 Xin Zhang
Xin Zhang Juan Gao2
Juan Gao2 Jinghong Ma
Jinghong Ma- 1Department of Neurology, Xuanwu Hospital of Capital Medical University, Beijing, China
- 2Department of Neurology, Baoding No.1 Central Hospital, Baoding, China
- 3Department of Neurology, The Fourth Affiliated Hospital of Harbin Medical University, Harbin, China
- 4Department of Geriatrics Center, Shenyang No.4 People's Hospital of China Medical University, Shenyang, China
Background: ANXA11 mutations were first reported to be associated with amyotrophic lateral sclerosis (ALS) in 2017. Several studies have investigated the prevalence of ANXA11 mutations in different populations, while less is known about the spectrum of phenotypes and the genotype–phenotype correlation with this gene mutation.
Case presentation: Here, we report a 74-year-old man who was initially diagnosed with progressive supranuclear palsy (PSP) because of repeated falls, slight upward gaze palsy, and mild cognitive dysfunction at the onset. He finally turned out to be ALS with more and more prominent limb weakness and atrophy, together with the evidence of chronic neurogenic change and ongoing denervation on electromyography. Brain magnetic resonance imaging showed extensive cortical atrophy. A missense mutation c.119A > G (p.D40G) on the ANXA11 gene was identified using whole-exome sequencing, which confirmed the diagnosis of ALS. We performed a systematic review of the literature about ALS-relevant cases with ANXA11 mutations and identified 68 affected subjects and 29 variants with the ANXA11 gene. We summarized the phenotypes of ANXA11 mutations and the clinical characteristics of nine patients harboring the ANXA11 p.D40G variant including our case.
Conclusions: The phenotype of ANXA11-related cases is heterogeneous, and most cases showed typical ALS, while some could also have the characteristics of frontotemporal dementia (FTD) and PSP, even inclusion body myopathies (hIBM) occurred in familial ALS (FALS). Our patient presented with ALS with a co-morbid PSP-like symptom (ALS-PSP) phenotype, which has not been reported. Except for our patient, the remaining eight patients with the ANXA11 p.D40G variant presented with a classical ALS phenotype without cognitive impairment.
Introduction
Amyotrophic lateral sclerosis (ALS) is a malignant neurodegenerative disorder with a substantial heritable component. About 60% of familial ALS (FALS) and 10% of sporadic ALS (SALS) have genetic variations (1, 2). To date, more than 30 genes have been reported to be associated with ALS (3). In European populations, the most common mutation was the C9orf72, followed by SOD1, while in Asians, the most common mutation was the SOD1 (4). Until 2017, ANXA11 mutations were first reported to be associated with ALS (5). There were a few reports about ANXA11 variation, and most of the reported cases presented as typical ALS phenotype, and some with frontotemporal dementia (FTD). Herein, we report a case of atypical ALS with ANXA11 gene mutation who first showed obvious extrapyramidal symptoms and repeated falls and was initially misdiagnosed as progressive supranuclear palsy (PSP). We performed a systematic review of the literature to investigate the genotype–phenotype correlation of ANXA11 and the clinical phenotypes with the ANXA11 p.D40G mutation.
Case presentation
A 74-year-old man presented to our clinic due to unsteady walking and repeated falls for 13 months, accompanied by a slightly drooped head and bradykinesia. At the same time, his daughter noticed that he would occasionally have forced laughter. Twelve months ago, he developed slurred speech and weakness in bilateral arms. Neurologic examinations revealed mild cognitive dysfunction (the Mini-Mental State Examination score was 23/30 and the Montreal Cognitive Assessment score was 22/30, with 11 years of education), dysarthria, slight upward gaze palsy, the muscle strength of bilateral arms was grade 5−/5, bradykinesia, slightly increased muscle tone in limbs except for the neck, postural instability, hyperreflexia, and positive Babinski signs bilaterally. Brain magnetic resonance imaging showed extensive cortical atrophy (Figure 1). Electromyography showed no abnormality. Within the next 9 months, his symptoms worsened rapidly, and he became wheelchair-bound and developed significantly slurred speech until mutism. At that time, physical examination showed a drooped head, mutism, muscle strength of extremities was grade 0, hypermyotonia, hyperreflexia, positive Babinski signs bilaterally, carpopedal contracture, and fasciculation in hands. Electromyography showed chronic neurogenic change and ongoing denervation. A missense mutation c.119A > G (p.D40G) of the ANXA11 gene was identified using whole-exome sequencing and verified by Sanger sequencing (Figure 2). In the early stages of the disease, our patient showed postural instability and slight supranuclear gaze palsy and was misdiagnosed as suggestive PSP in one of the best neurological hospitals in Beijing, later because of a gradual decline in cognitive function, FTD was also suggested by an experienced neurologist. As the disease progressed rapidly, he finally turned out to be ALS. Apart from the chronic obstructive pulmonary disease, his medical history was unremarkable. He denied a family history of ALS and related disorders. He is a retired worker living in Beijing for a long time, with no history of special chemical exposure. He was initially treated with levodopa (375 mg/day) and the dosage gradually increased to 750 mg/day but without significant improvement except that muscle rigidity improved to some extent. At the 6-month follow-up, which was 19 months after the onset of the disease, he was bedridden and had a tracheostomy because of repeated pneumonia.
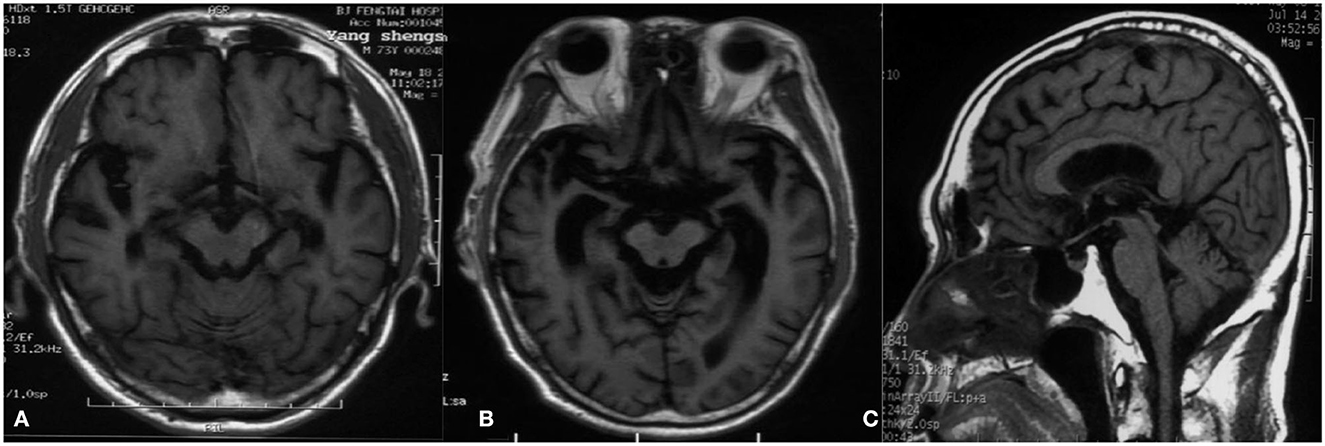
Figure 1. Magnetic resonance imaging. (A) T1-weighted image 2 months after the onset of the disease shows extensive cortical atrophy. (B) T1-weighted image 12 months after the onset of the disease reveals severe cortical atrophy, and the degree of atrophy gradually increased compared to A. (C) T1-weighted image 4 months after the onset of the disease demonstrates that the midbrain volume was comparatively well preserved compared with cortical atrophy.
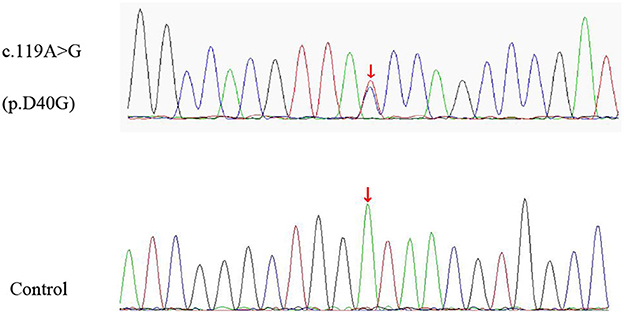
Figure 2. The missense variant of ANXA11 identified in our patient. Sequence chromatograms of polymerase chain reaction (PCR) show the heterozygous c.119A > G (p.D40G) variant (upper lane) compared with healthy control (lower lane).
Review of ALS-relevant cases with ANXA11 mutations
A literature review was performed by searching PubMed and China National Knowledge Infrastructure (CNKI) (from their inception until February 2022) using the keywords: “annexin A11,” “ANXA11,” “ALS,” and “amyotrophic lateral sclerosis.” Relevant articles describing any ALS-relevant case with ANXA11 mutations were selected. Nine articles were related to studies of interest. We identified 68 ALS-relevant cases with ANXA11 mutations including our case, and the phenotypes of each case are listed in Table 1 (5–13). As shown in Supplementary Figure S1, they came from China (32.35%, 22/68), Brazil (19.12%, 13/68), Korea (19.12%, 13/68), multicenter (including the United States, the UK, Italy, Spain, Germany, Ireland, Canada, the Netherlands, Belgium, and New Zealand, 17.65%, 12/68), and France (11.76%, 8/68). According to the previously reported cases, most (80.88%, 55/68) of the ANXA11 variants presented with typical ALS phenotype, and one (1.47%, 1/68) showed ALS with co-morbid FTD (ALS-FTD), while our patient (1.47%, 1/68) showed ALS with co-morbid PSP-like symptoms (ALS-PSP), which has not been reported. The phenotypes of ANXA11 mutations are summarized in Supplementary Figure S2. We identified 29 ANXA11 variants marked in Figure 3, and four variants (13.79%, 4/29) are considered to be pathogenic or likely pathogenic, including p.D40Y, p.D40G, p.G38R, and p.A58_Q187del (7–9, 11). We identified nine patients harboring the ANXA11 p.D40G variant including our case. As shown in Supplementary Table S1, except for our patients, they all presented with classical ALS phenotypes without cognitive impairment (5, 9, 11). The onset age of six patients (66.67%, 6/9) exceeded 70 years of age, and the initial symptoms of six patients (66.67%, 6/9) were bulbar dysfunction.
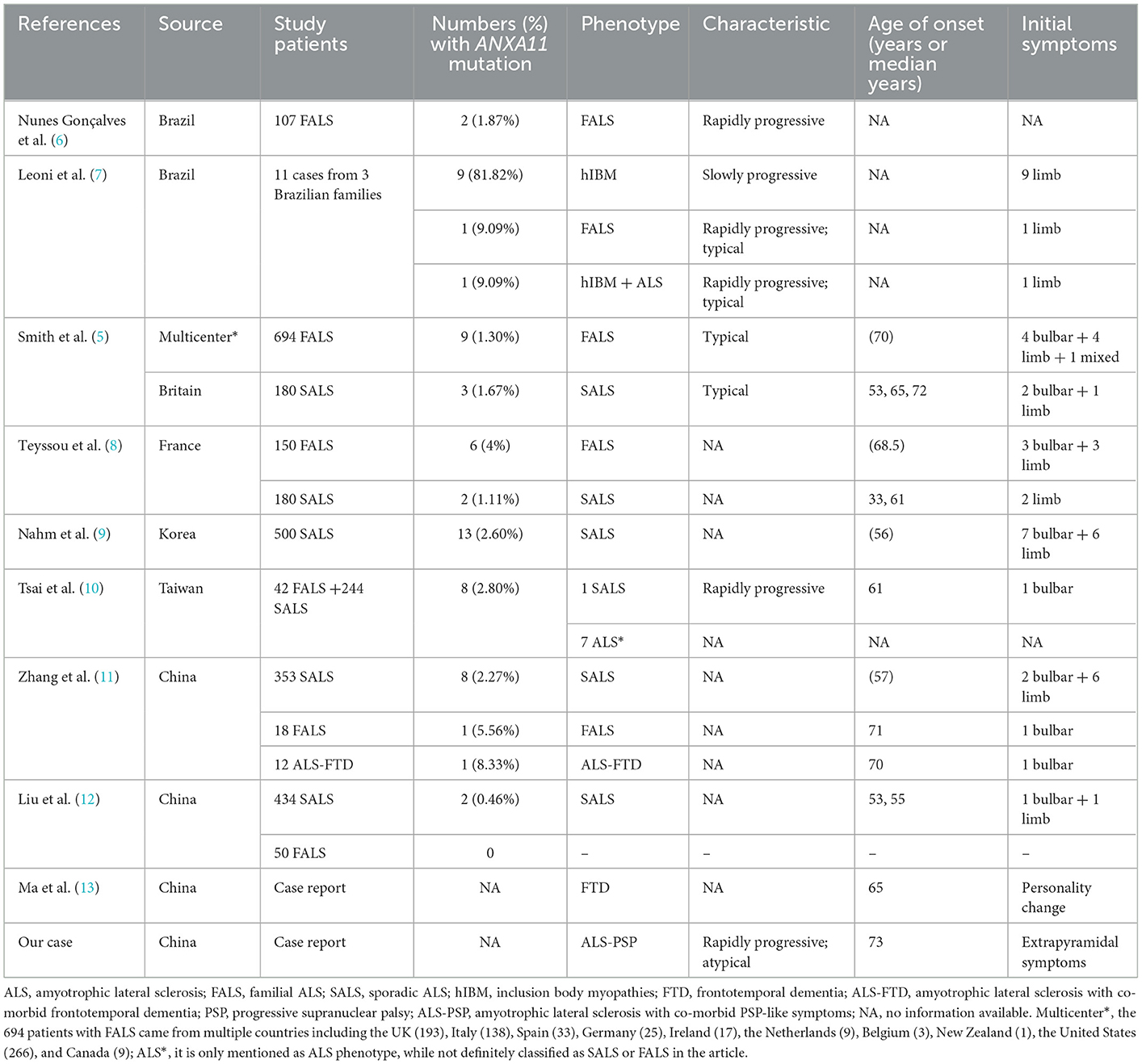
Table 1. Clinical phenotypes of 68 patients with ANXA11 mutations.
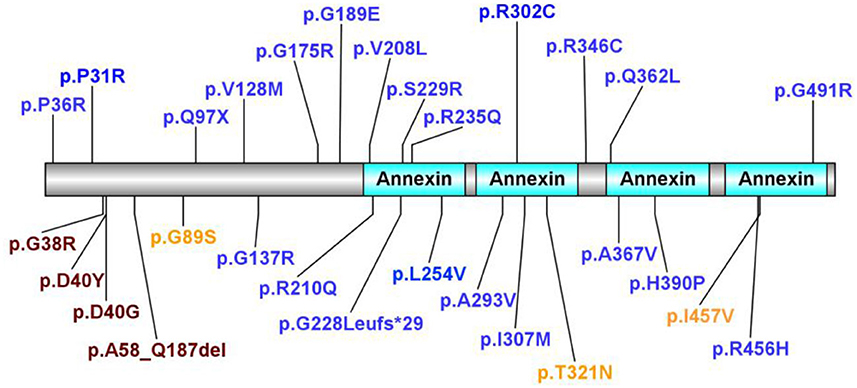
Figure 3. In total, 29 ANXA11 variants identified. In total, four variants considered to be pathogenic or likely pathogenic are marked in purple. Variants considered to be benign or likely benign are marked in orange. Variants of uncertain significance are marked in blue.
Discussion and conclusion
We report a case who initially presented with PSP-like symptoms, combined with prominent pyramidal features and cognitive impairment, and eventually developed extensive lower motor neuron damage. A missense mutation c.119A > G (p.D40G) of the ANXA11 gene was identified using whole-exome sequencing, which confirmed the diagnosis of ALS. This is the first reported case of ANXA11-related ALS (ANXA11-ALS) presented with ALS-PSP phenotype, and the patients with ANXA11 p.D40G-relevant ALS reported previously all presented with classical ALS phenotype without cognitive impairment.
ANXA11 mutations were first reported to be associated with ALS in 2017 (5). Thus far, there are 68 ALS-relevant cases with ANXA11 mutations including our case and 29 ANXA11 variants have been identified. Four variants are considered to be pathogenic or likely pathogenic, including p.D40Y, p.D40G, p.G38R, and p.A58_Q187del (7–9, 11). Most ANXA11-related ALS cases presented as typical ALS phenotype, while there was one Chinese patient with ALS with the ANXA11 p.P36R variant reported by Zhang et al. presenting as ALS-FTD phenotype (11), and some patients with the ANXA11 p.D40Y variant reported in Brazilian families presenting as inclusion body myopathy (hIBM) apart from ALS (7). This indicates the phenotypic heterogeneity with ANXA11 mutations even within the same pedigree. Our case is the first reported case with ALS-PSP phenotype, and except for our patient, cognitive impairment has not been reported in previous patients with ANXA11 p.D40G-relevant ALS.
Functional data showed that p.D40G is located in proximity to the calcyclin-binding region, and the variant could result in abnormal binding of calcyclin (5). Transfected human embryonic kidney cells expressing ANXA11 with the p.D40G mutation showed altered binding to calcyclin (5). Liao et al. (14) proved that p.D40G mutation could reduce the stability of the ANXA11 protein. Smith et al. (5) found that ANXA11-positive protein aggregates were abundant in spinal cord motor neurons and hippocampal neuronal axons in a patient with ALS carrying the p.D40G mutation.
Although there is no positive family history of this patient, we know that all ALS-associated genes and many other genes associated with related conditions show age-dependent penetrance, with the risk of disease manifestation increasing with age and some of the implicated genes are incompletely penetrant (15, 16), family size is becoming smaller and smaller, family members may die due to other causes before the onset of the disease, and these are all the reasons that the patients may show negative family history. In fact, about 10% of patients with SALS have gene mutations, and first-degree relatives of patients with SALS are at an 8-fold higher risk of developing the disease (1, 17). Thus, whether to take genetic testing only depending on the family history will miss some genetic variations in the clinical practice, especially with smaller families becoming the norm. In fact, the patient's only daughter also carries the same mutation as her father but does not show any clinical symptoms. She is 48 years old, so maybe her risk to have the disease will increase with aging but still has the opportunity to remain normal as Hardiman et al. (15) have indicated that familial forms of ALS are often characterized by < 50% penetrance. A meta-analysis aiming to determine the genetic features of ALS in the Chinese population showed that, in Chinese SALS, the highest mutation frequency was identified in the SOD1 gene (1.6%), followed by FUS (1.3%), SQSTM1 (1.0%), OPTN (0.9%), and CCNF (0.8%) (1). While in 2018, Zhang et al. (11) recruited 353 Chinese patients with SALS to investigate the genetic contribution of ANXA11 by Sanger Sequencing and concluded that ANXA11 mutation accounted for a mutant frequency of 2.3% in SALS. It seems that the ANXA11 mutation is the leading gene in Chinese SALS. In the same year, Tsai et al. (10) screened a cohort of 244 Taiwanese patients with SALS using the same method and found eight missense variants in the ANXA11 gene but only one variant was absent from population databases. Combined with these two studies, the mutant frequency of ANXA11 is about 1.4%. According to these data, SOD1 may be still the most common gene mutation in Chinese SALS, and ANXA11 followed as the second.
The classic phenotype of ALS is characterized by the degeneration of upper and lower motor neurons, while behavioral and cognitive impairments are also common symptoms. According to previous studies, approximately 50% of cases show various degrees of cognitive impairment, from mild to FTD (18, 19). Generally, C9orf72, TARDBP, and TBK1 variations often combine with cognitive impairment, while SOD1 is known as “Pure” ALS genes (20–23). Thus far, the clinical phenotype spectrum of ANXA11 is unclear. According to the previously reported cases, the ANXA11 variant can present with classical SALS, FALS, classic motor symptoms with co-morbid FTD, and even ALS, hIBM, and ALS plus hIBM in one pedigree simultaneously. Our patient carrying ANXA11 mutation presented with ALS-PSP phenotype, and this is the first reported case of this phenotype. Many genes associated with ALS are pleiotropic. For example, the mutation in valosin-containing protein (VCP) has been detected in family pedigrees with heterogeneous phenotypes such as ALS, FTD, hIBM, and Paget disease of bone (24). Hence, in 2013, Benatar et al. (25) proposed the concept of multisystem proteinopathy (MSP), which is an inherited pleiotropic degenerative disorder that can affect muscle, bone, and the nervous system. In 2015, Taylor et al. (26) proposed an operational definition of MSP, which is a combination of two or more phenotypes of hIBM, Paget disease of bone, ALS, or FTD. According to these criteria, Leoni et al. (7) proposed that ANXA11 should be considered as a gene associated with a novel type of MSP (MSP type 6), rather than just an ALS-related gene.
Our patient presented with prominent PSP-like symptoms initially, including repeated falls and abnormal eye movement, which is rare in ALS. In fact, there were a few studies that have addressed the patients with ALS accompanying extrapyramidal symptoms including both hyperkinetic and hypokinetic movement disorders (27–29). In 2019, Calvo et al. (29) recruited 101 patients with ALS and identified 31 patients (30.7%, 31/101) with the co-morbid parkinsonian disorder (ALS-PK), who showed bradykinesia (100%), axial symptoms (100%), rigidity (89.2%), tremor (57.1%), and cognitive impairment (35.7%). They detected four mutations in four of the 31 patients with ALS-PK, including C9orf72 (3.2%, 1/31), TARDBP (3.2%, 1/31), LRRK2 (3.2%, 1/31), and PARK2 (3.2%, 1/31). However, ANXA11-ALS who showed an atypical ALS with PSP-like symptoms has not been previously reported. In 2012, D'Ascenzo et al. (30) enrolled 16 patients with ALS with predominant upper motor neuron involvement and extrapyramidal-like features and found eight of them (50%, 8/16) showed a slight to a severe reduction in striatal dopamine transporter-positron emission tomography uptake. Unfortunately, due to poor physical condition, our patient could not complete the dopamine transporter-positron emission tomography. In 2019, a cohort of 97 autopsied cases of sporadic ALS was examined by Ito et al. (27). They identified 11 cases (11.3%, 11/97) who showed pallidonigroluysian degeneration (PNLD), and two patients with PNLD (18.2%, 2/11) developed extrapyramidal signs as the initial symptoms, while extrapyramidal signs were not observed in the remaining 86 cases without PNLD. Thus, they thought that PNLD accounted for the early development of extrapyramidal signs. By taking levodopa, our patient could improve his muscle rigidity to some extent, and thus, we speculated that the pallidonigroluysian system of this patient might be affected.
We report a case of sporadic ALS with PSP-like symptoms. Genetic testing confirmed the ANXA11 p.D40G variant. Through the literature review, we found 68 ALS-relevant patients with ANXA11 mutations, which presented with typical ALS, hIBM, FTD, or a combination of these phenotypes. Our ALS-PSP phenotype is first reported to be associated with ANXA11. According to the previous reports, 29 heterozygous nonsynonymous ANXA11 variants were identified, and p.D40Y, p.D40G, p.G38R, and p.A58_Q187del are identified as pathogenic or likely pathogenic. Most patients with ALS with the ANXA11 p.D40G variant presented with a classical ALS phenotype without cognitive impairment except for our patient. Thus far, due to the limited number of cases, the genotype–phenotype correlation in ANXA11-ALS is not clear. As we learn more about ANXA11 variation, we may have a deeper understanding of its variety and phenotype in the future.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethical Committee of Xuanwu Hospital of Capital Medical University. Written informed consent was obtained from the patient's only daughter for the publication of any potentially identifiable images or data included in this article.
Author contributions
XZ analyzed and interpreted the data and wrote the manuscript. JG, CC, ZZ, and PC analyzed and interpreted the data. JM designed and conceptualized the study, interpreted the data, and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The National Key R&D Program of China (No. 2021YFC2501200) provided financial support to conduct this research, including the study design, collection, analysis, interpretation of data, and manuscript writing.
Acknowledgments
We would like to express our sincere gratitude to our patient and his family for their cooperation in the preparation of this report.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1086264/full#supplementary-material
Supplementary Figure S1. Geographic distribution of 68 affected subjects with ANXA11 mutations. Multicenter*, nine patients with ANXA 11 mutations from 694 patients with FALS coming from multiple countries including the UK (193), Italy (138), Spain (33), Germany (25), Ireland (17), the Netherlands (9), Belgium (3), New Zealand (1), the United States (266), and Canada (9).
Supplementary Figure S2. The phenotypes of 68 cases with ANXA11 mutations. ALS, amyotrophic lateral sclerosis; SALS, sporadic ALS; FALS, familial ALS; hIBM, inclusion body myopathies; ALS*, it is only mentioned as ALS phenotype, while not definitely classified as SALS or FALS in the article; hIBM+ALS, hIBM with co-morbid ALS; FTD, frontotemporal dementia; ALS-FTD, ALS with co-morbid FTD; ALS-PSP, ALS with co-morbid PSP-like symptoms.
Supplementary Table S1. Clinical phenotype of the patients with ANXA11 p.D40G-relevant ALS.
Abbreviations
ALS, amyotrophic lateral sclerosis; PSP, progressive supranuclear palsy; FTD, frontotemporal dementia; hIBM, inclusion body myopathies; FALS, familial ALS; ALS-PSP, ALS with co-morbid PSP-like symptoms; SALS, sporadic ALS; ALS-FTD, ALS with co-morbid FTD; ANXA11-ALS, ANXA11-related ALS; MSP, multisystem proteinopathy; ALS-PK, ALS patients with co-morbid parkinsonian disorder; PNLD, pallidonigroluysian degeneration.
References
1. Wei Q, Chen X, Chen Y, Ou R, Cao B, Hou Y, et al. Unique characteristics of the genetics epidemiology of amyotrophic lateral sclerosis in China. Sci China Life Sci. (2019) 62:517–25. doi: 10.1007/s11427-018-9453-x
2. Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. (2014) 17:17–23. doi: 10.1038/nn.3584
3. Le Gall L, Anakor E, Connolly O, Vijayakumar UG, Duddy WJ, Duguez S. Molecular and cellular mechanisms affected in ALS. J Pers Med. (2020) 10:101. doi: 10.3390/jpm10030101
4. Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. (2017) 88:540–9. doi: 10.1136/jnnp-2016-315018
5. Smith BN, Topp SD, Fallini C, Shibata H, Chen HJ, Troakes C, et al. Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci Transl Med. (2017) 9:eaad9157. doi: 10.1126/scitranslmed.aad9157
6. Nunes Gonçalves JP, Leoni TB, Martins MP, Peluzzo TM, Dourado MET, Saute JAM, et al. Genetic epidemiology of familial ALS in Brazil. Neurobiol Aging. (2021) 102:227.e1–4. doi: 10.1016/j.neurobiolaging.2021.01.007
7. Leoni TB, Gonzalez-Salazar C, Rezende TJR, Hernandez ALC, Mattos AHB, Coimbra Neto AR, et al. A novel multisystem proteinopathy caused by a missense ANXA11 variant. Ann Neurol. (2021) 90:239–52. doi: 10.1002/ana.26136
8. Teyssou E, Muratet F, Amador MD, Ferrien M, Lautrette G, Machat S, et al. Genetic screening of ANXA11 revealed novel mutations linked to amyotrophic lateral sclerosis. Neurobiol Aging. (2021) 99:102e11–20. doi: 10.1016/j.neurobiolaging.2020.10.015
9. Nahm M, Lim SM, Kim YE, Park J, Noh MY, Lee S, et al. ANXA11 mutations in ALS cause dysregulation of calcium homeostasis and stress granule dynamics. Sci Transl Med. (2020) 12:eaax3993. doi: 10.1126/scitranslmed.aax3993
10. Tsai PC, Liao YC, Jih KY, Soong BW, Lin KP, Lee YC. Genetic analysis of ANXA11 variants in a Han Chinese cohort with amyotrophic lateral sclerosis in Taiwan. Neurobiol Aging. (2018) 72:188e1–2. doi: 10.1016/j.neurobiolaging.2018.07.002
11. Zhang K, Liu Q, Liu K, Shen D, Tai H, Shu S, et al. ANXA11 mutations prevail in Chinese ALS patients with and without cognitive dementia. Neurol Genet. (2018) 4:e237. doi: 10.1212/NXG.0000000000000237
12. Liu X, Wu C, He J, Zhang N, Fan D. Two rare variants of the ANXA11 gene identified in Chinese patients with amyotrophic lateral sclerosis. Neurobiol Aging. (2019) 74:235e9–12. doi: 10.1016/j.neurobiolaging.2018.09.020
13. Ma H, Wang S, Wang F, Gong M, Zhang Q, Song S, et al. Frontotemporal dementia with ANXA11 gene mutation: a case report and literature review. Chin J Neurol. (2020) 53:772–6. doi: 10.3760/cma.j.cn113694-20200317-00185
14. Liao D, Liao Q, Huang C, Bi F. Mutations of G38R and D40G cause amyotrophic lateral sclerosis by reducing Annexin A11 protein stability. Zhong Nan Da Xue Xue Bao Yi Xue Ban. (2018) 43:577–82. doi: 10.11817/j.issn.1672-7347.2018.06.001
15. Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. (2017) 3:17071. doi: 10.1038/nrdp.2017.71
16. Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol. (2017) 13:96–104. doi: 10.1038/nrneurol.2016.182
17. Hanby MF, Scott KM, Scotton W, Wijesekera L, Mole T, Ellis CE, et al. The risk to relatives of patients with sporadic amyotrophic lateral sclerosis. Brain. (2011) 134(Pt 12):3454–7. doi: 10.1093/brain/awr248
18. Phukan J, Elamin M, Bede P, Jordan N, Gallagher L, Byrne S, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry. (2012) 83:102–8. doi: 10.1136/jnnp-2011-300188
19. Montuschi A, Iazzolino B, Calvo A, Moglia C, Lopiano L, Restagno G, et al. Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J Neurol Neurosurg Psychiatry. (2015) 86:168–73. doi: 10.1136/jnnp-2013-307223
20. Ji AL, Zhang X, Chen WW, Huang WJ. Genetics insight into the amyotrophic lateral sclerosis/frontotemporal dementia spectrum. J Med Genet. (2017) 54:145–54. doi: 10.1136/jmedgenet-2016-104271
21. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
22. van der Zee J, Gijselinck I, Van Mossevelde S, Perrone F, Dillen L, Heeman B, et al. TBK1 mutation spectrum in an extended european patient cohort with frontotemporal dementia and amyotrophic lateral sclerosis. Hum Mutat. (2017) 38:297–309. doi: 10.1002/humu.23161
23. Ranganathan R, Haque S, Coley K, Shepheard S, Cooper-Knock J, Kirby J. Multifaceted genes in amyotrophic lateral sclerosis-frontotemporal dementia. Front Neurosci. (2020) 14:684. doi: 10.3389/fnins.2020.00684
24. Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. (2010) 68:857–64. doi: 10.1016/j.neuron.2010.11.036
25. Benatar M, Wuu J, Fernandez C, Weihl CC, Katzen H, Steele J, et al. Motor neuron involvement in multisystem proteinopathy: Implications for ALS. Neurology. (2013) 80:1874–80. doi: 10.1212/WNL.0b013e3182929fc3
26. Taylor JP. Multisystem proteinopathy Intersecting genetics in muscle, bone, and brain degeneration. Neurology. (2015) 85:658–60. doi: 10.1212/WNL.0000000000001862
27. Ito J, Shimizu H, Ohta K, Idezuka J, Tanaka H, Kondo H, et al. Amyotrophic lateral sclerosis with pallidonigroluysian degeneration: a clinicopathological study. Ann Neurol. (2020) 87:302–12. doi: 10.1002/ana.25652
28. Pupillo E, Bianchi E, Messina P, Chiveri L, Lunetta C, Corbo M, et al. Extrapyramidal and cognitive signs in amyotrophic lateral sclerosis: a population based cross-sectional study. Amyotroph Lateral Scler Frontotemp Degen. (2015) 16:324–30. doi: 10.3109/21678421.2015.1040028
29. Calvo A, Chio A, Pagani M, Cammarosano S, Dematteis F, Moglia C, et al. Parkinsonian traits in amyotrophic lateral sclerosis (ALS): a prospective population-based study. J Neurol. (2019) 266:1633–42. doi: 10.1007/s00415-019-09305-0
Keywords: amyotrophic lateral sclerosis, ANXA11, genotype, phenotype, progressive supranuclear palsy
Citation: Zhang X, Gao J, Chi C, Zhao Z, Chan P and Ma J (2023) An atypical ALS with PSP-like symptoms caused by ANXA11 p.D40G mutation: A case report and literature review. Front. Neurol. 14:1086264. doi: 10.3389/fneur.2023.1086264
Received: 01 November 2022; Accepted: 20 January 2023;
Published: 16 February 2023.
Edited by:
Chunyu Li, Sichuan University, ChinaReviewed by:
Juliana Vasques, Federal University of Rio de Janeiro, BrazilVerônica Marques Zembrzuski, Oswaldo Cruz Foundation (Fiocruz), Brazil
Copyright © 2023 Zhang, Gao, Chi, Zhao, Chan and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinghong Ma,  amluZ2hvbmdtYUAxNjMuY29t
amluZ2hvbmdtYUAxNjMuY29t