
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 08 February 2023
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1109782
This article is part of the Research Topic Updating Amyloid Neuropathy Knowledge: From Diagnosis to Treatment View all 5 articles
 Stefano Tozza1
Stefano Tozza1 Marco Luigetti2,3
Marco Luigetti2,3 Giovanni Antonini4
Giovanni Antonini4 Anna Mazzeo5
Anna Mazzeo5 Daniele Severi1
Daniele Severi1 Andrea Di Paolantonio3,6
Andrea Di Paolantonio3,6 Luca Leonardi4
Luca Leonardi4 Massimo Russo5
Massimo Russo5 Angela Romano2,3Francesca Forcina4
Angela Romano2,3Francesca Forcina4 Luca Gentile5Maria Nolano1,7Consalvo Mattia8,9
Luca Gentile5Maria Nolano1,7Consalvo Mattia8,9 Fiore Manganelli1*
Fiore Manganelli1*Introduction: Pain is a common symptom of hereditary transthyretin amyloidosis (ATTRv), however, its occurrence in late-onset ATTRv has not been investigated thoroughly. Our aim was to describe the pain experience and its impact on quality of life (QoL) in symptomatic patients and presymptomatic carriers harboring a transthyretin (TTR) gene mutation with a late-onset phenotype.
Materials and methods: Study participants (aged ≥18 years) were consecutively recruited from four Italian centers. Clinical disability was assessed using the Familial Amyloid Polyneuropathy (FAP) stage and Neuropathy Impairment Score (NIS). The Norfolk questionnaire evaluated QoL and the Compound Autonomic Dysfunction Test assessed autonomic involvement. Neuropathic pain was screened using the Douleur Neuropathique 4 (DN4) questionnaire, and pain intensity and its impact on daily activity were assessed using the Brief Pain Inventory severity and interference subscores. Data on the type of TTR mutation, presence of cardiomyopathy, treatment, and Body Mass Index (BMI) were collected.
Results: Overall, 102 subjects with TTR mutations (mean age ± SD 63.6 ± 13.5 years) were recruited, including 78 symptomatic patients (68.1 ± 10.9 years) and 24 presymptomatic carriers (49 ± 10.3 years). Pain was reported by 75.5% of all subjects, but was more frequent in symptomatic patients than in presymptomatic carriers (85.9 vs. 41.6%, respectively). Pain exhibited neuropathic features (DN4≥4) in 69.2% of symptomatic patients and in 8.3% of presymptomatic carriers. Subjects with neuropathic pain were older (p = 0.015) had worse FAP stage (p < 0.001), higher NIS scores (p < 0.001), greater autonomic involvement (p = 0.003), and a lower QoL (p < 0.001) than those without neuropathic pain. Neuropathic pain was associated with higher pain severity (p < 0.001) and had a significant negative impact on daily activities (p < 0.001) Neuropathic pain was not associated with gender, mutation type, TTR therapy, or BMI.
Conclusion: Approximately 70% of late-onset ATTRv patients complained of neuropathic pain (DN4≥4) that worsened as peripheral neuropathy progressed and increasingly interfered with daily activities and QoL. Notably, 8% of presymptomatic carriers complained of neuropathic pain. These results suggest that assessment of neuropathic pain may be useful to monitor disease progression and identify early manifestations of ATTRv.
Hereditary transthyretin amyloidosis (ATTRv; v for “variant”) is a progressive multisystem disorder caused by mutations in the transthyretin (TTR) gene. The mutated TTR tetramer dissociates in misfolded monomers that accumulate in tissues such as the heart and peripheral nervous system (PNS), leading, respectively, to cardiomyopathy and progressive axonal peripheral neuropathy (1). PNS involvement is the presenting complaint in most cases of ATTRv and causes an axonal length-dependent sensory-motor and autonomic polyneuropathy (2). According to the type of mutation and geographic area, two different ATTRv phenotypes exist (3): the early-onset phenotype ( ≤ 50 years) has a predominant small fiber involvement and is typical in endemic regions, while the late-onset phenotype (>50 years), which is typical of non-endemic areas such as Italy (4), has a progressive and prevalent large fiber damage (5).
Several symptoms are reported by patients. The most frequent symptoms are pain, numbness, fatigue, and weakness (6) that substantially impact on the functioning, well-being, and quality of life (QoL) of patients (7) and their relatives (8). Notably, health status is severely impaired by pain/discomfort in 70% of ATTRv patients (9).
Pain, including electric shock, tingling, pins and needles, burning, and itching sensations (10), is widely documented in the early-onset ATTRv phenotype, whereas it has not been thoroughly investigated in the late-onset phenotype. In fact, in the early-onset phenotype, in keeping with the preferential loss of small nerve fibers (11), the patients primarily notice pain and thermal sensation dysfunction as well as dysautonomia. Conversely, in late-onset ATTRv phenotype, the small nerve fiber involvement may remain under recognized if not properly investigated, as it is usually overwhelmed by motor and sensory impairment.
Consequently, the aim of this study was to describe the neuropathic pain experience, its frequency, and impact on QoL in symptomatic patients and presymptomatic carriers harboring a TTR gene mutation associated with the late-onset ATTRv phenotype.
This multicenter study was conducted in four Italian centers with experience in the diagnosis and management of ATTRv (University of Naples Federico II, Fondazione Policlinico Universitario A. Gemelli IRCCS of Rome, Sant'Andrea Hospital of Sapienza University of Rome, University of Messina). The study was approved by the local institutional ethics committee in each center.
Study participants (aged ≥18 years) were consecutively recruited and included both symptomatic patients and presymptomatic carriers with known pathogenic TTR mutations. Carriers, who had normal nerve conduction, were identified as carrying a TTR gene mutation in the setting of genetic familial counseling (12).
Subjects were excluded from study if they had concomitant disease (e.g., diabetes) in which pain could represent a relevant feature.
Age at enrolment, gender, mutation type, ATTRv therapy, pain killers, body mass index (BMI), and the presence of cardiomyopathy were recorded.
Symptomatic patients and presymptomatic carriers underwent an extensive neurological evaluation, and several scales were applied to assess disease stage (Familial Amyloid Polyneuropathy [FAP] stage) (13), neuropathic impairment (Neuropathy Impairment Score [NIS]) (14), autonomic involvement (Compound Autonomic Dysfunction Test [CADT]) (15), and QoL (Norfolk Quality of Life-Diabetic Neuropathy [QOL-DN]) (16).
The presence of pain and its distribution (generalized, distal at upper and lower limbs, at lower limbs or at upper limbs) was recorded. The Douleur Neuropathique 4 (DN4) questionnaire was used to establish if pain had neuropathic features (DN4 ≥4) (17). The Brief Pain Inventory (BPI) evaluated the severity of pain (BPI severity) and its impact on daily activity (BPI interference) (18).
Descriptive analyses were based on the mean±standard deviation (SD) for numerical variables and percentage for categorical data. After verifying the Gaussian distribution of variables through the Kolmogorov–Smirnov test, the Student's T-test was used to compare continuous variables between subjects with neuropathic pain (DN4 ≥4) and those without neuropathic pain (DN4 < 4). Pearson's chi-square test was used to compare categorical variables between these groups. When significant differences were detected (p < 0.05), post-hoc analyses of categorical variables through adjusted standardized residuals with Bonferroni-corrected p-values (p ≤ 0.006) were calculated in order to identify which cells of the contingency table contributed most to the significant effect (19). Analyses and graphics were performed using the statistical software IBM SPSS Statistic version 25.
In total, 102 subjects with TTR mutations (mean age ± SD 63.6 ± 13.5 years) were recruited, including 78 symptomatic patients (68.1 ± 10.9 years) and 24 presymptomatic carriers (49 ± 10.3 years). Most symptomatic patients had a disease severity of FAP 1 (sensory neuropathy) or FAP 2 (require assistance for walking). Demographic and clinical findings are summarized in Table 1.
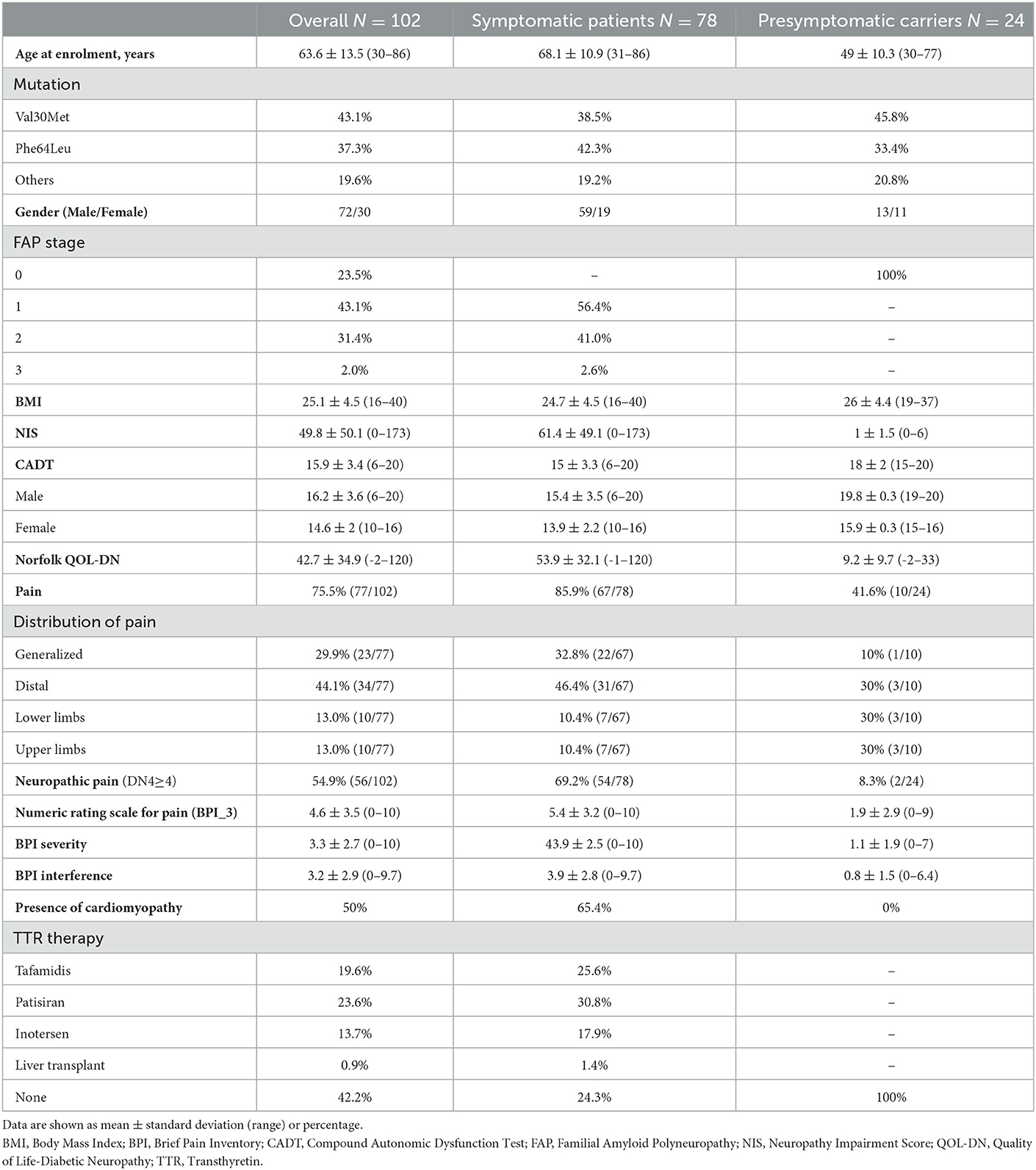
Table 1. Demographic and clinical findings.
Pain was reported by 75.5% of all subjects, but was more frequent in symptomatic patients than in presymptomatic carriers (85.9 vs. 41.6%, respectively). The distribution of pain was predominantly generalized or distal in symptomatic patients, whereas presymptomatic carriers reported distal pain or pain in the lower or upper limbs. Neuropathic pain (DN4 ≥4) was reported by 69.2% of symptomatic patients and 8.3% of presymptomatic carriers. These patients reported more frequently electric shock (66.7%) and less frequently burning (54.3%) and painful cold (52.6%) sensation. The pain was typically associated with tingling and numbness (91.2%) and less frequently with pins and needles (63.2%) and itching (31.6%) sensation. Reduced touch and pinprick sensation was present in the 82.5 and 68.4% respectively, whereas the allodynia phenomenon was reported only in the 49.1% of patients suffering from neuropathic pain.
Subjects with neuropathic pain were significantly older (66.5 vs. 60.1 years; p = 0.015) and had more severe disease (FAP 1–2 vs. 0–1, p < 0.001) and neuropathic impairment (NIS, 69.9 ± 49.5 vs. 22.6 ± 36.8; p < 0.001) than subjects without neuropathic pain (DN4 < 4). The presence of neuropathic pain was associated with significantly greater autonomic involvement (CADT; p = 0.003) and lower QoL (Norfolk QOL-DN; p < 0.001) compared with no neuropathic pain (Figure 1).
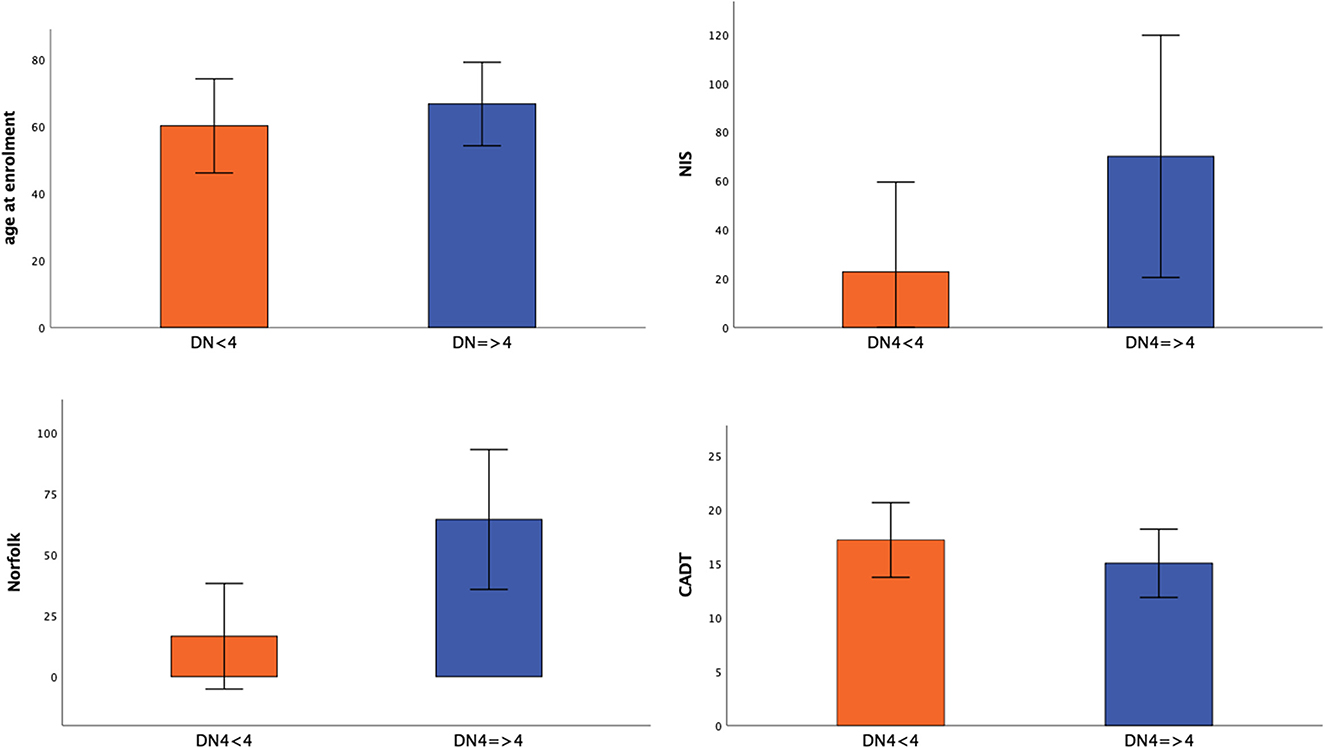
Figure 1. Significance differences between patients without (DN4 < 4) and with neuropathic pain (DN4 > 4). Mean value ± standard deviation of age at enrolment, NIS, Norfolk, CADT between patients without (DN4 < 4) and with neuropathic pain (DN4 > 4) was represented.
Significantly more subjects with neuropathic pain had cardiomyopathy than those without neuropathic pain (p = 0.005) (Table 2). The presence of neuropathic pain was associated with significantly higher pain severity (BPI severity; p < 0.001) and had a significant negative impact on daily activities (BPI interference; p < 0.001) (Figure 2).
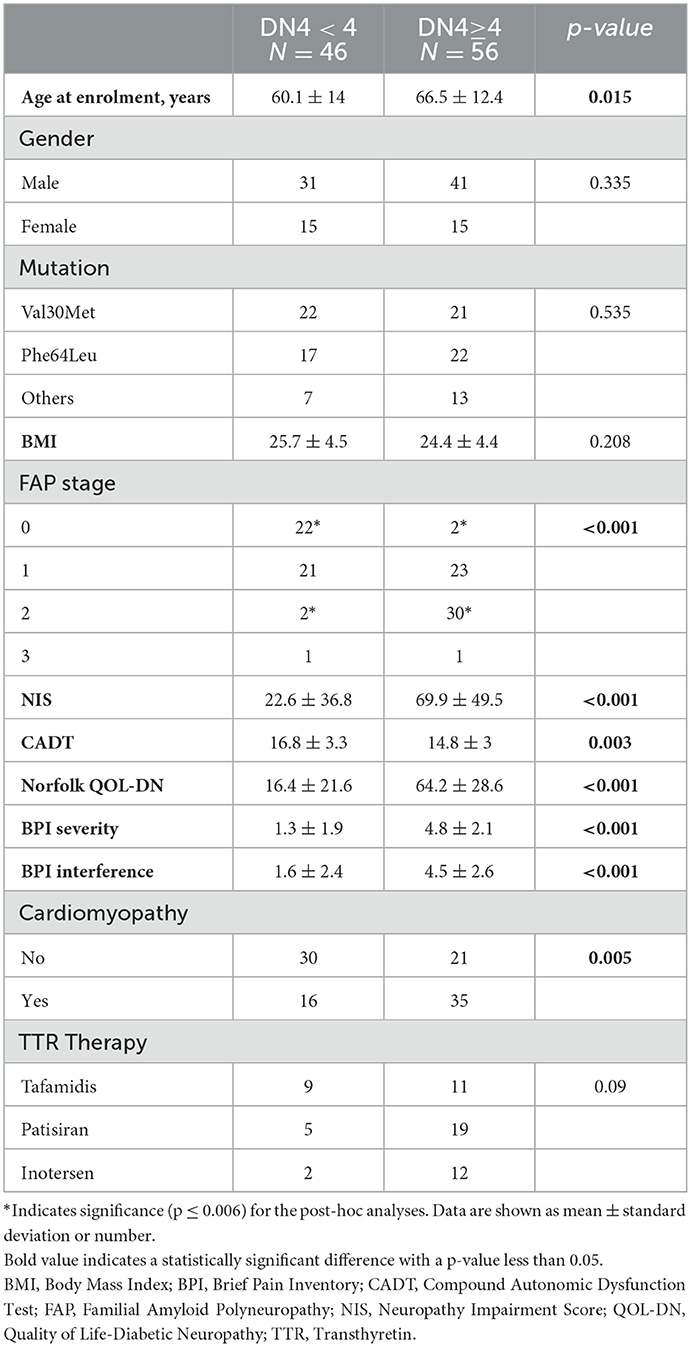
Table 2. Comparison of demographic and clinical data.
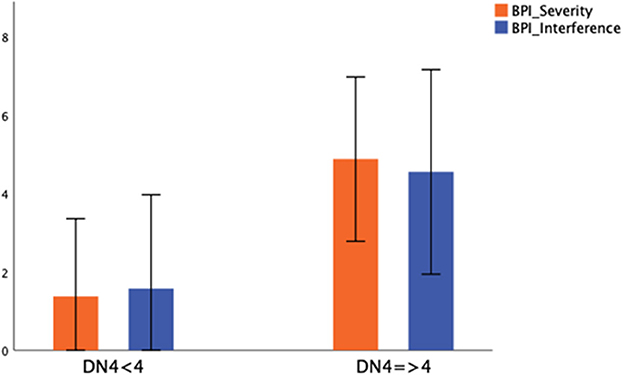
Figure 2. Severity and daily life interference in patients without (DN4 < 4) and with neuropathic pain (DN4 > 4). Mean value ± standard deviation of BPI Severity and BPI Interference between patients without (DN4 < 4) and with neuropathic pain (DN4 > 4) was represented.
Neuropathic pain was not associated with gender, mutation type, TTR therapy, or BMI (Table 2).
Neuropathic pain was treated in 82.4% of subjects, most frequently with anti-seizure drugs (63.8%) like pregabalin or gabapentin. Seven subjects were treated with NSAIDs, 3 with opioids, 1 with antidepressants, 1 with local steroidal injection, and 4 with combination therapy. Patients reported a satisfactory pain relief (>50% on the item BPI_8) in less than half of cases (45.9%), whereas 40.5% of patients referred persistent pain despite the therapy (< 30% on the item BPI_8).
Pain is a common symptom in ATTRv (6) and impacts patients' functioning, health status, and QoL (7). However, the frequency of neuropathic pain in the late-onset ATTRv phenotype is unknown.
To our knowledge, this is the first systematic assessment of pain in a large cohort of late-onset TTR-mutated subjects encompassing both symptomatic patients and presymptomatic carriers. We showed here that ~70% of symptomatic patients experienced neuropathic pain (DN4 ≥4) with a length-dependent distribution. The pain was mainly characterized by electric shock associated with tingling, numbness and reduced touch sensation. All these features are consistent with a prominent involvement of larger myelinated A-β fibers (20) as typically occurs in late-onset ATTRv (3).
When compared with subjects without neuropathic pain, those with neuropathic pain were older, exhibited a more severe disability (FAP stage and NIS), had a lower QoL (Norfolk questionnaire), had significant autonomic involvement (CADT), and were more likely to have cardiomyopathy. Accordingly, we propose that neuropathic pain in ATTRv worsens as disease progresses and increasingly impacts patients' QoL.
This suggests that a thorough assessment of pain is important to define an optimal treatment regimen. In addition, careful pain investigation may offer relevant information to clinicians for monitoring disease worsening or to assess multisystem involvement (e.g., cardiomyopathy).
Interestingly, we found that 8% (N = 2) of presymptomatic carriers reported neuropathic pain. Both subjects were young women (aged 33 and 40 years at enrolment) and carriers of the Glu89Gln variant. Neither reported dysautonomic (CADT=16) or neuropathic (NIS= 0) symptoms, whereas both complained of a mild impact in QoL (Norfolk = 33 and 10).
This finding raises the question of whether presymptomatic carriers who complain of neuropathic pain should instead be considered symptomatic patients and, consequently, be able to access the available therapies. Indeed, according to expert consensus (12), neuropathic pain, including spontaneous and evoked symptoms, could be considered as definitively related to the onset of symptomatic ATTR amyloidosis even without an instrumental demonstration of small nerve fiber impairment (e.g., quantitative sensory testing, skin biopsy). Nonetheless, the finding of pain, especially if neuropathic, should lead to further assessment for neuropathy beyond the conventional nerve conduction study.
The results of our study, therefore, suggest that it would be judicious to query the presence of neuropathic pain in presymptomatic relatives who carry a TTR gene mutation, as its presence might represent an early manifestation of ATTRv. In this perspective, it is largely documented that small nerve fiber damage (21–23) may precede, by many years, the signs and symptoms of neuropathy in the late-onset ATTRv phenotype.
Lastly, we found that almost one fifth of patients (17.6%) had not received any symptomatic treatment for neuropathic pain. Moreover, patients receiving painkillers did not achieve significant pain relief in about 40% of the cases. In the late-onset ATTRv phenotype, pain and its treatment could be overlooked if clinicians are more focused on motor disability.
In conclusion, clinicians should not neglect the pain experience when evaluating ATTRv patients or presymptomatic relatives who carry a TTR gene mutation. The DN4 questionnaire is a reliable screening tool to identify, with high specificity, the presence of neuropathic pain (17). As pain impacts QoL, individuals who experience pain have the right to appropriate pain relief. Assessment of neuropathic pain may be a useful strategy to monitor disease progression in symptomatic patients and may be suitable to identify early manifestation of the disease in presymptomatic relatives who carry a TTR gene mutation.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Local Ethical Board University of Naples Federico II of Naples. The patients/participants provided their written informed consent to participate in this study.
ST contributed to the acquisition, analysis and interpretation of data for the work, drafted, and revised the work. ML, GA, AM, and MN contributed to the acquisition, interpretation of data for the work, and revised the work. DS, AD, LL, MR, AR, FF, and LG contributed to the acquisition of data for the work and revised the work. CM contributed to the conception and design of the work and revised the work. FM contributed to the conception and design of the work, interpretation of data for the work, and drafted and revised the work. All authors gave final approval for the final version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Unconditional editorial support by SOBI.
Editorial assistance was provided by Melanie Gatt (Ph.D.), an independent medical writer, on behalf of Health Publishing and Services Srl.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Manganelli F, Fabrizi GM, Luigetti M, Mandich P, Mazzeo A, Pareyson D. Hereditary transthyretin amyloidosis overview. Neurol Sci. (2020) 43:595–604. doi: 10.1007/s10072-020-04889-2
2. Luigetti M, Romozzi M, Bisogni G, Cardellini D, Cavallaro T, Di Paolantonio A, et al. hATTR pathology: nerve biopsy results from Italian referral centers. Brain Sci. (2020) 10:780. doi: 10.3390/brainsci10110780
3. Tozza S, Severi D, Spina E, Iovino A, Aruta F, Ruggiero L, et al. The neuropathy in hereditary transthyretin amyloidosis: A narrative review. J Peripher Nerv Syst. (2021) 26:155–9. doi: 10.1111/jns.12451
4. Russo M, Obici L, Bartolomei I, Cappelli F, Luigetti M, Fenu S, et al. ATTRv amyloidosis Italian Registry: clinical and epidemiological data. Amyloid. (2020) 27:259–65. doi: 10.1080/13506129.2020.1794807
5. Waddington-Cruz M, Wixner J, Amass L, Kiszko J, Chapman D, Ando Y, et al. Characteristics of Patients with Late- vs. Early-Onset Val30Met Transthyretin Amyloidosis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Neurol Ther. (2021) 10:753–66. doi: 10.1007/s40120-021-00258-z
6. Lovley A, Raymond K, Guthrie SD, Pollock M, Sanchorawala V, White MK. Patient-reported burden of hereditary transthyretin amyloidosis on functioning and well-being. J Patient Rep Outcomes. (2021) 5:3. doi: 10.1186/s41687-020-00273-y
7. Yarlas A, Gertz MA, Dasgupta NR, Obici L, Pollock M, Ackermann EJ, et al. Burden of hereditary transthyretin amyloidosis on quality of life. Muscle Nerve. (2019) 60:169–75. doi: 10.1002/mus.26515
8. Magliano L, Obici L, Sforzini C, Mazzeo A, Russo M, Cappelli F, et al. Psychosocial burden and professional and social support in patients with hereditary transthyretin amyloidosis (ATTRv) and their relatives in Italy. Orphanet J Rare Dis. (2021) 16:163. doi: 10.1186/s13023-021-01812-6
9. Ines M, Coelho T, Conceicao I, Ferreira L, de Carvalho M, Costa J. Health-related quality of life in hereditary transthyretin amyloidosis polyneuropathy: a prospective, observational study. Orphanet J Rare Dis. (2020) 15:67. doi: 10.1186/s13023-020-1340-x
10. Ng Wing Tin S, Plante-Bordeneuve V, Salhi H, Goujon C, Damy T, Lefaucheur JP. Characterization of Pain in Familial Amyloid Polyneuropathy. J Pain. (2015) 16:1106–14. doi: 10.1016/j.jpain.2015.07.010
11. Di Stefano G, Di Lionardo A, Di Pietro G, Truini A. Neuropathic pain related to peripheral neuropathies according to the iasp grading system criteria. Brain Sci. (2020) 11:1. doi: 10.3390/brainsci11010001
12. Conceicao I, Damy T, Romero M, Galan L, Attarian S, Luigetti M, et al. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid. (2019) 26:3–9. doi: 10.1080/13506129.2018.1556156
13. Cakar A, Durmus-Tekce H, Parman Y. Familial Amyloid Polyneuropathy. Noro Psikiyatr Ars. (2019) 56:150–6. doi: 10.29399/npa.23502
14. Dyck PJB, Gonzalez-Duarte A, Obici L, Polydefkis M, Wiesman JF, Antonino I, et al. Development of measures of polyneuropathy impairment in hATTR amyloidosis: From NIS to mNIS + 7. J Neurol Sci. (2019) 405:116424. doi: 10.1016/j.jns.2019.116424
15. Denier C, Ducot B, Husson H, Lozeron P, Adams D, Meyer L, et al. A brief compound test for assessment of autonomic and sensory-motor dysfunction in familial amyloid polyneuropathy. J Neurol. (2007) 254:1684–8. doi: 10.1007/s00415-007-0617-5
16. Vinik EJ, Vinik AI, Paulson JF, Merkies IS, Packman J, Grogan DR, et al. Norfolk QOL-DN: validation of a patient reported outcome measure in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. (2014) 19:104–14. doi: 10.1111/jns5.12059
17. Spallone V, Morganti R, D'Amato C, Greco C, Cacciotti L, Marfia GA. Validation of DN4 as a screening tool for neuropathic pain in painful diabetic polyneuropathy. Diabet Med. (2012) 29:578–85. doi: 10.1111/j.1464-5491.2011.03500.x
18. Poquet N, Lin C. The Brief Pain Inventory (BPI). J Physiother. (2016) 62:52. doi: 10.1016/j.jphys.2015.07.001
19. García-pérez MA, Núñez-antón V. Cellwise residual analysis in two-way contingency tables. Educ Psychol Measure. (2003) 63:825–39. doi: 10.1177/0013164403251280
20. Truini A, Garcia-Larrea L, Cruccu G. Reappraising neuropathic pain in humans–how symptoms help disclose mechanisms. Nat Rev Neurol. (2013) 9:572–82.
21. Leonardi L, Galosi E, Vanoli F, Fasolino A, Di Pietro G, Luigetti M, et al. Skin biopsy and quantitative sensory assessment in an Italian cohort of ATTRv patients with polyneuropathy and asymptomatic carriers: possible evidence of early non-length dependent denervation. Neurol Sci. (2022) 43:1359–64. doi: 10.1007/s10072-021-05434-5
22. Luigetti M, Di Paolantonio A, Guglielmino V, Romano A. Cutaneous silent period in ATTRv carriers: a possible early marker of nerve damage? Neurol Sci. (2022) 43:6979–82. doi: 10.1007/s10072-022-06317-z
Keywords: ATTRv, hereditary amyloidosis, pain, quality of life, transthyretin
Citation: Tozza S, Luigetti M, Antonini G, Mazzeo A, Severi D, Di Paolantonio A, Leonardi L, Russo M, Romano A, Forcina F, Gentile L, Nolano M, Mattia C and Manganelli F (2023) Neuropathic pain experience in symptomatic and presymptomatic subjects carrying a transthyretin gene mutation. Front. Neurol. 14:1109782. doi: 10.3389/fneur.2023.1109782
Received: 28 November 2022; Accepted: 23 January 2023;
Published: 08 February 2023.
Edited by:
Chiara Terracciano, Gugliemo da Saliceto Hospital, ItalyReviewed by:
Paulo Victor Sgobbi Souza, Federal University of São Paulo, BrazilCopyright © 2023 Tozza, Luigetti, Antonini, Mazzeo, Severi, Di Paolantonio, Leonardi, Russo, Romano, Forcina, Gentile, Nolano, Mattia and Manganelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fiore Manganelli,  ZmlvcmVtYW5nYW5lbGxpQGdtYWlsLmNvbQ==
ZmlvcmVtYW5nYW5lbGxpQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.