 Vassilis Genoud1,2
Vassilis Genoud1,2 Denis Migliorini1,2,3,4*
Denis Migliorini1,2,3,4*- 1Department of Oncology, University Hospital of Geneva, Geneva, Switzerland
- 2Center for Translational Research in Onco-Haematology, University of Geneva, Geneva, Switzerland
- 3Brain Tumor and Immune Cell Engineering Laboratory, AGORA Cancer Research Center, Lausanne, Switzerland
- 4Swiss Cancer Center Léman (SCCL), Lausanne and Geneva, Geneva, Switzerland
Chimeric antigen receptor (CAR) T cell therapy represents a scientific breakthrough in the treatment of advanced hematological malignancies. It relies on cell engineering to direct the powerful cytotoxic T-cell activity toward tumor cells. Nevertheless, these highly powerful cell therapies can trigger substantial toxicities such as cytokine release syndrome (CRS) and immune cell-associated neurological syndrome (ICANS). These potentially fatal side effects are now better understood and managed in the clinic but still require intensive patient follow-up and management. Some specific mechanisms seem associated with the development of ICANS, such as cytokine surge caused by activated CAR-T cells, off-tumor targeting of CD19, and vascular leak. Therapeutic tools are being developed aiming at obtaining better control of toxicity. In this review, we focus on the current understanding of ICANS, novel findings, and current gaps.
1. Introduction
Chimeric antigen receptor (CAR) T cells have shown high efficacy in multiple hematological indications and are now widely implemented in many centers (1). Nevertheless, the management of patients receiving CAR-T cell therapies is beyond the clinical management of high-grade hematological diseases. Not all centers can offer this novel therapy to their patients, mainly due to the frequent need for intensive care unit management and high-end infrastructure to deliver cell-therapy products.
1.1. CAR-T cells
CARs are fusion proteins combining an antibody-based recognition part and intracellular activation and co-stimulation domains. Through cell engineering, human T cells can express these CARs, thereby conferring specificity to a cell surface antigen (Ag) of interest without major histocompatibility complex (MHC) restriction. CAR-T cells can target any type of cell depending on the epitope selected. The recognizing part of the receptor is composed of a single-chain variable fragment (scFv) derived from the variable portion of an antibody (Ab). Currently, CD-28 and 4-1BB are the two co-stimulatory domains approved in the clinic.
1.2. Secondary effects
CAR-T cells are exceptionally active cellular therapies and have brought unprecedented success to previously untreatable diseases. However, their high activity drives systemic toxicities, either cytokine release syndrome (CRS) or immune cell-associated neurologic syndrome (ICANS), sometimes referred to as neurotoxicity. These toxicities were not identified in mouse models but rapidly forced clinicians to adapt their management. As both are associated, we will introduce both CRS and ICANS, but the scope of this review is to focus on ICANS.
1.2.1. CRS
CRS is the most common CAR-T cell therapy-related toxicity, and it has been reported to develop in 30–100% of patients (2). It is characterized by clinical symptoms of hyperthermia and oxygenation or cardiovascular alterations. If promptly identified and well managed, it is most often fully reversible. Pathophysiological studies suggest that CRS results from pyroptotic cell death (3). As CAR-T cell cytotoxicity relies mainly on the release of granzyme B, it induces rapid activation of caspase 3 in target cells. This enzyme will cleave gasdermine E, which forms pores in the tumor cell membrane, leading to pyroptosis.
Consequently, gasdermine D in surrounding macrophages will be cleaved by caspase 1, leading to cytokine release by macrophages, inducing CRS (4). In the central nervous system (CNS), microglia cells have phagocytic functions and also express gasdermine D that can exacerbate neurotoxicity through pyroptosis (5). Because CAR-T cells release a high concentration of perforin or granzyme B compared with cytotoxic T lymphocytes, more immunogenic pyroptosis will be induced through the gasdermin pathway compared with the more common apoptosis pathway induced by cytotoxic T lymphocytes. Previous pre-clinical studies linked CRS severity to gasdermine cleavage (4). In other words, the CAR-T cell mechanism of toxicity indicates a more immunogenic cell death, leading to more potent activation of surrounding cells such as myeloid cells, amplifying the release of cytokines.
1.2.2. ICANS
ICANS is less common than CRS as only half of patients will experience this syndrome (2). It usually develops after CRS initiation, potentially illustrating a causal link. As for CRS, ICANS is generally reversible even though rare cases of fatal ICANS have been reported (< 1% of cases) (6). CD19 CAR-T cells are the most incriminated in the development of ICANS, and counterintuitively, intrathecal or intratumoral infusion of CAR-T cells for patients with glioblastoma does not induce ICANS (7, 8). Many advances have been made in understanding ICANS development, with multiple pre-clinical models and consensual grading of ICANS in patients (9). Since the pathophysiology is still poorly understood, we will address the most recent developments.
2. ICANS
2.1. Clinical presentation
ICANS may present with different symptoms, such as dysgraphia (6, 10), frontal lobe dysfunction (11), language disorders, or akinetic-mutism (9), and can rarely evolve into a seizure or fulminant cerebral edema (12, 13). It usually develops 4–6 days after CAR-T cell infusion and lasts 5–13 days (14, 15). Symptoms are fully reversible, but sometimes more prolonged toxicity can be observed (11, 16). Clinical workup includes laboratory measures, magnetic resonance imaging (MRI) studies, and electroencephalography (EEG). CAR-T cell kinetics of amplification and serum cytokine levels correlate with ICANS development (17, 18). Analysis of cerebrospinal fluid (CSF) is performed only in clinical trials and will be discussed later. MRI findings are most often unspecific (19), but studies described a potential specific pattern of edema in patients suffering from severe ICANS, primarily located in the bilateral thalami, supratentorial white matter, and brainstem region (10, 20). Abnormal EEG patterns can predict the development of clinical seizures in patients with ICANS (13, 21) and correlate with ICANS severity (22).
As ICANS was not anticipated from pre-clinical trials, it took some time to develop consensual grading. However, the American Society for Blood and Marrow Transplantation proposed a consensual definition and grading system (9) based on the clinical immune effector cell-associated encephalopathy score (6).
2.2. Pathophysiology
Better grading and reporting of ICANS in the clinic and the development of pre-clinical models of ICANS helped to decipher the mechanisms driving this syndrome. In particular, a CD19+ lymphoma xenograft model (23) and a humanized NSG mouse model (24) helped to improve our understanding. Nevertheless, they do not represent human cytokines and human hematopoietic cells and present xenograft vs. host reaction (25–28), thus limiting their direct translation.
Cytokines such as IL-6, IL-1, and TNF-α have been widely identified as at the root of CRS or ICANS, and their blockade in the clinic can limit CAR-T cell toxicities, as we will show later. Many other cytokines and cell subtypes are involved, and we will address them in the following sections.
2.2.1. Cytokines
During clinical studies, thorough analyses of serum and CSF identified IL1-6, IL-1, IFN-γ, TNF-α, and GM-CSF (20) as major contributors to the overactivation of the peripheral immune system.
IL-6, identified as the critical regulator of CRS in many clinical trials, harbors pro- and anti-inflammatory effects and is mainly produced by the myeloid lineage. It has an autocrine activity to promote macrophage maturation and activation, and its receptor is also widely expressed on immune cells and controls the acute phase of inflammation (18). IL-6 has been described as responsible for fatal CRS in pre-clinical models (23, 25) and for promoting macrophage activation (23). In patients, tocilizumab, a monoclonal IL-6 receptor (IL-6R) blocking Ab, can limit most CRS symptoms and reverse cytokine levels (29, 30).
Nevertheless, IL-6R blockade has no impact on limiting ICANS, contrasting with the blocking of the IL-1 axis (23), hinting at the primary role of IL-1 in ICANS physiopathology. Following CAR-T cell infusion, IL-1 elevates before IL-6 in the serum (25, 31). IL-1 further induces monocyte activation and neutrophil infiltration into the brain (31). IL-1 is also highly implicated in CRS, and the use of anakinra, an IL-1 receptor blocker, in a mouse model showed reduced symptoms and mortality, while not affecting CAR-T cell activity (25).
IFN-γ is secreted by activated CAR-T cells and macrophages and exerts fundamental anti-tumor and pro-inflammatory activity (32). CAR-T cells depend highly on IFN-γ, and its blocking will decrease CAR-T function.
TNF-α is another cytokine identified during CAR-T cell toxicity. It activates myeloid cell proliferation, migration, and production of cytokines (33). However, TNF-α also has a direct cytotoxic activity on target cells (34, 35). TNF-α blockade in the SCID-Beige model prevents IL-6 production by myeloid cells and limits CRS mortality, but also limits the efficacy of CAR-T cells (23).
GM-CSF essentially promotes differentiation and all effector functions of myeloid cells and has a central role in tissue inflammation (36, 37).
In conclusion, there is a clear link between serum cytokines and the development of constantly elevated ICANS in multiple studies with different constructs (13, 21, 30). Many cell types are implicated in producing these cytokines. However, a single-cell RNA sequencing study in mice identified that if many cell types produce IL-6, macrophages are the primary producer by far (23).
2.2.2. Myeloid cells
Activated CAR-T cells release IFN-γ, TNF-α, and GM-CSF (38), which are cytotoxic on tumor cells but will also activate myeloid cells. Macrophages are also activated by damage-associated molecular patterns such as ATP, HMGB1, histone H3, and other signals through Toll-like receptors resulting from tumor and surrounding cell death, and macrophages will, in turn, further release IL-6 and TNF-α (37). Interestingly, macrophages must be activated by functional CAR-T cells, as in patients not responding to CAR-T cell therapy, there is no CRS induction (39).
As GM-CSF can lead to IL-6 production, its blocking can be protective against the development of CRS and neurotoxicity without compromising CAR-T cell efficacy. Moreover, CAR-T cell KO for GM-CSF showed better cytotoxic activity (40). Conversely, monocyte ablation negatively affects CAR-T proliferation and expansion (25).
2.2.3. Endothelial cell activation and blood–brain barrier dysfunction
In the CNS, vascular exchanges with the parenchyma are highly controlled by endothelial cells (EC), which form with pericytes, smooth muscle cells, and astrocytes' end foots the blood–brain barrier (BBB) (41).
The EC permeability is regulated by the vascular endothelial growth factor (VEGF) (42) and angiopoietin (Ang)/tyrosine kinase with immunoglobulin-like and EGF-like domains (Tie) axis (43, 44) and is altered during CAR-T cell treatment (13). At constitutional levels, Ang1 is produced by platelets and perivascular cells and binds to Tie2 to stabilize the endothelium. Nevertheless, when activated by inflammatory cytokines, EC will release Ang2 and displace Ang1, leading to a vascular leak. Consequently, a higher Ang2/Ang1 ratio has been linked to higher ICANS severity (45), and Ang1 overexpression in mice preserved EC function and integrity (46). Adhesion molecules such as vascular cell adhesion protein 1 and intercellular adhesion molecule 1 are overexpressed during the high-inflammatory state caused by CAR-T cell treatment and facilitate leukocyte infiltration (33, 47).
Moreover, EC and exposed pericytes will produce IL-6 and VEGF in response to inflammation and, in particular excessive IFN-γ (48–51), affecting the BBB tight junctions and further worsening vascular leak (52, 53). In contact to excessive TNF-α, EC will also produce matrix metalloproteinase 2 and 9, further disrupting cell–matrix adhesion and contributing to increasing permeability (54), which can even lead to cerebral edema (55).
In a rhesus macaque pre-clinical model of ICANS, studying a CD20 CAR-T cell with 4-1BB co-stimulation domain, an increased concentration of cytokines was identified in the CSF. Moreover, a higher infiltration of CAR-T cells with increased expression of the integrin VLA4 was identified. Histological analysis revealed panencephalitis, with multifocal meningitis, and perivascular T-cell infiltration 8 days after infusion (56). In another model, IL-6R blockade did not ameliorate meningeal thickening and macrophage infiltration to the brain, but IL-1 blockade did (25). CAR-T cells with KO of GM-CSF led to decreased IL-6 levels and downregulated interactions with myeloid cells, leading to a restored endothelium permeability state (40, 57). In addition, in clinical trials, higher levels of CAR-T cells and cytokines in the CSF have been linked to higher-grade ICANS (21, 58, 59).
The thorough investigation of fatal brain edema cases during CAR-T cell therapy could identify different associated risk factors, such as younger age, higher CD8 ratio, higher IL-15 serum concentrations, and low platelets before infusion, as well as rapid expansion and higher IL-2 and TNF-α peak (60). Pathological examination of the brain revealed BBB disruption but no activated T cells in the CNS (60). In other words, many findings indicate the incriminating role of vascular leak due to BBB breakdown with elevated cytokine levels in the CNS. However, if CAR-T cells are also found in the CSF, it seems that they are not required for ICANS development.
2.2.4. Other factors associated with ICANS
2.2.4.1. Other soluble factors
Catecholamines are also elevated during CRS and ICANS (61). Adrenaline and noradrenaline have direct activation functions on CAR-T cells and will promote subsequent cytokine release (62). Therefore, by limiting this amplifying loop, CRS can be limited (61). Nevertheless, as severe CRS is defined by cardiovascular instability, limiting catecholamines could be detrimental.
Phosphorus has also been described to be associated with ICANS (63). The high metabolic activity during the CAR-T cell expansion phase may decrease phosphorus availability, and hypophosphatemia can lead to neurologic symptoms similar to ICANS. If a causality link is still to be proven, phosphorus supplementation would be easy to implement.
2.2.4.2. Disseminated intravascular coagulation
Similarities with disseminated intravascular coagulation and sepsis are present in severe CRS or ICANS with hypofibrinogenemia and increased fibrin degradation, leading to endothelial cell disruption (18). Elevated serum D-Dimers decreased fibrinogen, and platelets seem to indicate a thrombotic microangiopathy process that compromises the BBB (13, 14). Activated and impaired vascular integrity can expose tissue factors, and collagen fibers triggering coagulation pathways has been observed (64).
2.2.4.3. CNS infiltration of immune cells
Many studies identified CAR-T cells in the CSF of patients suffering from ICANS (48, 58, 65), with different phenotypes such as Th1, Th17, and regulatory T cells (25). Macrophages were also identified in the CNS in fatal CAR-T cell therapy cases, with parenchyma infiltration and expansion in the perivascular space (12, 66). A xenograft mouse model used GM-CSF neutralization to reduce CNS infiltration of CAR-T cells and myeloid (40), hinting at a potential therapeutic lead, even though there is no clear causality link between CAR-T cell CNS infiltration and ICANS development.
2.2.4.4. Astrocytes and microglia activation
Glial fibrillary acidic protein (GFAP), a validated marker for astroglial cells injury (67), and S100b, a marker of astrocyte activation and injury (68), were both elevated in the CSF of patients experiencing ICANS (69). Activated astrocytes are also described in inflammatory or degenerative diseases such as multiple sclerosis, Alzheimer's disease, and Parkinson's disease (70). IFN-γ has a direct toxic effect on astrocytes, leading to further CNS inflammation and immune infiltration (47, 71). Astrocytes also interact with neurons by producing glutamate (54). When exposed to IL-1, astrocytes show a decreased functionality in glutamate signaling (55). Neuron activity is also affected by excessive TNF-α exposure by altering glutamate balance (72), and high levels of IL-6 can alter neuron excitability, leading to EEG anomalies (73). Moreover, BBB disruption has been associated with increased epilepsy risk (74).
Microglia cells have phagocytic functions and are involved in cognitive functions and neurodegeneration (70, 75). When activated in the case of inflammation, they can secrete IL-1 and TNF-α, further worsening BBB permeability and neuron injury (72). Hence multiple CNS cell types suffer during ICANS, and some also contribute to amplifying the inflammatory state.
2.2.4.5. CNS targeting
CNS infiltration by tumor cells is associated with a dismal prognosis (76), but not all patients with CNS leukemia developed ICANS (21). Moreover, tumor cells expressing the targeted Ag are not required for ICANS development (77), and few patients developing ICANS have CNS involvement. Thus, the presence of direct on-tumor activity of CAR-T cells does not seem to be the driving mechanism of ICANS. A recent study suggests that mural cells of the endothelium in the CNS are CD19 positive and could be targeted by CD19 CAR-T cells in an off-tumor, on-target manner (78). This novel finding could also explain the relatively higher incidence of ICANS with CD19 CAR-T cells compared to other targets. However, CD19 is not the only targeted Ag expressed in the CNS. A single-cell RNA sequencing study confirmed the expression of CD22 in microglia cells (75), which exerts negative regulation of microglia phagocytosis (79). Nevertheless, CAR-T cells targeting CD22 do not seem to lead to a higher incidence of ICANS than other targets (75, 80); therefore, arguing against direct CAR-T cell Ag targets to initiate ICANS. We can hypothesize that targeting mural cells through the CD19 Ag would further intensify vascular leaks and all the consequences cited earlier.
Further analysis and direct comparison of different CAR constructs are still needed to confirm this. On-target, off-tumor toxicity is also suspected for BCMA CAR-T cells, evaluated for patients with multiple myeloma. Non-ICANS neurologic symptoms with parkinsonism have been observed in a subset of patients, and preliminary data seem to indicate BCMA expression in cells located in the basal ganglia (81). Further analyses are needed, but careful neurological follow-up is warranted while using BCMA CAR-T cells.
2.3. Management
2.3.1. IL-6 blockade
Tocilizumab is a monoclonal Ab targeting the receptor of IL-6, inducing rapid regression of CRS symptoms and cytokine levels. It is now incorporated in the standard management of CRS (2) and is generally rapidly introduced to better control the development of CRS. Nevertheless, if tocilizumab is potent to limit CRS severity and duration, it does not affect ICANS incidence and severity and is even reported to increase ICANS in some studies (58, 82). One hypothesis to explain the lack of efficacy of IL-6 receptor blockade could be that soluble IL-6 is increased in the serum by blocking the receptor and could cross the altered BBB. In contrast, tocilizumab as an Ab with a higher molecular weight would be limited to blocking the IL-6 receptor in the CNS therefore artificially displacing the detrimental effect on IL-6 from the periphery to the CNS. Because the studies associating tocilizumab with a higher incidence of ICANS had a small sample size and were not randomized, we cannot conclude on the strength of this association, but careful analysis should be prompted for tocilizumab safety use in CRS. One alternative to tocilizumab could be siltuximab as it binds to soluble IL-6 (83) and limits IL-6 increased levels in the CSF (6), but its clinical application would require further studies.
2.3.2. Corticosteroids
Contrasting with IL-6, blockade corticosteroids are recommended in managing ICANS (2) as they will induce rapid and profound systemic anti-inflammatory function by blocking cytokine signaling or adhesion molecules and even induce apoptosis of immune cells (84). However, clinical studies have observed CAR-T cell activity limitation with high-dose corticosteroids in patients with severe ICANS (85–87). More controlled administration of corticosteroids at an earlier setting to limit the length of exposure sometimes showed no detrimental effect on CAR-T cell's efficacy (30, 88, 89). If further studies are needed to describe the potentially detrimental effect of corticosteroids more robustly in CAR-T cells, its use in clinics will not be limited as it is, to date, the most recommended therapy to control ICANS (2). Alternatively, CAR-T cells can be engineered to resist the immunosuppressive effect of corticosteroids. Especially for GBM, where corticosteroids are a cornerstone for managing tumor symptoms, a CAR targeting the IL-13 receptor α2 was further engineered by disrupting the glucocorticoid receptor. Preliminary results seem to show maintained CAR-T cell activity even with a concomitant high dose of dexamethasone (90).
2.3.3. IL-1 blockade
Because IL-1 plays a central role in ICANS, its blockade could offer more precise control of the symptoms and prevent the too-wide anti-inflammatory effects of corticosteroids. Anakinra, an IL-1 receptor antagonist, can penetrate the BBB (56, 91) and is approved for immunological diseases such as Still's disease (92), rheumatoid arthritis (93), and macrophage activation syndrome (94). In recent mouse pre-clinical studies, anakinra better limited neurotoxicity and brain meningeal thickening compared to tocilizumab, without impairing CAR-T cell functionality (25, 31). Many clinical trials are evaluating the potential role of anakinra in limiting the incidence and severity of ICANS (95), and a preliminary study with eight patients observed ICANS control in 50% of them (96).
3. Discussion and perspectives
3.1. GM-CSF
As myeloid cells, and particularly macrophages, are hypothesized to be the critical cell subtype at the origin of CRS by producing IL-6, blocking their maturation and activation with GM-CSF neutralization have been evaluated. In a mouse model, the use of lenzilumab, an anti-GM-CSF Ab, showed reduced CAR-T cell and myeloid cell infiltration to the CNS without compromising CAR-T cell function and efficacy (40).
3.2. IFN-γ
As another approach, inhibition of IFN-γ production with Janus kinase (JAK) inhibitors such as ruxolitinib (97) or itacitinib (98), or with Bruton's tyrosine kinase (BTK) inhibitor ibrutinib (99), all limited CAR-T cell efficacy, even though they induced ICANS remission. A monoclonal IFN-γ blocking Ab emapalumab (100) is available, and may also help to better understand the role of IFN-γ in the development of ICANS.
3.3. Adhesion molecules
As VLA4 was described to be overexpressed on CAR-T cells infiltrating the CNS compared to non-CAR-T cells, the blocking Ab natalizumab (101) was evaluated in mice and could prevent CAR-T cell CNS infiltration and reduce inflammation.
3.4. CAR constructs
As CAR-T cell proliferation itself is a factor of therapy response but generates cytokine secretion, CAR-T cells are themselves a causative factor for related toxicities. Redesigning CAR constructs by modulating non-signaling domains, including hinge and transmembrane regions or the scFv, may limit the induction of toxicities (102, 103). Editing the scFv for less affinity or with fully human components could retain efficacy while inducing less severe CRS (29, 104). Co-stimulatory domains are also determinant, as CD28 is associated with earlier and potentially more severe CRS than with 4-1BB (30, 105). Nevertheless, reports seem to indicate that JCAR014, a CD19 CAR-T cell with 4-1BB co-stimulatory domain, induces fewer ICANS than other CD19 CAR-T cells (106). It is hypostatized that infusion of equal numbers of CD4 and CD8, which is controlled for JCAR014, could be responsible for this difference, indicating the CD4/CD8 infusion ratio as a determinant factor for ICANS development.
Controlling CAR-T cells already infused in patients through “kill switches” or reversible control of their activity is in development. Adding cell surface proteins such as EGFR or CD20 to target them with cetuximab or rituximab for destruction could be implemented (107), but it would lead to toxicity related to these therapies. Other strategies to embed suicide genes, such as apoptosis inducers with specific triggers toward CAR products, could allow them to induce their self-destruction with a precise signal (108), but CAR constructs are limited in length and may limit the addition of multiple additional systems.
4. Conclusion
ICANS are now better defined and reported, allowing a real-life grasp of their clinical implication. Improved understanding of mechanisms implicated during ICANS through experimental observations in clinics and with the development of pre-clinical models led to the development of many new strategies to tackle ICANS, as summarized in Table 1 and Figure 1. Nevertheless, some points are not fully understood such as the fact that CAR-T cell studies for solid tumors reported almost no ICANS events in ovary, sarcoma, and glioblastoma trials (7, 8, 109, 110).
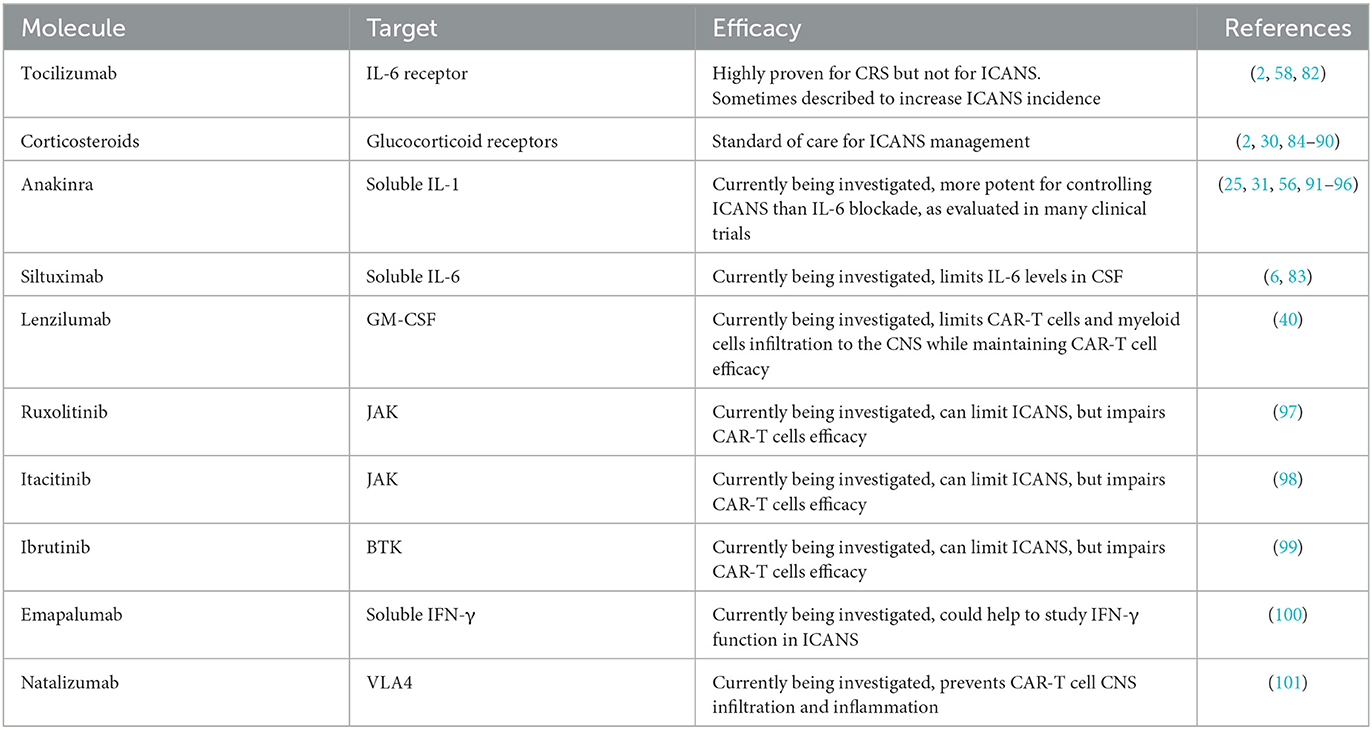
Table 1. Strategies for ICANS management.
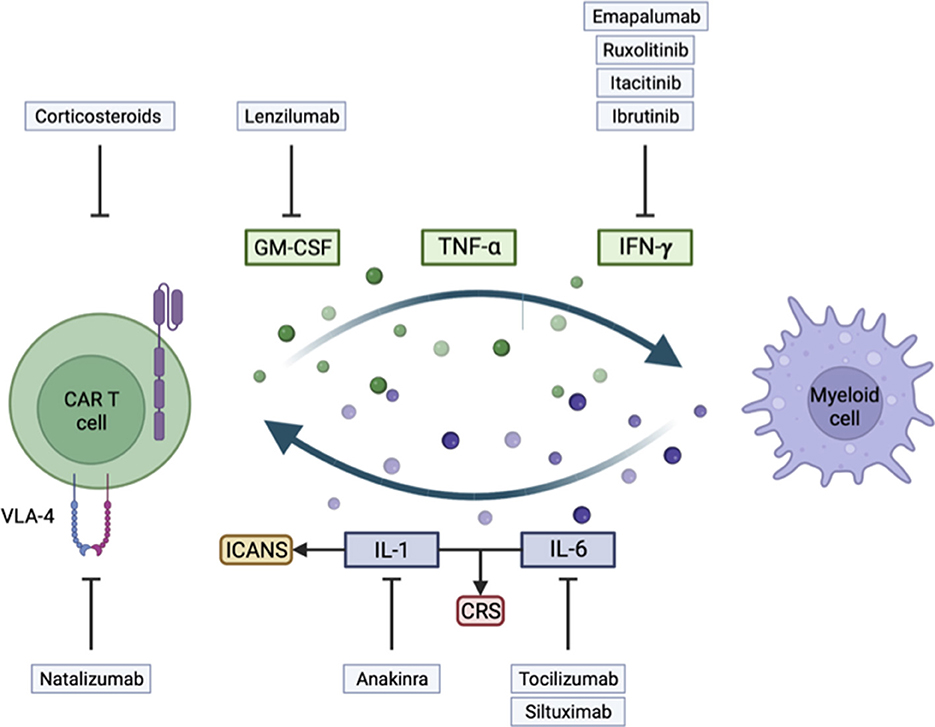
Figure 1. Illustration of key mechanisms involved in the development of immune effector cell-associated neurotoxicity syndrome (ICANS) and cytokine release syndrome (CRS), and their corresponding treatment strategy.
Better control of CAR-T cell toxicities would probably be the subsequent critical development for broader application, as studies are now evaluating the application of CAR therapies in non-cancerous diseases. We now see development in Ag-specific regulatory T cells to tackle autoimmune diseases (111–114) or CAR-T cells to control organ transplant tolerance (18). In such a situation, on a benefits/risk balance, the weight or risks would have to be well pondered, as high toxicities will be less acceptable.
With broader applications and new CAR subsets coming to the clinic, other toxicities will also be observed and will need further adaptation. We already foresee delayed toxicities with BCMA CAR-T cells inducing parkinsonism-like symptoms in some patients (81), which is not described with the most common CAR-T product to date targeting CD19. CD22 CAR-T cells also seem to drive toxicities at a later time (115) than CD19 CAR-T cells.
Altogether, careful monitoring of patients with current or future CAR-T cell therapies is warranted to allow prompt management and adaptation to unexpected toxicities, with more robust and anticipated management of CAR-T cells toxicities and ICANS in particular, broader application will be facilitated.
Author contributions
VG and DM wrote, edited, and approved the final version of the manuscript. DM supervised the writing. All authors contributed to the article and approved the submitted version.
Funding
Open access funding provided by University of Geneva. DM was funded by the ISREC Foundation, Innosuisse, Swiss National Science Foundation, Swiss Bridge Foundation, PHRT ETH Zurich Foundation, and Ligue Genevoise contre le cancer.
Conflict of interest
DM is an inventor of patents filed by the University of Pennsylvania and the University of Geneva in the field of cell and gene therapy.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Greenbaum U, Kebriaei P, Srour SA, Olson A, Bashir Q, Neelapu SS, et al. Chimeric antigen receptor T-cell therapy toxicities. Br J Clin Pharmacol. (2021) 87:2414–24. doi: 10.1111/bcp.14403
2. Hayden PJ, Roddie C, Bader P, Basak GW, Bonig H, Bonini C, et al. Management of adults and children receiving CAR-T cell therapy: 2021 best practice recommendations of the european society for blood and marrow transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE) and the European Haematology Association (EHA). Ann Oncol. (2021) 33:259–75. doi: 10.1016/j.annonc.2021.12.003
3. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. (2020). 368:6494. doi: 10.1126/science.aaz7548
4. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-mediated target cell pyroptosis by CAR-T cells triggers cytokine release syndrome. Sci Immunol. (2020) 5:eaax7969. doi: 10.1126/sciimmunol.aax7969
5. Zhang J, Su D, Liu Q, Yuan Q, Ouyang Z, Wei Y, et al. Gasdermin D-mediated microglial pyroptosis exacerbates neurotoxicity of aflatoxins B1 and M1 in mouse primary microglia and neuronal cultures. Neurotoxicology. (2022) 91:305–20. doi: 10.1016/j.neuro.2022.06.003
6. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy — assessment and management of toxicities. Nat Rev Clin Oncol. (2018) 15:47–62. doi: 10.1038/nrclinonc.2017.148
7. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor t-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
8. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. (2015) 7:275ra22. doi: 10.1126/scitranslmed.aaa4963
9. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow. (2019) 25:625–38. doi: 10.1016/j.bbmt.2018.12.758
10. Rubin DB, Danish HH, Ali AB Li K, Larose S, Monk AD, et al. Neurological toxicities associated with chimeric antigen receptor T-cell therapy. Brain. (2019) 142:1334–48. doi: 10.1093/brain/awz053
11. Pensato U, Muccioli L, Cani I, Janigro D, Zinzani PL, Guarino M, et al. Brain dysfunction in COVID-19 and CAR-T therapy: cytokine storm-associated encephalopathy. Ann Clin Transl Neurol. (2021) 8:968–79. doi: 10.1002/acn3.51348
12. Torre M, Solomon IH, Sutherland CL, Nikiforow S, DeAngelo DJ, Stone RM, et al. Neuropathology of a case with fatal CAR-T cell-associated cerebral edema. J Neuropathol Exp Neurol. (2018) 77:877–82. doi: 10.1093/jnen/nly064
13. Gust J, Hay KA, Hanafi L-A, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood–brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. (2017) 7:1404–19. doi: 10.1158/2159-8290.CD-17-0698
14. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. (2017) 377:2545–54. doi: 10.1056/NEJMoa1708566
15. Brown BD, Tambaro FP, Kohorst M, Chi L, Mahadeo KM, Tewari P, et al. Immune effector cell associated neurotoxicity (ICANS) in pediatric and young adult patients following chimeric antigen receptor (CAR) T-cell therapy: can we optimize early diagnosis? Front Oncol. (2021) 11:634445. doi: 10.3389/fonc.2021.634445
16. Maillet D, Belin C, Moroni C, Cuzzubbo S, Ursu R, Sirven-Villaros L, et al. Evaluation of mid-term (6-12 months) neurotoxicity in B-cell lymphoma patients treated with CAR-T cells: a prospective cohort study. Neuro Oncol. (2021) 23:1569–75. doi: 10.1093/neuonc/noab077
17. Hong R, Hu Y, Huang H. Biomarkers for chimeric antigen receptor T cell therapy in acute lymphoblastic leukemia: prospects for personalized management and prognostic prediction. Front Immunol. (2021) 12:627764. doi: 10.3389/fimmu.2021.627764
18. Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. (2022) 22:85–96. doi: 10.1038/s41577-021-00547-6
19. Strati P, Nastoupil LJ, Westin J, Fayad LE, Ahmed S, Fowler NH, et al. Clinical and radiologic correlates of neurotoxicity after axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. (2020) 4:3943–51. doi: 10.1182/bloodadvances.2020002228
20. Gust J, Ponce R, Liles WC, Garden GA, Turtle CJ. Cytokines in CAR-T cell-associated neurotoxicity. Front Immunol. (2020) 11:577027. doi: 10.3389/fimmu.2020.577027
21. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR-T cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. (2018) 8:958–71. doi: 10.1158/2159-8290.CD-17-1319
22. Maziarz RT, Schuster SJ, Romanov VV, Rusch ES Li J, Signorovitch JE, et al. Grading of neurological toxicity in patients treated with tisagenlecleucel in the JULIET trial. Blood Adv. (2020) 4:1440–7. doi: 10.1182/bloodadvances.2019001305
23. Giavridis T, Van Der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M, et al. cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. (2018) 24:731–8. doi: 10.1038/s41591-018-0041-7
24. Nijmeijer BA, Willemze R, Falkenburg JH. An animal model for human cellular immunotherapy: specific eradication of human acute lymphoblastic leukemia by cytotoxic T lymphocytes in NOD/scid mice. Blood. (2002) 100:654–60. doi: 10.1182/blood.V100.2.654
25. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR-T cells. Nat Med. (2018) 24:739–48. doi: 10.1038/s41591-018-0036-4
26. Siegler EL, Wang P. Preclinical models in chimeric antigen receptor-engineered T-cell therapy. Hum Gene Ther. (2018) 29:534–46. doi: 10.1089/hum.2017.243
27. Mastaglio S, Genovese P, Magnani Z, Ruggiero E, Landoni E, Camisa B, et al. NY-ESO-1 TCR single edited stem and central memory T cells to treat multiple myeloma without graft-versus-host disease. Blood. (2017) 130:606–18. doi: 10.1182/blood-2016-08-732636
28. Bondanza A, Valtolina V, Magnani Z, Ponzoni M, Fleischhauer K, Bonyhadi M, et al. Suicide gene therapy of graft-versus-host disease induced by central memory human T lymphocytes. Blood. (2006) 107:1828–36. doi: 10.1182/blood-2005-09-3716
29. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
30. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR-T cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
31. Gottschlich A, Endres S, Kobold S. Therapeutic strategies for targeting IL-1 in cancer. Cancers (Basel). (2021) 13:477. doi: 10.3390/cancers13030477
32. Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. (2013) 13:759–71. doi: 10.1038/nrc3611
33. Bradley JR. TNF-mediated inflammatory disease. J Pathol. (2008) 214:149–60. doi: 10.1002/path.2287
34. Pennica D, Nedwin GE, Hayflick JS, Seeburg PH, Derynck R, Palladino MA, et al. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature. (1984) 312:724–9. doi: 10.1038/312724a0
35. Michie J, Beavis PA, Freeman AJ, Vervoort SJ, Ramsbottom KM, Narasimhan V, et al. Antagonism of IAPs enhances CAR-T cell efficacy. Cancer Immunol Res. (2019) 7:183–92. doi: 10.1158/2326-6066.CIR-18-0428
36. Becher B, Tugues S, Greter M, GM-CSF. From growth factor to central mediator of tissue inflammation. Immunity. (2016) 45:963–73. doi: 10.1016/j.immuni.2016.10.026
37. Hao Z, Li R, Meng L, Han Z, Hong Z. Macrophage, the potential key mediator in CAR-T related CRS. Exp Hematol Oncol. (2020) 9:15. doi: 10.1186/s40164-020-00171-5
38. Spear P, Barber A, Rynda-Apple A, Sentman CL. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-gamma and GM-CSF. J Immunol. (2012) 188:6389–98. doi: 10.4049/jimmunol.1103019
39. Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
40. Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. (2019) 133:697–709. doi: 10.1182/blood-2018-10-881722
41. Profaci CP, Munji RN, Pulido RS, Daneman R. The blood-brain barrier in health and disease: Important unanswered questions. J Exp Med. (2020) 217:e20190062. doi: 10.1084/jem.20190062
42. Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. (2019) 176:1248–64. doi: 10.1016/j.cell.2019.01.021
43. Leligdowicz A, Richard-Greenblatt M, Wright J, Crowley VM, Kain KC. Endothelial activation: the Ang/Tie axis in sepsis. Front Immunol. (2018) 9:838. doi: 10.3389/fimmu.2018.00838
44. Page AV, Liles WC. Biomarkers of endothelial activation/dysfunction in infectious diseases. Virulence. (2013) 4:507–16. doi: 10.4161/viru.24530
45. Schwameis M, Schorgenhofer C, Assinger A, Steiner MM, Jilma B, VWF. excess and ADAMTS13 deficiency: a unifying pathomechanism linking inflammation to thrombosis in DIC, malaria, and TTP. Thromb Haemost. (2015) 113:708–18. doi: 10.1160/TH14-09-0731
46. Witzenbichler B, Westermann D, Knueppel S, Schultheiss HP, Tschope C. Protective role of angiopoietin-1 in endotoxic shock. Circulation. (2005) 111:97–105. doi: 10.1161/01.CIR.0000151287.08202.8E
47. Savarin C, Hinton DR, Valentin-Torres A, Chen Z, Trapp BD, Bergmann CC, et al. Astrocyte response to IFN-gamma limits IL-6-mediated microglia activation and progressive autoimmune encephalomyelitis. J Neuroinflammation. (2015) 12:79. doi: 10.1186/s12974-015-0293-9
48. Siegler EL, Kenderian SS. Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: insights into mechanisms and novel therapies. Front Immunol. (2020) 11:1973. doi: 10.3389/fimmu.2020.01973
49. Dalal PJ, Muller WA, Sullivan DP. Endothelial cell calcium signaling during barrier function and inflammation. Am J Pathol. (2020) 190:535–42. doi: 10.1016/j.ajpath.2019.11.004
50. Kang S, Kishimoto T. Interplay between interleukin-6 signaling and the vascular endothelium in cytokine storms. Exp Mol Med. (2021) 53:1116–23. doi: 10.1038/s12276-021-00649-0
51. Garbuzova-Davis S, Ehrhart J, Sanberg PR, Borlongan CV. Potential role of humoral IL-6 cytokine in mediating pro-inflammatory endothelial cell response in amyotrophic lateral sclerosis. Int J Mol Sci. (2018) 19:423. doi: 10.3390/ijms19020423
52. Chai Q, He WQ, Zhou M, Lu H, Fu ZF. Enhancement of blood-brain barrier permeability and reduction of tight junction protein expression are modulated by chemokines/cytokines induced by rabies virus infection. J Virol. (2014) 88:4698–710. doi: 10.1128/JVI.03149-13
53. Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta. (2009) 1788:864–71. doi: 10.1016/j.bbamem.2008.08.027
54. Hosomi N, Ban CR, Naya T, Takahashi T, Guo P, Song XY, et al. Tumor necrosis factor-alpha neutralization reduced cerebral edema through inhibition of matrix metalloproteinase production after transient focal cerebral ischemia. J Cereb Blood Flow Metab. (2005) 25:959–67. doi: 10.1038/sj.jcbfm.9600086
55. Rama Rao KV, Jayakumar AR, Tong X, Alvarez VM, Norenberg MD. Marked potentiation of cell swelling by cytokines in ammonia-sensitized cultured astrocytes. J Neuroinflammation. (2010) 7:66. doi: 10.1186/1742-2094-7-66
56. Galea J, Ogungbenro K, Hulme S, Greenhalgh A, Aarons L, Scarth S, et al. Intravenous anakinra can achieve experimentally effective concentrations in the central nervous system within a therapeutic time window: results of a dose-ranging study. J Cereb Blood Flow Metab. (2011) 31:439–47. doi: 10.1038/jcbfm.2010.103
57. Sachdeva M, Duchateau P, Depil S, Poirot L, Valton J. Granulocyte-macrophage colony-stimulating factor inactivation in CAR-T cells prevents monocyte-dependent release of key cytokine release syndrome mediators. J Biol Chem. (2019) 294:5430–7. doi: 10.1074/jbc.AC119.007558
58. Locke FL, Neelapu SS, Bartlett NL, Lekakis LJ, Jacobson CA, Braunschweig I, et al. Preliminary results of prophylactic tocilizumab after axicabtageneciloleucel (axi-cel; KTE-C19) treatment for patients with refractory, aggressive non-hodgkin lymphoma (NHL). Blood. (2017) 130:1547. doi: 10.1182/blood.V130.Suppl_1.1547.1547
59. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
60. DeAngelo DJ, Ghobadi A, Park JH, Dinner SN, Mannis GN, Lunning MA, et al. Clinical outcomes for the phase 2, single-arm, multicenter trial of JCAR015 in adult B-ALL (ROCKET Study). J Immunother Cancer. (2017).
61. Staedtke V, Bai RY, Kim K, Darvas M, Davila ML, Riggins GJ, et al. Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature. (2018) 564:273–7. doi: 10.1038/s41586-018-0774-y
62. Bao JY, Huang Y, Wang F, Peng YP, Qiu YH. Expression of alpha-AR subtypes in T lymphocytes and role of the alpha-ARs in mediating modulation of T cell function. Neuroimmunomodulation. (2007) 14:344–53. doi: 10.1159/000129670
63. Tang JP, Peters CW, Quiros C, Wang X, Klomhaus AM, Yamada RE, et al. Hypophosphatemia due to increased effector cell metabolic activity is associated with neurotoxicity symptoms in CD19-targeted CAR-T cell therapy. Cancer Immunol Res. (2022) 10:1433–40. doi: 10.1158/2326-6066.CIR-22-0418
64. Miao L, Zhang Z, Ren Z, Li Y. Reactions related to CAR-T cell therapy. Front Immunol. (2021) 12:663201. doi: 10.3389/fimmu.2021.663201
65. Deng Q, Han G, Puebla-Osorio N, Ma MCJ, Strati P, Chasen B, et al. Characteristics of anti-CD19 CAR-T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat Med. (2020) 26:1878–87. doi: 10.1038/s41591-020-1061-7
66. Danish H, Santomasso BD. Neurotoxicity biology and management. Cancer J. (2021) 27:126–33. doi: 10.1097/PPO.0000000000000507
67. Kim HJ, Tsao JW, Stanfill AG. The current state of biomarkers of mild traumatic brain injury. JCI Insight. (2018) 3:e97105. doi: 10.1172/jci.insight.97105
68. Sofroniew MV. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist. (2014) 20:160–72. doi: 10.1177/1073858413504466
69. Gust J, Finney OC Li D, Brakke HM, Hicks RM, Futrell RB, et al. Glial injury in neurotoxicity after pediatric CD19-directed chimeric antigen receptor T cell therapy. Ann Neurol. (2019) 86:42–54. doi: 10.1002/ana.25502
70. Wolf SA, Boddeke HW, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol. (2017) 79:619–43. doi: 10.1146/annurev-physiol-022516-034406
71. Ottum PA, Arellano G, Reyes LI, Iruretagoyena M, Naves R. Opposing roles of interferon-gamma on cells of the central nervous system in autoimmune neuroinflammation. Front Immunol. (2015) 6:539. doi: 10.3389/fimmu.2015.00539
72. Clark IA, Vissel B. Excess cerebral TNF causing glutamate excitotoxicity rationalizes treatment of neurodegenerative diseases and neurogenic pain by anti-TNF agents. J Neuroinflammation. (2016) 13:236. doi: 10.1186/s12974-016-0708-2
73. Conroy SM, Nguyen V, Quina LA, Blakely-Gonzales P, Ur C, Netzeband JG, et al. Interleukin-6 produces neuronal loss in developing cerebellar granule neuron cultures. J Neuroimmunol. (2004) 155:43–54. doi: 10.1016/j.jneuroim.2004.06.014
74. Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia. (2006) 47:1761–74. doi: 10.1111/j.1528-1167.2006.00817.x
75. Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer's disease. Nat Commun. (2020) 11:6129. doi: 10.1038/s41467-020-19737-2
76. Del Principe MI, Buccisano F, Soddu S, Maurillo L, Cefalo M, Piciocchi A, et al. Involvement of central nervous system in adult patients with acute myeloid leukemia: Incidence and impact on outcome. Semin Hematol. (2018) 55:209–14. doi: 10.1053/j.seminhematol.2018.02.006
77. Dyson A, Singer M. Animal models of sepsis: why does pre-clinical efficacy fail to translate to the clinical setting? Crit Care Med. (2009) 37:S30–7. doi: 10.1097/CCM.0b013e3181922bd3
78. Parker KR, Migliorini D, Perkey E, Yost KE, Bhaduri A, Bagga P, et al. Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell. (2020) 183:126–42 e17. doi: 10.1016/j.cell.2020.08.022
79. Pluvinage JV, Haney MS, Smith BAH, Sun J, Iram T, Bonanno L, et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature. (2019) 568:187–92. doi: 10.1038/s41586-019-1088-4
80. Dorovini-Zis K, Schmidt K, Huynh H, Fu W, Whitten RO, Milner D, et al. The neuropathology of fatal cerebral malaria in malawian children. Am J Pathol. (2011) 178:2146–58. doi: 10.1016/j.ajpath.2011.01.016
81. Van Oekelen O, Aleman A, Upadhyaya B, Schnakenberg S, Madduri D, Gavane S, et al. Neurocognitive and hypokinetic movement disorder with features of parkinsonism after BCMA-targeting CAR-T cell therapy. Nat Med. (2021) 27:2099–103. doi: 10.1038/s41591-021-01564-7
82. Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, Kakehi T. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood. (2008) 112:3959–64. doi: 10.1182/blood-2008-05-155846
83. Chen F, Teachey DT, Pequignot E, Frey N, Porter D, Maude SL, et al. Measuring IL-6 and sIL-6R in serum from patients treated with tocilizumab and/or siltuximab following CAR-T cell therapy. J Immunol Methods. (2016) 434:1–8. doi: 10.1016/j.jim.2016.03.005
84. Timmermans S, Souffriau J, Libert C, A. General Introduction to Glucocorticoid Biology. Front Immunol. (2019) 10:1545. doi: 10.3389/fimmu.2019.01545
85. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR-T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. (2014) 6:224ra25. doi: 10.1126/scitranslmed.3008226
86. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. (2013) 5:177ra38. doi: 10.1126/scitranslmed.3005930
87. Strati P, Ahmed S, Furqan F, Fayad LE, Lee HJ, Iyer SP, et al. Prognostic impact of corticosteroids on efficacy of chimeric antigen receptor T-cell therapy in large B-cell lymphoma. Blood. (2021) 137:3272–6. doi: 10.1182/blood.2020008865
88. Topp M, Van Meerten T, Houot R, Minnema MC, Milpied N, Lugtenburg PJ, et al. Earlier steroid use with axicabtagene ciloleucel (Axi-Cel) in patients with relapsed/refractory large B cell lymphoma. Blood. (2019) 134:243. doi: 10.1182/blood-2019-126081
89. Strati P, Furqan F, Westin J, Fayad L, Ahmed S, Lee HJ, et al. Prognostic impact of dose, duration, and timing of corticosteroid therapy in patients with large B-cell lymphoma treated with standard of care axicabtagene ciloleucel (Axi-cel). Am Soc Clinical Oncol. (2020). doi: 10.1200/JCO.2020.38.15_suppl.8011
90. Brown CE, Rodriguez A, Palmer J, Ostberg JR, Naranjo A, Wagner JR, et al. Off-the-shelf, steroid-resistant, IL13Ralpha2-specific CAR-T cells for treatment of glioblastoma. Neuro Oncol. (2022) 24:1318–30. doi: 10.1093/neuonc/noac024
91. Gutierrez EG, Banks WA, Kastin AJ. Blood-borne interleukin-1 receptor antagonist crosses the blood-brain barrier. J Neuroimmunol. (1994) 55:153–60. doi: 10.1016/0165-5728(94)90005-1
92. Vastert SJ, Jamilloux Y, Quartier P, Ohlman S, Osterling Koskinen L, Kullenberg T, et al. Anakinra in children and adults with Still's disease. Rheumatology (Oxford). (2019) 58:vi9–vi22. doi: 10.1093/rheumatology/kez350
93. Ramirez J, Canete JD. Anakinra for the treatment of rheumatoid arthritis: a safety evaluation. Expert Opin Drug Saf. (2018) 17:727–32. doi: 10.1080/14740338.2018.1486819
94. Sonmez HE, Demir S, Bilginer Y, Ozen S. Anakinra treatment in macrophage activation syndrome: a single center experience and systemic review of literature. Clin Rheumatol. (2018) 37:3329–35. doi: 10.1007/s10067-018-4095-1
95. ClinicalTrials.gov. Available online at: https://clinicaltrials.gov/ct2/results?cond=CAR-T&term=Anakinra&cntry=&state=&city=&dist= (accessed November 10, 2022).
96. Strati P, Ahmed S, Kebriaei P, Nastoupil LJ, Claussen CM, Watson G, et al. Clinical efficacy of anakinra to mitigate CAR-T cell therapy-associated toxicity in large B-cell lymphoma. Blood Adv. (2020) 4:3123–7. doi: 10.1182/bloodadvances.2020002328
97. Pan J, Deng B, Ling Z, Song W, Xu J, Duan J, et al. Ruxolitinib mitigates steroid-refractory CRS during CAR-T therapy. J Cell Mol Med. (2021) 25:1089–99. doi: 10.1111/jcmm.16176
98. Huarte E, O'Connor RS, Peel MT, Nunez-Cruz S, Leferovich J, Juvekar A, et al. Itacitinib (INCB039110), a JAK1 Inhibitor, reduces cytokines associated with cytokine release syndrome induced by CAR-T cell therapy. Clin Cancer Res. (2020) 26:6299–309. doi: 10.1158/1078-0432.CCR-20-1739
99. Ruella M, Kenderian SS, Shestova O, Klichinsky M, Melenhorst JJ, Wasik MA, et al. Kinase inhibitor ibrutinib to prevent cytokine-release syndrome after anti-CD19 chimeric antigen receptor T cells for B-cell neoplasms. Leukemia. (2017) 31:246–8. doi: 10.1038/leu.2016.262
100. Henter JI, von Bahr Greenwood T, Bergsten E. Emapalumab in primary hemophagocytic lymphohistiocytosis. N Engl J Med. (2020) 383:596–8. doi: 10.1056/NEJMc2020754
101. Khoy K, Mariotte D, Defer G, Petit G, Toutirais O, Le Mauff B. Natalizumab in multiple sclerosis treatment: from biological effects to immune monitoring. Front Immunol. (2020) 11:549842. doi: 10.3389/fimmu.2020.549842
102. Brudno JN, Lam N, Vanasse D, Shen YW, Rose JJ, Rossi J, et al. Safety and feasibility of anti-CD19 CAR-T cells with fully human binding domains in patients with B-cell lymphoma. Nat Med. (2020) 26:270–80. doi: 10.1038/s41591-019-0737-3
103. Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A safe and potent anti-CD19 CAR-T cell therapy. Nat Med. (2019) 25:947–53. doi: 10.1038/s41591-019-0421-7
104. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR-T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. (2019) 25:1408–14. doi: 10.1038/s41591-019-0549-5
105. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
106. Gauthier J, Gazeau N, Hirayama AV, Hill JA, Wu V, Cearley A, et al. Impact of CD19 CAR-T cell product type on outcomes in relapsed or refractory aggressive B-NHL. Blood. (2022) 139:3722–31. doi: 10.1182/blood.2021014497
107. Paszkiewicz PJ, Frassle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, et al. Targeted antibody-mediated depletion of murine CD19 CAR-T cells permanently reverses B cell aplasia. J Clin Invest. (2016) 126:4262–72. doi: 10.1172/JCI84813
108. Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, et al. Inducible caspase-9 selectively modulates the toxicities of cd19-specific chimeric antigen receptor-modified T cells. Mol Ther. (2017) 25:580–92. doi: 10.1016/j.ymthe.2017.01.011
109. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. (2006) 12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183
110. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified t cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. (2015) 33:1688–96. doi: 10.1200/JCO.2014.58.0225
111. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. (2017) 17:931–43. doi: 10.1111/ajt.14185
112. Rosado-Sanchez I, Levings MK. Building a CAR-Treg: going from the basic to the luxury model. Cell Immunol. (2020) 358:104220. doi: 10.1016/j.cellimm.2020.104220
113. Dawson NAJ, Rosado-Sanchez I, Novakovsky GE, Fung VCW, Huang Q, McIver E, et al. Functional effects of chimeric antigen receptor co-receptor signaling domains in human regulatory T cells. Sci Transl Med. (2020) 12:557. doi: 10.1126/scitranslmed.aaz3866
114. Boroughs AC, Larson RC, Choi BD, Bouffard AA, Riley LS, Schiferle E, et al. Chimeric antigen receptor co-stimulation domains modulate human regulatory T cell function. JCI insight. (2019) 4:8. doi: 10.1172/jci.insight.126194
Keywords: immune effector cell-associated neurotoxicity syndrome (ICANS), neurotoxicity, cellular therapies, chimeric antigen receptor (CAR) T cells, cytokine release syndrome (CRS)
Citation: Genoud V and Migliorini D (2023) Novel pathophysiological insights into CAR-T cell associated neurotoxicity. Front. Neurol. 14:1108297. doi: 10.3389/fneur.2023.1108297
Received: 25 November 2022; Accepted: 03 February 2023;
Published: 08 March 2023.
Edited by:
Leonard Verhagen Metman, Rush University, United StatesReviewed by:
Manish Malviya, Memorial Sloan Kettering Cancer Center, United StatesUmberto Pensato, Humanitas Research Hospital, Italy
Copyright © 2023 Genoud and Migliorini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Denis Migliorini, ZGVuaXMubWlnbGlvcmluaUB1bmlnZS5jaA==