 Thérèse Boyle
Thérèse Boyle Suran L. Fernando
Suran L. Fernando James Drummond3,4
James Drummond3,4 John Parratt
John Parratt- 1Clinical Immunology and Allergy, Royal North Shore Hospital, St Leonards, NSW, Australia
- 2Immunology Laboratory, Royal North Shore Hospital, St Leonards, NSW, Australia
- 3Faculty of Medicine and Health, The University of Sydney, Camperdown, NSW, Australia
- 4Department of Neuroradiology, Royal North Shore Hospital, St Leonards, NSW, Australia
- 5Department of Neurology, Royal North Shore Hospital, St Leonards, NSW, Australia
Background: Tumefactive demyelinating lesions (TDLs) are defined as lesions >2 cm on MRI of the brain. They are identified in a range of demyelinating diseases including massive demyelination due to Marburg's acute MS, Schilder's Disease, Balo's concentric sclerosis, and Tumefactive MS. Apart from the rare demyelinating variants which are often diagnosed histologically, there are no detailed data to phenotype TDLs.
Methods: We describe the clinical and radiological features of four similar patients with very large TDLs (>4 cm), that are not consistent with the rare demyelinating variants and may represent a distinct phenotype.
Results: All patients presented with hemiplegia and apraxia. The mean age at onset was 37 years with an equal sex distribution. All patients were diagnosed with Tumefactive demyelination based on MRI and CSF analysis, precluding the need for brain biopsy. All responded to potent immunotherapy (including high dose corticosteroids, plasma exchange, rituximab, and/or cyclophosphamide). The mean lag from diagnosis to treatment was 1 day. The median EDSS at presentation was six and recovery to a median EDSS of two occurred over 6 months.
Conclusion: We propose that Tumefactive lesions larger than 4 cm are termed “Giant demyelinating lesions” (GDLs) not only on the basis of size, but a rapid and fulminant demyelinating presentation leading to acute, severe neurological disability that is, nonetheless, responsive to immunotherapy. Further clinical studies are required to ratify this proposed phenotype, establish the immunological profile and best treatment for such patients.
Introduction
Various terms have been proposed for demyelinating lesions according to their size. Small lesions are described as <3.5 mm (1), whilst large demyelinating lesions are defined as >1 cm (2). MS diagnostic criteria require lesions to be >3 mm in long axis, though those <3 mm with demyelinating characteristics or topography are still considered abnormal (3). The largest lesions, thus far described, are >2 cm in size and historically mimic neoplasms leading to the term tumefactive demyelinating lesions (TDLs). TDLs are detected in at least 1–2/1,000 cases of MS but also occur in acute disseminated encephalomyelitis (ADEM), neuromyelitis optica spectrum disorder (NMO-SD) and the rare variants of MS including Marburg's acute MS, Balo's concentric sclerosis (BCS), and Schilder's disease (4–8).
Marburg's acute MS was first described by Otto Marburg in 1906 and is characterized by large (commonly cerebral hemisphere) lesions with death typically occurring within a year from symptom onset. In 1912, Paul Schilder described a form of demyelinating disease in children and young adults characterized by large cerebral hemisphere lesions with development of new, progressively larger lesions. However, there is debate as to whether this is a unique phenotype or a misdiagnosis of other conditions such as adrenoleukodystrophy (4). Baló's concentric sclerosis (BCS) was named after the Hungarian pathologist József Baló (1895–1979), following his description in 1928 of a demyelinating condition characterized by concentric layers of demyelination and remyelination on MRI or histopathology (9). Balo's lesions can vary in size and occur in the cerebral hemispheres but also basal ganglia, pons, cerebellum, and very infrequently the spinal cord and optic nerves (4).
The demyelinating variants such as Marburg's MS have a particularly aggressive and fulminant course due to their large multifocal lesion volume and extensive axonal transections (10–13). Whilst immunosuppressive treatment has reduced mortality, the morbidity of aggressive Tumefactive demyelination remains high (14, 15). By comparison, smaller TDLs occurring in Tumefactive MS often confer a good prognosis. There is paucity of literature describing the characteristics and prognosis of patients presenting with very large lesions that do not exhibit the phenotypic features of one of the rare demyelinating variants. Furthermore, treatment recommendations for TDLs vary and there are no consensus guidelines (16–18) although it is recognized that Marburg's MS and BCS typically require more aggressive treatment including cyclophosphamide (18–21).
We describe four patients who presented with an acute, monophasic and rapidly evolving neurological syndrome due to very large demyelinating lesions which were >4 cm in size. Their clinical presentations, radiological features and response to treatment are sufficiently homogenous that we propose the term giant demyelinating lesions (GDLs) for the causative abnormalities found on MRI, and describe the outcomes of these remarkable cases.
Case description and diagnostic assessment
Four patients presented to a tertiary center. One patient presented in 2014, whilst the other three presented between February 2019 and June 2020. All patients suffered from rapidly progressive left hemiparesis with apraxia. None of the patients had acute cognitive changes or encephalopathy. The mean age was 37 years (range 19–55 years) at symptom onset with an equal sex distribution. One patient is Italian ancestrally (migrated from Italy) and the other three patients, Australian. None of the patients were taking regular medication at the time of presentation. The clinical characteristics including medical and family history are summarized in Table 1 and the investigations in Table 2.
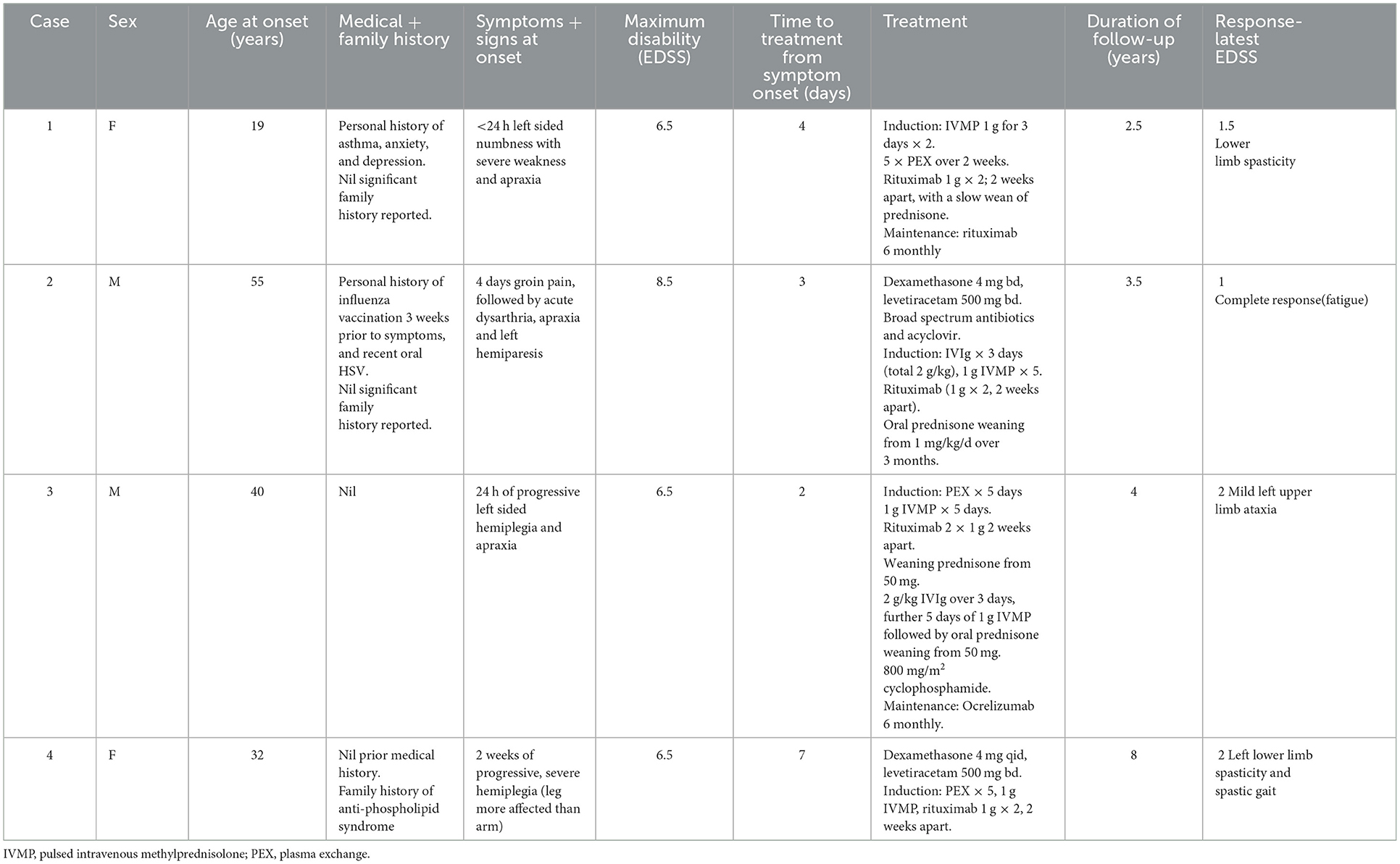
Table 1. Patient clinical characteristics, diagnosis, treatment, and response.
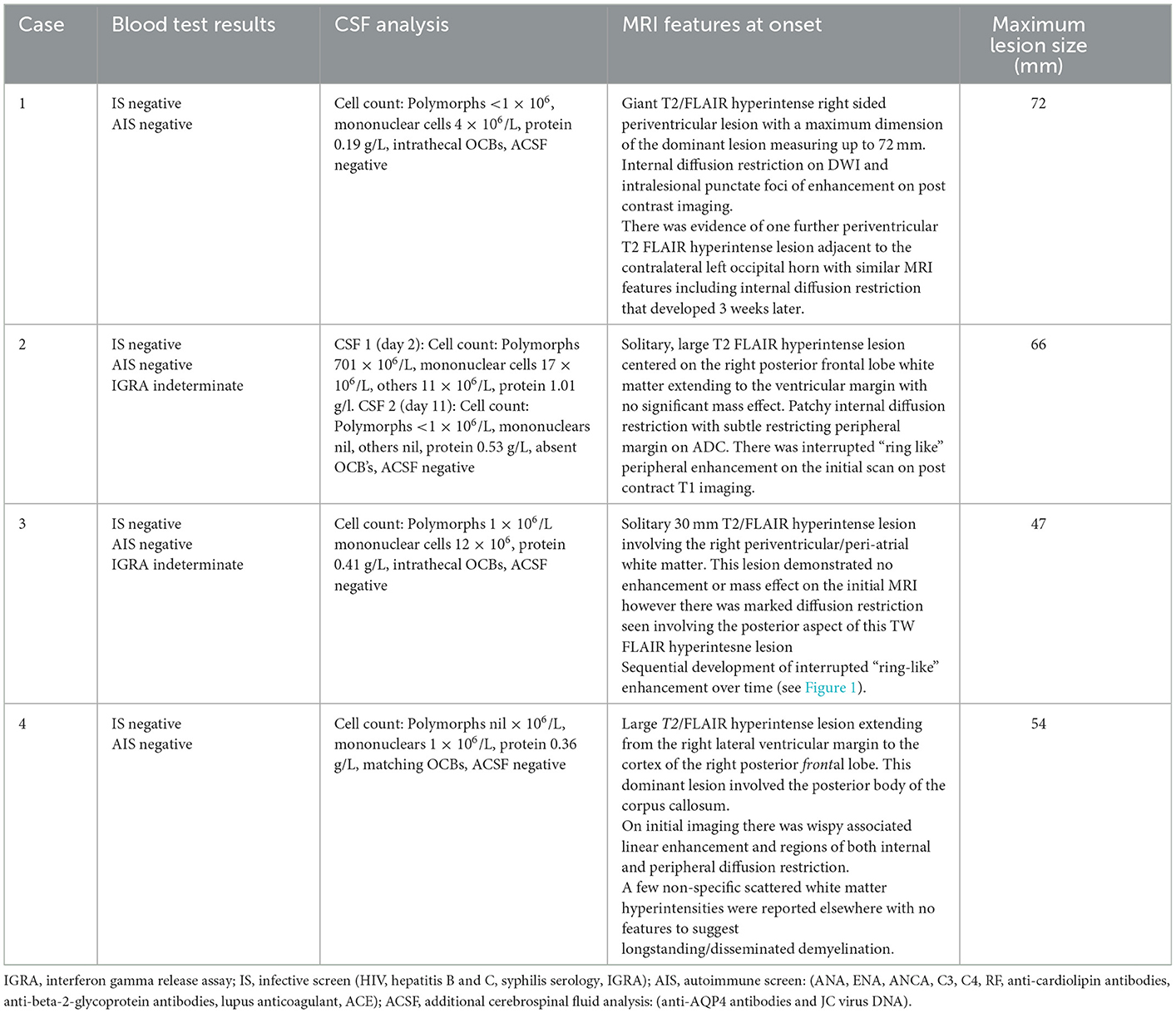
Table 2. Patient investigations.
All the patients were assessed with CT and CT angiography followed by serial MRI scans (multiple pre and post gadolinium sequences including FLAIR and SWI, Table 2). There was no evidence of hemorrhagic demyelination or vasculitis on any scan. MRI brain demonstrated TDLs in all cases (Figures 1A–D), and the mean size of the lesions at maximum was 59.75 mm (Table 2). Spinal demyelination was absent. All of the lesions demonstrated common radiological features; they were centered on white matter, extended from the lateral ventricular margin to the cerebral cortex in the frontal and parietal lobes and had minimal mass effect. All lesions demonstrated T2/FLAIR hyperintensity with T1 hypointensity and variable patterns of diffusion restriction and gadolinium enhancement (Figure 1). One patient exhibited a Balo-like lesion with a subsequent second much smaller lesion in the contralateral hemisphere (Figure 1A). There was substantial radiological improvement in all cases, with case 1 and 2 showing near-complete resolution of the GDLs (Figures 1A, B).
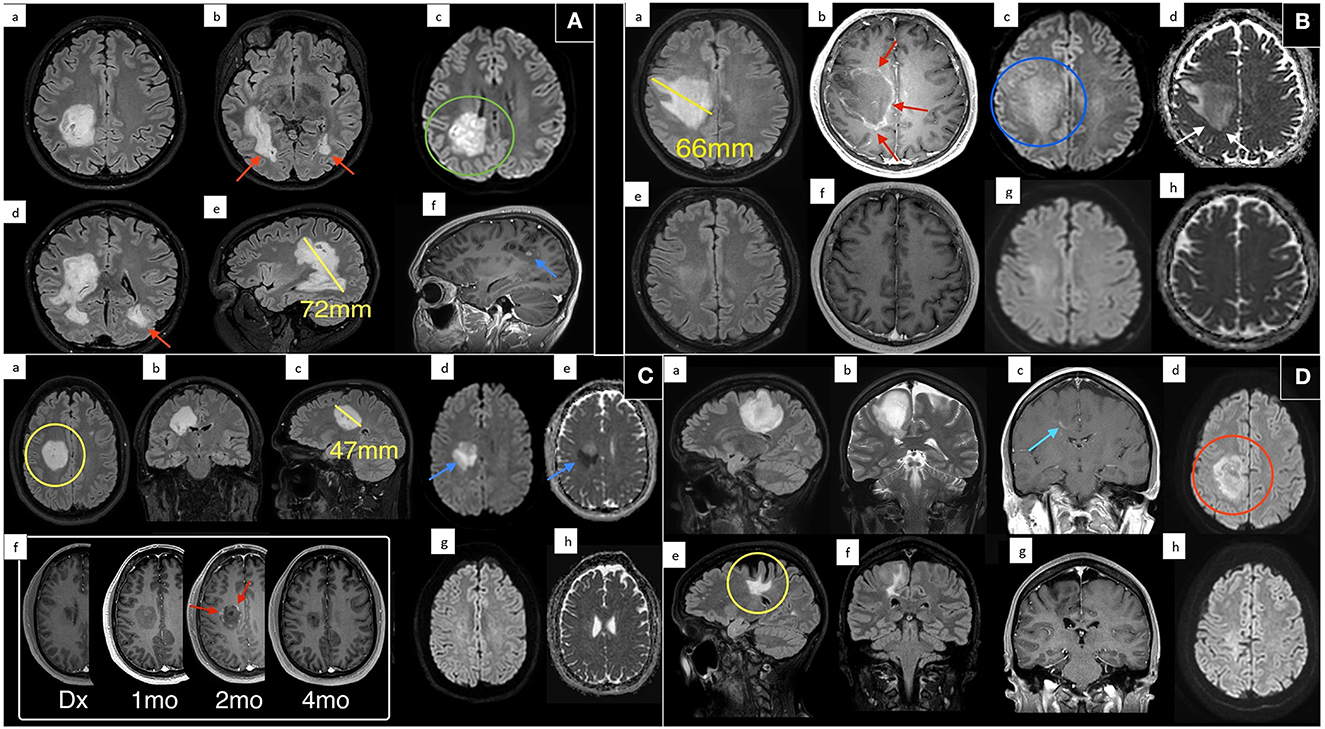
Figure 1. Magnetic resonance imaging of each patient at presentation and various treatment time points. (A) Case 1: MRI following presentation demonstrates a giant hyperintense confluent right sided periventricular lesion on axial T2 FLAIR (a/b), with coronal oblique T2 FLAIR reconstruction (d), and sagittal oblique (e) reconstruction. Maximum dimension of the dominant lesion measuring up to 72 mm on sagittal imaging. Evidence of internal diffusion hyperintensity (green circle) on axial DWI (c) and internal punctate foci of enhancement (blue arrow) on sagittal T1 post contrast (f). There was evidence of further periventricular T2 FLAIR hyperintense lesion adjacent to the contralateral left occipital horn (red arrows). (B) Case 2: MRI at presentation (a–d) and 4 months follow up (e–h). Scan at diagnosis demonstrates a large solitary T2 FLAIR (a) hyperintense lesion centered on the right frontal white matter with no significant mass effect. This lesion measured up to 66 mm with peripheral enhancement (red arrows) on post contrast T1 imaging (b) with patchy internal diffusion restriction (blue circle) on DWI (c), and subtle peripheral restricting peripheral margin (white arrow) on ADC (d). Near complete resolution on follow up imaging with minimal residual T2 FLAIR hyperintensity (e). (C) Case 3: MRI at presentation (a–e), multiple follow up times points post contrast T1 imaging (f), and 4 month follow up DWI (g) and ADC (h). On multiplanar T2 FLAIR (a–c) there is a large periventricular lesion (yellow circle) measuring up to 47 mm with marked associated diffusion restriction (blue arrows) on DWI (d) and ADC (e). No enhancement was demonstrated on scan at diagnosis (c) however the patient progressively developed classic interrupted peripheral enhancement (red arrows). On delayed 4 month follow up scan enhancement and diffusion restriction had resolved (g/h). (D) Case 4: MRI at presentation with sagittal T2 FLAIR (a), coronal T2 (b), coronal T1 post contrast (c), and DWI (d) demonstrates a large T2/FLAIR hyperintensity extending from the periventricular margin to the cortex of the right posterior frontal lobe. There was wispy associated linear enhancement (blue arrow) at diagnosis on post contrast T1 imaging (c) and regions of internal and peripheral diffusion restriction (red circle) on DWI (d). Six year follow up scan (e–h) shows marked reduction in FLAIR signal, some associated cortical atrophy and volume loss (yellow circle) and resolution of previous enhancement and diffusion restriction.
The CSF was generally bland with a mononuclear pleocytosis in only one case. Two patients had intrathecal synthesis of oligoclonal bands, one had matched serum and CSF bands and the CSF was normal in Case 2 (Table 2). All patients were negative for anti-aquaporin-4, anti-myelin oligodendrocyte glycoprotein (MOG) and anti-neuronal antibodies in CSF and/or serum.
Based on the severity of the clinical presentation and results of investigations, treatment was prioritized in all cases. Neurosurgical opinion was sought for case 3 though biopsy was deferred due to subsequent rapid improvement following additional treatment. Other diagnoses were considered but high grade Glioma was excluded based on radiological features (CT and MRI) and CSF abnormalities in most cases, neurosarcoidosis principally on MRI, infection from the incongruent clinical presentation (absence of fever and obtundation) and bland CSF, and lymphoma principally on MRI findings and in one patient of concern; hypometabolism of the lesions on FDG-PET.
All patients were treated with intravenous methylprednisolone (IVMP) and plasma exchange, followed by rituximab. One patient had cyclophosphamide as an adjunctive treatment (outlined in Table 1; Figure 2). Patients also received supportive care with physiotherapy to aid recovery. The clinical response to treatment, as measured by the Extended Disability Status Scale (EDSS) is shown in Figure 2. The median maximum EDSS was 6.5 and severe neurological disability developed quickly reaching this point at a mean of 9 days after onset. However, all patients recovered steadily with aggressive treatment reaching a median EDSS of 2 (after a mean follow-up duration of 4.5 years). Patients 1 and 3 required maintenance treatment, whilst patients 2 and 4 were treated with induction treatment alone. All patients were monitored in the outpatient setting with clinical assessment including EDSS and repeat MRI every 3–6 months. All patients were adherent with treatment and incurred no adverse effects on clinical assessment and regular laboratory tests. The diagnosis for all patients remains unspecified Tumefactive demyelination albeit patient 1 had radiological features of BCS and patient 2 had a precipitant and onset similar to ADEM. Patient 3 likely has Tumefactive MS, though remains in remission following an unusual monophasic presentation with a TDL at the onset. All patients exhibited very large lesions, with similar characteristics that suggest that these cases could be termed GDLs.
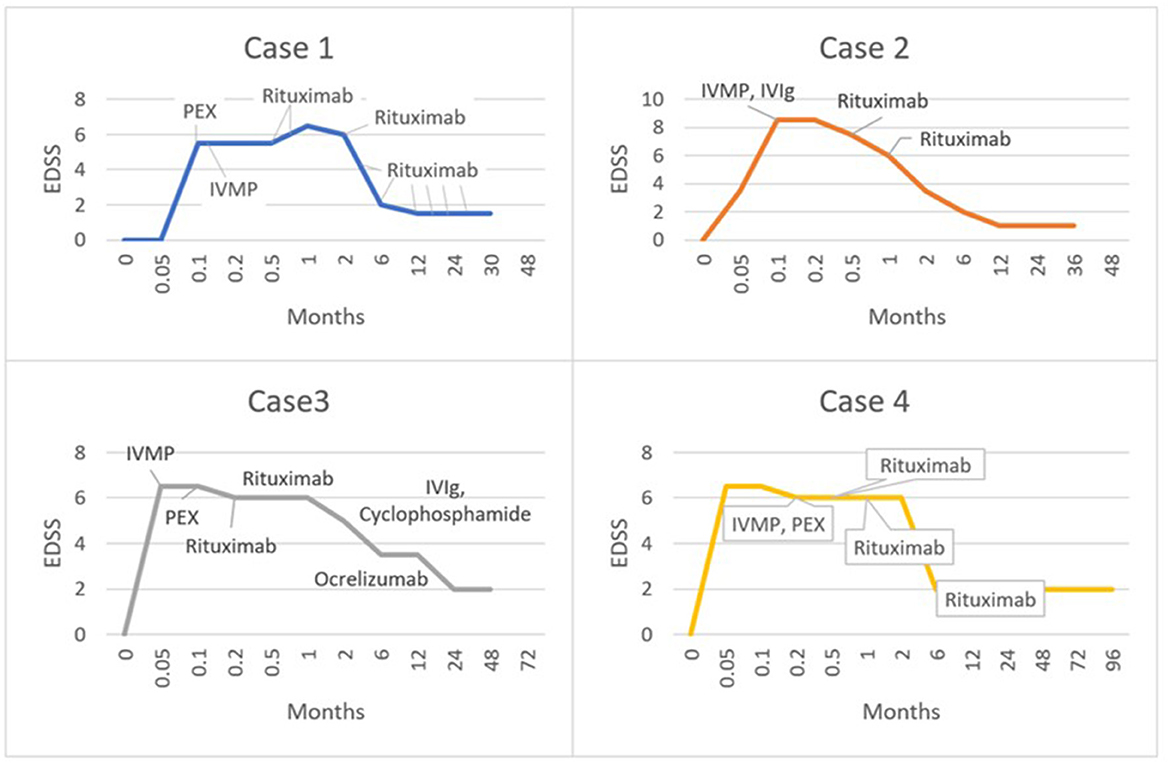
Figure 2. Graphs illustrating the clinical response of each patient according to timing and nature of treatment, as represented by the Extended Disability Status Scale (EDSS).
Discussion
Tumefactive demyelinating lesions are identified principally on MRI and specific diagnostic criteria have been proposed, but not validated or widely implemented (22). The radiographic features that are suggestive of demyelination include incomplete rim enhancement, a peripheral margin of diffusion restriction, absence of mass effect and generally, lack of cortical involvement (23, 24). MS is one of the causes while other pathologies including infection, autoimmune diseases and malignancy can be reasonably excluded using the clinical history, autoimmune serology, microbiology, CSF-analysis and additional imaging (11, 14, 25–30). It is seldom necessary to undertake a brain biopsy to facilitate the diagnosis of Tumefactive demyelination (13, 31).
Distinguishing lesions >4 cm as GDLs identifies a group of patients at high risk of neurological morbidity. While GDLs clearly share many features of TDLs it is our view that the distinctions of much lower incidence (in our experience), greater size and severity, speed of evolution and cortical involvement are sufficient to categorize this group as a particular and rare entity (32). Based on clinical and radiological data, GDLs grow rapidly from the ventricular surface and the leading edge of the lesion does not stop until the cerebral cortex is involved, sometimes to the level of the pia. They are quickly destructive as shown by rapid clinical deterioration, the development of chronic T1 hypointense changes in the core of the lesion, and lasting, focal cerebral atrophy. The diagnosis in each case here was made based on features suggestive of demyelination radiologically and CSF findings, particularly the presence of oligoclonal bands (OCBs) (27, 33).
It can be reasonably assumed, in the absence of histopathology, that the lesions described in our case series are demyelinating based on two main findings: the rapid evolution of neurological deficits implying conduction block and the radiological similarities to TDLs that have been biopsied in some series (12, 34). One of the patients met criteria for multiple sclerosis at presentation, with a couple of trivial demyelinating lesions elsewhere implying a common mechanism with that disease, but another patient exhibited MRI features of Balo's phenomenon, whilst a third might have been considered an ADEM variant because of preceding viral infection and vaccination, with remarkable recovery. However, the Balo's type GDL responded to plasma exchange and rituximab which is inconsistent with the proposed mechanism of that condition; a primary loss or destruction of oligodendrocytes in the absence of humoral immunological factors (4, 35), and ADEM is characterized by multifocal small perivenous demyelinating lesions (36, 37). Indeed, the lesions described in our case series do not fit well with any previously described demyelinating condition (14, 27).
The pathogenesis of TDLs is complex and probably involves multiple genetic and immunological mechanisms (15, 38, 39). It is interesting that T-cell dysregulation as seen in HIV infection, nivolumab and natalizumab treatment may be the substrate for TDLs in some cases, and similarly T-cell subset shifting through treatment with fingolimod (17, 18, 40, 41). This might lead to dysregulation of B-cell and plasma cell immune responses by altering the immunoregulatory T-cell phenotype. Occasional reports have identified IgG and complement deposition in TDLs (42–44) and a number of reports citing the therapeutic effects of plasma exchange and rituximab imply B-cell and IgG-mediated mechanisms are also important (17, 27, 40, 41). Proven antibody mediated diseases such as NMO-SD are associated with TDLs (4). Humoral abnormalities were shown in all but one of our patients. The absence of OCBs in case 2 may reflect the timing of collection during the disease course, and does not exclude an antibody mediated process with a consistent clinical-MRI phenotype (45). Notably, the patient responded to PEX and Rituximab. The pathogenesis of ADEM is also unclear though it is typically triggered by an infection or vaccination with subsequent molecular mimicry as the proposed mechanism of disease (46).
By inference as a subgroup or variant of TDLs, GDLs exhibit features consistent with a humoral pathogenesis, and all the patients responded in some way to plasma exchange or intravenous immunoglobulin. Of course, the treatment was not exclusive and intravenous methylprednisolone would have reduced granulocyte functions and cytokine release within the lesion (47). Moreover, one patient required cyclophosphamide as an adjunctive treatment to rituximab, attenuating T-cell responses as well as diminishing antigen presentation from B-cell ablation (16, 48). In case studies PEX (18, 40, 41), rituximab and cyclophosphamide are beneficial and safe (16–18, 49, 50). Mitoxantrone and natalizumab are variably effective (18), but the latter can cause TDLs in NMO-SD (19).
We acknowledge that TDLs are quite variable in their etiology and pathological mechanisms and recent work indicates that the treatment algorithm should be adjusted for phenotypic differences and age. For example, Tzanetakos et al proposed treatment is tailored to patients depending on whether they exhibit Balo's phenomenon (19, 20). There have been no clinical trials to date or formal treatment guidelines for TDLs and, by and large, an escalation approach is recommended from high dose corticosteroids to PEX followed by rituximab and/or cyclophosphamide according to response, with consideration of brain biopsy for refractory cases (19). In our series, time and brain were conserved with the inception of corticosteroid therapy and plasma exchange whilst the investigations were continuing to complete the diagnosis. Patients were treated quickly and aggressively with the intention of maximizing the effect of rituximab (an anti-CD20 B-cell depleting monoclonal antibody) through a leaky blood brain barrier early in the course of the disease. We think the rapidity of B-cell suppression may have influenced the disability outcomes in these patients which were excellent.
Patients with TDLs have a variable prognosis, and one-third develop MS over a period of up to 5 years (4). There is lack of consensus as to the maintenance therapy for TDLs (10, 18). Adverse prognostic factors include lesion size >5 cm, infiltrating lesions and older age (10, 11, 51). Patients with established MS who develop TDLs, or who subsequently meet criteria should receive conventional recommended MS treatment but with caution when switching from natalizumab or fingolimod and where the pathogenesis is uncertain, B-lymphocyte suppression may have broader efficacy (35). Those with TDLs who do not meet criteria for MS should be treated according to the severity of the disease and treatment response. To this point, two patients in our series continue B-cell immunosuppressive treatment and two have ceased therapy without any relapses. None of the patients have developed new lesions over a prolonged period of follow up.
This is the first report comprising patients with very large demyelinating lesions that do not fit classical descriptions of multiple sclerosis or the rare demyelinating variants. We propose these patients have a distinct phenotype and are unified by the presence of giant demyelinating lesions (GDLs). We suggest using this criterion, with other clinical and laboratory data, to lead to rapid diagnosis and treatment.
However, we acknowledge the significant limitations of a case series design and the small sample size. Further collective work is required to better elucidate the outcomes and response to treatment in patients with GDLs. Brain biopsies were not performed in our cohort of patients as their disease responded to treatment and because imaging and ancillary tests were consistent with a demyelinating process. Whilst this reduces iatrogenic risk and shortens the time to maximum potency treatment, histopathological study would elucidate the nature of the inflammatory infiltrate allowing for more targeted or tailored treatment regimens. In time, perhaps genetic testing may also have utility in this rare condition (32, 52).
Finally, characterizing specific groups of patients with tumefactive demyelinating lesions may facilitate a search for biomarkers and pathomechanisms similar to the successes recently realized in autoimmune encephalitis (53, 54) and Myelin Oligodendrocyte Glycoprotein Antibody Disease (55, 56). Our clinical and treatment data imply GDLs are mediated, at least in part, by humoral mechanisms and further efforts should be made in this field to facilitate precision phenotyping and accurate treatment (57).
Patient perspective
All four patients expressed their initial concern regarding possible irreversibility of this unusual condition and therefore relief from effective and well-tolerated treatment with functional recovery and ability to return to work. All patients are highly supportive of ongoing research on this condition.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
JP and SF designed the study. JP, SF, AF, and TB were the primary carers whilst all authors participated in case management. The manuscript was written by TB and all authors commented on versions of the manuscript. All authors read and approved the final manuscript.
Funding
This study was funded by Immunology Trust Royal North Shore Hospital/NSW Health Pathology, MS Research Australia and the Joe and Clara Vucetic Foundation as salary support.
Acknowledgments
The authors acknowledge the funding sources and all other health professionals involved in the patients care.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wang L, Lai HM, Thompson AJ, Miller DH. Survey of the distribution of lesion size in multiple sclerosis: implication for the measurement of total lesion load. J Neurol Neurosurg Psychiatry. (1997) 63:452–5. doi: 10.1136/jnnp.63.4.452
2. Dale RC, de Sousa C, Chong WK, Cox TC, Harding B, Neville BG. Acute disseminated encephalomyelitis, multiphasic disseminated encephalomyelitis and multiple sclerosis in children. Brain. (2000) 123 Pt 12:2407–22. doi: 10.1093/brain/123.12.2407
3. Filippi M, Preziosa P, Banwell BL, Barkhof F, Ciccarelli O, De Stefano N, et al. Assessment of lesions on magnetic resonance imaging in multiple sclerosis: practical guidelines. Brain. (2019) 142:1858–75. doi: 10.1093/brain/awz144
4. Frederick MC, Cameron MH. Tumefactive demyelinating lesions in multiple sclerosis and associated disorders. Curr Neurol Neurosci Rep. (2016) 16:26. doi: 10.1007/s11910-016-0626-9
5. Poser S, Luer W, Bruhn H, Frahm J, Bruck Y, Felgenhauer K. Acute demyelinating disease. Classification and non-invasive diagnosis. Acta Neurol Scand. (1992) 86:579–85. doi: 10.1111/j.1600-0404.1992.tb05490.x
6. Kantorová E, Marcinek J, Zelenák K, Kantor K, Michalik J, Sivák Š, et al. Tumefactive demyelination of the spinal cord: a case report. Spinal Cord. (2015) 53:877–80. doi: 10.1038/sc.2015.52
7. Makary MS, Kirsch CF. Tumefactive demyelinating disease with isolated spinal cord involvement. Acta Radiol Short Rep. (2014) 3:2047981614539324. doi: 10.1177/2047981614539324
8. Štourač P, Kolčava J, Kerkovský M, Koprivová T, Kren L, Bednarík J. Progressive tumefactive demyelination as the only result of extensive diagnostic work-up: a case report. Front Neurol. (2021) 12:701663. doi: 10.3389/fneur.2021.701663
9. Jarius S, Würthwein C, Behrens JR, Wanner J, Haas J, Paul F, et al. Baló's concentric sclerosis is immunologically distinct from multiple sclerosis: results from retrospective analysis of almost 150 lumbar punctures. J Neuroinflammation. (2018) 15:22. doi: 10.1186/s12974-017-1043-y
10. Di Gregorio M, Torri Clerici VLA, Fenu G, Gaetani L, Gallo A, Cavalla P, et al. Defining the course of tumefactive multiple sclerosis: a large retrospective multicentre study. Eur J Neurol. (2021) 28:1299–307. doi: 10.1111/ene.14672
11. Mando R, Muallem E, Meka SG, Berghea R. A blind spot in the diagnostic field: the challenging diagnosis of tumefactive multiple sclerosis. Case Rep Neurol Med. (2018) 2018:6841291. doi: 10.1155/2018/6841291
12. Brod SA, Lindsey JW, Nelson F. Tumefactive demyelination: clinical outcomes, lesion evolution and treatments. Mult Scler J Exp Transl Clin. (2019) 5:2055217319855755. doi: 10.1177/2055217319855755
13. Lucchinetti CF, Gavrilova RH, Metz I, Parisi JE, Scheithauer BW, Weigand S, et al. Clinical and radiographic spectrum of pathologically confirmed tumefactive multiple sclerosis. Brain. (2008) 131(Pt 7):1759–75. doi: 10.1093/brain/awn098
14. Sarbu N, Shih RY, Jones RV, Horkayne-Szakaly I, Oleaga L, Smirniotopoulos JG. White matter diseases with radiologic-pathologic correlation. Radiographics. (2016) 36:1426–47. doi: 10.1148/rg.2016160031
15. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol. (2017) 13:25–36. doi: 10.1038/nrneurol.2016.187
16. Fereidan-Esfahani M, Tobin WO. Cyclophosphamide in treatment of tumefactive multiple sclerosis. Mult Scler Relat Disord. (2021) 47:102627. doi: 10.1016/j.msard.2020.102627
17. Sempere AP, Feliu-Rey E, Sanchez-Perez R, Nieto-Navarro J. Rituximab for tumefactive demyelination refractory to corticosteroids and plasma exchange. J Neurol Neurosurg Psychiatry. (2013) 84:1338. doi: 10.1136/jnnp-2013-305456
18. Vakrakou AG, Tzanetakos D, Argyrakos T, Koutsis G, Evangelopoulos M-E, Andreadou E, et al. Recurrent fulminant tumefactive demyelination with marburg-like features and atypical presentation: therapeutic dilemmas and review of literature. Front Neurol. (2020) 11:536. doi: 10.3389/fneur.2020.00536
19. Algahtani H, Shirah B, Alassiri A. Tumefactive demyelinating lesions: a comprehensive review. Mult Scler Relat Disord. (2017) 14:72–9. doi: 10.1016/j.msard.2017.04.003
20. Tzanetakos D, Vakrakou AG, Tzartos JS, Velonakis G, Evangelopoulos ME, Anagnostouli M, et al. Heterogeneity of Balo's concentric sclerosis: a study of eight cases with different therapeutic concepts. BMC Neurol. (2020) 20:400. doi: 10.1186/s12883-020-01971-2
21. Garell PC, Menezes AH, Baumbach G, Moore SA, Nelson G, Mathews K, et al. Presentation, management and follow-up of Schilder's disease. Pediatr Neurosurg. (1998) 29:86–91. doi: 10.1159/000028695
22. Neuroimmunology Group of Neurology Branch of Chinese Medical Association, Neuroimmunology Committee of Chinese Society for Immunology, Immunology Society of Chinese Stroke Association. Chinese guidelines for the diagnosis and management of tumefactive demyelinating lesions of central nervous system. Chin Med J. (2017) 130:1838–50. doi: 10.4103/0366-6999.211547
23. Taylor B, Patel MP, Peters KB. When tumefactive demyelination is truly a tumor: case report of a radiographic misdiagnosis. CNS Oncol. (2021) 10:CNS69. doi: 10.2217/cns-2020-0028
24. Suh CH, Kim HS, Jung SC, Choi CG, Kim SJ. MRI findings in tumefactive demyelinating lesions: a systematic review and meta-analysis. AJNR Am J Neuroradiol. (2018) 39:1643–9. doi: 10.3174/ajnr.A5775
25. Agarwal A, Pinho M, Raj K, Yu FF, Bathla G, Achilleos M, et al. Neurological emergencies associated with COVID-19: stroke and beyond. Emerg Radiol. (2020) 27:747–54. doi: 10.1007/s10140-020-01837-7
26. Trip SA, Miller DH. Imaging in multiple sclerosis. J Neurol Neurosurg Psychiatry. (2005) 76(Suppl. 3):iii11–8. doi: 10.1136/jnnp.2005.073213
27. Hardy TA, Chataway J. Tumefactive demyelination: an approach to diagnosis and management. J Neurol Neurosurg Psychiatry. (2013) 84:1047–53. doi: 10.1136/jnnp-2012-304498
28. Shiraishi W, Umemura T, Nakayama Y, Yamada Y, Shijo M, Hashimoto T. Case report: paraneoplastic tumefactive demyelination associated with seminoma. Front Neurol. (2022) 13:946180. doi: 10.3389/fneur.2022.946180
29. Kotsenas AL, Morris JM, Wald JT, Parisi JE, Campeau NG. Tumefactive cerebral amyloid angiopathy mimicking CNS neoplasm. Am J Roentgenol. (2013) 200:50–6. doi: 10.2214/AJR.12.8500
30. Abdoli M, Freedman MS. Neuro-oncology dilemma: tumour or tumefactive demyelinating lesion. Mult Scler Relat Disord. (2015) 4:555–66. doi: 10.1016/j.msard.2015.07.013
31. Hardy TA. Pseudotumoral demyelinating lesions: diagnostic approach and long-term outcome. Curr Opin Neurol. (2019) 32:467–74. doi: 10.1097/WCO.0000000000000683
32. Abozaid GM, Kerr K, McKnight A, Al-Omar HA. Criteria to define rare diseases and orphan drugs: a systematic review protocol. BMJ Open. (2022) 12:e062126. doi: 10.1136/bmjopen-2022-062126
33. Nakayama M, Naganawa S, Ouyang M, Jones KA, Kim J, Capizzano AA, et al. A review of clinical and imaging findings in tumefactive demyelination. Am J Roentgenol. (2021) 217:186–97. doi: 10.2214/AJR.20.23226
34. Smith KJ. Conduction properties of central demyelinated and remyelinated axons, and their relation to symptom production in demyelinating disorders. Eye. (1994) 8:224–37. doi: 10.1038/eye.1994.51
35. Maia C, Novo A, Sousa M, Brás P, Brito O, Rebelo O, et al. Tumefactive demyelinating lesions spectrum disorders and the potential role of contemporary disease modifying treatments: a case report. Mult Scler Relat Disord. (2021) 47:102669. doi: 10.1016/j.msard.2020.102669
36. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. (2018) 17:162–73. doi: 10.1016/S1474-4422(17)30470-2
37. Karussis D. The diagnosis of multiple sclerosis and the various related demyelinating syndromes: a critical review. J Autoimmun. (2014) 48–9:134–42. doi: 10.1016/j.jaut.2014.01.022
38. Canto E, Oksenberg JR. Multiple sclerosis genetics. Mult Scler. (2018) 24:75–9. doi: 10.1177/1352458517737371
39. van Langelaar J, Rijvers L, Smolders J, van Luijn MM. B and T cells driving multiple sclerosis: identity, mechanisms and potential triggers. Front Immunol. (2020) 11:760. doi: 10.3389/fimmu.2020.00760
40. Rahmlow MR, Kantarci O. Fulminant demyelinating diseases. Neurohospitalist. (2013) 3:81–91. doi: 10.1177/1941874412466873
41. Weinshenker BG, O'Brien PC, Petterson TM, Noseworthy JH, Lucchinetti CF, Dodick DW, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol. (1999) 46:878–86. doi: 10.1002/1531-8249(199912)46:6<878::AID-ANA10>3.0.CO;2-Q
42. Breitkopf K, Aytulun A, Forster M, Kraus B, Turowski B, Huppert D, et al. Case report: a case of severe clinical deterioration in a patient with multiple sclerosis. Front Neurol. (2020) 11:782. doi: 10.3389/fneur.2020.00782
43. Jarius S, Konig FB, Metz I, Ruprecht K, Paul F, Bruck W, et al. Pattern II and pattern III MS are entities distinct from pattern I MS: evidence from cerebrospinal fluid analysis. J Neuroinflamm. (2017) 14:171. doi: 10.1186/s12974-017-0929-z
44. Vakrakou AG, Brinia M-E, Svolaki I, Argyrakos T, Stefanis L, Kilidireas C. Immunopathology of tumefactive demyelinating lesions-from idiopathic to drug-related cases. Front Neurol. (2022) 13:868525. doi: 10.3389/fneur.2022.868525
45. Sechi E, Flanagan EP. Antibody-mediated autoimmune diseases of the CNS: challenges and approaches to diagnosis and management. Front Neurol. (2021) 12:673339. doi: 10.3389/fneur.2021.673339
46. Clare E. Acute disseminated encephalomyelitis. JAAPA. (2022) 35:32–5. doi: 10.1097/01.JAA.0000832604.33045.9d
47. Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. (2011) 335:2–13. doi: 10.1016/j.mce.2010.04.005
48. Lee DSW, Rojas OL, Gommerman JL. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat Rev Drug Discov. (2021) 20:179–99. doi: 10.1038/s41573-020-00092-2
49. Kaegi C, Wuest B, Schreiner J, Steiner UC, Vultaggio A, Matucci A, et al. Systematic review of safety and efficacy of rituximab in treating immune-mediated disorders. Front Immunol. (2019) 10:1990. doi: 10.3389/fimmu.2019.01990
50. Sánchez P, Meca-Lallana V, Barbosa A, Manzanares R, Palmí I, Vivancos J. Tumefactive demyelinating lesions of 15 patients: clinico-radiological features, management and review of the literature. J Neurol Sci. (2017) 381:32–8. doi: 10.1016/j.jns.2017.08.005
51. Plowman RS, Varma H. Prognostic factors in Tumefactive demyelinating lesions: a retrospective study. J Neurol Sci. (2021) 428:117591. doi: 10.1016/j.jns.2021.117591
52. Chen X-Y, Chen Y, Fang W-H, Wu Z-Y, Wang D-L, Xu Y-W, et al. Integrative and comparative single-cell analysis reveals transcriptomic difference between human tumefactive demyelinating lesion and glioma. Commun Biol. (2022) 5:941. doi: 10.1038/s42003-022-03900-0
53. Giannoccaro MP, Crisp SJ, Vincent A. Antibody-mediated central nervous system diseases. Brain Neurosci Adv. (2018) 2:2398212818817497. doi: 10.1177/2398212818817497
54. Prüss H. Autoantibodies in neurological disease. Nat Rev Immunol. (2021) 21:798–813. doi: 10.1038/s41577-021-00543-w
55. Loos J, Pfeuffer S, Pape K, Ruck T, Luessi F, Spreer A, et al. MOG encephalomyelitis: distinct clinical, MRI and CSF features in patients with longitudinal extensive transverse myelitis as first clinical presentation. J Neurol. (2020) 267:1632–42. doi: 10.1007/s00415-020-09755-x
56. Linington C, Bradl M, Lassmann H, Brunner C, Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol. (1988) 130:443–54.
Keywords: case series, case report, tumefactive demyelinating lesion (TDL), giant demyelinating lesions, Extended Disability Status Scale
Citation: Boyle T, Fernando SL, Drummond J, Fontes A and Parratt J (2023) Phenotyping variants of tumefactive demyelinating lesions according to clinical and radiological features—A case series. Front. Neurol. 14:1092373. doi: 10.3389/fneur.2023.1092373
Received: 08 November 2022; Accepted: 18 January 2023;
Published: 03 February 2023.
Edited by:
Judith M. Greer, The University of Queensland, AustraliaReviewed by:
Emma C. Tallantyre, Cardiff University, United KingdomAbdorreza Naser Moghadasi, Tehran University of Medical Sciences, Iran
Copyright © 2023 Boyle, Fernando, Drummond, Fontes and Parratt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thérèse Boyle,  dGhlcmVzZWJveWxlQGhvdG1haWwuY29t
dGhlcmVzZWJveWxlQGhvdG1haWwuY29t