
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 09 March 2023
Sec. Epilepsy
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1092887
This article is part of the Research TopicEpilepsy – Case Report Collection 2022View all 13 articles
 Ikhlas Ben Ayed1,2Olfa Jallouli3,4
Ikhlas Ben Ayed1,2Olfa Jallouli3,4 Yoshiko Murakami5
Yoshiko Murakami5 Amal Souissi1
Amal Souissi1 Salma Mallouli3,4
Salma Mallouli3,4 Amal Bouzid1
Amal Bouzid1 Fatma Kamoun3,4Ines Elloumi1
Fatma Kamoun3,4Ines Elloumi1 Fakher Frikha1
Fakher Frikha1 Abdelaziz Tlili6,7
Abdelaziz Tlili6,7 Sarah Weckhuysen8
Sarah Weckhuysen8 Taroh Kinoshita5,9
Taroh Kinoshita5,9 Chahnez Charfi Triki3,4Saber Masmoudi1*
Chahnez Charfi Triki3,4Saber Masmoudi1*Pathogenic germline variants in the PIGT gene are associated with the “multiple congenital anomalies–hypotonia-seizures syndrome 3” (MCAHS3) phenotype. So far, fifty patients have been reported, most of whom suffer from intractable epilepsy. Recently, a comprehensive analysis of a cohort of 26 patients with PIGT variants has broadened the phenotypical spectrum and indicated that both p.Asn527Ser and p.Val528Met are associated with a milder epilepsy phenotype and less severe outcomes. Since all reported patients are of Caucasian/Polish origin and most harbor the same variant (p.Val528Met), the ability to draw definitive conclusions regarding the genotype–phenotype correlation remains limited. We report a new case with a homozygous variant p.Arg507Trp in the PIGT gene, detected on clinical exome sequencing. The North African patient in question displays a predominantly neurological phenotype with global developmental delay, hypotonia, brain abnormalities, and well-controlled epileptic seizures. Homozygous and heterozygous variants in codon 507 have been reported to cause PIGT deficiency without biochemical confirmation. In this study, FACS analysis of knockout HEK293 cells that had been transfected with wild-type or mutant cDNA constructs demonstrated that the p.Arg507Trp variant leads to mildly reduced activity. Our result confirm the pathogenicity of this variant and strengthen recently reported evidence on the genotype–phenotype correlation of the PIGT variant.
It is estimated that 10–20% of all membrane proteins are post-translationally modified at their C-terminus by glycosylphosphatidylinositol (GPI), a complex glycophospholipid that anchors over 150 proteins to the cell surface. The transfer of the GPI anchor to proteins is catalyzed by the GPI transamidase (GPI-TA) complex (1, 2). Phosphatidylinositol glycan anchor biosynthesis class T (PIGT) is a highly conserved 578-amino-acid protein. PIGT interacts with other components of the GPI-TA complex (PIGK, PIGS, PIGT, PIGU, and PGAA1) and appears to be the most important protein in the formation of this complex and the stability of the other components (3, 4). Disorders caused by germline pathogenic variants in PareT are referred to as multiple congenital anomalies–hypotonia-seizures syndrome 3 (MCAHS3, OMIM#615398). This syndrome is characterized by congenital hypotonia, global developmental delay (GDD) or intellectual disability (ID), and infantile-onset epilepsy with various types of epileptic EEG abnormalities (focal sharp-slow wave, focal sharp spike/polyspikes wave, and generalized polyspikes-wave complexes) (5, 6). Other congenital anomalies involving dysmorphic facial features, cerebral and cerebellar atrophy, and defects in the skeletal, ophthalmological, cardiac, and genitourinary systems are also reported (7–9).
Up to now, 50 patients with PIGT deficiency, most of them Caucasian, have been diagnosed with 17 pathogenic variants, including 10 missense, one non-sense, four frameshifts, and two splice sites. The largest extant genotype–phenotype study, reviewed by Bayat et al. reports on 26 patients with the p.Val528Met variant in either homozygous or compound heterozygous state and on a single patient who had the p.Asn527Ser variant (10, 11). These patients presented with moderate to severe GDD and later onset of epilepsy, and generally became seizure-free on monotherapy. Since all reported patients thus far have been of Caucasian/Polish origin and most harbor the same variant (p.Val528Met), the ability to draw definitive conclusions regarding the genotype–phenotype correlation remains limited. Here, we provide additional findings supporting this correlation by reporting a new case from North Africa, of a patient carrying the PIGT p.Arg507Trp variant located in the C-terminal region and associated with GDD and partially tractable epilepsy. Functional studies using PIGT knockout HEK293 cells showed mildly decreased activity of the GPI-TA complex, supporting the pathogenicity of the variant and providing further evidence of an association between the localization of variants in the C-terminal region and the moderate epilepsy phenotype (seizure-free or partially seizure-free on treatment).
We reviewed the patient's personal history regarding progress of the pregnancy and childbirth, psychomotor milestone achievement, and neurological examinations. We also reviewed the history of epileptic seizures (type, age of onset, response to antiseizure medication), electroencephalographic data (background, epileptiform discharges, epileptic seizure if recorded), and brain imaging data, as well as the etiological assessment.
For the genetic study, DNA was isolated from peripheral blood and clinical exome sequencing was performed using the TruSight™ One Sequencing Panel. Libraries were prepared and data analysis was performed as previously described (12). The generated VCF file was annotated using the VarAft application (13). A virtual gene panel was applied using the Genetic Epilepsy Syndromes panel from Genomics England PanelApp (https://panelapp.genomicsengland.co.uk/panels/). The variant-filtering process consisted of the following four steps: (i) selection of variants localized in exons or bordering introns (±12 bp); (ii) exclusion of variants with a frequency above 1% in general populations; (iii) selection of variants predicted as pathogenic by CADD; and (iv) selection of variants predicted as pathogenic by the UMD-Predictor tool. In-silico pathogenicity was also evaluated using the VarCards tool (http://varcards.biols.ac.cn/). The preferentially selected variants were validated and familial co-segregation was analyzed via Sanger sequencing.
The study protocol was approved by the local medical ethics committee of South Tunisia (Accession number 28/2019). Written informed consent was obtained from the patient's parents.
PIGT-knockout HEK293 cells were generated and transfected, as described previously, with human wild-type or mutant PIGT cDNA cloned into pTA, a weak promoter (TA promoter-driven expression vector) that helps with the detection of mild partial loss of function (14). Two days later, restoration of GPI-AP (CD59, DAF, and CD16) expression was measured via flow cytometry. Cells were stained with PE-conjugated anti-human CD16 antibody (3G8, Biolegend) along with mouse anti-human CD59 (5H8) and mouse anti-human DAF (IA10) antibodies, followed by PE-conjugated second antibody. Levels of expressed wild-type and p.Arg507Trp mutant HA-tagged PIGT in pME-vector transfected cells were analyzed by western blotting using an anti-HA antibody (C29F4, Cell Signaling Tec, Danvers, MA, USA). Levels of protein expression were normalized to luciferase activity for transfection efficiencies and to expression of GAPDH for loading controls.
The proband, a Libyan girl born in 2017, was the first child of healthy consanguineous parents (Figure 1A). She was born at full term, and pregnancy and delivery were uneventful with normal menstruation. Global hypotonia and horizontal nystagmus were noted from the neonatal period. At the age of 4 months, the patient presented with multiple (two to three) daily seizures with asymmetric adduction movement of the 4 limbs and head deviation. The ictal sleep EEG showed asymmetric tonic spasm (Figure 1B). The interictal sleep EEG showed organized EEG with the presence of rare spindles and diffuse spike-wave discharges (Figure 1C). The patient presented with a global psychomotor delay with hypotonia, a severe speech delay, and walking impairment. Brain MRI, performed at age 14 months, revealed severe atrophy of the cerebral hemispheres with slight cerebellar hypoplasia and enlargement of lateral ventricles (Figure 1D). Metabolic screening was normal. She was treated with corticosteroid for 2 weeks (hydrocortisone 15 mg/kg/day) without improvement; hence, she was switched to vigabatrin (100 mg/kg/day) in association with valproate (30 mg/kg/day). Her overall antiepileptic drug response was good, but she still experienced seizures once every 2 weeks.
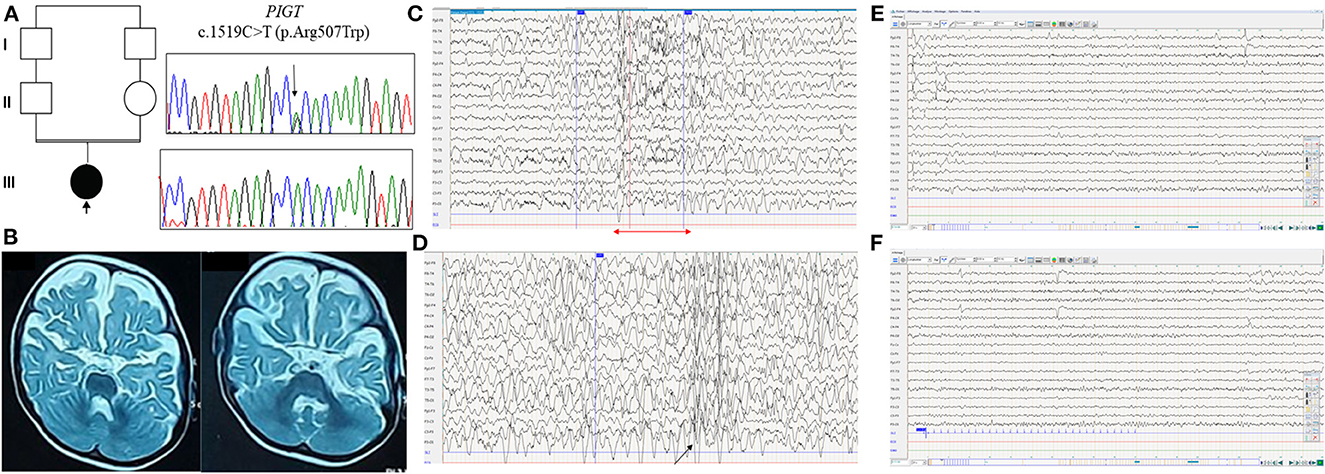
Figure 1. Clinical and molecular findings in the Libyan family with PIGT variant c.1519C>T; p.Arg507Trp. (A) Pedigree of the reported family with electropherograms of the identified homozygous PIGT variant (III.1) compared with heterozygous carrier sequences (II.1 and II.2). (B) Brain MRI T2-weighted axial images reveal severe atrophy of the cerebral hemispheres with slight cerebellar hypoplasia. (C) EEG recording, obtained when the patient was 2 years old, with the following parameters: 20 electrodes, 7 μv/mm, 0.03 s, 15, 50 Hz. The EEG shows slow spike-wave discharge followed by diffuse fast rhythms (red arrow). (D) Sleep EEG shows moderately organized EEG with the presence of rare spindles and diffuse spike-wave discharges (black arrow). (E, F) EEG showing normal background rhythm without photosensitivity.
Based on clinical data acquired during the most recent follow-up at the age of 2 years 6 months, our patient showed severe motor and cognitive retardation: she cannot hold her head up or sit, she has no eye tracking, with wandering movements of the eyeballs, expressive language is completely absent, and she has axial hypotonia with spastic tetraparesis. She also has dysmorphic features, including a broad forehead, acquired microcephaly, deep-set eyes, epicanthus, and high-arched palate. At the most recent follow-up, the patient had normal sleep EEG with normal background rhythm without photosensitivity (Figures 1E, F).
A total of 9,542 common single-nucleotide variants (SNVs) and indels were detected. The filtering process, as previously described, resulted in identification of 13 homozygous and 118 heterozygous variants. Based on their predicted effect on protein function and the likelihood of the autosomal recessive mode of inheritance, we evaluated the resulting variants, under the assumption that the disease gene would be more likely to cause an epileptic and/or a DD phenotype. Initially, only homozygous variants were considered and the PIGT variant (NM_015937.6):c.1519C>T (p.Arg507Trp) was selected as the candidate variant for the patient's phenotype. For the purpose of further investigation, we reevaluated the filtering process considering an autosomal dominant mode of inheritance; however, no relevant variants were detected. The PIGT variant was verified via Sanger sequencing, and both asymptomatic parents were found to be heterozygous (Figure 1A). The identified missense variant (rs146484791) was reported with a frequency of 0.0023% in the gnomAD database and has not been previously reported in the homozygous state. In-silico pathogenicity prediction using the VarCards tool showed a deleterious effect on the PIGT protein (damaging score 0.83), with a damaging CADD score of 35. An alternative variant, p.Arg507Gln, was found to have been reported in the heterozygous state with p.Val528Met in two patients with PIGT deficiency (10, 11).
Rescue experiments using variant cDNA driven by the weak promoter pTA, performed on PIGT-knockout HEK293 cells, indicated that the p.Arg507Trp variant results in a mild reduction in the amounts of CD59, CD16, and DAF anchored to the cell membrane (Figure 2A). Western blot analysis showed that expression of the mutant protein was not decreased but was even higher than expression of the wild-type protein (Figure 2B).
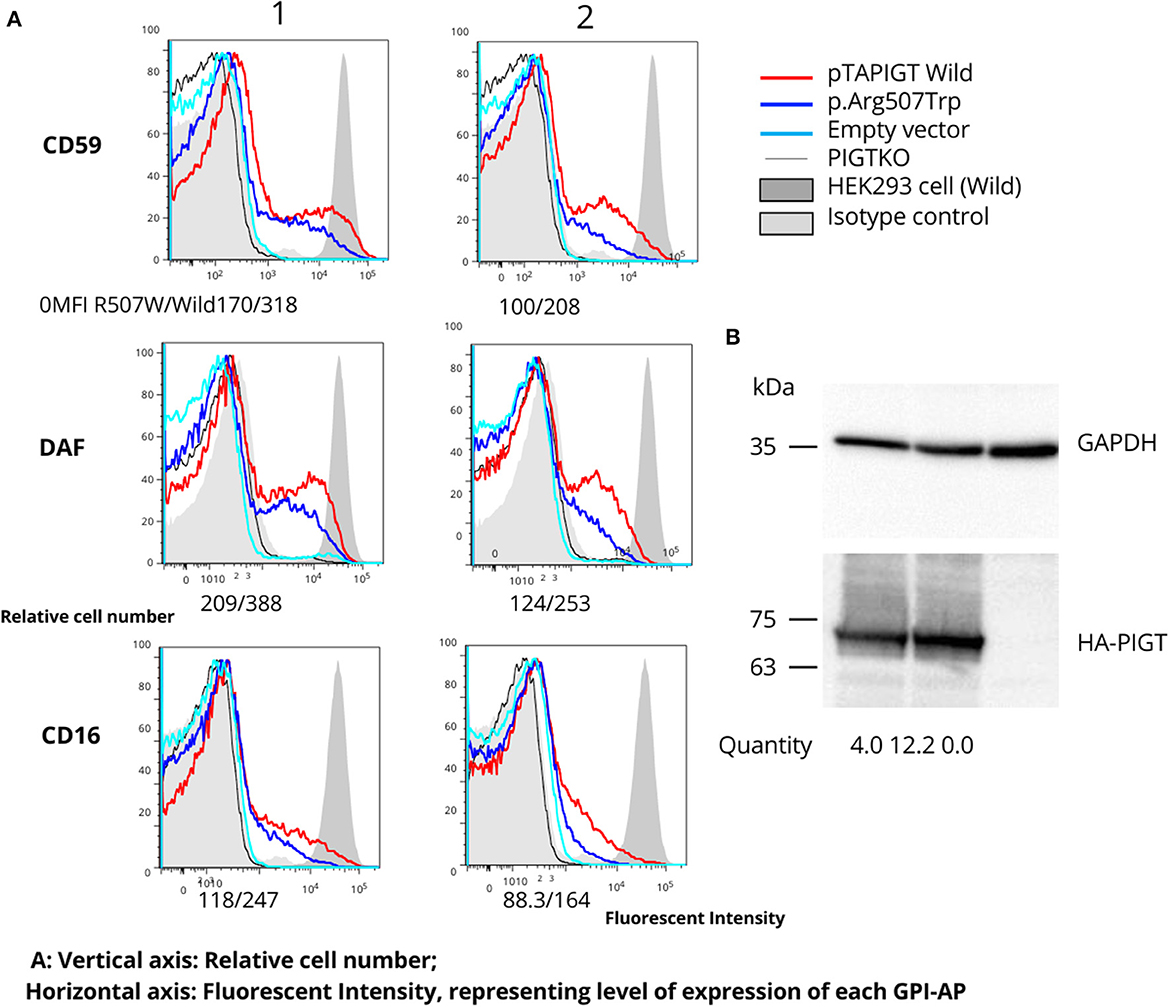
Figure 2. Follow-up functional studies on the p.Arg507Trp variant. (A) PIGT knockout HEK293 cells were transfected with variable amounts of p.Arg507Trp-variant PIGT or wild-type PIGT expressing plasmids driven by the weak promoter (pTA; 1: 4 ug; 2: 0.8 ug). Restoration of expression of GPI-APs (CD59, DAF, and CD16) was measured using flow cytometry. The p.Arg507Trp variant could not restore GPI-APs at a similar level to wild-type PIGT. The vertical axis represents the relative cell number; the horizontal axis represents fluorescent intensity, reflecting the level of expression of each GPI-AP. (B) Levels of expressed wild-type and p.Arg507Trp mutant HA-tagged PIGT in pME vector-transfected cells were analyzed by western blotting using an anti-HA antibody. After normalization to luciferase activity and GAPDH, expression of the mutant protein appeared to be similar to that of the wild-type protein.
Taking these findings together, we classified the Arg507Trp as a likely pathogenic variant in accordance with the ACMG recommendations: PM2 (variant not found in gnomAD genomes with good coverage of gnomAD genomes = 29.9; pathogenic, supporting), PM5 (alternative pathogenic variant Arg507Gln had been already reported (10, 11); pathogenic, moderate), and PS3 (well-established in vitro functional studies supportive of a damaging effect of the gene product; pathogenic, strong).
Clinical exome sequencing is now used routinely as a tool to diagnose many different inborn errors of metabolism (IEMs) in patients with developmental and epileptic encephalopathy (DEE), such as congenital disorders of glycosylation. However, despite the tremendous progress that has been made in the development of both sequencing technology and bioinformatics in recent years, it should be noted that other diagnostic tests are still necessary to confirm or reject a diagnosis with certainty in cases where this approach results in the identification of genetic variants of uncertain clinical significance (VOUS). The variant p.Arg507Trp in the PIGT gene, identified in the homozygous state in this study, has previously been reported in a compound heterozygous state with Val528Met in two Russian patients (11). In addition, an alternative variant in the same location (Arg507Gln) has been reported in a Polish patient (10). According to ACMG classification, this variant is still classified as a VOUS, since it has not been biologically confirmed. The functional experiments performed here using mutant HEK293 cells provide evidence to support the pathogenicity of this variant; this evidence is sufficient to reclassify this variant as likely pathogenic. On the other hand, the milder decreased activity is consistent with the moderate phenotype of our patient. Here we should highlight that one of the limitations of our study is the brief period of follow-up of our patient, who is only at the age of 2 years at the time of writing. However, we were able to identify our patient's phenotype specifically as a moderate epileptic one in comparison to the majority of PIGT-deficiency patients, who often suffer from severe drug-resistant epilepsy with convulsive status epilepticus, sometimes with neonatal–infantile onset. Together with other recent studies (10, 11), our study also serves to confirm the genotype–phenotype correlation for PIGT that was first described in 2019. Bayat et al. report on 24 patients harboring the p.Val528Met variant in either homozygous or compound heterozygous state and one patient with the p.Asn527Ser variant, all of whom presented with a less severe epilepsy phenotype with considerably later onset of epilepsy; there was also no premature mortality in any of these patients (11). In Table 1, we present a comparison of the clinical findings for our patient with those of previously reported patients with the p.Arg507Trp variant. All patients presenting with the p.Arg507Trp variant in either homozygous or compound heterozygous state, including our patient, have been found to exhibit GDD and became free or partially free of epileptic seizures following treatment with CBZ/VPA. Additionally, a comparison of the phenotypes of patients harboring both p.Asn527Ser and p.Val528Met variants with that of our patient revealed a similar spectrum of symptoms regarding the onset of seizures (between 4 months and 5.5 years), seizure types (focal to bilateral tonic–clonic seizures as the predominant type), degree of cognitive and developmental outcome (moderate to severe), and response to antiepileptic drugs. Therefore, we conclude that missense variants p.Asn527Ser, p.Val528Met, and p.Arg507Trp, located in the C-terminal region, are associated with an epileptic phenotype with comparatively better outcome.
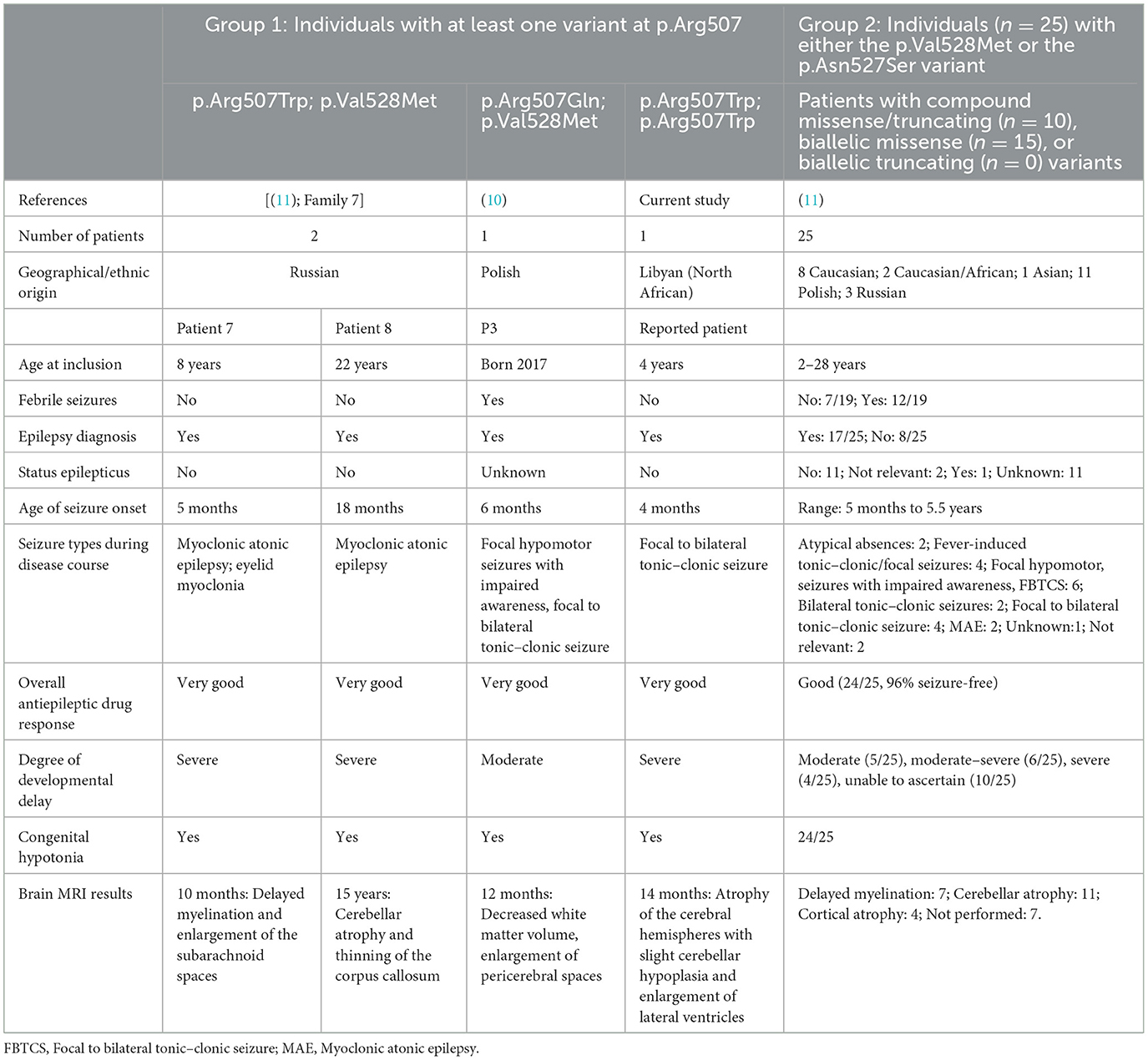
Table 1. Brief clinical overview of all individuals with PIGT deficiency associated with moderate seizure phenotype of variants within the C-terminal region (PIGT: NM_015937.6) for whom clinical information has been published.
The existing literature was also searched to identify all missense variants described as occurring with a severe phenotype, including severe epilepsy with frequently recurrent episodes of convulsive status epilepticus, abnormal interictal EEG, and severe drug-resistant epilepsy. In total, 10 missense variants (either in homozygous or in compound heterozygous state) were collected; these have been observed in 23 patients belonging to 14 families of various ethnicities (Table 2). Interestingly, we note that all of these variants occur in the N-terminal region of the PIGT protein (Figure 3). Hence, we suggest that the phenotypic variability of PIGT deficiency could be related to the position of the affected residue and its effect on the function or structure of the PIGT protein, as well as the interaction with components of the GPI-TA complex. It has been shown that PIGT is covalently linked to the majority of PIGK via a functionally important disulfide bond between PIGT-Cys182 and PIGK-Cys92 in the GPI-TA complex. In addition, PIGT and PIGS are linked to one other, and PIGT has an additional function in stabilizing GPI-TA by linking PIGS to GAA1 and PIGK (3, 4). In the case of the homozygous p.Thr183Pro and p.Glu184Lys variants, substitutions occur in the residue directly adjacent to Cys182, which is responsible for the PIGT–PIGK disulfide bond. In the p.Thr183Pro variant, the threonine is substituted by a proline, which is an inflexible residue common in very tight protein junctions (5). In the p.Glu184Lys variant, glutamate (negatively charged) is substituted by a positively charged lysine. These two substitutions could affect the biochemical environment around Cys182, possibly affecting the stability of the disulfide bond leading to loss of function in PIGT and the GPI-TA complex. Considering the available experimental data, a recent study has additionally suggested that the luminal part of PIGT (73-427 aa) consists of a β-propeller domain with a central hole that regulates the access of substrate protein C-termini to the active site of the cysteine protease PIGK (19). Therefore, missense variants located in the β-propeller domain could affect the interaction of PIGT with the active site of PIGK, leading to the observed severe phenotype.
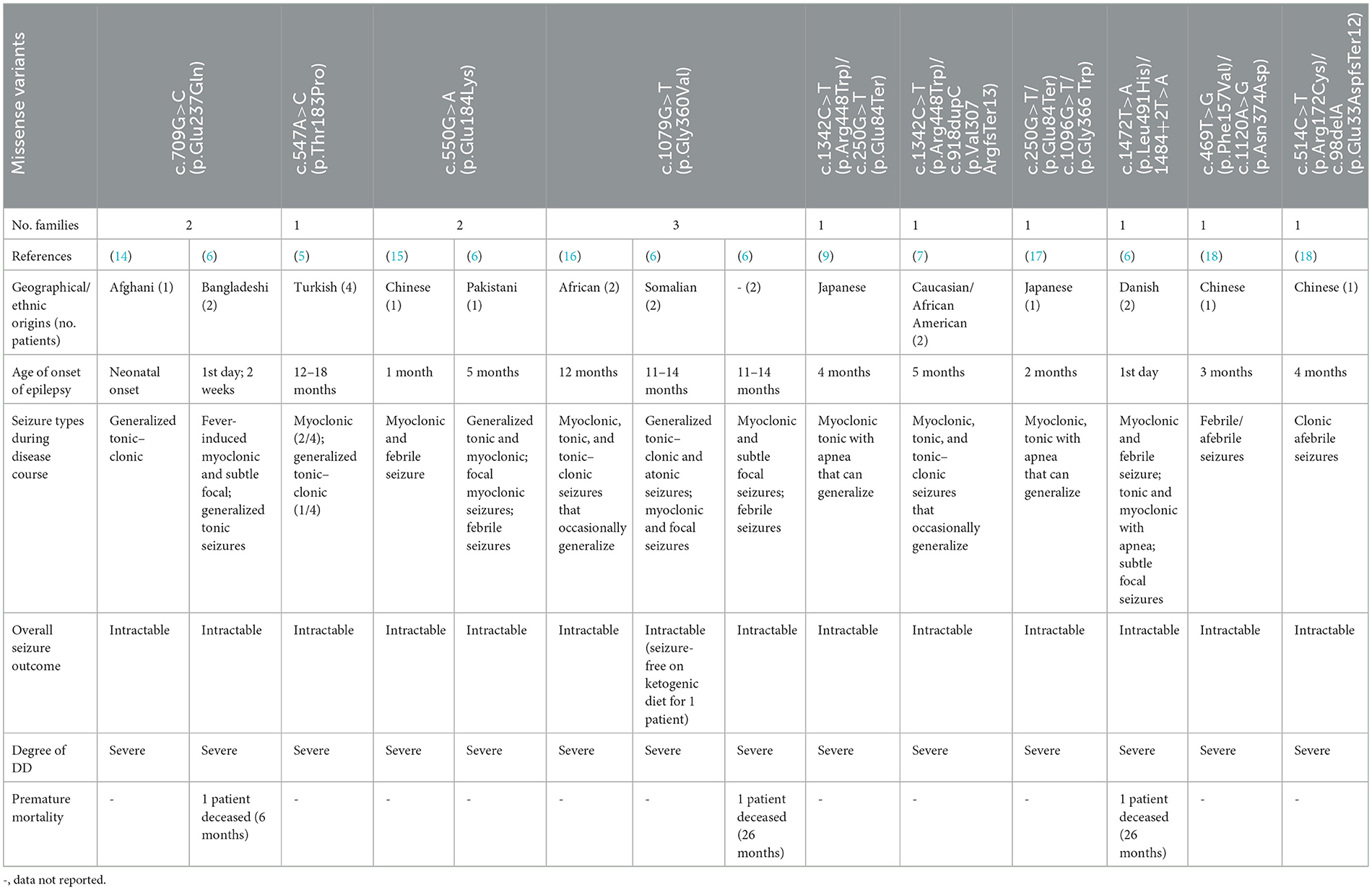
Table 2. Published clinical findings in individuals harboring a severe phenotype of PIGT deficiency associated with missense variants.

Figure 3. Domain map of the PIGT protein depicting the locations of milder and severe pathogenic variants.
Concerning variants occurring in the C-terminal region on the cytoplasmic side (p.Val528Met, p.Asn527Ser, and p.Arg507Trp), we suggest that these variants would not affect the PIGT-GPI-APs interaction, instead representing residual activity, which might explain the moderate phenotype. This hypothesis can be supported by the results of our rescue experiments using variant cDNA driven by the weak promoter pTA on PIGT-knockout HEK293 cells. Specifically, p.Arg507Trp variant did not restore the GPI-APs to a similar level as observed with wild-type PIGT, suggesting decreased activity of the variant. In addition, the same effect has also been demonstrated for the p.Val528Met variant, although this effect was only observed when the pTK promoter was used (14).
Our study details a new case of the p.Arg507Trp variant in the C-terminal region, associated with a moderate epileptic phenotype. This supports the previously reported genotype–phenotype correlation of PIGT deficiency (10, 11), which will be useful for future genetic counseling.
The datasets presented in this article are not readily available because of ethical/privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
The study protocol was approved by the Local Medical Ethics Committee of South of Tunisia (Accession number 28/2019). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
IB conceived the study, wrote the manuscript, and analyzed and interpreted the NGS data. OJ, SMal, and FK analyzed and interpreted all clinical data. AT, AS, and AB conducted the clinical exome sequencing. YM performed the functional study. IE performed a segregation analysis. FF and AS assisted in writing the manuscript. SMas, SW, TK, and CT revised and edited the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the Tunisian Ministry of Higher Education and Research (Grant/Award Number: LR15CBS07). It was also partially supported by the SEED project (the European Union's Horizon 2020 Research and Innovation Program) under Grant Agreement Number: 856592.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Fujita M, Kinoshita T. GPI-anchor remodeling: Potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim Biophys Acta. (2012) 1821:1050–8. doi: 10.1016/j.bbalip.2012.01.004
2. Kinoshita T, Fujita M. Biosynthesis of GPI-anchored proteins: Special emphasis on GPI lipid remodeling. J Lipid Res. (2016) 57:6–24. doi: 10.1194/jlr.R063313
3. Ohishi K, Inoue N, Kinoshita T. PIG-S and PIG-T, essential for GPI anchor attachment to proteins, form a complex with GAA1 and GPI8. EMBO J. (2001) 20:4088–98. doi: 10.1093/emboj/20.15.4088
4. Ohishi K, Nagamune K, Maeda Y, Kinoshita T. Two subunits of glycosylphosphatidylinositol transamidase, GPI8 and PIG-T, form a functionally important intermolecular disulfide bridge. J Biol Chem. (2003) 278:13959–67. doi: 10.1074/jbc.M300586200
5. Kvarnung M, Nilsson D, Lindstrand A, Korenke GC, Chiang SCC, Blennow E, et al. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet. (2013) 50:521–8. doi: 10.1136/jmedgenet-2013-101654
6. Bayat A, Knaus A, Juul AW, Dukic D, Gardella E, Charzewska A, et al. PIGT-CDG, a disorder of the glycosylphosphatidylinositol anchor: Description of 13 novel patients and expansion of the clinical characteristics. Genet Med. (2019) 21:2216–23. doi: 10.1038/s41436-019-0512-3
7. Lam C, Golas GA, Davids M, Huizing M, Kane MS, Krasnewich DM, et al. Expanding the clinical and molecular characteristics of PIGT-CDG, a disorder of glycosylphosphatidylinositol anchors. Mol Genet Metab. (2015) 115:128–40. doi: 10.1016/j.ymgme.2015.04.007
8. Kohashi K, Ishiyama A, Yuasa S, Tanaka T, Miya K, Adachi Y, et al. Epileptic apnea in a patient with inherited glycosylphosphatidylinositol anchor deficiency and PIGT mutations. Brain Dev. (2018) 40:53–7. doi: 10.1016/j.braindev.2017.06.005
9. Nakashima M, Kashii H, Murakami Y, Kato M, Tsurusaki Y, Miyake N, et al. Novel compound heterozygous PIGT mutations caused multiple congenital anomalies-hypotonia-seizures syndrome 3. Neurogenetics. (2014) 15:193–200. doi: 10.1007/s10048-014-0408-y
10. Jezela-Stanek A, Szczepanik E, Mierzewska H, Rydzanicz M, Rutkowska K, Knaus A, et al. Evidence of the milder phenotypic spectrum of c. 1582G >A PIGT variant: Delineation based on seven novel Polish patients. Clin Genet. (2020) 98:468–76. doi: 10.1111/cge.13822
11. Bayat A, Pandziwiat M, Obersztyn E, Goldenberg P, Zacher P, Döring JH, et al. Deep-phenotyping the less severe spectrum of PIGT deficiency and linking the gene to myoclonic atonic seizures. Front Genet. (2021) 12:663643. doi: 10.3389/fgene.2021.663643
12. Ayed IB, Ouarda W, Frikha F, Kammoun F, Souissi A, Said MB, et al. SRD5A3-CDG: 3D structure modeling, clinical spectrum, and computer-based dysmorphic facial recognition. Am J Med Genet A. (2021) 185:1081–90. doi: 10.1002/ajmg.a.62065
13. Desvignes J-P, Bartoli M, Delague V, Krahn M, Miltgen M, Béroud C, et al. VarAFT: A variant annotation and filtration system for human next-generation sequencing data. Nucleic Acids Res. (2018) 46:W545–53. doi: 10.1093/nar/gky471
14. Pagnamenta AT, Murakami Y, Taylor JM, Anzilotti C, Howard MF, Miller V, et al. Analysis of exome data for 4293 trios suggests GPI-anchor biogenesis defects are a rare cause of developmental disorders. Eur J Hum Genet. (2017) 25:669–79. doi: 10.1038/ejhg.2017.32
15. Yang L, Peng J, Yin XM, Pang N, Chen C, Wu TH, et al. Homozygous PIGT mutation lead to multiple congenital anomalies-hypotonia seizures syndrome 3. Front Genet. 9:153. doi: 10.3389/fgene.2018.00153
16. Skauli N, Wallace S, Chiang SCC, Barøy T, Holmgren A, Stray-Pedersen A, et al. Novel PIGT Variant in two brothers: Expansion of the multiple congenital anomalies-hypotonia seizures syndrome 3 phenotype. Genes (Basel). (2016) 7:108.
17. Kohashi K, Ishiyama A, Yuasa S, Tanaka T, Miya K, Adachi Y, et al. Epileptic apnea in a patient with inherited glycosylphosphatidylinositol anchor deficiency and PIGT mutations. Brain Dev. (2018) 40:53–7.
18. Jiao X, Xue J, Gong P, Bao X, Wu Y, Zhang Y, et al. Analyzing clinical and genetic characteristics of a cohort with multiple congenital anomalies-hypotonia-seizures syndrome (MCAHS). Orphanet J Rare Dis. (2020) 15:78.
Keywords: PIGT gene, global developmental delay, epilepsy, genotype phenotype correlation, GPI transamidase
Citation: Ben Ayed I, Jallouli O, Murakami Y, Souissi A, Mallouli S, Bouzid A, Kamoun F, Elloumi I, Frikha F, Tlili A, Weckhuysen S, Kinoshita T, Triki CC and Masmoudi S (2023) Case report: Functional analysis of the p.Arg507Trp variant of the PIGT gene supporting the moderate epilepsy phenotype of mutations in the C-terminal region. Front. Neurol. 14:1092887. doi: 10.3389/fneur.2023.1092887
Received: 20 December 2022; Accepted: 01 February 2023;
Published: 09 March 2023.
Edited by:
Fernando Cendes, State University of Campinas, BrazilReviewed by:
Xena Giada Pappalardo, University of Catania, ItalyCopyright © 2023 Ben Ayed, Jallouli, Murakami, Souissi, Mallouli, Bouzid, Kamoun, Elloumi, Frikha, Tlili, Weckhuysen, Kinoshita, Triki and Masmoudi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saber Masmoudi, c2FiZXIubWFzbW91ZGlAY2JzLnJucnQudG4=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.