 Yaqian Huang
Yaqian Huang Manli Lu1†
Manli Lu1† Yongjun Cao
Yongjun Cao Xia Zhang
Xia Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 09 March 2023
Sec. Stroke
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1118843
This article is part of the Research Topic Vascular Immunity and Ischemic Stroke View all 15 articles
As a specific lymphatic marker and a key ligand of C-type lectin-like receptor 2 (CLEC-2), podoplanin (Pdpn) is involved in various physiological and pathological processes such as growth and development, respiration, blood coagulation, lymphangiogenesis, angiogenesis, and inflammation. Thrombotic diseases constitute a major cause of disability and mortality in adults, in which thrombosis and inflammation play a crucial role. Recently, increasing evidence demonstrates the distribution and function of this glycoprotein in thrombotic diseases such as atherosclerosis, ischemic stroke, venous thrombosis, ischemic-reperfusion injury (IRI) of kidney and liver, and myocardial infarction. Evidence showed that after ischemia, Pdpn can be acquired over time by a heterogeneous cell population, which may not express Pdpn in normal conditions. In this review, the research progresses in understanding the roles and mechanisms of podoplanin in thromobotic diseases are summarized. The challenges of podoplanin-targeted approaches for disease prognosis and preventions are also discussed.
Podoplanin (Pdpn), named according to its expression in renal podocytes, is a type I transmembrane glycoprotein containing a large number of O-glycoside chains, which makes it a member of mucin-type proteins. Due to its expression in human and several mammal species in various cells and tissues, it has many different names. In human it is also called gp36 and T1α (1), however, in mice which is also known as Aggrus, OTS-8, gp38, and antigen PA2.26 (2–4). Pdpn is mainly involved in growth and development, respiration, blood coagulation, lymphangiogenesis, angiogenesis, and inflammation (5–7). Especially the interaction with its receptor C-type lectin-like receptor 2 (CLEC-2) has been shown to play an important role in thromboinflammation (8, 9). Pdpn expression is upregulated in both epithelial and mesenchymal cell compartments during thrombosis and inflammation, and a growing body of evidence indicates its prominence in these pathologies of thrombotic diseases.
Pdpn consists of a heavily O-glycosylated ectodomain, a hydrophobic membrane spanning domain, and a short cytoplasmic tail (CT) of only nine amino acids. Besides C-type lectin-like receptor 2 (CLEC-2), there are variable proteins interacting with Pdpn, such as CCL21, galectin-8, and heat-shock protein A9 (HSPA9) binding its ectodomain; CD9 and CD44 interacting with its transmenbrane domain; and ezrin, radixin, and moesin (ERM) binding to its CT. Through these interactions, Pdpn exerts various functions like platelet aggregation/activation, platelet biogenesis, immune surveillance, cytoskeleton rearrangement, and epithelial-mesenchymal transitions (EMTs) by protein–protein interactions for the lack of obvious enzymatic motifs (10–12) (Figure 1). Mostly, Pdpn is expressed on various cells, or at plasma membrane extensions, such as microvilli, filopodia, and ruffles, linking to the actin cytoskeleton to rearrange cytoskeleton and regulate cell motility. A fraction of Pdpn is localized in detergent-resistant membrane domains or raft platforms regulated by its CT and transmembrane domains, which appears to be necessary for Pdpn-mediated EMT and cell migration (13, 14). Besides, a soluble form of Pdpn (sPdpn) has recently been detected and investigated (15, 16). Cells ectopically or endogenously expressing Pdpn has been found to release extracellular vesicles (EVs) that contain Pdpn mRNA and protein. Pdpn incorporates into membrane shed microvesicles (MVs) and endosomal-derived exosomes (EXOs), and immunoelectron microscopy revealed its colocalization with the classical EV marker CD63 (15). Ovarian cancer cells express Pdpn themselves and also release Pdpn-rich EVs, both causing platelet aggregation, leading to venous thrombosis (16). Those Pdpn-EXO may contribute to sPdpn in circulating body fluid for Pdpn+ microparticles were detected in human body fluids including plasma and other liquids, which were quantitated using surface plasmon resonance, immunohistochemistry, and a double-antibody sandwich ELISA (17–20).
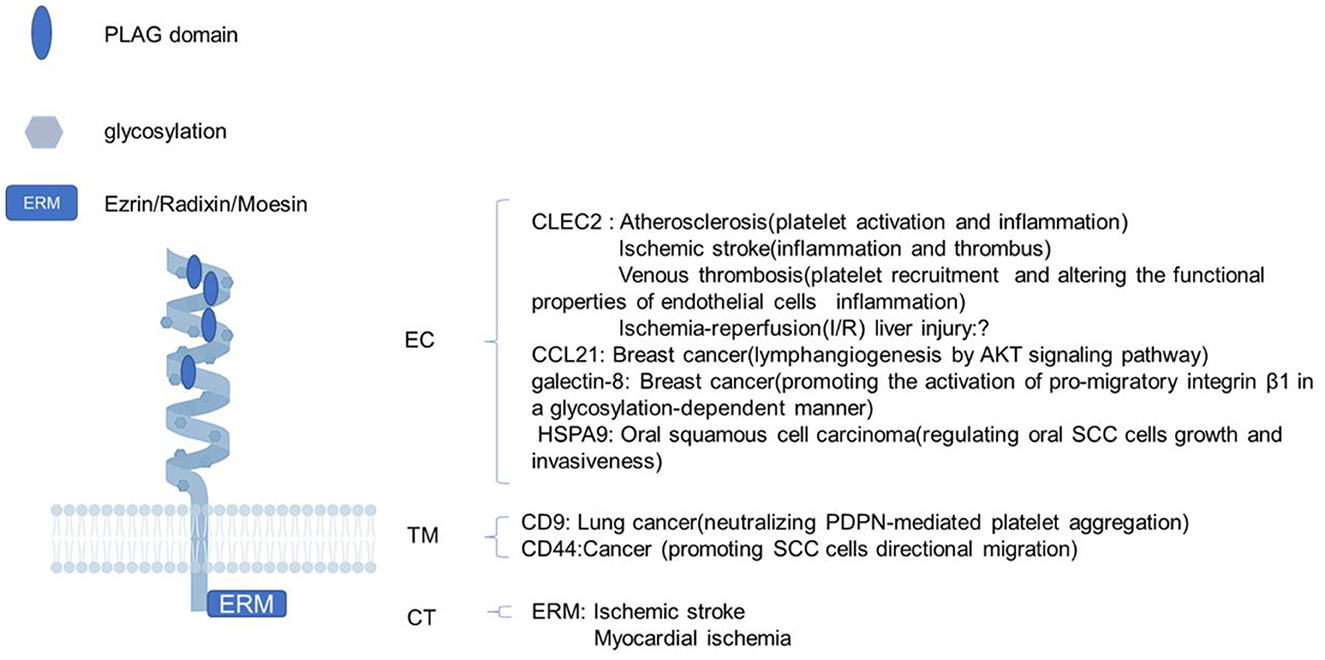
Figure 1. The structure of podoplanin and its functions with interacting proteins. Schematic representation of the molecular structure of podoplanin with a heavily glycosylated extracellular domain, a single transmembrane domain, and a short 9-amino acid cytoplasm. The ligands and biological processes during which the identified molecules interacting with podoplanin are presented. EC, ectodomain; TM, transmembrane region; CT, cytosolic domain; PLAG, platelet aggregation-stimulating.
The Pdpn research was originally started from the cloning of highly metastatic NL-17 subclone from mouse colon 26 cancer cell lines and the establishment of 8F11 monoclonal antibody (mAb) that could neutralize NL-17-induced platelet aggregation and hematogenous metastasis. Pdpn was identified as the antigen of 8F11 mAb, whose ectopic expression brought cells the platelet-aggregating abilities and hematogenous metastasis phenotypes. From the 8F11 mAb recognition epitopes, Pdpn is found to contain tandemly repeated, highly conserved motifs, designated platelet aggregation-stimulating (PLAG) domains, which are associated with the CLEC-2 binding (21). Pdpn was discovered for the first time in rat and mice lungs, and on the surface of stromal cells in lymph nodes (LNs) in mice, which has been found to be expressed in a wide variety of cells later, such as lymphatic endothelial cells, tumor cells, osteocytes, choroid plexus epithelial cells, glial cells, and cancer-associated fibroblasts for its pleiotropic functions (7, 22).
Among the many protein ligands of Pdpn, CLEC-2, and ERM proteins are studied comprehensively. CLEC-2 is a main receptor for Pdpn. The PLAG3 and PLAG4 domains of Pdpn are required for its binding to CLEC-2 (23, 24). The combination of CLEC-2 with the PLAG domains in the extracellular domain of Pdpn induces platelet activation and regulates inflammation through the Src, Syk, and SLP-76 kinase pathway (25, 26). Additionally, the interaction of Pdpn with CLEC-2 enhanced the interaction between Pdpn and ERM proteins and CD44, which activated Rho GTPase signaling pathway (27, 28). Both the interaction of Pdpn with CLEC-2 and with ERM are the two main pathways of cytoskeleton reorganization and inflammation regulation, which have been demonstrated to contribute to the occurrence and development of thrombotic diseases (29, 30). Studies show that Pdpn plays an important role in the functional regulation of immune cells. Following inflammatory or ischemic stimulation, Pdpn expression was upregulated in macrophages, microglia, and other immune cells, which influenced their motility and functionally phenotype transformation (31–33).
Atherosclerosis is usually considered as a chronic inflammatory disease, which is the main root cause of thrombotic diseases characterized by lipid deposition in parts of the artery accompanied by smooth muscle cell (SMC) and fibrous matrix proliferation. Unstable atherosclerotic plaque rupture and following thrombus formation, or vascular stenosis lead to arteriosclerotic cardiovascular disease (ASCVD) resulting in high rate of mortality in the population (34). Platelet activation and aggression has a well-established role in the development and manifestation of atherosclerosis (35–37). Both CLEC-2 and Pdpn have been shown to bind to atherosclerotic lesions. CLEC-2 co-localized with vascular SMCs, while Pdpn was localized to SMCs and macrophages (38). Besides, Pdpn expression in SMCs and macrophages increased with atherosclerotic progression. However, in a rat model similar to the plaque erosion in human which contains relatively few inflammatory cells and more SMCs compared with plaque rupture, Pdpn was found to be overexpressed in endothelial cells, not in SMCs. Further exploration showed that vascular endothelial growth factor (VEGF)-A, which is expressed in SMCs, macrophages, and endothelial cells in the advanced atherosclerotic lesions, induced Pdpn expression. Therefore, it is speculated that VEGF-A from superficial SMCs stimulates endothelial Pdpn expression, which interacts with CLEC-2 to induce platelet aggregation and thrombus formation (39). The results remind us that at different stages of atherosclerosis, Pdpn expression varies in different cells and plays different roles. This partly might be explained by the fact that inflammatory stimulation upregulated Pdpn expression in macrophages, and Pdpn was expressed on inflammatory but not tissue-resident macrophages (31). Toll-like receptor (TLR) stimulation and some inflammatory cytokines activates Pdpn expression. Additionally, in advanced atherosclerotic plaque, Pdpn was detected in a membranous or cytoplasmic staining pattern, suggesting Pdpn may contribute to atherosclerosis development in both CLEC-2-dependent and independent manners (38). Pdpn is expressed in stromal myofibroblasts, which contribute to cell migration and invasion, suggesting a role of Pdpn in vascular remodeling and atherosclerotic progression in atherosclerotic plaques. On the other hand, inflammatory cytokines in plaque progression promote Pdpn expression in stromal cells and endothelial cells. Besides, adventitial lymphatics in the arterial walls protect against atherosclerosis, which are important in reverse cholesterol transport from atherosclerotic lesions (40). Pdpn was specifically associated with lymphatic endothelium number of adventitial lymphatics of human internal carotid artery, which demonstrated Pdpn may participate in atherosclerosis via regulating functions and regeneration of adventitial lymphatic vessels in atherosclerotic lesions (41). In a disturbed blood flow (d-flow) model, monocyte Pdpn was upregulated by d-flow, and the myeloid-specific Pdpn deletion mitigated the subendothelial accumulation of platelets and monocytes/macrophages, which ameliorated vascular inflammation (42) (Table 1).

Table 1. Pdpn in thrombotic diseases.
Much evidence confirmed the role of Pdpn in the development and manifestation of atherosclerosis mainly through inflammation and lymphatic vessel functional regulation pathways. CLEC-2 is the important partner for the role of Pdpn in atherosclerosis, however, other receptors and signaling pathways need to be explored.
Ischemic stroke is one of the most common thrombotic diseases, caused by a blood clot occluding one or multiple cerebral arteries, which means rapid recanalization of the occluded blood vessel is necessary for the treatment of acute ischemic stroke (AIS). However, even recanalization is successful, symptoms can still aggravate. This is called ischemia/reperfusion (I/R) injury, in which thrombotic and inflammatory pathways play a crucial role. Thus, ischemic stroke is recognized as a thromboinflammation disease (85). The Pdpn/CLEC-2 axis is thought to be a major regulator of thrombo-inflammatory disorders (86, 87). Therefore, we previously conducted a prospective observational study, including 352 AIS patients and 112 healthy controls. The results showed that plasma CLEC-2 (pCLEC-2) levels were associated with stroke progression and poor prognosis at 90 days. During 1 year follow-up, pCLEC-2 levels were also predictive for higher incidence of death and vascular events (43, 44). Further we examined the mechanism of Pdpn/CLEC-2 axis in cerebral ischemia injury using a mouse middle cerebral artery occlusion (MCAO) model. In this study, the expression of CLEC-2 and Pdpn increased after ischemia/reperfusion (I/R) injury and anti-Pdpn antibody pretreatment reduced infarct volume and attenuated the neurological deficits with a significant decrease of IL-18 and IL-1β, indicating a possible role of the Pdpn/CLEC-2 axis in the regulation of inflammation in ischemic stroke via modulating NLRP3 inflammasome (45). An upregulated Pdpn expression in reactive astrocytes in the ischemic model was observed, which might be a part of compensatory response to ischemic brain injury. This implied a remarkable role of Pdpn in astrocytes in ischemic brain injury, and cellular interactions among astrocytes, neurons, and microglia await to be elucidated further (20). Qian et al. reported the molecular mechanism of Pdpn neutralization inhibiting I/R-induced microglial activation using transcriptome sequencing analysis and found numerous inflammation-related signaling pathways were regulated by the anti-Pdpn treatment (46). Some upper proteins such as TRPM7 kinase might downregulate CLEC-2 to protect mice from acute ischemic disease without developing intracranial hemorrhage, which could provide us some clues on the mechanism of Pdpn/CLEC-2 axis in ischemic stroke (88). Both vascular and neurovascular interaction mechanisms may be involved, awaiting to be elucidated. Moreover, the interaction of the CT of Pdpn with the ERM protein family activates Rho GTPases. RhoA/ROCK signaling pathway in astrocytes is suggested to be crucial in neurogenesis and angiogenesis after cerebral ischemia, which indicates the crosstalk among podoplanin, ERM protein family, and astrocytes in ischemic stroke needs to be further studied (Table 1).
Pdpn contributes to the cerebral ischemia injury mainly through thrombosis and inflammation pathways. Its expression is upregulated after brain ischemia in various kinds of cells, some of which may not express Pdpn in normal conditions. However, the exact cellular interactions, vascular and neurovascular interaction mechanisms, and molecular signaling pathways remains to be elucidated.
Deep vein thrombosis (DVT) is a type of blood clot within deep veins, which is one of the most common venous thromboembolic disorders with a high mortality. Its underlying mechanisms still remain unclear, however, recent evidence has demonstrated that immune cells and inflammatory processes are involved in DVT initiation besides blood coagulation disorder (89). DVT is rich in red cells and fibrin, the formation of which involves the interaction of von Willebrand factor (vWF), platelets, neutrophils, and mast cells (47–49). In a murine DVT model of inferior vena cava (IVC) stenosis, it has been demonstrated that general inducible deletion of CLEC-2 or platelet-specific deficiency in CLEC-2 are protected against DVT. Also, anti-Pdpn antibody treatment resulted in a reduction of thrombus formation (50). The mechanisms have been suspected that the interaction of CLEC-2 in platelets and overexpressed Pdpn in the IVC wall induced venous thrombus formation. Highly distorted flow caused by IVC stenosis and following hypoxia led to upregulated Pdpn expression (51). However, Pdpn upregulation cannot only be a cause for thrombosis but might also be triggered by thrombus formation, which indicates both mechanisms may operate in parallel forming a positive feedback (Table 1). A recent study has demonstrated a role of CLEC-2 in cerebral venous thrombosis (CVT), an unusual manifestation of venous thrombosis. The results showed antibody (INU1-fab)-induced cooperative signaling of CLEC-2 and GPIIb/IIIa triggered a CVT-like thrombotic syndrome in mice. The authors speculated that INU1-fab alters the conformation of CLEC-2 and facilitates its interaction with an unknown ligand enriched in cerebral veins (90). Thus, Pdpn, a main ligand of CLEC-2 for platelet activation, was thought to be a candidate partner, as it is obviously upregulated in different inflammatory tissues including the brain, and can be shed from the cell surface to circulate in plasma (20, 91). However, it needs to be further explored.
A crucial role of the interaction between CLEC-2 and Pdpn in venous thrombosis has been revealed. Upregulated Pdpn expression was observed in DVT. The exact cellular expression and molecular signaling pathway remains to be uncovered, especially for CVT. Also, whether there are interactions between Pdpn and other receptors in venous thrombosis needs to be explored.
Moreover, Pdpn-associated platelet activation has been demonstrated to contribute to cancer-associated thrombosis, which are based on the upregulation of Pdpn on the cell surface of brain tumor cells. CATS trial reported that Pdpn overexpression was strongly associated with the amount of intratumoral thrombotic vessels and increased VTE risk in cancer patients. Platelet counts were lower and plasma D-dimer levels were higher in those with Pdpn-expressing brain tumors (52). Increased Pdpn expression in glioma cells coincides with the development of venous thrombo-embolism, which is correlated with laboratory evidence of coagulation activation by elevated D-dimer levels (54). CLEC-2-Pdpn interaction has been suggested to stimulate cancer-associated thrombosis in which thromboinflammation plays a crucial role. One hand, thromboinflammation induces ectopic podoplanin expression in vascular endothelial cells or macrophages; on the other hand, CLEC-2 depletion reduces levels of plasma inflammatory cytokines (55). Anti-Pdpn antibody treatment inhibited platelet activation in vitro and decreased the incidence of VTE in mice (56). In oral squamous cell carcinoma and ovarian cancer, the same results have been reported (16, 57). Hypermethylation of CpG islands in the Pdpn promoter was regulated by mutant isocitrate dehydrogenase (IDH) in glioma, which resulted in decreased Pdpn expression (58). Indeed, combination of IDH1 mutation and Pdpn expression in brain tumors can help identify patients at high risk of VTE (53, 59). Further exploration found Pdpn was released with exosome-like EVs shed from cells (60). Additionally, in a mouse model of systemic Salmonella Typhimurium infection, Pdpn was upregulated in monocyets and Kupffer cells (KCs) and its combination with CLEC-2 promoted the formation of infection-driven thrombosis in the liver (61, 92) (Table 1). Different forms of Pdpn participate in the formation of cancer-associated thrombosis, to which pdpn-mediated thrombosis, inflammation, and intratumoral vessel generation contributes. Besides CLEC-2, there may be other partners interacting with Pdpn in cancer-associated thrombosis.
Ischemia-reperfusion injury (IRI) is one of the most common causes of acute kidney injury (AKI), a serious and often deadly condition. Kidney IRI accounts for almost 50% of AKI cases, which is mediated by free radicals and reactive oxygen species (ROS) after periods of disrupted blood flow (68). Pdpn was named according to its expression in podocytes, mainly along their urinary surfaces, indicating a potentially functional role of Pdpn in kidney IRI (62). In a mouse model of kidney IRI, decreased Pdpn expression in the glomerulus and increased expression in the tubulointerstitial compartment of the kidney shortly after IRI was demonstrated. And the intensity of Pdpn in the tubulointerstitial compartment increased with the severity of ischemia, and the distribution of its expression changed over time (68). Moreover, an increase in the urine Pdpn-to-creatinine ratio was found to correlate with the onset of renal IRI. The researchers speculated that Pdpn was shed from the podocytes in an extracellular-vesicle form and expelled into the urine, which might be internalizated by the proximal tubule epithelium. Another hypothesis was spindle-shaped cells expressing Pdpn in the interstitium of the medulla might migrate to kidney from another organ, playing an important role in neovascularization during processes of kidney IRI. However, the exact mechanisms need to be further explored. Pdpn expression was significantly decreased in the renal glomerulus of diabetic kidney disease mice with an underlying chronic renal ischemia (70).
During the process, the activation of NF-κB signaling pathway in podocytes downregulated the expression of Pdpn, leading to increased podocyte apoptosis. Moreover, rapamycin, a kind of mTOR inhibitor, had a controversial role in the treatment of acute ischemic kidney injury. Some studies indicated a damage-promoting role of rapamycin during kidney IR injury (64, 65), while some reported a protective role of rapamycin against kidney IR injury (63, 65, 66). However, there was evidence on correlation between phosphorylated mTOR expression and Pdpn expression in esophageal squamous cell carcinoma and traumatic brain injury, which indicated Pdpn might participated in kidney IRI via mTOR pathway, awaiting to be explored (67, 71). Immune responses are involved in the pathophysiology of ischemic acute kidney injury (AKI) (93). In some immune diseases of kidney such as rescentic glomerulonephritis (GN), membrane Pdpn on fibroblastic reticular cells (FRCs) may play an important role in the pathogenesis. The effect of treatment with anti-Pdpn antibody was similar to that of FRC depletion by decreasing T-cell activation in the lymph node (LN), resulting in reduction of kidney injury (69). Fibroblastic reticular cells also maintain the integrity of high endothelial venules (HEVs) through interactions between Pdpn on the FRCs and CLEC-2 on platelets (9). Anti-Pdpn treatment led to disorganization of laminin fibers in the kidney LN, which was associated with remarkably reduced expansion of the lymphatic vasculature (69). Therefore, it is hypothesized that Pdpn on FRCs may contribute to ischemic kidney injury by immune regulation, which may be the future research contents (Table 1).
The role of Pdpn in kidney IRI remains unclear. Its mechanisms are complex. Both sPdpn and cellular form participate in the pathogenesis, in which NF-κB and mTOR signaling pathways have been implicated. Moreover, the role of Pdpn on FRCs in activation of T-cells and maintenance of the integrity of HEVs in kidney IRI needs to be explored.
Myocardial ischemia (MI) is the commonest cardiovascular disease and one of the major causes of morbidity and mortality worldwide, in the pathogenesis of which inflammation and following heart tissue evolution play an important role. In the process, the growth and expansion of cardiac lymphatic vasculature in response to MI, is crucial for the transportation of extravasated proteins and lipids, inflammatory, and immune responses, as well as fluid balance (75, 76, 79). Therefore, Pdpn as a specific lymphatic marker, is thought to be vital in the cardiac development as well as the pathogenesis of MI. The function of Pdpn is crucial for epicardiac development and myocardial differentiation and its knockout shows a hypoplastic myocardium, atrioventricular valve abnormalities, and coronary artery abnormalities, which is partly correlated with reduced epithelial-mesenchymal transformation (EMT) caused by down-regulation of Pdpn (72, 74). Moreover, Pdpn deficiency results in hypoplastic sinus venosus myocardium including the sinoatrial node, which is also related to abnormal EMT due to up-regulated E-cadherin and down-regulated RhoA controlled by Pdpn (73). In the adult heart, Pdpn-positive cells only constitute <5% of the myocardial small cell population, which is only expressed by cardiac lymphatic endothelial cells in homeostatic conditions (94). However, after myocardial infarction (MI), Pdpn is upregulated in a heterogeneous cell population such as PDGFRα-, PDGFRβ-, and CD34-positive cells, besides lymphatic endothelial cells. Therefore, researchers thought Pdpn might be a sign of activation of a cohort of progenitor cells in different phases of post-ischemic myocardial wound repair. Inhibition of Pdpn by Pdpn-neutralizing antibodies reduces inflammation post-MI without full suppression leading to heart function and scar composition improvement. The increase of Pdpn-positive cells last from the acute (2 days) to the chronic phase of MI (2 weeks to 1 month) (82), which indicates a vital role of Pdpn in inflammation and wound repair after MI. Cimini et al. identified Pdpn as a potential cellular mediator of the lymphangiogenic and fibrogenic responses during different stages of myocardial wound repair after infarction (82). After injury, Pdpn is co-expressed by four kinds of cells such as PDGFRα-, PDGFRβ-, CD34-positive cells, and lymphatic endothelial cells which are responsible for regeneration, fibrosis, and inflammatory processes of the same pathologies. In the process, inflammation was thought to contribute to the recruitment of Pdpn-bearing LYVE-1-negative cells to the site of myocardial repair or the activation of Pdpn expression in responsive cell cohorts, which started the myocardial wound repair after infarction. At different stages of MI, Pdpn is expressed on various kinds of cells, for example, PDGFRα-positive cells during the whole process and PDGFRβ and CD34-positive cells at later stages of infarct healing in the mature scar. This means Pdpn plays multiple roles in the pathogenesis of MI. Cimini et al. reported the inhibition of the interaction between Pdpn and CLEC-2 expressing immune cells in the heart improved the cardiac performance, regeneration, and angiogenesis. In the model, Pdpn neutralizing antibody treatment induced recruitment of anti-inflammatory monocytes/macrophages and increased expression of anti-inflammatory cytokines (95).
Pericytes with PDGFR-β is very connected with Pdpn expression and transplantation of allogenic pericytes improves myocardial vascularization after MI resulting from the regulation of the endothelium in angiogenesis (81, 96). While mesenchymal stem cells (MSCs) expressing PDGFRα in the heart showed cardiomyocyte, endothelial, and smooth muscle lineage potential (77). In vitro differentiation of cardiac PDGFRα-positive cells brings out a lot of SMCs and endothelial cells only, indicating a predominant role of Pdpn in cardiac MSCs PDGFRα-positive cells in the vascular and mesenchymal compartments. CD34+telocytes expressed Pdpn after 15 days of MI, which supports cardiac growth, regeneration, renovation of connective tissue, and repair due to the unique communication with cardiac stem and progenitor cells (78, 80). Moreover, Pdpn expression significantly enhanced the migration of mesenchymal stromal cells (MSCs) and Pdpn-expressing MSCs extended processes into the endothelial cell layer, which could interact with circulating platelets (83) (Table 1).
In conclusion, cardiac ischemic injury induces upregulated and ectopic expression of Pdpn. The interaction of Pdpn and CLEC-2 or ERM proteins may participate in post-MI inflammatory response and cardiac repair through inflammation regulation, cytoskeleton reorganization, and lymphangiogenic and fibrogenic responses. The exact mechanisms remain unclear. And the interaction of Pdpn with other partners in cardiac ischemic injury needs to be further explored.
Hepatic I/R injury is usually associated with surgical procedures, trauma, liver transplantation, or resection as a consequence of interrupted blood supply to the liver, which leads to liver dysfunction and failure, as well as multiple organ failure (97, 98). Kupffer cells (KCs) and platelets were reported as two main roles in the procedure (99–101). Nakata et al. revealed Pdpn expression in the cytosol of hepatocytes in the post-ischemic liver and KC depletion weakened the Pdpn expression, which suggested that activated KCs regulate the expression of Pdpn in hepatocytes after I/R without clear mechanisms (84). Moreover, the authors demonstrated in the acute phase of hepatic I/R injury, the binding of CLEC-2 on the cell surface of platelets to Pdpn in hepatocytes activated platelets in the hepatic sinusoid (84). Therefore, the crosstalk among podoplanin, KCs, and platelets in hepatic I/R injury needs to be further studied (Table 1).
Pdpn, as an important glyprotein, has multiple interacting proteins in various tissues and organs, demonstrating its pleiotropic functions, especially a role in thrombosis and inflammation. Thrombosis and inflammation contribute to the pathogenesis of thrombotic diseases, such as atherosclerosis, ischemic stroke, venous thrombosis, acute kidney and liver ischemic injury, and myocardial ischemia. Evidence showed that after ischemia, Pdpn can be acquired over time by a heterogeneous cell population such as SMCs, endothelial cells, astrocytes, pericytes, MSCs, telocytes, and so on, which may not express Pdpn in normal conditions. However, the exact mechanisms of Pdpn in such ischemic diseases have not clearly been demonstrated. Pdpn in different cells plays different roles such as thrombosis, inflammation, vascularization, lymphagiogenesis, growth, and regeneration. However, many issues remain to be elucidated further; for instance, cell/stage-specific effects of Pdpn and according molecular mechanisms, and the relevance of anti-Pdpn treatment on ischemic diseases, especially ischemic stroke, venous thrombosis, and myocardial ischemia. The solutions to these issues can provide a new target of treating thrombotic diseases from bench to clinical translation.
YH: writing-original draft, visualization, and data curation. ML and YW: data curation, visualization, and resources. CZ: validation, investigation, and resources. YC: writing—review, editing, and funding acquisition. XZ: conceptualization, methodology, project administration, writing—review, editing, funding acquisition, and supervision. All authors have carefully read and confirmed the final manuscript. All authors contributed to the article and approved the submitted version.
The study was supported by the opening project of Jiangsu Key Laboratory of Neurological and Psychiatric Diseases (KJS2030), Key Talent Project of Nuclear Technology Medical Application of the Second Affiliated Hospital of Soochow University (XKTJ-HRC2021006), the China National Natural Fund Project (No. 82171296), and Discipline Construction Program of the Second Affiliated Hospital of Soochow University (XKTJ-TD202004).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Zimmer G, Oeffner F, Von Messling V, Tschernig T, Gröness HJ, Klenk HD, et al. Cloning and characterization of gp36, a human mucin-type glycoprotein preferentially expressed in vascular endothelium. Biochem J. (1999) 341(Pt 2):277–84. doi: 10.1042/bj3410277
2. Toyoshima M, Nakajima M, Yamori T, Tsuruo T. Purification and characterization of the platelet-aggregating sialoglycoprotein gp44 expressed by highly metastatic variant cells of mouse colon adenocarcinoma 26. Cancer Res. (1995) 55:767–73.
3. Nose K, Saito H, Kuroki T. Isolation of a gene sequence induced later by tumor-promoting 12-O-tetradecanoylphorbol-13-acetate in mouse osteoblastic cells (MC3T3-E1) and expressed constitutively in ras-transformed cells. Cell Growth Differ. (1990) 1:511–8.
4. Gandarillas A, Scholl FG, Benito N, Gamallo C, Quintanilla M. Induction of PA2.26, a cell-surface antigen expressed by active fibroblasts, in mouse epidermal keratinocytes during carcinogenesis. Mol Carcinog. (1997) 20:10–8. doi: 10.1002/(sici)1098-2744(199709)20:1<10::aid-mc3>3.0.co;2-m
5. Lee JY, Park C, Cho YP, Lee E, Kim H, Kim P, et al. Podoplanin-expressing cells derived from bone marrow play a crucial role in postnatal lymphatic neovascularization. Circulation. (2010) 122:1413–25. doi: 10.1161/CIRCULATIONAHA.110.941468
6. Tsukiji N, Inoue O, Morimoto M, Tatsumi N, Nagatomo H, Ueta K, et al. Platelets play an essential role in murine lung development through Clec-2/podoplanin interaction. Blood. (2018) 132:1167–79. doi: 10.1182/blood-2017-12-823369
7. Quintanilla M, Montero-Montero L, Renart J, Martín-Villar E. Podoplanin in inflammation and cancer. Int J Mol Sci. (2019) 20:707. doi: 10.3390/ijms20030707
8. Boulaftali Y, Hess PR, Getz TM, Cholka A, Stolla M, Mackman N, et al. Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest. (2013) 123:908–16. doi: 10.1172/JCI65154
9. Herzog BH, Fu J, Wilson SJ, Hess PR, Sen A, McDaniel JM, et al. Podoplanin maintains high endothelial venule integrity by interacting with platelet CLEC-2. Nature. (2013) 502:105–9. doi: 10.1038/nature12501
10. Kaneko MK, Kato Y, Kameyama A, Ito H, Kuno A, Hirabayashi J, et al. Functional glycosylation of human podoplanin: glycan structure of platelet aggregation-inducing factor. FEBS Lett. (2007) 581:331–6. doi: 10.1016/j.febslet.2006.12.044
11. Martín-Villar E, Scholl FG, Gamallo C, Yurrita MM, Muñoz-Guerra M, Cruces J, et al. Characterization of human PA226 antigen (T1alpha-2, podoplanin), a small membrane mucin induced in oral squamous cell carcinomas. Int J Cancer. (2005) 113:899–910. doi: 10.1002/ijc.20656
12. Scholl FG, Gamallo C, Vilaró S, Quintanilla M. Identification of PA226 antigen as a novel cell-surface mucin-type glycoprotein that induces plasma membrane extensions and increased motility in keratinocytes. J Cell Sci. (1999) 112:4601–13. doi: 10.1242/jcs.112.24.4601
13. Barth K, Bläsche R, Kasper M. T1alpha/podoplanin shows raft-associated distribution in mouse lung alveolar epithelial E10 cells. Cell Physiol Biochem. (2010) 25:103–12. doi: 10.1159/000272065
14. Fernández-Muñoz B, Yurrita MM, Martín-Villar E, Carrasco-Ramírez P, Megías D, Renart J, et al. The transmembrane domain of podoplanin is required for its association with lipid rafts and the induction of epithelial-mesenchymal transition. Int J Biochem Cell Biol. (2011) 43:886–96. doi: 10.1016/j.biocel.2011.02.010
15. Carrasco-Ramírez P, Greening DW, Andrés G, Gopal SK, Martín-Villar E, Renart J, et al. Podoplanin is a component of extracellular vesicles that reprograms cell-derived exosomal proteins and modulates lymphatic vessel formation. Oncotarget. (2016) 7:16070–89. doi: 10.18632/oncotarget.7445
16. Sasano T, Gonzalez-Delgado R, Muñoz NM, Carlos-Alcade W, Cho MS, Sheth RA, et al. Podoplanin promotes tumor growth, platelet aggregation, and venous thrombosis in murine models of ovarian cancer. J Thromb Haemost. (2022) 20:104–14. doi: 10.1111/jth.15544
17. Roca E, Lacroix R, Judicone C, Laroumagne S, Robert S, Cointe S, et al. Detection of EpCAM-positive microparticles in pleural fluid: a new approach to mini-invasively identify patients with malignant pleural effusions. Oncotarget. (2016) 7:3357–66. doi: 10.18632/oncotarget.6581
18. Sankiewicz A, Guszcz T, Mena-Hortelano R, Zukowski K, Gorodkiewicz E. Podoplanin serum and urine concentration in transitional bladder cancer. Cancer Biomark. (2016) 16:343–50. doi: 10.3233/CBM-160572
19. Bai B, Ma W, Wang K, Ha S, Wang JB, Tan BX, et al. Detection of D2-40 monoclonal antibody-labeled lymphatic vessel invasion in esophageal squamous cell carcinoma and its clinicopathologic significance. Cancer Biol Med. (2013) 10:81–5. doi: 10.7497/j.issn.2095-3941.2013.02.003
20. Zhao X, Pan Y, Ren W, Shen F, Xu M, Yu M, et al. Plasma soluble podoplanin is a novel marker for the diagnosis of tumor occurrence and metastasis. Cancer Sci. (2018) 109:403–11. doi: 10.1111/cas.13475
21. Takemoto A, Miyata K, Fujita N. Platelet-activating factor podoplanin: from discovery to drug development. Cancer Metastasis Rev. (2017) 36:225–34. doi: 10.1007/s10555-017-9672-2
22. Renart J, Carrasco-Ramírez P, Fernández-Muñoz B, Martín-Villar E, Montero L, Yurrita MM, et al. New insights into the role of podoplanin in epithelial-mesenchymal transition. Int Rev Cell Mol Biol. (2015) 317:185–239. doi: 10.1016/bs.ircmb.2015.01.009
23. Kato Y, Kaneko MK, Kunita A, Ito H, Kameyama A, Ogasawara S, et al. Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Sci. (2008) 99:54–61. doi: 10.1111/j.1349-7006.2007.00634.x
24. Nagae M, Morita-Matsumoto K, Kato M, Kaneko MK, Kato Y, Yamaguchi Y, et al. A platform of C-type lectin-like receptor CLEC-2 for binding O-glycosylated podoplanin and nonglycosylated rhodocytin. Structure. (2014) 22:1711–21. doi: 10.1016/j.str.2014.09.009
25. Suzuki-Inoue K, Fuller GL, García A, Eble JA, Pöhlmann S, Inoue O, et al. A novel Syk-dependent mechanism of platelet activation by the C-type lectin receptor CLEC-2. Blood. (2006) 107:542–9. doi: 10.1182/blood-2005-05-1994
26. Suzuki-Inoue K, Inoue O, Ozaki Y. Novel platelet activation receptor CLEC-2: from discovery to prospects. J Thromb Haemost. (2011) 9(Suppl 1):44–55. doi: 10.1111/j.1538-7836.2011.04335.x
27. Bourne JH, Beristain-Covarrubias N, Zuidscherwoude M, Campos J, Di Y, Garlick E, et al. CLEC-2 prevents accumulation and retention of inflammatory macrophages during murine peritonitis. Front Immunol. (2021) 12:693974. doi: 10.3389/fimmu.2021.693974
28. Martín-Villar E, Megías D, Castel S, Yurrita MM, Vilaró S, Quintanilla M. Podoplanin binds ERM proteins to activate RhoA and promote epithelial-mesenchymal transition. J Cell Sci. (2006) 119(Pt 21):4541–53. doi: 10.1242/jcs.03218
29. Zhang Z, Zhang N, Yu J, Xu W, Gao J, Lv X, et al. The role of podoplanin in the immune system and inflammation. J Inflamm Res. (2022) 15:3561–72. doi: 10.2147/JIR.S366620
30. Cheok YY, Tan GMY, Fernandez KC, Chan YT, Lee CYQ, Cheong HC, et al. Podoplanin drives motility of active macrophage via regulating filamin C during Helicobacter pylori infection. Front Immunol. (2021) 12:702156. doi: 10.3389/fimmu.2021.702156
31. Kerrigan AM, Navarro-Nuñez L, Pyz E, Finney BA, Willment JA, Watson SP, et al. Podoplanin-expressing inflammatory macrophages activate murine platelets via CLEC-2. J Thromb Haemost. (2012) 10:484–6. doi: 10.1111/j.1538-7836.2011.04614.x
32. Fei M, Wang H, Zhou M, Deng C, Zhang L, Han Y. Podoplanin influences the inflammatory phenotypes and mobility of microglia in traumatic brain injury. Biochem Biophys Res Commun. (2020) 523:361–7. doi: 10.1016/j.bbrc.2019.12.003
33. Nylander AN, Ponath GD, Axisa PP, Mubarak M, Tomayko M, Kuchroo VK, et al. Podoplanin is a negative regulator of Th17 inflammation. JCI Insight. (2017) 2:e92321. doi: 10.1172/jci.insight.92321
34. Torres N, Guevara-Cruz M, Velázquez-Villegas LA, Tovar AR. Nutrition and atherosclerosis. Arch Med Res. (2015) 46:408–26. doi: 10.1016/j.arcmed.2015.05.010
35. Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. (2007) 357:2482–94. doi: 10.1056/NEJMra071014
36. Marx C, Novotny J, Salbeck D, Zellner KR, Nicolai L, Pekayvaz K, et al. Eosinophil-platelet interactions promote atherosclerosis and stabilize thrombosis with eosinophil extracellular traps. Blood. (2019) 134:1859–72. doi: 10.1182/blood.2019000518
37. Wang N, Tall AR. Cholesterol in platelet biogenesis and activation. Blood. (2016) 127:1949–53. doi: 10.1182/blood-2016-01-631259
38. Inoue O, Hokamura K, Shirai T, Osada M, Tsukiji N, Hatakeyama K, et al. Vascular smooth muscle cells stimulate platelets and facilitate thrombus formation through platelet CLEC-2: implications in atherothrombosis. PLoS ONE. (2015) 10:e0139357. doi: 10.1371/journal.pone.0139357
39. Furukoji E, Yamashita A, Nakamura K, Hirai T, Asada Y. Podoplanin expression on endothelial cells promotes superficial erosive injury and thrombus formation in rat carotid artery: implications for plaque erosion. Thromb Res. (2019) 183:76–9. doi: 10.1016/j.thromres.2019.10.015
40. Kutkut I, Meens MJ, McKee TA, Bochaton-Piallat ML, Kwak BR. Lymphatic vessels: an emerging actor in atherosclerotic plaque development. Eur J Clin Invest. (2015) 45:100–8. doi: 10.1111/eci.12372
41. Drozdz K, Janczak D, Dziegiel P, Podhorska M, Patrzałek D, Ziółkowski P, et al. Adventitial lymphatics of internal carotid artery in healthy and atherosclerotic vessels. Folia Histochem Cytobiol. (2008) 46:433–6. doi: 10.2478/v10042-008-0083-7
42. Tang C, Wang L, Sheng Y, Zheng Z, Xie Z, Wu F, et al. CLEC-2-dependent platelet subendothelial accumulation by flow disturbance contributes to atherogenesis in mice. Theranostics. (2021) 11:9791–804. doi: 10.7150/thno.64601
43. Zhang X, Zhang W, Wu X, Li H, Zhang C, Huang Z, et al. Prognostic significance of plasma CLEC-2 (C-Type Lectin-Like Receptor 2) in patients with acute ischemic stroke. Stroke. 2018:STROKEAHA118022563. doi: 10.1161/STROKEAHA.118.022563
44. Wu X, Zhang W, Li H, You S, Shi J, Zhang C, et al. Plasma C-type lectin-like receptor 2 as a predictor of death and vascular events in patients with acute ischemic stroke. Eur J Neurol. (2019) 26:1334–40. doi: 10.1111/ene.13984
45. Meng D, Ma X, Li H, Wu X, Cao Y, Miao Z, et al. A role of the podoplanin-CLEC-2 axis in promoting inflammatory response after ischemic stroke in mice. Neurotox Res. (2021) 39:477–88. doi: 10.1007/s12640-020-00295-w
46. Qian S, Qian L, Yang Y, Cui J, Zhao Y. Transcriptome sequencing analysis revealed the molecular mechanism of podoplanin neutralization inhibiting ischemia/reperfusion-induced microglial activation. Ann Transl Med. (2022) 10:638. doi: 10.21037/atm-22-1952
47. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
48. Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Köllnberger M, et al. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. (2011) 117:1400–7. doi: 10.1182/blood-2010-05-287623
49. Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. (2012) 10:136–44. doi: 10.1111/j.1538-7836.2011.04544.x
50. Payne H, Ponomaryov T, Watson SP, Brill A. Mice with a deficiency in CLEC-2 are protected against deep vein thrombosis. Blood. (2017) 129:2013–20. doi: 10.1182/blood-2016-09-742999
51. Kolenda J, Jensen SS, Aaberg-Jessen C, Christensen K, Andersen C, Brünner N, et al. Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma-derived spheroids. J Neurooncol. (2011) 103:43–58. doi: 10.1007/s11060-010-0357-8
52. Mir Seyed Nazari P, Riedl J, Pabinger I, Ay C. The role of podoplanin in cancer-associated thrombosis. Thromb Res. (2018) 164(Suppl 1):S34–9. doi: 10.1016/j.thromres.2018.01.020
53. Mir Seyed Nazari P, Riedl J, Preusser M, Posch F, Thaler J, Marosi C, et al. Combination of isocitrate dehydrogenase 1 (IDH1) mutation and podoplanin expression in brain tumors identifies patients at high or low risk of venous thromboembolism. J Thromb Haemost. (2018) 16:1121–7. doi: 10.1111/jth.14129
54. Riedl J, Preusser M, Nazari PM, Posch F, Panzer S, Marosi C, et al. Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood. (2017) 129:1831–9. doi: 10.1182/blood-2016-06-720714
55. Suzuki-Inoue K. Platelets and cancer-associated thrombosis: focusing on the platelet activation receptor CLEC-2 and podoplanin. Blood. (2019) 134:1912–8. doi: 10.1182/blood.2019001388
56. Wang X, Liu B, Xu M, Jiang Y, Zhou J, Yang J, et al. Blocking podoplanin inhibits platelet activation and decreases cancer-associated venous thrombosis. Thromb Res. (2021) 200:72–80. doi: 10.1016/j.thromres.2021.01.008
57. Lee HY, Yu NY, Lee SH, Tsai HJ, Wu CC, Cheng JC, et al. Podoplanin promotes cancer-associated thrombosis and contributes to the unfavorable overall survival in an ectopic xenograft mouse model of oral cancer. Biomed J. (2020) 43:146–62. doi: 10.1016/j.bj.2019.07.001
58. Sun C, Xiao L, Zhao Y, Shi J, Yuan Y, Gu Y, et al. Wild-type IDH1 and mutant IDH1 opposingly regulate podoplanin expression in glioma. Transl Oncol. (2020) 13:100758. doi: 10.1016/j.tranon.2020.100758
59. Watanabe J, Natsumeda M, Okada M, Kanemaru Y, Tsukamoto Y, Oishi M, et al. Podoplanin expression and IDH-wildtype status predict venous thromboembolism in patients with high-grade gliomas in the early postoperative period. World Neurosurg. (2019) 128:e982–8. doi: 10.1016/j.wneu.2019.05.049
60. Tawil N, Bassawon R, Meehan B, Nehme A, Montermini L, Gayden T, et al. Glioblastoma cell populations with distinct oncogenic programs release podoplanin as procoagulant extracellular vesicles. Blood Adv. (2021) 5:1682–94. doi: 10.1182/bloodadvances.2020002998
61. Zwicker JI. Risking thromboembolism: podoplanin and glioma. Blood. (2017) 129:1742–3. doi: 10.1182/blood-2017-02-763524
62. Breiteneder-Geleff S, Matsui K, Soleiman A, Meraner P, Poczewski H, Kalt R, et al. Podoplanin, novel 43-kd membrane protein of glomerular epithelial cells, is down-regulated in puromycin nephrosis. Am J Pathol. (1997) 151:1141–52.
63. Weichhart T, Säemann MD. The multiple facets of mTOR in immunity. Trends Immunol. (2009) 30:218–26. doi: 10.1016/j.it.2009.02.002
64. Kezic A, Becker JU, Thaiss F. The effect of mTOR-inhibition on NF-κB activity in kidney ischemia-reperfusion injury in mice. Transplant Proc. (2013) 45:1708–14. doi: 10.1016/j.transproceed.2013.02.110
65. Kezic A, Thaiss F, Becker JU, Tsui TY, Bajcetic M. Effects of everolimus on oxidative stress in kidney model of ischemia/reperfusion injury. Am J Nephrol. (2013) 37:291–301. doi: 10.1159/000348496
66. Zhang C, Zheng L, Li L, Wang L, Li L, Huang S, et al. Rapamycin protects kidney against ischemia reperfusion injury through recruitment of NKT cells. J Transl Med. (2014) 12:224. doi: 10.1186/s12967-014-0224-z
67. Chuang WY, Chang YS, Chao YK, Yeh CJ, Ueng SH, Chang CY, et al. Phosphorylated mTOR expression correlates with podoplanin expression and high tumor grade in esophageal squamous cell carcinoma. Int J Clin Exp Pathol. (2015) 8:12757–65.
68. Kasinath V, Yilmam OA, Uehara M, Yonar M, Jiang L, Li X, et al. Urine podoplanin heralds the onset of ischemia-reperfusion injury of the kidney. Am J Physiol Renal Physiol. (2019) 316:F957–65. doi: 10.1152/ajprenal.00538.2018
69. Kasinath V, Yilmam OA, Uehara M, Jiang L, Ordikhani F, Li X, et al. Activation of fibroblastic reticular cells in kidney lymph node during crescentic glomerulonephritis. Kidney Int. (2019) 95:310–20. doi: 10.1016/j.kint.2018.08.040
70. Yu J, Zong GN, Wu H, Zhang KQ. Podoplanin mediates the renoprotective effect of berberine on diabetic kidney disease in mice. Acta Pharmacol Sin. (2019) 40:1544–54. doi: 10.1038/s41401-019-0263-3
71. Gao C, Wang H, Wang T, Luo C, Wang Z, Zhang M, et al. Platelet regulates neuroinflammation and restores blood-brain barrier integrity in a mouse model of traumatic brain injury. J Neurochem. (2020) 154:190–204. doi: 10.1111/jnc.14983
72. Mahtab EA, Wijffels MC, Van Den Akker NM, Hahurij ND, Lie-Venema H, Wisse LJ, et al. Cardiac malformations and myocardial abnormalities in podoplanin knockout mouse embryos: correlation with abnormal epicardial development. Dev Dyn. (2008) 237:847–57. doi: 10.1002/dvdy.21463
73. Mahtab EA, Vicente-Steijn R, Hahurij ND, Jongbloed MR, Wisse LJ, DeRuiter MC, et al. Podoplanin deficient mice show a RhoA-related hypoplasia of the sinus venosus myocardium including the sinoatrial node. Dev Dyn. (2009) 238:183–93. doi: 10.1002/dvdy.21819
74. Douglas YL, Mahtab EA, Jongbloed MR, Uhrin P, Zaujec J, Binder BR, et al. Pulmonary vein, dorsal atrial wall and atrial septum abnormalities in podoplanin knockout mice with disturbed posterior heart field contribution. Pediatr Res. (2009) 65:27–32. doi: 10.1203/PDR.0b013e31818bc11a
75. Cui Y. Impact of lymphatic vessels on the heart. Thorac Cardiovasc Surg. (2010) 58:1–7. doi: 10.1055/s-0029-1240553
76. Loukas M, Abel N, Tubbs RS, Grabska J, Birungi J, Anderson RH. The cardiac lymphatic system. Clin Anat. (2011) 24:684–91. doi: 10.1002/ca.21104
77. Noseda M, Harada M, McSweeney S, Leja T, Belian E, Stuckey DJ, et al. PDGFRα demarcates the cardiogenic clonogenic Sca1+ stem/progenitor cell in adult murine myocardium. Nat Commun. (2015) 6:6930. doi: 10.1038/ncomms7930
78. Popescu LM, Curici A, Wang E, Zhang H, Hu S, Gherghiceanu M. Telocytes and putative stem cells in ageing human heart. J Cell Mol Med. (2015) 19:31–45. doi: 10.1111/jcmm.12509
79. Aspelund A, Robciuc MR, Karaman S, Makinen T, Alitalo K. Lymphatic system in cardiovascular medicine. Circ Res. (2016) 118:515–30. doi: 10.1161/CIRCRESAHA.115.306544
80. Díaz-Flores L, Gutiérrez R, Díaz-Flores L Jr, Goméz MG, Sáez FJ, Madrid JF. Behaviour of telocytes during physiopathological activation. Semin Cell Dev Biol. (2016) 55:50–61. doi: 10.1016/j.semcdb.2016.01.035
81. Caporali A, Martello A, Miscianinov V, Maselli D, Vono R, Spinetti G. Contribution of pericyte paracrine regulation of the endothelium to angiogenesis. Pharmacol Ther. (2017) 171:56–64. doi: 10.1016/j.pharmthera.2016.10.001
82. Cimini M, Cannatá A, Pasquinelli G, Rota M, Goichberg P. Phenotypically heterogeneous podoplanin-expressing cell populations are associated with the lymphatic vessel growth and fibrogenic responses in the acutely and chronically infarcted myocardium. PLoS ONE. (2017) 12:e0173927. doi: 10.1371/journal.pone.0173927
83. Wakai T, Fissore RA. Constitutive IP(3)R1-mediated Ca(2+) release reduces Ca(2+) store content and stimulates mitochondrial metabolism in mouse GV oocytes. J Cell Sci. (2019) 132:jcs225441. doi: 10.1242/jcs.225441
84. Nakata Y, Kono H, Akazawa Y, Hirayama K, Wakana H, Fukushima H, et al. Role of podoplanin and Kupffer cells in liver injury after ischemia-reperfusion in mice. Surg Today. (2022) 52:344–53. doi: 10.1007/s00595-021-02378-3
85. Schuhmann MK, Stoll G, Bieber M, Vögtle T, Hofmann S, Klaus V, et al. CD84 links T cell and platelet activity in cerebral thrombo-inflammation in acute stroke. Circ Res. (2020) 127:1023–35. doi: 10.1161/CIRCRESAHA.120.316655
86. Rayes J, Watson SP, Nieswandt B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J Clin Invest. (2019) 129:12–23. doi: 10.1172/JCI122955
87. Martin EM, Zuidscherwoude M, Morán LA Di Y, García A, Watson SP. The structure of CLEC-2: mechanisms of dimerization and higher-order clustering. Platelets. (2021) 32:733–43. doi: 10.1080/09537104.2021.1906407
88. Gotru SK, Chen W, Kraft P, Becker IC, Wolf K, Stritt S, et al. TRPM7 kinase controls calcium responses in arterial thrombosis and stroke in mice. Arterioscler Thromb Vasc Biol. (2018) 38:344–52. doi: 10.1161/ATVBAHA.117.310391
89. Budnik I, Brill A. Immune factors in deep vein thrombosis initiation. Trends Immunol. (2018) 39:610–23. doi: 10.1016/j.it.2018.04.010
90. Stegner D, Göb V, Krenzlin V, Beck S, Hemmen K, Schuhmann MK, et al. Foudroyant cerebral venous (sinus) thrombosis triggered through CLEC-2 and GPIIb/IIIa dependent platelet activation. Nat Cardiovasc Res. (2022) 1:132–41. doi: 10.1038/s44161-021-00017-1
91. Lax S, Rayes J, Wichaiyo S, Haining EJ, Lowe K, Grygielska B, et al. Platelet CLEC-2 protects against lung injury via effects of its ligand podoplanin on inflammatory alveolar macrophages in the mouse. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L1016–l29. doi: 10.1152/ajplung.00023.2017
92. Hitchcock JR, Cook CN, Bobat S, Ross EA, Flores-Langarica A, Lowe KL, et al. Inflammation drives thrombosis after Salmonella infection via CLEC-2 on platelets. J Clin Invest. (2015) 125:4429–46. doi: 10.1172/JCI79070
93. Kinsey GR, Sharma R, Okusa MD. Regulatory T cells in AKI. J Am Soc Nephrol. (2013) 24:1720–6. doi: 10.1681/ASN.2013050502
94. Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D'Antoni ML, Debuque R, et al. Revisiting cardiac cellular composition. Circ Res. (2016) 118:400–9. doi: 10.1161/CIRCRESAHA.115.307778
95. Lee PY, Nelson-Maney N, Huang Y, Levescot A, Wang Q, Wei K, et al. High-dimensional analysis reveals a pathogenic role of inflammatory monocytes in experimental diffuse alveolar hemorrhage. JCI Insight. (2019) 4:e129703. doi: 10.1172/jci.insight.129703
96. Alvino VV, Fernández-Jiménez R, Rodriguez-Arabaolaza I, Slater S, Mangialardi G, Avolio E, et al. Transplantation of allogeneic pericytes improves myocardial vascularization and reduces interstitial fibrosis in a swine model of reperfused acute myocardial infarction. J Am Heart Assoc. (2018) 7:e006727. doi: 10.1161/JAHA.117.006727
97. Huguet C, Gavelli A, Bona S. Hepatic resection with ischemia of the liver exceeding one hour. J Am Coll Surg. (1994) 178:454–8.
98. Lemasters JJ, Thurman RG. Reperfusion injury after liver preservation for transplantation. Annu Rev Pharmacol Toxicol. (1997) 37:327–38. doi: 10.1146/annurev.pharmtox.37.1.327
99. Colletti LM, Cortis A, Lukacs N, Kunkel SL, Green M, Strieter RM. Tumor necrosis factor up-regulates intercellular adhesion molecule 1, which is important in the neutrophil-dependent lung and liver injury associated with hepatic ischemia and reperfusion in the rat. Shock. (1998) 10:182–91. doi: 10.1097/00024382-199809000-00006
100. Devey L, Ferenbach D, Mohr E, Sangster K, Bellamy CO, Hughes J, et al. Tissue-resident macrophages protect the liver from ischemia reperfusion injury via a heme oxygenase-1-dependent mechanism. Mol Ther. (2009) 17:65–72. doi: 10.1038/mt.2008.237
Keywords: podoplanin, thrombotic, inflammation, CLEC-2, platelet activation, epithelial-mesenchymal transition
Citation: Huang Y, Lu M, Wang Y, Zhang C, Cao Y and Zhang X (2023) Podoplanin: A potential therapeutic target for thrombotic diseases. Front. Neurol. 14:1118843. doi: 10.3389/fneur.2023.1118843
Received: 08 December 2022; Accepted: 22 February 2023;
Published: 09 March 2023.
Edited by:
Xia Wang, University of New South Wales, AustraliaReviewed by:
Lianchun Wang, University of South Florida, United StatesCopyright © 2023 Huang, Lu, Wang, Zhang, Cao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunyuan Zhang, emN5c2VsZkBzaW5hLmNvbQ==; Yongjun Cao, eW9uZ2p1bmNhb0AxMjYuY29t; Xia Zhang, c3RlZmVuY2UxMDI1QDE2My5jb20=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.