONE. (2013) 8:e53250. doi: 10.1371/journal.pone.0053250
48. Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic interactions between Aβ, Tau and α-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. (2010) 30:7281–9. doi: 10.1523/JNEUROSCI.0490-10.2010
49. Cook C, Petrucelli L. Aβ puts Alpha in synuclein. Neuron. (2020) 105:205–6. doi: 10.1016/j.neuron.2020.01.001
50. Larson ME, Sherman MA, Greimel S, Kuskowski M, Schneider JA, Bennett DA, et al. Soluble α-synuclein is a novel modulator of Alzheimer's disease pathophysiology. J Neurosci. (2012) 32:10253–66. doi: 10.1523/JNEUROSCI.0581-12.2012
51. Vergallo A, Bun RS, Toschi N, Baldacci F, Zetterberg H, Blennow K, et al. Association of CSF alpha-synuclein with total and phosphor-tau181 protein concentrations and brain amyloid load in cognitively normal subjective memory complainers stratified by Alzheimer's disease biomarkers. Alzheimers Dement. (2018) 14:1623–31. doi: 10.1016/j.jalz.2018.06.3053
52. Monge-García V, García-Ayllón MS, Sáez-Valero J, Sánchez-Payá J, Navarrete-Rueda F, Manzanares-Robles J, et al. Relation between alpha-synuclein and CSF core biomarkers of Alzheimer's disease. Medicina. (2021) 57:954. doi: 10.3390/medicina57090954
53. Garcia-Alloza M, Gil-Bea FJ, Diez-Ariza M, Chen CP, Francis PT, Lasheras B, et al. Cholinergic-serotoninergic imbalance contributes to cognitive and behavioural symptoms in Alzheimer's disease. Neuropsychologia. (2005) 43:442–9. doi: 10.1016/j.neuropsychologia.2004.06.007
54. Lai MK, Lai OF, Keene J, Esiri MM, Francis PT, Hope T, et al. Psychosis of Alzheimer's disease is associated with elevated muscarinic M2 binding in the cortex. Neurology. (2001) 57:805–11. doi: 10.1212/WNL.57.5.805
55. Tsang SW, Francis PT, Esiri MM, Wong PT, Chen CP, Lai MK. Loss of [3H]4-DAMP binding to muscarinic receptors in the orbitofrontal cortex of Alzheimer's disease patients with psychosis. Psychopharmacology. (2008) 198:251–9. doi: 10.1007/s00213-008-1124-9
56. Ballard C, Piggott M, Johnson M, Cairns N, Perry R, McKeith I, et al. Delusions associated with elevated muscarinic binding in dementia with Lewy bodies. Ann Neurol. (2000) 48:868–76. doi: 10.1002/1531-8249(200012)48:6<868::AID-ANA7>3.0.CO;2-0
57. Zubenko GS, Moossy J, Martinez AJ, Rao G, Claassen D, Rosen J, et al. Neuropathological and neurochemical correlates of psychosis in primary dementia. Arch Neurol. (1991) 48:619–24. doi: 10.1001/archneur.1991.00530180075020
58. Marcos B, García-Alloza M, Gil-Bea FJ, Chuang TT, Francis PT, Chen CP, et al. Involvement of an altered 5-HT-[6] receptor function in behavioral symptoms of Alzheimer's disease. J Alzheimers Dis. (2008) 14:43–50. doi: 10.3233/JAD-2008-14104
59. Widner H. Core Methodology. In:Kompoliti K, Verhagen L, , editors. Clinical Rating Scales In Encyclopedia of Movement Disorders. New York: Elsewier. (2010) p. 58–64.
60. Thomas AJ, Mahin-Babaei F, Saidi M, Lett D, Taylor JP, Walker L, et al. Improving the identification of dementia with Lewy bodies in the context of an Alzheimer's-type dementia. Alzheimers Res Ther. (2018) 10: 27.
61. Bougea A, Stefanis L, Paraskevas GP, Emmanouilidou E, Efthymiopoulou E, Vekrelis K, et al. Neuropsychiatric symptoms and α-synuclein profile of patients with Parkinson's disease dementia, dementia with Lewy bodies and Alzheimer's disease. J Neurol. (2018) 256:2295–301. doi: 10.1007/s00415-018-8992-7
62. Spies PE, Melis RJ, Sjögren MJ, Rikkert MG, Verbeek MM. CSF α-synuclein does not discriminate between dementia disorders. J Alzheimers Dis. (2009) 16:363–9. doi: 10.3233/JAD-2009-0955
63. Reesink FE, Lemstra AW, van Dijk KD, Berendse HW, van de Berg WD, Klein M, et al. CSF α-synuclein does not discriminate dementia with Lewy bodies from Alzheimer's disease. J Alzheimers Dis. (2010) 22:87–95. doi: 10.3233/JAD-2010-100186
64. Kapaki E, Paraskevas GP, Emmanouilidou E, Vekrellis K. The diagnostic value of CSF α-synuclein in the differential diagnosis of dementia with Lewy bodies vs. normal subjects and patients with Alzheimer's disease. PLoS ONE. (2013) 8:e81654. doi: 10.1371/journal.pone.0081654
65. Oeckl P, Metzger F, Nagl M, von Arnim CA, Halbgebauer S, Steinacker P, et al. Alpha, beta and gamma-synuclein quantification in CSF by multiple reaction monitoring reveals increased concentrations in Alzheimer's disease and Creutzfeldt-Jakob disease but no alterations in synucleinopathies. Mol Cell Proteomics. (2016) 15:3126–38. doi: 10.1074/mcp.M116.059915
66. Molinuevo JL, Ayton S, Batrla R, Bednar MM, Bittner T, Cummings J, et al. Current state of Alzheimer's fluid biomarkers. Acta Neuropathol. (2018) 136:821–53. doi: 10.1007/s00401-018-1932-x
67. Van Hulle C, Jonaitis EM, Betthauser TJ, Batrla R, Wild N, Kollmorgen G, et al. An examination of a novel multipanel of CSF biomarkers in the Alzheimer's disease clinical and pathological continuum. Alzheimers Dement. (2020) 17:431–45. doi: 10.1002/alz.12204
68. McGrowder DA, Miller F, Vaz K, Nwokocha C, Wilson-Clarke C, Anderson-Cross M, et al. Cerebrospinal fluid biomarkers of Alzheimer's disease: current evidence and future perspectives. Brain Sci. (2021) 11:215. doi: 10.3390/brainsci11020215
69. Beatino MF, de Luca C, Campese N, Belli E, Piccarducci R, Giampietri L, et al. α-synuclein as an emerging pathophysiological biomarker of Alzheimer's disease. Expert Rev Mol Diagn. (2022) 6:1–15. doi: 10.1080/14737159.2022.2068952
70. Barba L, Rumeileh SA, Bellomo G, Paoletti FP, Halbgebauer S, Oeckl P, et al. Cerebrospinal fluid β-synuclein as a synaptic biomarker for preclinical Alzheimer's disease. J Neurol Neurosurg Psychiatry. (2023) 94:83–86. doi: 10.1136/jnnp-2022-329124
71. Halbgebauer S, Rumeileh SA, Oeckl P, Steinacker P, Roselli F, Wiesner D, et al. Blood β-synuclein and neurofilament light chain during the course of prion disease. Neurology. (2022) 98:e1434–e1445. doi: 10.1212/WNL.0000000000200002
72. del Campo M, Mollenhauer B, Bertolotto A, Engelborghs S, Hampel H, Simonsen AH, et al. Recommendations to standardize preanalytical confounding factors in Alzheimer's and Parkinson's disease CSF biomarkers: an update. Biomark Med. (2012) 6:419–30. doi: 10.2217/bmm.12.46
73. Fairfoul G, McGuire LI, Pal S, Ironside JW, Neumann J, Christie S, et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol. (2016) 3:812–8. doi: 10.1002/acn3.338
74. Groveman BR, Orrù CD, Hughson AG, Raymond LD, Zanusso G, Ghetti B, et al. Rapid and ultra-sensitive quantification of disease-associated α-synuclein seeds in brain and CSF by αSyn RT-QuIC. Acta Neuropathol Commun. (2018) 6:7. doi: 10.1186/s40478-018-0508-2
75. Campese N, Beatino MF, Del Gamba C, Belli E, Giampietri L, Del Prete E, et al. Ultrasensitive techniques and protein misfolding amplification assays for biomarker-guided reconceptualization of Alzheimer's and other neurodegenerative diseases. Expert Rev Neurother. (2021) 21:949–67. doi: 10.1080/14737175.2021.1965879
Keywords: psychosis, Alzheimer, alpha-synuclein, CSF, biomarkers
Citation: Monge-García S, García-Ayllón M-S, Sánchez-Payá J, Gasparini-Berenguer R, Cortés-Gómez M-Á, Sáez-Valero J and Monge-Argilés J-A (2023) Validity of CSF alpha-synuclein to predict psychosis in prodromal Alzheimer's disease. Front. Neurol. 14:1124145. doi: 10.3389/fneur.2023.1124145
Received: 14 December 2022; Accepted: 18 April 2023;
Published: 24 May 2023.
Edited by:
Jian He, Shanghai Jiao Tong University, ChinaReviewed by:
Samir Abu Rumeileh, Martin Luther University of Halle-Wittenberg, GermanyCopyright © 2023 Monge-García, García-Ayllón, Sánchez-Payá, Gasparini-Berenguer, Cortés-Gómez, Sáez-Valero and Monge-Argilés. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José-Antonio Monge-Argilés, bW9uZ2Vfam9zQGd2YS5lcw==; María-Salud García-Ayllón, bXMuZ2FyY2lhQHVtaC5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.